

Abstract
We have developed a high-throughput method for analyzing the methylation status of hundreds of preselected genes simultaneously and have applied it to the discovery of methylation signatures that distinguish normal from cancer tissue samples. Through an adaptation of the GoldenGate genotyping assay implemented on a BeadArray platform, the methylation state of 1536 specific CpG sites in 371 genes (one to nine CpG sites per gene) was measured in a single reaction by multiplexed genotyping of 200 ng of bisulfite-treated genomic DNA. The assay was used to obtain a quantitative measure of the methylation level at each CpG site. After validating the assay in cell lines and normal tissues, we analyzed a panel of lung cancer biopsy samples (N = 22) and identified a panel of methylation markers that distinguished lung adenocarcinomas from normal lung tissues with high specificity. These markers were validated in a second sample set (N = 24). These results demonstrate the effectiveness of the method for reliably profiling many CpG sites in parallel for the discovery of informative methylation markers. The technology should prove useful for DNA methylation analyses in large populations, with potential application to the classification and diagnosis of a broad range of cancers and other diseases.
DNA methylation is widespread and plays a critical role in the regulation of gene expression in development, differentiation, and diseases such as multiple sclerosis, diabetes, schizophrenia, aging, and cancers (Li et al. 1993; Laird and Jaenisch 1996; Egger et al. 2004). Methylation in particular gene regions, for example in promoters, can inhibit gene expression (Jones and Laird 1999; Baylin and Herman 2000; Jones and Baylin 2002). Recent work has shown that the gene silencing effect of methylated regions is accomplished through the interaction of methylcytosine binding proteins with other structural components of chromatin (Razin 1998), which, in turn, makes the DNA inaccessible to transcription factors through histone deacetylation and chromatin structure changes (Bestor 1998).
Changes in DNA methylation are recognized as one of the most common forms of molecular alteration in human neoplasia (Baylin and Herman 2000; Balmain et al. 2003; Feinberg and Tycko 2004). Hypermethylation of CpG islands located in the promoter regions of tumor suppressor genes is now firmly established as the most frequent mechanism for gene inactivation in cancers (Esteller 2002; Herman and Baylin 2003). In contrast, a global hypomethylation of genomic DNA (Feinberg and Vogelstein 1983) and loss of IGF2 imprinting were observed in tumor cells (Cui et al. 2003; Sakatani et al. 2005); and a correlation between hypomethylation and increased gene expression was reported for many oncogenes (Feinberg and Vogelstein 1983; Hanada et al. 1993). In addition, monitoring global changes in DNA methylation has been applied to molecular classification of cancers (Huang et al. 1999; Costello et al. 2000). Most recently, gene hypermethylation was associated with clinical risk groups for neuroblastoma (Alaminos et al. 2004), as well as with hormone receptor status and response to tamoxifen in breast cancer (Widschwendter et al. 2004b; Martens et al. 2005). Therefore, it should be feasible to use methylation markers to classify and predict different kinds or stages of cancer, cancer therapeutic outcomes, and patient survival. However, the analysis of large sample sets will be required in order to discover such associations. Analysis of methylation on this scale—many target sites per sample and many samples per study—still represents a significant challenge (Jones and Baylin 2002; Dennis 2003; Rakyan et al. 2004).
Here we describe an adaptation of a high-throughput single nucleotide polymorphism (SNP) genotyping system (Fan et al. 2003) to DNA methylation detection, based on genotyping of bisulfite-converted genomic DNA. This technology combines a miniaturized bead-based array platform, a high level of assay multiplexing, and scalable automation for sample handling and data processing. We used this technology to analyze methylation profiles of 1536 CpG sites from 371 genes in cell lines and in lung cancers and normal tissues, and have identified a subset of candidate biomarkers that need to be validated in a prospective study.
Results
Implementation of the methylation profiling assay on the SNP genotyping platform
We adapted the GoldenGate SNP genotyping assay (Fan et al. 2003) for DNA methylation detection. Nonmethylated cytosines (C) are converted to uracil (U) when treated with bisulfite, while methylated cytosines remain unchanged (Wang et al. 1980). Because the hybridization behavior of uracil is similar to that of thymine (T), the detection of the methylation status of a particular cytosine can be carried out following bisulfite treatment by using a genotyping assay for a C/T polymorphism.
In this study, we designed assays for 1536 CpG sites from the 5′-regulatory region of 371 genes (one to nine CpG sites per gene). These genes (Supplemental Table 1) were selected based on their biological relevance. They include tumor suppressor genes and oncogenes; genes that are indirectly involved in cancer development, for example, DNA repair genes; metastasis-inhibitor genes; genes regulated by various signaling pathways, and/or responsible for altered cell growth, differentiation and apoptosis; genes considered to be targets for oncogenic transformation; genes of innate host defense; genes involved in surfactant function of the lung; imprinted genes; and previously reported differentially methylated genes (Adorjan et al. 2002; Esteller 2002; Tsou et al. 2002).
The assay procedure is similar to that described previously for standard SNP genotyping (Fan et al. 2003), except that four oligonucleotides, two allele-specific oligonucleotides (ASOs), and two locus-specific oligonucleotides (LSOs) are required for each assay site rather than three (see the legend to Fig. 1). Briefly, bisulfite-treated, biotinylated genomic DNA (gDNA) was immobilized on paramagnetic beads. Pooled query oligonucleotides were annealed to the gDNA under a controlled hybridization program, and then washed to remove excess or mishybridized oligonucleotides. Hybridized oligonucleotides were then extended and ligated to generate amplifiable templates. Requiring the joining of two fragments to create a PCR template in this scheme provided an additional level of locus specificity. It is unlikely that any incorrectly hybridized ASOs and LSOs will be adjacent, and therefore should not be able to ligate after ASO extension. A PCR reaction was performed with fluorescently labeled universal PCR primers. The methylation status of an interrogated CpG site was determined by calculating β, which is defined as the ratio of the fluorescent signal from the methylated allele to the sum of the fluorescent signals of both methylated and unmethylated alleles (see Methods for details). The β-value provides a continuous measure of levels of DNA methylation in samples, ranging from 0 in the case of completely unmethylated sites to 1 in completely methylated sites.
Figure 1.

DNA methylation assay scheme. (A) Bisulfite conversion of DNA. (B) For each CpG site, two pairs of probes were designed to interrogate either the top or bottom strand: an allele-specific oligonucleotide (ASO) and locus-specific oligonucleotide (LSO) probe pair for the methylated state of the CpG site and a corresponding ASO-LSO pair for the unmethylated state. Each ASO consists of a 3′-portion that hybridizes to the bisulfite-converted genomic DNA, with the 3′-base complementary to either the “C” or “T” allele of the targeted CpG site, and a 5′-portion that incorporates a universal PCR primer sequence P1 or P2. The LSOs consist of three parts: At the 5′-end is a CpG locus-specific sequence; in the middle is an address sequence, complementary to a corresponding capture sequence on the array; and at the 3′-end is a universal PCR priming site (P3). Pooled assay oligonucleotides were first annealed to bisulfite-converted genomic DNA. An allele-specific primer extension step was then carried out; ASOs were extended only if their 3′-base was complementary to their cognate CpG site in the gDNA template. Allele-specific extension was followed by ligation of the extended ASOs to their corresponding LSOs, to create PCR templates. The ligated products were then amplified by PCR using common primers P1, P2, and P3′, and hybridized to a microarray bearing the complementary address sequences. P1 and P2 were fluorescently labeled, each with a different dye, and associated with the “T” (unmethylated) allele or the “C” (methylated) allele, respectively.
Development of internal controls for the methylation assay
In order to develop a robust methylation detection method, we needed a set of reliable controls. We used plasmids pUC19 and pACYC184 and phage φX174 as internal control DNAs. These DNAs (unmethylated, in vitro methylated, or mixed at a 1:1 ratio) were spiked into 200 ng of human genomic DNA at a 1:1 molar ratio (at ∼2–4 pg of plasmid DNA/1 μg of gDNA, depending on the plasmid size), and were used in every experiment to monitor both bisulfite conversion efficiency and accuracy of methylation detection (Supplemental Fig. 1).
To calibrate quantitative measurements of methylation, we used “methylated” and “unmethylated” genomic templates. The unmethylated templates were generated by genome-wide amplification of human genomic DNA using a Repli-g DNA amplification kit (Molecular Staging). After amplification, endogenous DNA methylation was diluted at least 100- to 1000-fold, effectively rendering the amplified genomic DNA “unmethylated.” The methylated templates were generated by in vitro methylation using a CpG-methylase, SssI (New England BioLabs).
We obtained very low methylation values for most of the 1536 CpG sites in the unmethylated reference DNA sample as expected, with ∼95% of the CpG sites showing methylation levels lower than 50% (data not shown). In contrast, we found that ∼93% of the CpG sites were methylated to >50% of completion in the in vitro methylated reference DNA. In general, these results indicate that our assay is specific, and faithfully reports the methylation status of most of the targeted CpG sites in tested samples. Some degree of cross-hybridization may explain those outlier CpG sites where elevated methylation was observed in the unmethylated reference DNA and the sites that were undermethylated in the in vitro methylated reference DNA, as may be expected by the reduced complexity of bisulfite-converted DNA. All such unvalidated sites were still included and monitored in the assay reactions, but excluded from consideration for methylation marker development (see below).
Bisulfite conversion efficiency
Methylation detection in bisulfite-converted DNA is based on the different sensitivity of cytosine and 5-methylcytosine to deamination by bisulfite (Wang et al. 1980). Under acidic conditions, cytosine undergoes conversion to uracil, while methylated cytosine remains unreactive. An effective bisulfite conversion protocol is a necessary prerequisite for a robust methylation profiling assay. Incomplete conversion of cytosine to uracil can result in appearance of false-positive methylation signals, and can reduce the overall quality of the assay data. To estimate the conversion efficiency, we analyzed 7097 cytosines in 173 independent DNA fragments derived from eight genomic regions by bisulfite sequencing (Frommer et al. 1992). To avoid ambiguity, only the cytosines from non-CpG sites were counted. The sequence data indicated that the DNA conversion rate was 99.7% with our current protocol (see Methods for details).
Methylation assay reproducibility
For each sample, 1 μg of genomic DNA was used for each bisulfite conversion, and 20% of the converted DNA (corresponding to 200 ng of starting gDNA) was then used to assay the 1536 CpG sites simultaneously on an array. Technical replicates were done for each sample using the same converted DNA. We obtained highly reproducible DNA methylation profiles between these technical replicates (Fig. 2, left and center panels), with an average R2 of 0.98 ± 0.02 when the β-values were compared (Supplemental Table 2). When β-values for matching normal and carcinoma clinical samples were compared, differential methylation was readily detected (Fig. 2, right panel).
Figure 2.

Methylation assay reproducibility and differential methylation detection. Comparison of methylation profiles between lung cancer and matching normal tissue. The β-value (i.e., the methylation ratio measured for all 1536 CpG sites) obtained from one replicate experiment is plotted against that obtained from another technical replicate experiment. The left and center panels show the reproducibility of replicated assays on DNAs derived from lung adenocarcinoma G12022 (center) and its matching normal tissue (left). The right panel shows the comparison between the normal and the matching adenocarcinoma samples.
Methylation status of housekeeping genes located on the X chromosome
DNA methylation is involved in transcriptional inactivation of genes on one of the two X chromosomes in female somatic cells (i.e., X chromosome silencing), which compensates for the dosage of functional X-linked genes between male and female (Carrel and Willard 2005). We measured the methylation status of six X-linked housekeeping genes—EFNB1, ELK1, FMR1, G6PD, GPC3, and GLA—together with the rest of the 371 genes in male and female genomic DNAs. In general, methylation levels of these genes correlated well with the gender of the sample source, that is, no or very low methylation was detected in male DNA samples and hemimethylation was detected in female DNA samples.
We then selected the best 18 “gender-specific” CpG sites from these X-chromosomal genes (with β < 0.1 in male genomic DNA) to estimate the assay's ability for detecting difference of methylation levels between samples. We diluted female genomic DNA into male genomic DNA at ratios of 5:95, 10:90, 20:80, and 50:50, prior to bisulfite conversion. Two independent sets of mixtures were made and measured in parallel (Fig. 3A); and four replicates were done for each mixture. We observed that the standard deviation of the β-value obtained for all the 1536 CpG sites across the four replicates was <0.06 in 99% of cases, and the average slope of β versus the expected methylation level for the selected X-chromosomal sites was equal to 1. Therefore, we estimate that our method can discriminate levels of methylation (β-values) that differ by as little as 0.17 (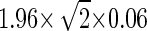 ). Since the tails of their respective signal distributions did not have large overlapping areas (<16% assuming Gaussian error), there is a high probability that the response produced at one methylation level will be significantly different from the expected signal produced at another. It is worth noting that the standard deviation of these measurements was not uniform across the range of β-values. It had a parabolic shape with the maximum peak around β = 0.5. Therefore, the absolute performance of the assay depends on the methylation level itself. For example, for 16 out of the 18 CpG sites, methylation levels from 5:95 and 0:100 mixtures could be unambiguously distinguished from each other (i.e., the maximum β-value in the 0:100 mixtures was less than the minimum β-value in the 5:95 mixtures) (Fig. 3B). This indicates that our assay can detect as little as 2.5% methylation for well-performing CpG sites in the optimal range of the response curve.
). Since the tails of their respective signal distributions did not have large overlapping areas (<16% assuming Gaussian error), there is a high probability that the response produced at one methylation level will be significantly different from the expected signal produced at another. It is worth noting that the standard deviation of these measurements was not uniform across the range of β-values. It had a parabolic shape with the maximum peak around β = 0.5. Therefore, the absolute performance of the assay depends on the methylation level itself. For example, for 16 out of the 18 CpG sites, methylation levels from 5:95 and 0:100 mixtures could be unambiguously distinguished from each other (i.e., the maximum β-value in the 0:100 mixtures was less than the minimum β-value in the 5:95 mixtures) (Fig. 3B). This indicates that our assay can detect as little as 2.5% methylation for well-performing CpG sites in the optimal range of the response curve.
Figure 3.

Methylation detection in gDNA mixtures. (A) Female genomic DNA was diluted with male genomic DNA at ratios of 5:95, 10:90, 20:80, and 50:50. Two sets of mixtures were made and measured: M1 (male NA10923)/F1 (female NA10924) and M2 (male NA07033)/F2 (female NA06999). Methylation levels of six X chromosome-linked genes were calculated as the average of several (one to five) CpG sites for each gene. (B) Methylation profiles of individual CpG sites (only four are shown). Error bars represent the standard deviation of β-values calculated from four replicate experiments, done with the first set of mixtures (NA10923/NA10924).
Methylation profiling in cancer cell lines
To demonstrate the applicability of our method for studying DNA methylation in cancer, we applied the assay developed for the 1536 CpG sites to a panel of 17 colon, breast, lung, and prostate cancer cell lines, as well as seven DNA samples derived from different normal tissues. Sixteen CpG sites distinguishing cancer from normal samples were selected based on a Mann-Whitney test (P < 0.00001) and an additional filter of mean change of methylation level >0.34 (0.17 × 2). Forty-eight CpG sites distinguishing individual cancer types were selected based on a Kruskal-Wallis test (P < 0.035) and a standard deviation across cancer samples >0.34. This gave us a balanced list of 16 cancer-specific markers and 48 cancer type-specific markers. Using these markers, all cancer samples were correctly separated from normal samples by hierarchical clustering, with Ward's linkage method and correlation-based distance metric. This separation of cancer samples was not sensitive to the choice of distance metric or linkage method (data not shown). Figure 4 shows the differential methylation profiles in normal versus cancer samples as well as specific methylation signatures that were obtained for individual cancer types. In general, our data correlate well with previous cell line methylation profiling results (Paz et al. 2003; Melnikov et al. 2005). For example, GSTP1 was completely methylated in the LNCaP prostate cancer cell line and semimethylated in the PC3 cell line (data not shown), as previously reported (Singal et al. 2001). The overall methylation level in colon cancer cell lines appears to be higher as compared to the other cell lines, also consistent with previous results (Paz et al. 2003).
Figure 4.

Methylation profiling in cancer cell lines. Seven DNA samples derived from different normal tissues and 17 colon, breast, lung, and prostate cancer cell lines were profiled. All cancer samples were correctly separated from normal samples using agglomerative clustering based on 64 cancer-specific methylation makers, and highly specific methylation signatures were obtained for each cancer type. Green, yellow, and red colors represent low, medium, and high methylation levels, respectively.
Validation of microarray data by methylation-specific PCR
Methylation-specific PCR (MSP) has been widely used to monitor the methylation status of individual genes (Herman et al. 1996; Eads et al. 2000). We used MSP to confirm the methylation status of CpG sites from five genes that were identified by our microarray analysis as showing distinct methylation profiles in normal lung tissue and lung cancer cell lines. MSP primers that are specific to either methylated or unmethylated DNA were designed to target corresponding CpG sites within the promoter regions of CFTR, DBC1, DLK1, EYA4, and NPY genes (Table 1). In addition, a pair of primers recognizing a non-CpG-containing region of the β-actin gene (ACTB) was used to measure DNA input. The specificity of each set of MSP primers was first tested using in vitro methylated and unmethylated reference DNAs described above. MSP conditions were optimized to maximize the discrimination between the two methylation states. Bisulfite-treated genomic DNAs derived from one normal lung tissue and six lung cancer cell lines were analyzed using real-time MSP. The methylated and unmethylated alleles in each genomic DNA sample were amplified in separate reactions. Each reaction was performed in duplicate, and the average of the crossover threshold (Ct) values was used to calculate the concentration of the methylated or unmethylated allele at the target site. Of the 35 MSP data points, 34 were highly concordant with the methylation status determined by the genotyping-based microarray analysis, with a Spearman correlation coefficient r = 0.89 (Fig. 5). These results confirm the overall validity of our method.
Table 1.
Methylation-specific PCR primer sequence and amplicon size
| Primer set | Forward primer, 5′ → 3′ | Reverse primer, 5′ → 3′ | Size (bp) |
|---|---|---|---|
| ACTB | TGGTGATGGAGGAGGTTTAGTAAGT | AACCAATAAAACCTACTCCTCCCTTAA | 130 |
| CFTR-M | GTTTTGGGTTTGGCGGATTTTGACGC | CCCGCAAATAAACGACAATCGCGAC | 142 |
| CFTR-U | GGTTTTGGGTTTGGTGGATTTTGATGT | CATCCCACAAATAAACAACAATCACAAC | 146 |
| DBC1-M | ACGCGATCCCTTTAAATACTCGTACG | GAGGAGAGACGGGAGGTCGTTTCG | 131 |
| DBC1-U | ATATACACAATCCCTTTAAATACTCATACA | GGAGGAGAGATGGGAGGTTGTTTTG | 136 |
| DLK1-M | TCCGAAAAAATCTACGACCCAAATTCG | AGACGGGTATAGGTATTTCGCGAGC | 132 |
| DLK1-U | TCTCCAAAAAAATCTACAACCCAAATTCA | GTTAGATGGGTATAGGTATTTTGTGAGT | 137 |
| EYA4-M | TACGGAGATTACGGCGGCGTTATTC | AACGCGAAAAACGACGACGCGCGA | 150 |
| EYA4-U | GGGTATGGAGATTATGGTGGTGTTATTT | CCAAAAACACAAAAAACAACAACACACAA | 158 |
| NPY-M | TTAAAACCCTCTAACCGAAAACTTCCG | ACGATTAGCGCGGTATTTTCGTCGG | 131 |
| NPY-U | CTTTAAAACCCTCTAACCAAAAACTTCCA | TTTTTATGATTAGTGTGGTATTTTTGTTGG | 138 |
Figure 5.

Comparison of methylation-specific PCR and array-based methylation data. MSP was used to confirm the methylation status of CpG sites within the promoter regions of five genes that showed distinct methylation profiles in one normal lung tissue and six lung cancer cell lines from array-based methylation analysis (1: normal lung tissue; 2: NCI-H69; 3: NCI-H526; 4: NCI-H358; 5: NCI-H1299; 6: NCI-H1395; and 7: NCI-H2126). The methylation level is represented as bars, blue for microarray data and red for MSP data.
Methylation marker identification in lung adenocarcinomas
We measured the methylation status of the 1536 CpG sites in 23 lung adenocarcinoma and 23 normal lung tissue samples. We used a data matrix of β-values to identify CpG sites that showed differential methylation in cancers. Using a Mann-Whitney test, we first compared 11 normal samples to 11 adenocarcinoma samples (a training set of samples obtained from Philipps-University of Marburg, Germany). We used a false discovery rate (FDR) approach (Benjamini and Hochberg 1995) to select a list of differentially methylated CpG sites. At an FDR = 0.001 cutoff, we identified 207 differentially methylated sites. To select markers that had the largest difference between cancer and normal tissues, we applied an additional filter that required a minimum difference of 0.15 in β between the two groups. We thus obtained a list of 55 differentially methylated markers for lung adenocarcinoma (Supplemental Table 3). Among these markers, more were hypermethylated in adenocarcinoma, including the genes ASCL2, CDH13, HOXA11, HOXA5, NPY, RUNX3, TERT, and TP73, which were selected for validation by bisulfite sequencing (see below). Of these genes, methylation of CDH13 (Toyooka et al. 2001; Ogama et al. 2004), HOXA5 (Chen et al. 2003), RUNX3 (Li et al. 2004), and TP73 (Lomas et al. 2004) is known to be associated with tumor progression in various types of cancer. The human telomerase reverse transcriptase gene (TERT) was shown to be inactivated in most differentiated cells, but reactivated in the majority of cancer cells (Liu et al. 2004). However, a recent study (Widschwendter et al. 2004a) reported methylation of TERT in cervical cancer and its correlation with poor prognosis. One of the markers on the list, neuropeptide Y (NPY), which was shown to be hypermethylated in 19 out of the 23 analyzed adenocarcinoma samples and had no or very low methylation in the normal samples, was not previously reported as a cancer marker. NPY may influence lipid metabolism and is potentially associated with hypertension (Tomaszewski et al. 2004). It would be interesting to test if NPY plays any role in lung surfactant-related function.
Clustering of independent sample sets based on the identified methylation markers
We used agglomerative nesting with the Ward method and correlation-based distance to cluster the training set samples (the German samples mentioned above) based on the methylation profiles of the 55 selected markers. Cancer samples were clearly distinguishable from normal samples with one error—cancer sample G12029 coclustered with the normal samples (Fig. 6A).
Figure 6.

Cluster analysis of lung adenocarcinoma samples. (A) Eleven cancer and 11 normal tissue samples were used as a training set to identify a list of 55 CpG sites that are differentially methylated in cancer versus normal tissues with high confidence level (adjusted P-value < 0.001) and significant change in absolute methylation level (|Δβ| > 0.15). Cancer sample G12029 was mistakenly coclustered with normal samples. (B) The selected 55 CpG sites were used for classification of an independent test sample set of 12 lung adenocarcinoma and 12 normal tissue samples, collected from a different institute. All the normal and cancer samples were separated into two distinguished groups with a single error—cancer sample D12162 was coclustered with normal samples. Normal samples are underlined in green, cancer in red. The asterisks indicate misclassified samples.
To assess the power of the selected methylation markers for reliable classification of prospective cancer and normal samples, we clustered an independent test set of samples based on the methylation profiles of these markers. This test set contained 12 normal and 12 adenocarcinoma samples, collected from The Pennsylvania State University College of Medicine Tumor Bank. We obtained a 100% specificity (12/12) and 92% sensitivity (11/12). The specificity was calculated as (TN, true negative; FP, false positive; TP, true positive; FN, false negative) TN/(TN + FP), and the sensitivity was calculated as TP/(TP + FN). One cancer sample D12162 was coclustered with the normal samples in this test set (Fig. 6B). This analysis indicates that the differential methylation pattern for the identified markers was preserved in the two completely unrelated training and test sample sets and that methylation profiling of these markers allows the identification of cancer samples with high specificity and sensitivity.
Methylation marker validation by bisulfite sequencing
We validated eight CpG sites that showed elevated methylation in the adenocarcinoma samples using bisulfite sequencing (Frommer et al. 1992). We chose this method over other methylation detection methods for several reasons: (1) this would provide another validation of our method, in addition to the MSP method; (2) we had only limited amount of DNA from the clinical samples, and bisulfite sequencing required less input DNA as compared to other methods; and (3) we wanted to use the bisulfite sequencing data to estimate the bisulfite conversion rate (see above).
PCR primers were designed flanking the CpG sites of interest (Table 2). DNAs from two normal and four adenocarcinoma samples were treated with bisulfite, and regions of interest were amplified by PCR. PCR fragments were cloned, and individual colonies were picked for sequencing (see Methods for details). Twelve cloned fragments were sequenced for each CpG site in selected samples. In all cases we observed an increase in methylation in cancer samples compared to normal samples. Even though the absolute levels of methylation detected by the two different methods were somewhat different (Table 3), a strong correlation was obtained between these two data sets, with a Spearman correlation coefficient r = 0.70. Overall, these results suggest that our assay can reliably detect methylation differences in clinical samples for more than 1000 CpG sites and that the assay can be used for both marker discovery and validation.
Table 2.
Bisulfite sequencing primer sequence and amplicon size
| Primer set | Forward primer, 5′ → 3′ | Reverse primer, 5′ → 3′ | Size (bp) |
|---|---|---|---|
| ASCL2 | GTAGTTTATTTTTATTTTTAGTAGATTAA | AAAACCAAAATCTCAACCAATC | 260 |
| CDH13 | ATTTTTTGGAAAAGTGGAATTAGTT | CCAAATAAATCAACAACAACATCAC | 241 |
| HOXA11 | TAGTTTTTGTGTTTTTGTTTTTGT | ATAACTTAATTACACTCTCTCATTCATAAT | 272 |
| HOXA5 | GTTGTAGGGAGTTTGGGTTTATT | CCTAAAAAATCTTCATCACAAAATC | 102 |
| NPY | GAGAAGGGGTAGAAGTTTTTGAAAT | ATCTCCTACCAACAAAACTACCAAC | 267 |
| RUNX3 | TTTTGTAGTTATTGTTGTTTTTTTT | CAAATTTCAAAATCTTACAAACCTC | 187 |
| TERT | TTTGAGAATTTGTAAAGAGAAATGA | AATATAAAAACCCTAAAAACAAATAC | 290 |
| TP73 | AGTAAATAGTGGGTGAGTTATGAAGATG | TACACCAAACCCTAACTAAAAAACC | 287 |
Table 3.
Methylation results generated from microarray analysis and bisulfite sequencing
| CpG site | Sample ID | % methylation, bisulfite sequencing | % methylation, array analysis |
|---|---|---|---|
| ASCL2_1038 | nD12209 | 0.00 | 0.09 |
| D12155 | 0.67 | 0.79 | |
| CDH13_1358 | nD12195 | 0.00 | 0.11 |
| D12170 | 0.40 | 0.71 | |
| HOXA11_802 | nD12209 | 0.17 | 0.09 |
| D12155 | 0.92 | 0.92 | |
| HOXA5_576 | nD12209 | 0.20 | 0.60 |
| D12163 | 0.75 | 0.75 | |
| NPY_1009 | nD12209 | 0.00 | 0.12 |
| D12165 | 1.00 | 0.88 | |
| RUNX3_368 | nD12195 | 0.90 | 0.27 |
| D12170 | 1.00 | 0.67 | |
| TERT_900 | nD12209 | 0.00 | 0.60 |
| D12165 | 0.44 | 0.92 | |
| TP73_377 | nD12195 | 0.09 | 0.05 |
| D12170 | 0.46 | 0.62 |
Discussion
DNA methylation detection methods include methylation-specific enzyme digestion (Singer-Sam et al. 1990), bisulfite DNA sequencing (Frommer et al. 1992; Dupont et al. 2004), methylation-specific PCR (MSP) (Herman et al. 1996) and MethyLight (Eads et al. 2000; Cottrell et al. 2004), methylation-sensitive single nucleotide primer extension (MS-SnuPE) (Gonzalgo and Jones 1997), restriction landmark genomic scanning (RLGS) (Kawai et al. 1994; Akama et al. 1997), MALDI mass spectrometry (Tost et al. 2003), and differential methylation hybridization (DMH) (Huang et al. 1999). However, none of these methods combines random access to specific sequences in the genome with high throughput and low cost, which is needed for analyzing methylation profiles at high resolution in large sample sets. In addition, many of these methods are insensitive to low levels of methylation changes in diseased tissues, for example, 10% or 20% hypermethylation.
In this study, we developed and characterized a highly reproducible and multiplexed method for high-throughput quantitative measurement of DNA methylation. The method provides not just a discrete measure of positive versus negative DNA methylation, but a continuous measure of levels of DNA methylation. For a 17% difference in absolute methylation level (e.g., 10% vs. 27%), signals are expected to have largely nonoverlapping distributions. The assay can detect as little as 2.5% of methylation for some CpG sites. Unlike restriction enzyme-based methods, assay probes can be specifically designed for most of the CpG sites in the genome, and assay oligonucleotides can be designed to interrogate either the Watson or Crick strand or both at each CpG site (Fig. 1). Assay results are read out on a universal array. As a result, gene (or CpG) sets can be refined iteratively, if desired, because no custom arrays need to be developed. The method can detect changes in methylation status at up to 1536 different CpG sites simultaneously using only 200 ng of genomic DNA.
We applied this technology to the high-throughput discovery and validation of potential biomarkers of lung cancer. Lung cancer is the second most common cancer among both men and women and is the leading cause of cancer death in both genders. There is no established early detection test for the disease, and only 15% of lung cancer cases are diagnosed when the disease is localized. The ability to accurately detect malignant cells in a wide range of clinical specimens including sputum, blood, or tissue has significant implications for screening high-risk individuals for this cancer. In this study, we first analyzed the methylation status of 1536 CpG sites (derived from 371 genes) in 11 lung adenocarcinomas and 11 matching normal tissue samples. A panel of 55 adenocarcinoma-specific methylation markers was identified by combining P-value and magnitude of change thresholds (Fig. 6A). Furthermore, we validated the adenocarcinoma markers in an independent sample set (N = 24) with high sensitivity and specificity (Fig. 6B). These results demonstrate the utility of our method for marker identification and validate the robustness of the markers identified.
Because methylation detection interrogates genomic DNA, rather than RNA or protein, it offers several technological advantages in a clinical diagnostic setting: (1) readily available source materials, particularly important for prognostic research, because DNA can be more reliably extracted than RNA from archived biological samples for study; (2) capability for multiplexing, allowing simultaneous measurement of multiple targets to improve assay specificity; (3) easy amplification of assay products to achieve high sensitivity; and (4) the ability to detect a positive signal in tumors that arises from methylation inactivation of one allele of tumor-suppressor genes (Balmain et al. 2003). Detecting the appearance of a positive signal should be a more robust and reliable measurement than detecting a twofold gene expression change at the mRNA level in these tumors. We are currently modifying our method and prospectively collecting three types of samples from lung cancer patients at the time of bronchoscopy: bronchoaveolar lavage (BAL) fluid, sputum, and whole blood, with the aim of developing a highly multiplexed, sensitive, and minimally invasive methylation analysis system that can be applied to early and noninvasive diagnosis of lung cancer, and to monitor cancer progression and response to treatment. In general, this technology should prove useful for comprehensive DNA methylation analyses in large populations, with potential application to the classification and diagnosis of a broad range of cancers and other diseases.
Methods
Assay probe design
A 1.5-kb sequence from the 5′-regulatory region was extracted for each target gene based on human genome RefSeq build 34, version 3 (released on March 10, 2004). CpG islands of interest from this 1.5-kb region were selected and “bisulfite-converted” computationally. We adapted an automated SNP genotyping assay probe design program (Fan et al. 2003) for this methylation study. For each CpG site, four probes were designed: two allele-specific oligonucleotides (ASO) and two locus-specific oligonucleotides (LSO). Each ASO-LSO oligonucleotide pair corresponded to either the methylated or unmethylated state of the CpG site (Fig. 1). The gap size between the ASO and LSO oligonucleotides varied from 1 base to 20 bases, which allowed difficult sequences or ambiguous bases in CpG islands of interest to be avoided. This flexibility is particularly important for methylation studies because of a decrease in sequence complexity as a result of bisulfite treatment. If other CpG sites were present in close vicinity of the target CpG site, we made the assumption that they had the same methylation status as the site of interest. This design hypothesis was based on previously reported bisulfite sequencing results, in which a majority (>90%) of the adjacent CpG sites was shown to be co-methylated or co-demethylated (Bird 2002; Grunau et al. 2002; Tost et al. 2003; Rakyan et al. 2004). This assumption was also confirmed by our own bisulfite sequencing results. It is worth pointing out that this design strategy is used in methylation-specific PCR primer design (Herman et al. 1996) and other microarray-based DNA methylation analysis (Adorjan et al. 2002). While there were many CpG sites within each CpG island, we only selected those for which robust assays could be designed. The sequence information for the 1536 designed CpG sites is included in Supplemental Table 4.
DNA samples for methylation analysis
DNA from breast cancer cell lines MCF-7, MDA-MB-435, MDA-MB-468, and T-47D; colon cancer cell lines Fet, HT29, HCT116, LS174, and SW480; and prostate cancer cell lines PC3 and LNCaP was extracted using a modified Trizol method according to the manufacturer's recommendations (Invitrogen). DNA from lung cancer cell lines NCI-H69 (HTB-119D), NCI-H526 (CRL-5807D), NCI-H358 (CRL-5811D), NCI-H1299 (CRL-5803D), NCI-H1395 (CRL-5868D), and NCI-H2126 (CCL-256D) was purchased from ATCC. DNA from normal lung, ovary, breast, colon, and prostate tissues was purchased from Clinomics Biosciences. DNA samples NA06999, NA07033, NA10923, and NA10924 were purchased from the Coriell Institute for Medical Research.
Samples of lung tissue classified as cancerous and samples adjacent to the cancerous tissue but classified as normal were used in this study, under Human Subjects Institutional Review Board approved protocols. After pathological classification upon resection, the tissues were frozen and stored at –80°C. Twenty-two samples (the training set) were obtained from Philipps-University of Marburg, Germany, and 24 samples (the test set) were from The Pennsylvania State University College of Medicine Tumor Bank. Specifically, 23 lung adenocarcinoma and 23 normal tissues were used, of which 11 were matched pairs (Supplemental Table 2). The samples were pulverized under liquid nitrogen. DNA was extracted from the tissue powder by the QIAamp DNA Mini Kit (Qiagen) according to the manufacturer's instructions. The DNA was eluted from the column with dH2O, and stored at –80°C until use.
Plasmid DNA controls
Plasmids pUC19 and pACYC184 and phage φX174 were used as control DNAs. Unmethylated plasmid DNAs were purchased from New England BioLabs and then methylated in vitro using SssI (CpG) methylase (NEB). The completion of in vitro methylation was checked by restriction enzyme digestion using a methylation-sensitive enzyme, HpaII, and its isoschisomer, MspI, which is not sensitive to methylation. When assayed on an agarose gel, no digestion was detected in methylated pUC19, pACYC184, and φX174 after incubation with HpaII for 2 h at 37°C, while the unmethylated DNAs were completely digested. Both methylated and unmethylated DNAs were completely digested by MspI.
Bisulfite conversion of DNA and methylation assay
The EZ DNA methylation kit (Zymo Research) was used for bisulfite conversion of all DNA samples used in this study, according to the manufacturer's recommendations. For each conversion, 1 μg of genomic DNA was used. Bisulfite-converted genomic DNA from one conversion was then used for up to five array experiments. After bisulfite treatment, the remaining assay steps were identical to the GoldenGate genotyping assay (Fan et al. 2003), using Illumina-supplied reagents and conditions (Fan et al. 2006). Single-stranded PCR products were prepared by denaturation, then hybridized to a Sentrix Array Matrix (Fan et al. 2003). The array hybridization was conducted under a temperature gradient program, and arrays were imaged using a BeadArray Reader scanner (Barker et al. 2003). Image processing and intensity data extraction software were as described previously (Galinsky 2003).
BeadArray technology
Microarrays were assembled by loading pools of glass beads (3 μm in diameter) derivatized with oligonucleotides onto the etched ends of fiber-optic bundles (Barker et al. 2003). About 50,000 optical fibers were hexagonally packed to form an ∼1.4-mm diameter bundle. The fiber optic bundles were assembled into a 96-array matrix (Sentrix Array Matrix), which matched the dimensions of standard microtiter plates. This arrangement allowed simultaneous processing of 96 samples using standard robotics (Fan et al. 2003). Because the beads were positioned randomly, a decoding process was carried out to determine the location and identity of each bead in every array location (Gunderson et al. 2004). Decoding is a part of array manufacture and provides quality control for all elements of every array.
Methylation data analysis
Each methylation data point is represented by fluorescent signals from the M (methylated) and U (unmethylated) alleles. Background intensity computed from a set of negative controls was subtracted from each analytical data point. The ratio of fluorescent signals was then computed from the two alleles β = (max(M, 0))/(|U| + |M| + 100). The β-value reflects the methylation level of each CpG site. An absolute value was used in the denominator of the formula, as a compensation for any “negative signals” that may arise from global background subtraction (i.e., oversubtraction; a constant bias of 100 was added to regularize β when both U and M values were small). For cluster analysis, a matrix of correlation coefficients between calculated methylation signals was computed. Agglomerative nesting was applied using the Agnes function in the R package with Ward's method.
Methylation-specific PCR
After bisulfite treatment, the methylation status of particular CpG sites in genomic DNA was analyzed by methylation-specific PCR (Herman et al. 1996). In brief, the bisulfite-converted genomic DNA was amplified by real-time quantitative PCR using two sets of locus-specific MSP primers, which recognize methylated or unmethylated DNA, respectively. The MSP primers (Table 1) were designed using CpGWare, software provided by Chemicon. Real-time PCR analysis was performed on an ABI Prism 7900HT Sequence Detection System (Applied Biosystems).
The PCR reaction was performed using a 384-well optical tray in a final volume of 10 μL. The reaction mixture consists of 5 μL of 2× SYBR Green PCR master mix (Applied Biosystems) and 250 nM each primer and bisulfite-converted DNA template (∼50 ng, measured prior to bisulfite treatment). The real-time PCR cycling conditions were as follows: 50°C for 2 min, 95°C for 12 min, followed by 40 cycles at 95°C for 20 sec, 56°C for 30 sec, and 72°C for 1 min. After PCR, a thermal melt profile was performed to examine the homogeneity of PCR amplicons. Each DNA sample was analyzed in duplicate, and the mean was used for further analysis. The difference of the threshold cycle number (the Ct-values) between the methylated and unmethylated alleles, ΔCt = Ct (unmethylated) – Ct (methylated), was first determined. The percentage of methylated DNA, designated as the methylation level “c,” can be correlated to the ΔCt value through the equation ΔCt = log2[c/(1 – c)] (Zeschnigk et al. 2004; Martens et al. 2005). The resulting methylation level thereby equals 2ΔCt/(1 + 2ΔCt).
Bisulfite sequencing
Methylation status of selected CpG sites was examined by bisulfite sequencing. Primers were designed flanking the CpG sites of interest (Table 2). The primer landing sites did not contain CpG dinucleotides, and therefore the nucleotide sequences remained unchanged after bisulfite treatment. As a result, the methylated and unmethylated alleles would be equally amplified in the same reaction with the designed primer pair. The PCR-amplified fragments were cloned into the pCR4-TOPO vector (Invitrogen) followed by transformation into Escherichia coli TOP10-competent cells (Invitrogen). Transformants containing recombinant plasmids were selected by blue/white colony screening. PCR inserts were directly amplified from the white colonies in the reaction mixture (35 μL) containing 3.5 μL of GeneAmp 10× PCR buffer (Applied Biosystems), 1.5 units of AmpliTaq Gold (Applied Biosystems), 1.5 mM MgCl2, 200 nM dNTP, and 200 nM each of M13 forward (5′-GTAAAACGACGGCCAGT-3′) and reverse primer (5′-CAGGAAACAGCTATGAC-3′). The reaction was subjected to the following cycling conditions: 94°C for 10 min, followed by 35 cycles of 94°C for 30 sec, 50°C for 30 sec, 72°C for 30 sec, and a final cycle of 72°C for 5 min. The PCR products were sequenced by Agencourt Bioscience Corporation.
Acknowledgments
We thank Sean Hu, Joanne Yeakley, and Jessica Wang-Rodriguez for helpful discussions and critical reading of the manuscript, and Jerry Kakol for assistance with the initial probe design. We also thank an anonymous reviewer for reading the manuscript with extraordinary care and making several critical suggestions that improved the quality of the paper. We thank Daniel Beard for his assistance in obtaining and classifying the lung tissue samples from The Pennsylvania State University College of Medicine Tumor Bank. This work was supported in part by grants from the National Institutes of Health (2 R44 CA097851-02 to J.-B.F. and R37-NL-34788 to J.F.), and American Lung Association Research Award RG-066-N to Z.L.
[Supplemental material is available online at www.genome.org.]
Article published online ahead of print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.4410706.
Freely available online through the Genome Research Open Access option.
References
- Adorjan, P., Distler, J., Lipscher, E., Model, F., Muller, J., Pelet, C., Braun, A., Florl, A.R., Gutig, D., Grabs, G., et al. 2002. Tumour class prediction and discovery by microarray-based DNA methylation analysis. Nucleic Acids Res. 30 e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akama, T.O., Okazaki, Y., Ito, M., Okuizumi, H., Konno, H., Muramatsu, M., Plass, C., Held, W.A., and Hayashizaki, Y. 1997. Restriction landmark genomic scanning (RLGS-M)-based genome-wide scanning of mouse liver tumors for alterations in DNA methylation status. Cancer Res. 57 3294–3299. [PubMed] [Google Scholar]
- Alaminos, M., Davalos, V., Cheung, N.K., Gerald, W.L., and Esteller, M. 2004. Clustering of gene hypermethylation associated with clinical risk groups in neuroblastoma. J. Natl. Cancer Inst. 96 1208–1219. [DOI] [PubMed] [Google Scholar]
- Balmain, A., Gray, J., and Ponder, B. 2003. The genetics and genomics of cancer. Nat. Genet. 33 Suppl: 238–244. [DOI] [PubMed] [Google Scholar]
- Barker, D.L., Theriault, G., Che, D., Dickinson, T., Shen, R., and Kain, R. 2003. Self-assembled random arrays: High-performance imaging and genomics applications on a high-density microarray platform. Proc. SPIE 4966 1–11. [Google Scholar]
- Baylin, S.B. and Herman, J.G. 2000. DNA hypermethylation in tumorigenesis: Epigenetics joins genetics. Trends Genet. 16 168–174. [DOI] [PubMed] [Google Scholar]
- Benjamini, Y. and Hochberg, Y. 1995. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Statist. Soc. B57 289–300. [Google Scholar]
- Bestor, T.H. 1998. Gene silencing. Methylation meets acetylation. Nature 393 311–312. [DOI] [PubMed] [Google Scholar]
- Bird, A. 2002. DNA methylation patterns and epigenetic memory. Genes & Dev. 16 6–21. [DOI] [PubMed] [Google Scholar]
- Carrel, L. and Willard, H.F. 2005. X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature 434 400–404. [DOI] [PubMed] [Google Scholar]
- Chen, C.M., Chen, H.L., Hsiau, T.H., Hsiau, A.H., Shi, H., Brock, G.J., Wei, S.H., Caldwell, C.W., Yan, P.S., and Huang, T.H. 2003. Methylation target array for rapid analysis of CpG island hypermethylation in multiple tissue genomes. Am. J. Pathol. 163 37–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costello, J.F., Fruhwald, M.C., Smiraglia, D.J., Rush, L.J., Robertson, G.P., Gao, X., Wright, F.A., Feramisco, J.D., Peltomaki, P., Lang, J.C., et al. 2000. Aberrant CpG-island methylation has non-random and tumour-type-specific patterns. Nat. Genet. 24 132–138. [DOI] [PubMed] [Google Scholar]
- Cottrell, S.E., Distler, J., Goodman, N.S., Mooney, S.H., Kluth, A., Olek, A., Schwope, I., Tetzner, R., Ziebarth, H., and Berlin, K. 2004. A real-time PCR assay for DNA-methylation using methylation-specific blockers. Nucleic Acids Res. 32 e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui, H., Cruz-Correa, M., Giardiello, F.M., Hutcheon, D.F., Kafonek, D.R., Brandenburg, S., Wu, Y., He, X., Powe, N.R., and Feinberg, A.P. 2003. Loss of IGF2 imprinting: A potential marker of colorectal cancer risk. Science 299 1753–1755. [DOI] [PubMed] [Google Scholar]
- Dennis, C. 2003. Epigenetics and disease: Altered states. Nature 421 686–688. [DOI] [PubMed] [Google Scholar]
- Dupont, J.M., Tost, J., Jammes, H., and Gut, I.G. 2004. De novo quantitative bisulfite sequencing using the pyrosequencing technology. Anal. Biochem. 333 119–127. [DOI] [PubMed] [Google Scholar]
- Eads, C.A., Danenberg, K.D., Kawakami, K., Saltz, L.B., Blake, C., Shibata, D., Danenberg, P.V., and Laird, P.W. 2000. MethyLight: A high-throughput assay to measure DNA methylation. Nucleic Acids Res. 28 E32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egger, G., Liang, G., Aparicio, A., and Jones, P.A. 2004. Epigenetics in human disease and prospects for epigenetic therapy. Nature 429 457–463. [DOI] [PubMed] [Google Scholar]
- Esteller, M. 2002. CpG island hypermethylation and tumor suppressor genes: A booming present, a brighter future. Oncogene 21 5427–5440. [DOI] [PubMed] [Google Scholar]
- Fan, J.-B., Oliphant, A., Shen, R., Kermani, B., Garcia, F., Gunderson, K., Hansen, M., Steemers, F., Butler, S.L., Deloukas, P., et al. 2003. Highly parallel SNP genotyping. Cold Spring Harbor Symp. Quant. Biol. 68 69–78. [DOI] [PubMed] [Google Scholar]
- Fan, J.-B., Gunderson, K., Bibikova, M., Yeakley, J.M., Chen, J., Garcia, E.W., Lebruska, L., Laurent, M., Shen, R., and Barker, D. 2006. Genotyping and gene expression profiling using universal bead arrays, Methods Enzymol. (in press). [DOI] [PubMed]
- Feinberg, A.P. and Tycko, B. 2004. The history of cancer epigenetics. Nat. Rev. Cancer 4 143–153. [DOI] [PubMed] [Google Scholar]
- Feinberg, A.P. and Vogelstein, B. 1983. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature 301 89–92. [DOI] [PubMed] [Google Scholar]
- Frommer, M., McDonald, L.E., Millar, D.S., Collis, C.M., Watt, F., Grigg, G.W., Molloy, P.L., and Paul, C.L. 1992. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc. Natl. Acad. Sci. 89 1827–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galinsky, V.L. 2003. Automatic registration of microarray images. II. Hexagonal grid. Bioinformatics 19 1832–1836. [DOI] [PubMed] [Google Scholar]
- Gonzalgo, M.L. and Jones, P.A. 1997. Rapid quantitation of methylation differences at specific sites using methylation-sensitive single nucleotide primer extension (Ms-SNuPE). Nucleic Acids Res. 25 2529–2531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunau, C., Renault, E., and Roizes, G. 2002. DNA methylation database “MethDB”: A user guide. J. Nutr. 132 2435S–2439S. [DOI] [PubMed] [Google Scholar]
- Gunderson, K.L., Kruglyak, S., Graige, M.S., Garcia, F., Kermani, B.G., Zhao, C., Che, D., Dickinson, T., Wickham, E., Bierle, J., et al. 2004. Decoding randomly ordered DNA arrays. Genome Res. 14 870–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanada, M., Delia, D., Aiello, A., Stadtmauer, E., and Reed, J.C. 1993. bcl-2 gene hypomethylation and high-level expression in B-cell chronic lymphocytic leukemia. Blood 82 1820–1828. [PubMed] [Google Scholar]
- Herman, J.G. and Baylin, S.B. 2003. Gene silencing in cancer in association with promoter hypermethylation. N. Engl. J. Med. 349 2042–2054. [DOI] [PubMed] [Google Scholar]
- Herman, J.G., Graff, J.R., Myohanen, S., Nelkin, B.D., and Baylin, S.B. 1996. Methylation-specific PCR: A novel PCR assay for methylation status of CpG islands. Proc. Natl. Acad. Sci. 93 9821–9826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, T.H., Perry, M.R., and Laux, D.E. 1999. Methylation profiling of CpG islands in human breast cancer cells. Hum. Mol. Genet. 8 459–470. [DOI] [PubMed] [Google Scholar]
- Jones, P.A. and Baylin, S.B. 2002. The fundamental role of epigenetic events in cancer. Nat. Rev. Genet. 3 415–428. [DOI] [PubMed] [Google Scholar]
- Jones, P.A. and Laird, P.W. 1999. Cancer epigenetics comes of age. Nat. Genet. 21 163–167. [DOI] [PubMed] [Google Scholar]
- Kawai, J., Hirose, K., Fushiki, S., Hirotsune, S., Ozawa, N., Hara, A., Hayashizaki, Y., and Watanabe, S. 1994. Comparison of DNA methylation patterns among mouse cell lines by restriction landmark genomic scanning. Mol. Cell. Biol. 14 7421–7427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird, P.W. and Jaenisch, R. 1996. The role of DNA methylation in cancer genetic and epigenetics. Annu. Rev. Genet. 30 441–464. [DOI] [PubMed] [Google Scholar]
- Li, E., Beard, C., and Jaenisch, R. 1993. Role for DNA methylation in genomic imprinting. Nature 366 362–365. [DOI] [PubMed] [Google Scholar]
- Li, Q.L., Kim, H.R., Kim, W.J., Choi, J.K., Lee, Y.H., Kim, H.M., Li, L.S., Kim, H., Chang, J., Ito, Y., et al. 2004. Transcriptional silencing of the RUNX3 gene by CpG hypermethylation is associated with lung cancer. Biochem. Biophys. Res. Commun. 314 223–228. [DOI] [PubMed] [Google Scholar]
- Liu, L., Saldanha, S.N., Pate, M.S., Andrews, L.G., and Tollefsbol, T.O. 2004. Epigenetic regulation of human telomerase reverse transcriptase promoter activity during cellular differentiation. Genes Chromosomes Cancer 41 26–37. [DOI] [PubMed] [Google Scholar]
- Lomas, J., Aminoso, C., Gonzalez-Gomez, P., Eva Alonso, M., Arjona, D., Lopez-Marin, I., de Campos, J.M., Isla, A., Vaquero, J., Gutierrez, M., et al. 2004. Methylation status of TP73 in meningiomas. Cancer Genet. Cytogenet. 148 148–151. [DOI] [PubMed] [Google Scholar]
- Martens, J.W., Nimmrich, I., Koenig, T., Look, M.P., Harbeck, N., Model, F., Kluth, A., Bolt-de Vries, J., Sieuwerts, A.M., Portengen, H., et al. 2005. Association of DNA methylation of phosphoserine aminotransferase with response to endocrine therapy in patients with recurrent breast cancer. Cancer Res. 65 4101–4117. [DOI] [PubMed] [Google Scholar]
- Melnikov, A.A., Gartenhaus, R.B., Levenson, A.S., Motchoulskaia, N.A., and Levenson Chernokhvostov, V.V. 2005. MSRE-PCR for analysis of gene-specific DNA methylation. Nucleic Acids Res. 33 e93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogama, Y., Ouchida, M., Yoshino, T., Ito, S., Takimoto, H., Shiote, Y., Ishimaru, F., Harada, M., Tanimoto, M., and Shimizu, K. 2004. Prevalent hyper-methylation of the CDH13 gene promoter in malignant B cell lymphomas. Int. J. Oncol. 25 685–691. [PubMed] [Google Scholar]
- Paz, M.F., Fraga, M.F., Avila, S., Guo, M., Pollan, M., Herman, J.G., and Esteller, M. 2003. A systematic profile of DNA methylation in human cancer cell lines. Cancer Res. 63 1114–1121. [PubMed] [Google Scholar]
- Rakyan, V.K., Hildmann, T., Novik, K.L., Lewin, J., Tost, J., Cox, A.V., Andrews, T.D., Howe, K.L., Otto, T., Olek, A., et al. 2004. DNA methylation profiling of the human major histocompatibility complex: A pilot study for the human epigenome project. PLoS Biol. 2 e405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razin, A. 1998. CpG methylation, chromatin structure and gene silencing—A three-way connection. EMBO J. 17 4905–4908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakatani, T., Kaneda, A., Iacobuzio-Donahue, C.A., Carter, M.G., de Boom Witzel, S., Okano, H., Ko, M.S., Ohlsson, R., Longo, D.L., and Feinberg, A.P. 2005. Loss of imprinting of Igf2 alters intestinal maturation and tumorigenesis in mice. Science 307 1976–1978. [DOI] [PubMed] [Google Scholar]
- Singal, R., van Wert, J., and Bashambu, M. 2001. Cytosine methylation represses glutathione S-transferase P1 (GSTP1) gene expression in human prostate cancer cells. Cancer Res. 61 4820–4826. [PubMed] [Google Scholar]
- Singer-Sam, J., LeBon, J.M., Tanguay, R.L., and Riggs, A.D. 1990. A quantitative HpaII-PCR assay to measure methylation of DNA from a small number of cells. Nucleic Acids Res. 18 687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomaszewski, M., Charchar, F.J., Lacka, B., Pesonen, U., Wang, W.Y., Zukowska-Szczechowska, E., Grzeszczak, W., and Dominiczak, A.F. 2004. Epistatic interaction between β2-adrenergic receptor and neuropeptide Y genes influences LDL-cholesterol in hypertension. Hypertension 44 689–694. [DOI] [PubMed] [Google Scholar]
- Tost, J., Schatz, P., Schuster, M., Berlin, K., and Gut, I.G. 2003. Analysis and accurate quantification of CpG methylation by MALDI mass spectrometry. Nucleic Acids Res. 31 e50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyooka, K.O., Toyooka, S., Virmani, A.K., Sathyanarayana, U.G., Euhus, D.M., Gilcrease, M., Minna, J.D., and Gazdar, A.F. 2001. Loss of expression and aberrant methylation of the CDH13 (H-cadherin) gene in breast and lung carcinomas. Cancer Res. 61 4556–4560. [PubMed] [Google Scholar]
- Tsou, J.A., Hagen, J.A., Carpenter, C.L., and Laird-Offringa, I.A. 2002. DNA methylation analysis: A powerful new tool for lung cancer diagnosis. Oncogene 21 5450–5461. [DOI] [PubMed] [Google Scholar]
- Wang, R.Y., Gehrke, C.W., and Ehrlich, M. 1980. Comparison of bisulfite modification of 5-methyldeoxycytidine and deoxycytidine residues. Nucleic Acids Res. 8 4777–4790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widschwendter, A., Muller, H.M., Hubalek, M.M., Wiedemair, A., Fiegl, H., Goebel, G., Mueller-Holzner, E., Marth, C., and Widschwendter, M. 2004a. Methylation status and expression of human telomerase reverse transcriptase in ovarian and cervical cancer. Gynecol. Oncol. 93 407–416. [DOI] [PubMed] [Google Scholar]
- Widschwendter, M., Siegmund, K.D., Muller, H.M., Fiegl, H., Marth, C., Muller-Holzner, E., Jones, P.A., and Laird, P.W. 2004b. Association of breast cancer DNA methylation profiles with hormone receptor status and response to tamoxifen. Cancer Res. 64 3807–3813. [DOI] [PubMed] [Google Scholar]
- Zeschnigk, M., Bohringer, S., Price, E.A., Onadim, Z., Masshofer, L., and Lohmann, D.R. 2004. A novel real-time PCR assay for quantitative analysis of methylated alleles (QAMA): Analysis of the retinoblastoma locus. Nucleic Acids Res. 32 e125. [DOI] [PMC free article] [PubMed] [Google Scholar]