

Abstract
Allelic variation of gene expression is common in humans, and is of interest because of its potential contribution to variation in heritable traits. To identify human genes with allelic expression differences, we genotype DNA and examine mRNA isolated from the white blood cells of 12 unrelated individuals using oligonucleotide arrays containing 8406 exonic SNPs. Of the exonic SNPs, 1983, located in 1389 genes, are both expressed in the white blood cells and heterozygous in at least one of the 12 individuals, and thus can be examined for differential allelic expression. Of the 1389 genes, 731 (53%) show allele expression differences in at least one individual. To gain insight into the regulatory mechanisms governing allelic expression differences, we analyze a set of 60 genes containing exonic SNPs that are heterozygous in three or more samples, and for which all heterozygotes display differential expression. We find three patterns of allelic expression, suggesting different underlying regulatory mechanisms. Exonic SNPs in three of the 60 genes are monoallelically expressed in the human white blood cells, and when examined in families show expression of only the maternal copy, consistent with regulation by imprinting. Approximately one-third of the genes have the same allele expressed more highly in all heterozygotes, suggesting that their regulation is predominantly influenced by cis-elements in strong linkage disequilibrium with the assayed exonic SNP. The remaining two-thirds of the genes have different alleles expressed more highly in different heterozygotes, suggesting that their expression differences are influenced by factors not in strong linkage disequilibrium with the assayed exonic SNP.
The correlations between DNA variation and human phenotypic differences, such as height, weight, and susceptibility to certain diseases, are not well understood. While there is evidence that both coding (Koschinsky et al. 2001; Kim et al. 2003; Fondon III and Garner 2004) and regulatory (Prokunina et al. 2002; Tokuhiro et al. 2003) polymorphisms contribute to the observed variation in complex human traits, their relative contributions remain to be determined. Expression differences between alleles of the same gene have been observed in several species, including humans (Yan et al. 2002; Bray et al. 2003; Lo et al. 2003; Schadt et al. 2003; Pastinen et al. 2004), rats (Hubner et al. 2005), mice (Cowles et al. 2002; Schadt et al. 2003; Doss et al. 2005; Oliver et al. 2005), maize (Schadt et al. 2003), and yeast (Brem et al. 2002; Ronald et al. 2005), by comparing the relative abundance of mRNA transcripts isolated from cells obtained from normal individuals. Natural variation in the expression levels of many genes shows familial aggregation in humans (Cheung et al. 2003; Lo et al. 2003; Schadt et al. 2003; Morley et al. 2004) and simple segregation patterns in yeast (Brem et al. 2002), suggesting that a significant fraction of allelic expression differences are hereditary in nature. Differential allelic expression is of interest because of the possibility that the differences contribute to phenotypic variation between individuals.
Oligonucleotide arrays have been used previously to screen genes for allele-specific expression in yeast (Ronald et al. 2005) and humans (Lo et al. 2003). In these studies, the relative expression levels of the two alleles are determined by examining mRNA isolated from individuals who are heterozygous for an exonic single nucleotide polymorphism (SNP) in the gene. In the human study, Lo et al. (2003) used the Affymetrix HuSNP array, which contains 1063 exonic SNPs. Studies using the same methodology but other technologies have either focused on individual SNPs (Prokunina et al. 2002; Tokuhiro et al. 2003; Knight et al. 2004), or been limited to tens or hundreds of exonic SNPs (Yan et al. 2002; Lo et al. 2003; Pastinen et al. 2004; Ronald et al. 2005). Our work describes an oligonucleotide array specifically designed to analyze 8406 exonic SNPs in 4102 genes, which corresponds to ∼20% of all human genes (International Human Genome Sequencing Consortium 2004), for differential allelic expression. Use of this oligonucleotide array in combination with our experimental and analytical techniques provides an effective tool for identifying differentially expressed exonic SNP alleles.
Results
Genome-wide allelic expression analysis in human white blood cells
We performed a genome-wide analysis to determine the prevalence and characteristics of allele-specific expression in human white blood cells. DNA and RNA were extracted from the white blood cells of 12 unrelated individuals chosen at random from the Stanford Blood Center. High-density oligonucleotide arrays were designed to assay the allele-specific expression of 8406 exonic SNPs, in 4102 genes, in each of the individuals in a high-throughput manner. The arrays are generated by the tiling of 25-bp oligonucleotide probes, such that each SNP is queried by 80 distinct 25-bp probes (Fig. 1). Genomic DNA and cDNA samples from the same individual were amplified with PCR primers specific for intervals surrounding each SNP. The PCR products were then labeled, and hybridized to the high-density oligonucleotide arrays. We extracted the fluorescence intensities for all 80 probes corresponding to each SNP allele, and estimated the concentration of each allele in the DNA and cDNA samples. We then used the estimates to genotype the SNPs in each genomic DNA sample and to quantify the ratio of reference to alternate SNP alleles in the cDNA samples. Each experiment was performed in duplicate, with a total of four arrays being hybridized for each individual (two hybridized with cDNA and two with genomic DNA).
Figure 1.

Layout of the high-density oligonucleotide arrays used for differential allelic expression analysis. (A) Each exonic SNP is interrogated by 80 distinct probes (25-mers), which consist of four sets of 20 features, corresponding to the forward and reverse strands of the reference and alternate SNP alleles (the set corresponding to the reverse strand of the reference allele is shaded). Each set of 20 features consists of five groups of four features. The five groups vary by their relationship to the position of the SNP, with the center of the 25-bp features being offset by –2, –1, 0, 1, or 2 bases from the SNP. The four features within a group differ only at the central nucleotide position to provide one perfect match probe and three mismatch probes. The arrangement of these four features in squares on the arrays is shown with the indicated nucleotides being those at the center position of each probe. Note that at the 0-offset position, the central nucleotide is also the SNP, meaning that the features tiled for the reference allele are identical to those tiled for the alternate allele. This results in an additional perfect match feature for the reference allele, bringing the total number of perfect match features to six. (B) Fluorescence images of identical arrays hybridized to three different DNA samples, the first homozygous for the reference SNP allele (GG), the second heterozygous (GA), and the third homozygous for the alternate allele (AA). (C) The sequences of the five perfect match probes for the set of 20 features corresponding to the reverse strand of the reference SNP allele (left) and the five perfect match probes for the set of 20 features corresponding to the reverse strand of the alternate allele (right) for the particular SNP being queried in B. The SNP alleles are shown in bold.
Exonic SNPs were considered to be expressed in white blood cells if transcripts were detected in at least nine of the 12 individuals examined, and were considered to be differentially expressed if the allele frequency fold ratio (reference allele/alternate allele) in heterozygotes was ≥1.5 or ≤0.67 (i.e., the apparent reference allele frequency in the RNA, P, was ≥0.6 or ≤0.4) (Fig. 2A). Of the 8406 exonic SNPs examined, 3349 were expressed in the white blood cells, and 1983 of these were heterozygous in at least one individual and could therefore be examined for differential expression (Table 1). The 1983 heterozygous exonic SNPs are located in 1389 genes, with 401 of the genes containing multiple exonic SNPs and five of the exonic SNPs located in multiple RefSeq gene transcripts (http://www.ncbi.nlm.nih.gov/RefSeq). More than 50% of the 1389 assayable genes showed differential allelic expression in at least one individual. The false-positive and false-discovery rates are dependent on the fold-ratio threshold used for defining alleles as differentially expressed. For the fold ratio used in this study, ≥1.5, we estimate the rate of false-positive differential expression in the heterozygote data as 2.5%, and the false-discovery rate as 11.6%. Increasing the fold-ratio threshold from ≥1.5 to ≥2.0 would decrease the estimated false-discovery rate by ∼50%, whereas decreasing the fold-ratio threshold to ≥1.2 would substantially increase the estimated false-discovery rate.
Figure 2.

Expression differences between exonic SNP alleles. SNP alleles that are heterozygous in an individual (those with a DNA reference allele frequency of 0.5) are considered differentially expressed when the reference allele in mRNA has a frequency of ≤0.4 or ≥0.6 (white background), which corresponds to an allele ratio of ≥1.5. (A) DNA and RNA reference allele frequencies for exonic SNPs for individual 9. In this individual, there were 591 heterozygous exonic SNPs, and 108 (18%) of these were differentially expressed. (B) Heterozygous SNP alleles display three distinct expression patterns in the 12 unrelated individuals: monoallelic expression for ss24225694 in ZNF463; differential expression with the same allele expressed at a higher level in each heterozygous individual for ss24290540 in FLJ21069 and ss23668266 in C1orf38; and inconsistent favoring of alleles for ss24515622 in D21S2056E.
Table 1.
Number of exonic SNPs and genes that are differentially expressed in at least one of the 12 samples
| Assayeda | Expressedb | Heterozygousc | Differentially expressedd | |
|---|---|---|---|---|
| Number of RefSeq SNPs | 8406 | 3349 | 1983 | 895 (45%) |
| Number of RefSeq genes | 4102 | 2035 | 1389 | 731 (53%) |
The number of working assays.
Expressed in at least nine individuals.
Expressed in at least nine individuals and heterozygous in at least one individual.
Expressed, heterozygous SNPs and genes that are differentially expressed (allele ratio ≥1.5) in at least one individual.
The allelic expression data for each of the 1983 exonic SNPs are shown in Supplemental Table 1, with data for 13 exonic SNPs specifically discussed in this manuscript shown in Table 2. In these tables we provide the allele frequency fold ratios for each of the heterozygotes. On average, each individual had 502 heterozygous exonic SNPs, and of these, 22% were differentially expressed (Table 3). We report fold ratios that fall between 0.1 and 10, but because of limitations on the technology's ability to reliably determine extreme fold ratios, we report the rest as either ≥10 or ≤0.1. As an example of the distribution of allele frequencies for expressed genes in an individual, Figure 2A shows the RNA reference allele frequencies plotted against DNA reference allele frequencies for all the exonic SNPs for individual #9.
Table 2.
Ratio of reference allele frequency to alternate allele frequency in heterozygous samplesa

Data are shown for genes discussed in the text.
a The fold ratio of reference allele frequency/alternate allele frequency is given for heterozygotes that pass quality control filters. Blank cells indicate that data did not pass quality control filters. Fold ratios for differentially expressed SNPs (≥1.5) are shown as bold and red. Owing to limits of technology, we describe extreme ratios as ≥10 or ≤0.1. r = homozygous reference allele. a = homozygous alternate allele.
b The gene nomenclature used throughout the publication is from NCBI's Build 34.1 of the human genome.
c SNP identifiers used by NCBI's dbSNP for SNPs submited by Perlegen.
Table 3.
Number of SNPs and genes that are differentially expressed in each individual
| Individual | Heterozygous SNPs | No. of genesa,b | SNPs differentially expressedb,c | Genes differentially expressedb,c |
|---|---|---|---|---|
| 1 | 495 | 422 | 125 | 117 |
| 2 | 524 | 430 | 96 | 92 |
| 3 | 478 | 395 | 111 | 108 |
| 4 | 421 | 368 | 139 | 133 |
| 5 | 498 | 412 | 128 | 125 |
| 6 | 419 | 359 | 81 | 80 |
| 7 | 504 | 418 | 110 | 105 |
| 8 | 587 | 480 | 101 | 94 |
| 9 | 591 | 474 | 108 | 101 |
| 10 | 477 | 400 | 90 | 86 |
| 11 | 569 | 463 | 119 | 113 |
| 12 | 466 | 391 | 119 | 109 |
| Average | 502 | 418 | 111 (22%) | 105 (25%) |
Only SNPs and genes expressed in nine or more individuals are considered.
Genes containing at least one heterozygous SNP.
SNPs and genes do not map 1 to 1. A small number of SNPs map to more than one gene, and many genes contain more than one SNP.
Number of SNPs and genes with an allele ratio ≥1.5 in the indicated individual.
Validation
To validate our approach for studying allelic expression differences, we first examined the reproducibility of the observed differences between RNA preparations isolated from the same cells at different times as well as the effect of varying input cDNA concentration in the PCR reaction. Independently isolated RNA preparations were assayed using the high-density oligonucleotide arrays, and a regression of the resulting SNP data had an R2 of 0.98. Additionally, a regression of the SNP data obtained by varying input cDNA concentrations between 0.4 ng/μL and 2 ng/μL into the PCR reaction had an R2 of 0.99. These data suggest that our sample preparation methodology contributes surprisingly little to the observed allelic differences, and that the data obtained for a given SNP are highly reproducible.
We next examined the consistency of allelic expression estimates across multiple informative SNPs within the same gene and individual. There were 1321 such pairwise comparisons, and when the 1.5-fold allele frequency ratio threshold was used to define differentially expressed alleles, 1001 (75.8%) of them agreed. Given that 19.5% (22% observed – 2.5% false-positive rate) of the exonic SNPs are estimated to be differentially expressed, 68.6% of the SNP pairs are expected to agree by chance. Thus, the observed number of SNP pairs in agreement is greater than that expected by chance but low considering the high reproducibility of the allelic expression results observed for a given SNP. We decided to analyze the concordance of SNP pairs as a function of distance to determine if SNP pairs in close proximity to each other on the mRNA transcript were more likely to agree with each other than those spaced farther apart. This analysis was performed using Δp, the estimated magnitude of the difference between the reference allele frequencies in the cDNA sample and the DNA sample (see Methods and Supplemental Fig. 1), to avoid dependence on a particular choice of threshold for differential expression. The Pearson's correlation (R) between Δp estimates for the entire set of 1321 SNP pairs was 0.26 (P = 3.0 × 10–22). However, the 207 SNP pairs separated by <200 bp had an R of 0.44 (P = 4.7 × 10–11), and the 260 SNP pairs separated by <300 bp had an R of 0.42 (P = 2.1 × 10–12). Thus, SNP pairs with shorter distances between them in the transcript are much more likely to have similar differential expression fold-ratio values than SNP pairs spaced farther apart. This is likely due to many reasons, including that differentially regulated splice variants and incorrect gene annotations are more likely to result in disagreements between SNPs spaced farther apart than those in close proximity. Therefore, the finding that SNP pairs in the same gene within the same individual have relatively low agreement, 75.8%, is in part explained by biological reasons, but also suggests that our assay detects differential expression of different exonic SNPs with varying sensitivity.
As a final validation of the array methodology, we compared our allelic expression results with those obtained by real-time PCR analysis for seven randomly chosen exonic SNPs, for a total of 22 comparisons (Table 4). When using the 1.5-fold allele frequency ratio cutoff to define differentially expressed alleles, the results of the two technologies agreed 82% of the time. In 13 of the comparisons the exonic SNP alleles differentially expressed in the array analysis also showed differential expression by real-time PCR, in five comparisons exonic SNP alleles showed nearly equal expression in both techniques, and in four comparisons the techniques disagreed. For two of the four comparisons with results that disagreed, the fold ratios were in the correct direction and close in value, but the 1.5-fold threshold for differential expression was only reached using one of the technologies. Thus, the two technologies were significantly discrepant in only two of the 22 comparisons. Linear regression on the log fold ratios from the two techniques gave a correlation coefficient R2 of 0.707 (P = 9.3 × 10–7). Thus, while they correlated well in terms of their ability to identify differentially expressed genes, the fold ratios provided by the two technologies matched less closely.
Table 4.
Ratio of reference allele frequency to alternative allele frequencya as measured by array hybridization and real-time PCRb

aDifferentially expressed SNPs (≥1.5) are shown as bold and red.
bLinear regression on the ratios from the two methods gave a correlation coefficient, R2, of 0.707 (P = 9.317 × 10-7).
cThe comparisons were made in samples that were heterozygous for the exonic SNPs of each of the listed genes. Thus, the specific samples assayed for each gene varied.
dSNP identifiers used by NCBI's dbSNP for SNPs submitted by Perlegen.
eEstimated ratio of reference allele frequency to alternate allele frequency based on Δp from oligonucleotide array analysis, as described in Methods.
fEstimated ratio of reference allele frequency to alternate allele frequency based on ΔCt (Ct of reference allele — Ct of alternate allele) from real-time PCR experiments. Frequency (reference) = 1/(1 + 2EΔCt), as described by Germer et al. (2000).
gThese SNPs are not listed in Supplemental Tables 1 or 2 because they were not expressed in at least nine individuals.
These validation data show that when we determine that exonic SNP alleles are differentially expressed, those results are reproducible both between replicates on the array platform and across different platforms. The exact fold ratios of differential expression for exonic SNPs are not consistent across platforms, suggesting that they are not accurately determined by our assay. Additionally, our assay appears to detect differential expression of different exonic SNPs with varying sensitivity.
Allelic expression patterns reveal underlying molecular regulatory mechanisms
To gain insights into the underlying regulatory mechanisms responsible for allelic expression differences, we focused on the most highly informative exonic SNPs: those that are heterozygous in three or more samples and for which all heterozygotes display differential expression. In order not to exclude exonic SNPs that were differentially expressed in all individuals from consideration just because they missed the fold-ratio cutoff of ≥1.5 in some expressing heterozygotes, we relaxed our criteria to include those with fold ratios of ≥1.3. This allowed us to include exonic SNPs such as ss23604831 in MS4A7, which had a clear pattern of differential expression in all six heterozygotes, but a fold ratio in one individual (#3) that did not reach the ≥1.5 threshold (Table 2). A total of 61 differentially expressed exonic SNPs located in 61 genes were used to identify allele-specific expression trends because they met the following criteria: allele expression fold ratios of ≥1.3 in all heterozygotes, with at least one individual having a fold ratio of ≥1.5.
Examining the differential expression of the 61 exonic SNPs, we observed three distinct patterns: (1) monoallelic expression (defined here as a fold ratio of ≤0.1 or ≥10) in each of the expressing heterozygotes; (2) differential expression (not monoallelic) in each of the expressing heterozygotes, with the same allele being expressed at higher levels in each heterozygote; and (3) differential expression in each of the heterozygotes, with different alleles being expressed at higher levels in different heterozygotes. Data from one SNP, ss24102685 in MS4A6E, were rejected because of the apparent detection of the reference SNP allele in individuals homozygous for the alternate allele SNP (Supplemental Table 2), bringing the number of genes being analyzed to 60 (Supplemental Table 3).
Exonic SNPs in three of the 60 genes (5%) showed monoallelic expression in each of the expressing heterozygotes, ss23480954 in FLJ33071, ss38338836 in PRIM2A, and ss24225694 in ZNF463 (Fig. 2B), with data from three, four, and five heterozygotes respectively (Table 2). Monoallelic expression is consistent with genomic imprinting, an epigenetic phenomenon in which the expression of alleles is dependent on their parental origin, and generally results in the silencing of one allele (Wrzeska and Rejduch 2004; Wilkins 2005). Because it is the parental origin of an allele, rather than the allele itself, that determines which allele will be expressed in progeny, a characteristic of imprinted genes is random favoring of alleles in unrelated individuals, as seen with ss24225694 in ZNF463 (Fig. 2B). Below we describe additional experimental evidence that is also consistent with the regulation of FLJ33071, PRIM2A, and ZNF463 by genomic imprinting.
Assuming that exonic SNP alleles in mRNA isolated from the white blood cells of a single individual have been exposed to the same trans-acting factors, any expression variation seen between alleles using our approach must involve cis-acting factor(s), whether or not trans-factors are also involved. We propose that non–monoallelically expressed genes that consistently express a particular allele at a higher level than the other are likely to be regulated primarily by cis-factors in strong linkage disequilibrium with the assayed exonic SNP. Of the 57 exonic SNPs that did not show monoallelic expression, 31 are in genes that were differentially expressed with the same allele favored in each of the expressing heterozygotes. These include genes such as C1orf38 and FLJ21069, which were differentially expressed in each of eight and six expressing heterozygotes, respectively (Table 2; Fig. 2B).
The number of exonic SNP alleles expected to have the same allele consistently expressed more highly by chance alone varies with the number of heterozygotes expressing the exonic SNP. For example, the chances of having five or more heterozygotes favoring the same allele are substantially lower than the chances of having only three heterozygotes favoring the same allele. The results show that the 31 exonic SNPs observed with this allele-specific expression pattern is much higher than the ∼12 expected by chance (Table 5), and suggests that the observed differential allelic expression of roughly 19 of the corresponding genes (32%) is due to underlying cis-regulatory polymorphisms in strong linkage disequilibrium with the exonic SNP. The 31 genes showing potential allele-specific expression are uniformly distributed across the genome, with no bias toward specific chromosomal locations (Supplemental Table 4).
Table 5.
Differentially expressed exonic SNPs regulated by variants in linkage disequilibrium
|
No. of heterozygous samples (from total of 12 individuals)
|
No. of differentially expressed SNPs (allele ratio ≥1.3 in all heterozygous samples)a
|
Exonic SNPs favoring expression of one alleleb(not imprinted)
|
|
|---|---|---|---|
| No. observed | No. expected by chance | ||
| 3 | 34 | 18 | 8.50 |
| 4 | 12 | 6 | 1.50 |
| 5 | 7 | 3 | 0.44 |
| 6 | 3 | 3 | 0.09 |
| 7 | 0 | 0 | 0.00 |
| 8 | 1 | 1 | 0.01 |
| Total | 57 | 31c | 11.54 |
Allele ratio ≥1.5 in at least one sample, allele ratio ≥1.3 in all heterozygous samples.
Exonic SNPs for which the same allele is expressed higher in each of the heterozygous samples, excluding the three genes we consider likely to be regulated by imprinting.
The 31 genes containing these 31 exonic SNPs are listed in Supplemental Table 4.
Genes with allelic expression differences influenced by regulatory factors not in strong linkage disequilibrium with the assayed exonic SNP would be expected to have different alleles expressed at higher levels in different heterozygotes. An example of this is exonic SNP ss24515622 in the D21S2056E gene, which was expressed in five heterozygotes (Table 2; Fig. 2B). For this exonic SNP, all five heterozygotes met the ≥1.5 threshold for differential expression, with one allele favored in three of the heterozygotes and the other allele favored in the remaining two. A total of 26 of the 60 genes examined displayed similar inconsistent favoring of alleles and 12 of the ones displaying allele-specific expression are expected to do so by chance. Thus of the 60 examined genes, the observed allelic expression differences of 38 (63%) are likely to be influenced by factors not in strong linkage disequilibrium with the assayed exonic SNP.
Candidate genes for regulation by genomic imprinting
We experimentally examined the inheritance patterns of FLJ33071, PRIM2A, and ZNF463 to further investigate whether or not their expression is consistent with imprinting. Children heterozygous for the monoallelically expressed exonic SNPs of the three genes were identified from two large CEPH families, pedigrees 1344 and 1362 from the Coriell Institute for Medical Research (http://locus.umdnj.edu/ccr/) (Table 6). We obtained lymphoblast cell lines for each of the children who were heterozygous for at least one exonic SNP, isolated mRNA, and determined the extent of differential allelic expression using real-time PCR.
Table 6.
Analysis of candidate imprinted genes in CEPH families
|
Perlegen ssIDa
|
CEPH pedigree number
|
Genotypes of parents
|
Genotypes of heterozygous children
|
Allele ratiob
|
||
|---|---|---|---|---|---|---|
| Gene | Father | Mother | ||||
| FLJ33071 | 24480254 | 1344 | AA | AG | AG | 0.22 |
| Ref allele = A | AG | 0.22 | ||||
| AG | 0.22 | |||||
| AG | 0.22 | |||||
| 1362 | GG | AA | AG | ≥10 | ||
| AG | ≥10 | |||||
| AG | ≥10 | |||||
| AG | ≥10 | |||||
| AG | ≥10 | |||||
| AG | ≥10 | |||||
| PRIM2A | 38338836 | 1344 | TA | AA | AT | ≤0.1 |
| Ref allele = T | AT | ≤0.1 | ||||
| AT | ≤0.1 | |||||
| 1362 | TT | AA | AT | ≤0.1 | ||
| AT | ≤0.1 | |||||
| AT | ≤0.1 | |||||
| AT | ≤0.1 | |||||
| AT | ≤0.1 | |||||
| AT | ≤0.1 | |||||
| ZNF463 | 24225694 | 1344 | GG | GG | None | None |
| Ref allele = A | 1362 | AG | GG | AG | ≤0.1 | |
| AG | ≤0.1 | |||||
| AG | ≤0.1 | |||||
| AG | ≤0.1 | |||||
| AG | ≤0.1 | |||||
SNP identifiers used by NCBI's dbSNP for SNPs submitted by Perlegen.
Ratio of reference allele frequency/alternate allele frequency.
For FLJ33071, the maternally inherited allele of exonic SNP ss24480254 was predominantly expressed over the other allele in all heterozygous children in both pedigrees: the G SNP allele in pedigree 1344 and the A SNP allele in pedigree 1362. Additionally, there is a second exonic SNP (ss24480254) in FLJ33071 that was monoallelically expressed in two of the 12 unrelated white blood cell samples (Table 2). These data are consistent with the regulation of FLJ33071 by imprinting, with the expressed allele being inherited maternally.
For the PRIM2A exonic SNP ss38338836, the maternally derived allele (A) was monoallelically expressed in all heterozygous children in both pedigrees. Thus, the gene is monoallelically expressed in both the 12 original white blood cell samples and the two CEPH pedigrees. Consistent with imprinting as the regulatory mechanism governing expression, the exonic SNP alleles in the PRIM2A gene are randomly favored: in the two CEPH pedigrees, the A SNP allele is expressed (Table 6), and in the white blood cell samples, the T SNP allele is expressed.
For ZNF463, the maternally derived allele for exonic SNP ss24225694 was monoallelically expressed in all five heterozygous children in pedigree 1362. Pedigree 1344 had no heterozygous children and thus provided no information. In the 12 unrelated individuals, monoallelic expression of this SNP allele is randomly favored (Fig. 2B), which is consistent with imprinting. Two additional ZNF463 SNPs (ss23813114 and ss23813115), in the same exon as SNP ss24225694, also display monoallelic expression in heterozygous individuals (Table 2). However, unlike these three monoallelically expressed SNPs, which are all in the 3′-exon of the gene, three SNPs (ss24225691, ss24719563, and ss38338978) in the 5′-untranslated region of ZNF463 have biallelic expression in the white blood cell samples (Table 2). Determining the reason for this discrepancy would require further investigation. However, plausible explanations include the presence of alternative or multiple transcripts in the ZNF463 genomic interval that have not yet been identified and annotated.
There are no previous reports of imprinting for FLJ33071, PRIM2A, and ZNF463. Although our data strongly suggest that the expression of these three genes is regulated by imprinting in white blood cells, it is important to note that definitive validation would require the observation of parental inheritance of allele expression in at least three generations in large families, with switching of expressed alleles in different generations, dependent on the parental origin.
Discussion
We have analyzed the genetic basis of allele-specific expression differences in human white blood cells by comparing the relative levels of exonic SNP alleles within mRNA samples isolated from unrelated individuals. Of the 60 genes classified on the basis of their differential allelic expression patterns, approximately one-third are likely to be regulated predominantly by cis-elements in strong linkage disequilibrium with the assayed exonic SNP, and two-thirds are likely to have their regulation strongly influenced by elements not in linkage disequilibrium with the assayed exonic SNP. Our expression data suggesting that three out of the 60 genes are regulated by imprinting in human white blood cells is surprising, given that there are only ∼50 human genes with evidence of imprinting and parent-of-origin effects in the Imprinted Gene Catalogue (http://igc.otago.ac.nz/home.html), and it has generally been thought that the number of imprinted genes in mammals is low. Our results suggest that experiments using exonic SNPs for genotyping and expression analysis across multiple tissues at different developmental stages may result in the identification of many more genes regulated by genomic imprinting.
Methods
Exonic SNP selection and primer design
From a genome-wide collection of human single nucleotide polymorphisms (SNPs) discovered in an independent study by Perlegen Sciences (Hinds et al. 2005), we identified SNPs that were located within annotated RefSeq gene transcripts (NCBI Build 34.1). For inclusion in the study, these exonic SNPs were required to map to a single location in the human genome. Furthermore, the exonic SNPs were required to be >25 nt away from an intron–exon boundary, so that they could be amplified from both DNA and cDNA samples using a single set of PCR primer pairs. Primers were designed using Oligo 6 (Molecular Biology Insights), and fulfilled the following requirements: the amplicon was 50 to 200 bp in length; the PCR primers were between 17 and 22 nucleotides in length; and the primer pairs were unique in the human genome, based on a BLAST analysis, to ensure specific hybridization. Primer pairs were successfully designed for 8406 exonic SNPs that met the above requirements. The SNPs were located in 4102 RefSeq genes.
Calculation of p̂: Estimation of reference and alternate SNP allele frequencies
Oligonucleotides designed to assay the 8406 exonic SNPs were tiled on high-density arrays. The arrays were designed such that each SNP was interrogated by 80 distinct 25-bp probes (features), as shown in Figure 1. The fluorescence intensities of the reference and alternate perfect-match features on an array correlate with the concentration of the corresponding SNP allele in the DNA or cDNA sample. In heterozygous genomic DNA samples, the two alleles of an SNP are present in equal concentrations, but in heterozygous cDNA samples, allelic expression differences can lead to different concentrations of the two SNP alleles. We estimated the allele frequency in the samples, p̂, as the background adjusted proportion of the reference allele intensity in the total (reference allele plus alternate allele) intensity. p̂ was computed from ratios of trimmed means of intensities of the perfect-match (PM) features, after subtracting a measure of background computed from trimmed means of intensities of the mismatch (MM) features (Hinds et al. 2004a,b).
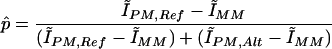 |
where
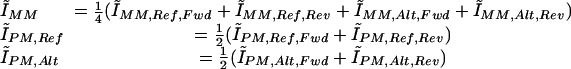 |
The Ĩ terms denote trimmed mean intensities for a set of features identified by the subscript. For example, ĨPM,Ref,Fwd is the trimmed mean intensity for perfect-match probes for the forward strand of the reference allele of the SNP. The trimmed means are arithmetic means of the intensity measurements calculated after discarding the highest and lowest 25% of values. As the arrays contain six perfect match features for each strand of each allele (e.g., the forward strand of the reference allele), this is achieved by sorting on the basis of intensity, discarding the highest and lowest intensity measurements, and giving the next highest and lowest intensity measurements half-weight. Thus, the corresponding trimmed mean intensity is obtained as:
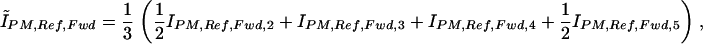 |
where the numeric subscripts 2–5 indicate the intensity-ordered rank of each measurement within the set of six perfect-match probes. This estimate of p̂ was used to determine genotypes in the DNA samples and differential allelic expression in the RNA samples, as discussed below.
Quality control filters for SNP assays
We used two quality control metrics (Hinds et al. 2004a,b, 2005) to assess the reliability of the intensity measurements for each SNP in array scans performed both for the determination of diploid genotypes in DNA samples and for the determination of allele frequency in cDNA samples. The first metric, “conformance,” indicates the presence of specific target DNAs or cDNAs for that SNP. The second metric, “signal-to-background ratio,” measures the relative amounts of specific and nonspecific binding. Cutoffs were applied to both of the metrics, and SNP feature sets that failed on either metric were discarded from further analysis (see below). Multiple previous experiments have shown that the use of these filters leads to high-quality data.
The conformance for a particular allele was defined as the fraction of feature groups in which the perfect-match feature was brighter than the three corresponding mismatch features. For each SNP allele, there are 10 such feature groups, five for the forward strand and five for the reverse strand. Conformance was computed independently for the reference and alternate SNP allele feature sets, and the larger of the two values was used. SNP measurements having conformance <0.9 were discarded from further evaluations.
The signal S, the background B, and the signal-to-background ratio R were calculated from intensity measurements for both alleles in the following manner:
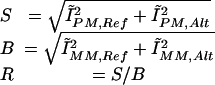 |
SNP measurements having R < 1.5 were discarded from further evaluations.
Determination of genotypes in DNA samples by clustering intensities
Individual genotypes for each SNP were determined by clustering the intensity measurements of all 24 DNA samples (12 individuals × 2 replicates), in the two-dimensional space defined by background-adjusted trimmed mean intensities of the perfect-match features for the reference and alternate alleles (Hinds et al. 2004a,b, 2005). After discarding SNP measurements with conformance <0.9 and signal-to-background ratio <1.5, we used a K-means algorithm to assign the measurements to clusters representing the three possible distinct diploid genotypes, homozygous-reference, heterozygous, and homozygous-alternate. Instead of estimating the background intensity from a single scan, we determined an optimal background value for each SNP that minimized the variance within the assigned genotype clusters. The K-means and background optimization steps were iterated until cluster membership and background estimates converged. To determine the appropriate number of genotype clusters, we repeated the analysis for one, two, and three clusters, and selected the most likely solution, considering likelihoods of the data and the cluster parameters. The data likelihood was determined using a normal mixture model for the distribution of intensities around the cluster means. The model likelihood was calculated using a prior distribution of expected positions for the homozygous-reference, heterozygous, and homozygous-alternate cluster centers, based on empirical data from multiple previous studies.
Determination of differential allelic expression using arrays
We computed a single p̂DNA value for each of the three genotypes (p̂R,DNA, p̂H,DNA, p̂A,DNA) by averaging the p̂DNA values across all the DNA samples that were homozygous-reference, heterozygous, and homozygous-alternate, respectively. p̂R,DNA, p̂H,DNA, and p̂A,DNA corresponded to reference allele frequencies of 1.0, 0.5, and 0.0, respectively. Owing to differential allelic expression in cDNA samples, the estimated reference SNP allele frequency p̂cDNA, calculated by averaging across cDNA replicates, could range from 0 to 1 in heterozygotes. We calculated the reference SNP allele frequency p̂cDNA in a given cDNA sample by linearly interpolating between the calculated values (p̂R,DNA, p̂H,DNA, p̂A,DNA) and the known corresponding reference SNP allele frequencies (1.0, 0.5, 0.0), respectively (Supplemental Fig. 1).
Thus, when the p̂cDNA value for a heterozygous SNP lay between p̂H,DNA and p̂R,DNA (or when no sample was typed as homozygous for the alternate allele), the frequency of the reference allele transcript in the cDNA sample, pcDNA, was determined as:
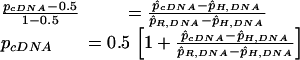 |
When the p̂cDNA value for a heterozygous SNP lay between p̂A,DNA and p̂H,DNA, the frequency of the reference allele transcript in the cDNA sample, pcDNA, was determined as:
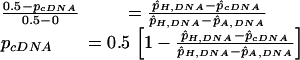 |
The difference (Δp) in the reference allele frequency between the cDNA (pcDNA) and the DNA (0.5) for heterozygotes is:
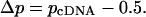 |
The ratio (fRef/Alt,cDNA) between the reference and alternate allele concentrations is:
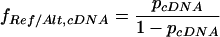 |
We report fold ratios that fall between 0.1 and 10, but because of limitations on the technology's ability to reliably determine extreme fold ratios, we report the rest as either ≥10 or ≤0.1.
Only transcripts for which the exonic SNPs passed the quality thresholds for conformance and signal-to-background ratios in at least 75% of samples (nine of the 12 individuals) were included in the study. The requirement for expression in 75% of the samples was chosen arbitrarily, to ensure that we focused on SNPs expressed in a preponderance of samples. The standard error, SE, in the estimate of Δp was determined by propagation of the errors in estimating p̂R,DNA, p̂H,DNA, and p̂cDNA. Data were excluded from the study if SE > 0.1, except where explicitly noted. We also excluded data from the study if intensities for the cDNA replicates for a sample were very different from the intensities for the corresponding DNA measurements, rendering a comparison between p̂cDNA and p̂H,DNA unreliable. The discrepancy between average signal intensities for the cDNA and DNA assays was quantified via the signal ratio ρ:
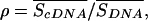 |
where the numerator is the average signal over the cDNA replicates for a sample and the denominator is the average signal over all DNA measurements that share the sample's genotype. Data were excluded from further consideration if ρ > 2.5 or ρ < 0.4. These thresholds in the SE and ρ were used to exclude spurious signals, and their particular values were picked by examining the data for homozygous SNPs, in which it was found that measurements failing these criteria accounted for only 2.4% of the data, but included 8.2% of the cases where |Δp| > 0.2.
Supplemental Table 2 provides the raw data for all SNPs that passed conformance and signal-to-background quality control filters, were expressed in at least nine samples, and were genotyped as heterozygous in at least one sample.
Estimation of false-positive and false-discovery rates
For homozygous exonic SNPs, the relative frequencies of reference and alternate alleles in DNA and cDNA samples must be identical. Therefore, the observed distribution of estimated allele frequency differences between DNA and cDNA samples for homozygous SNPs represented the noise in our assay and was used to estimate the rate of false-positive differential expression in the heterozygous SNP data. The rate of differential expression detected at a threshold t was estimated from the fraction of the heterozygous SNP data for which |Δp| > t. Differences in the distribution of the SE in the estimate of Δp between the homozygous and heterozygous SNP data were normalized to prevent an underestimation of the false-positive rate. The |Δp| data were separately divided for heterozygous SNPs and for both forms of homozygous SNPs combined (RR and AA) into five bins based on the value of the SE: (0–<0.02, 0.02–<0.04, 0.04–<0.06, 0.06–<0.08, 0.08–0.1). The false-positive rate was determined for each bin from the homozygous SNP data, and was given by the fraction of the data in the bin for which |Δp| > t. The overall false-positive rate in the heterozygous SNP data was estimated as a weighted mean of the binwise false-positive rates, with weights given by the fractions of the heterozygous SNP data that fell into each bin. The false-discovery rate was estimated from the ratio of the false-positive rate to the rate at which differential expression was detected. Based on an examination of the dependence of the false-discovery rate on allele ratio, we used a threshold t of 0.1 (allele ratio = 1.5) in this study; the corresponding false-positive rate was 2.5%, and the false-discovery rate was 11.6%.
Rates of differential expression and false discovery were also estimated by comparing the distribution of |Δp|/SE in homozygous and heterozygous exonic SNP assays, retaining the data with SE > 0.1. The statistic |Δp|/SE exceeded 2 in 35.3% of the heterozygous exonic SNPs assayed; the corresponding false-positive rate was estimated as 15.9% from the data for homozygous SNPs assayed. The corrected rate of differential expression estimated in this manner, 19.4%, was close to the estimate of 19.5% (22.0% [observed on average in each individual; see Table 3] – 2.5% [false-positive rate]), for a fold-ratio threshold of 1.5.
Effect of cDNA concentration in PCR step on allelic expression data
We tested the effects that input cDNA concentrations in the PCR reaction had on the allelic expression data. Using cDNA from a single preparation, we set up PCR with three different concentrations: 0.4 ng/μL, 0.8 ng/μL, and 2 ng/μL. The PCR products were labeled and hybridized to the exonic SNP arrays, and p̂cDNA values for 96 SNPs were determined. The p̂cDNA values of the 96 SNPs for the three different input concentrations were compared using the ANOVA single-factor test and had an average variance of 0.0005 (P = 1.7 × 10–175). The correlation coefficient of the p̂cDNA values for the 96 SNPs between the 0.8 ng/μL and 2 ng/μL samples was 0.99, and the correlation coefficient of the p̂cDNA values for the 96 SNPs between the 0.4 ng/μL and 2 ng/μL samples was also 0.99. Thus, varying input cDNA concentrations into the PCR reaction between 0.4 ng/μL and 2 ng/μL had little effect on the p̂cDNA values and thus on estimates of allelic expression differences. We used 0.4 ng/μL for our studies (see Supplemental Methods for details).
Correlation of allelic expression data from multiple sample preparations
We also tested the reproducibility of the p̂cDNA values when using RNA preparations isolated from the same cells at different times. We were unable to perform this analysis for the same samples used in the study as the white blood cells were collected from anonymous donors and thus could be obtained from each individual only once. For this reason, we used two lymphoblastoid cell lines obtained from Coriell, XGM10860 and Y-GM12560, and for each cell line independently isolated RNA twice. For sample XGM10860, p̂cDNA values for 4817 SNPs were compared between the two separate RNA preparations, and the correlation coefficient was 0.98. For sample Y-GM12560, p̂cDNA values for 4777 SNPs were compared, and the correlation coefficient between the two separate RNA preparations was also 0.98. These results indicate that RNA isolated at different time points from the same sample has very similar p̂cDNA values and thus estimates of allelic expression differences.
Acknowledgments
We thank Joe P. Karbowski, Patrick Chu, and Rhode S. Vergara for high-throughput PCR and array hybridization; Geoff B. Nilsen, Wade A. Barrett, and Michael Jen for designing the high-density arrays and for excellent assistance with data analysis; Andrew P. Kloek, David A. Hinds, Nila Patil, Karel Konvicka, and Katherine S. Pollard for helpful discussions; and Jerry Meek for assistance with creating figures. This publication was made possible by Grant Number 5 R44 HG002638-03 from NHGRI (to K.A.F.). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NHGRI.
[Supplemental material is available online at www.genome.org.]
Article published online ahead of print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.4559106.
References
- Bray, N.J., Buckland, P.R., Owen, M.J., and O'Donovan, M.C. 2003. Cis-acting variation in the expression of a high proportion of genes in human brain. Hum. Genet. 113: 149–153. [DOI] [PubMed] [Google Scholar]
- Brem, R.B., Yvert, G., Clinton, R., and Kruglyak, L. 2002. Genetic dissection of transcriptional regulation in budding yeast. Science 296: 752–755. [DOI] [PubMed] [Google Scholar]
- Cheung, V.G., Conlin, L.K., Weber, T.M., Arcaro, M., Jen, K.Y., Morley, M., and Spielman, R.S. 2003. Natural variation in human gene expression assessed in lymphoblastoid cells. Nat. Genet. 33: 422–425. [DOI] [PubMed] [Google Scholar]
- Cowles, C.R., Hirschhorn, J.N., Altshuler, D., and Lander, E.S. 2002. Detection of regulatory variation in mouse genes. Nat. Genet. 32: 432–437. [DOI] [PubMed] [Google Scholar]
- Doss, S., Schadt, E.E., Drake, T.A., and Lusis, A.J. 2005. Cis-acting expression quantitative trait loci in mice. Genome Res. 15: 681–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fondon III, J.W. and Garner, H.R. 2004. Molecular origins of rapid and continuous morphological evolution. Proc. Natl. Acad. Sci. 101: 18058–18063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germer, S., Holland, M.J., and Higuchi, R. 2000. High-throughput SNP allele-frequency determination in pooled DNA samples by kinetic PCR. Genome Res. 10: 258–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinds, D.A., Seymour, A.B., Durham, K., Banerjee, P., Ballinger, D.G., Milos, P.M., Cox, D.R., Thompson, J.F., and Frazer, K.A. 2004a. Application of pooled genotyping to scan candidate regions for association with HDL cholesterol levels. Hum. Genomics 1: 421–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinds, D.A., Stokowski, R.P., Patil, N., Konvicka, K., Kershenobich, D., Cox, D.R., and Ballinger, D.G. 2004b. Matching strategies for genetic association studies in structured populations. Am. J. Hum. Genet. 74: 317–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinds, D.A., Stuve, L.L., Nilsen, G.B., Halperin, E., Eskin, E., Ballinger, D.G., Frazer, K.A., and Cox, D.R. 2005. Whole-genome patterns of common DNA variation in three human populations. Science 307: 1072–1079. [DOI] [PubMed] [Google Scholar]
- Hubner, N., Wallace, C.A., Zimdahl, H., Petretto, E., Schulz, H., Maciver, F., Mueller, M., Hummel, O., Monti, J., Zidek, V., et al. 2005. Integrated transcriptional profiling and linkage analysis for identification of genes underlying disease. Nat. Genet. 37: 243–253. [DOI] [PubMed] [Google Scholar]
- International Human Genome Sequencing Consortium. 2004. Finishing the euchromatic sequence of the human genome. Nature 431: 931–945. [DOI] [PubMed] [Google Scholar]
- Kim, U.K., Jorgenson, E., Coon, H., Leppert, M., Risch, N., and Drayna, D. 2003. Positional cloning of the human quantitative trait locus underlying taste sensitivity to phenylthiocarbamide. Science 299: 1221–1225. [DOI] [PubMed] [Google Scholar]
- Knight, J.C., Keating, B.J., and Kwiatkowski, D.P. 2004. Allele-specific repression of lymphotoxin-α by activated B cell factor-1. Nat. Genet. 36: 394–399. [DOI] [PubMed] [Google Scholar]
- Koschinsky, M.L., Boffa, M.B., Nesheim, M.E., Zinman, B., Hanley, A.J., Harris, S.B., Cao, H., and Hegele, R.A. 2001. Association of a single nucleotide polymorphism in CPB2 encoding the thrombin-activable fibrinolysis inhibitor (TAF1) with blood pressure. Clin. Genet. 60: 345–349. [DOI] [PubMed] [Google Scholar]
- Lo, H.S., Wang, Z., Hu, Y., Yang, H.H., Gere, S., Buetow, K.H., and Lee, M.P. 2003. Allelic variation in gene expression is common in the human genome. Genome Res. 13: 1855–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morley, M., Molony, C.M., Weber, T.M., Devlin, J.L., Ewens, K.G., Spielman, R.S., and Cheung, V.G. 2004. Genetic analysis of genome-wide variation in human gene expression. Nature 430: 743–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver, F., Christians, J.K., Liu, X., Rhind, S., Verma, V., Davison, C., Brown, S.D., Denny, P., and Keightley, P.D. 2005. Regulatory variation at glypican-3 underlies a major growth QTL in mice. PLoS Biol. 3: e135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastinen, T., Sladek, R., Gurd, S., Sammak, A., Ge, B., Lepage, P., Lavergne, K., Villeneuve, A., Gaudin, T., Brandstrom, H., et al. 2004. A survey of genetic and epigenetic variation affecting human gene expression. Physiol. Genomics 16: 184–193. [DOI] [PubMed] [Google Scholar]
- Prokunina, L., Castillejo-Lopez, C., Oberg, F., Gunnarsson, I., Berg, L., Magnusson, V., Brookes, A.J., Tentler, D., Kristjansdottir, H., Grondal, G., et al. 2002. A regulatory polymorphism in PDCD1 is associated with susceptibility to systemic lupus erythematosus in humans. Nat. Genet. 32: 666–669. [DOI] [PubMed] [Google Scholar]
- Ronald, J., Akey, J.M., Whittle, J., Smith, E.N., Yvert, G., and Kruglyak, L. 2005. Simultaneous genotyping, gene-expression measurement, and detection of allele-specific expression with oligonucleotide arrays. Genome Res. 15: 284–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schadt, E.E., Monks, S.A., Drake, T.A., Lusis, A.J., Che, N., Colinayo, V., Ruff, T.G., Milligan, S.B., Lamb, J.R., Cavet, G., et al. 2003. Genetics of gene expression surveyed in maize, mouse and man. Nature 422: 297–302. [DOI] [PubMed] [Google Scholar]
- Tokuhiro, S., Yamada, R., Chang, X., Suzuki, A., Kochi, Y., Sawada, T., Suzuki, M., Nagasaki, M., Ohtsuki, M., Ono, M., et al. 2003. An intronic SNP in a RUNX1 binding site of SLC22A4, encoding an organic cation transporter, is associated with rheumatoid arthritis. Nat. Genet. 35: 341–348. [DOI] [PubMed] [Google Scholar]
- Wilkins, J.F. 2005. Genomic imprinting and methylation: Epigenetic canalization and conflict. Trends Genet. 21: 356–365. [DOI] [PubMed] [Google Scholar]
- Wrzeska, M. and Rejduch, B. 2004. Genomic imprinting in mammals. J. Appl. Genet. 45: 427–433. [PubMed] [Google Scholar]
- Yan, H., Yuan, W., Velculescu, V.E., Vogelstein, B., and Kinzler, K.W. 2002. Allelic variation in human gene expression. Science 297: 1143. [DOI] [PubMed] [Google Scholar]