

Abstract
An important aspect of understanding a biological pathway is to delineate the transcriptional regulatory mechanisms of the genes involved. Two important tasks are often encountered when studying transcription regulation, i.e., (1) the identification of common transcriptional regulators of a set of coexpressed genes; (2) the identification of genes that are regulated by one or several transcription factors. In this study, a systematic and statistical approach was taken to accomplish these tasks by establishing an integrated model considering all of the promoters and characterized transcription factors (TFs) in the genome. A promoter analysis pipeline (PAP) was developed to implement this approach. PAP was tested using coregulated gene clusters collected from the literature. In most test cases, PAP identified the transcription regulators of the input genes accurately. When compared with chromatin immunoprecipitation experiment data, PAP's predictions are consistent with the experimental observations. When PAP was used to analyze one published expression-profiling data set and two novel coregulated gene sets, PAP was able to generate biologically meaningful hypotheses. Therefore, by taking a systematic approach of considering all promoters and characterized TFs in our model, we were able to make more reliable predictions about the regulation of gene expression in mammalian organisms.
Gene expression is largely regulated by transcription factors (TFs) that recognize specific sequences, called cis-regulatory elements or TF-binding sites, in promoters. One of the ultimate goals of biological research is to construct the entire regulatory network of an organism (Covert et al. 2004). Clinically, a comprehensive understanding of transcriptional regulation in a specific pathological process may lead to new therapeutic strategies or discoveries of new drug targets. Computational approaches tackle this problem by modeling TF-binding sites using position weight matrices and searching for these sites in noncoding DNA sequences. Position weight matrices of many characterized transcription factors are available in databases such as TRANSFAC (Matys et al. 2003) and JASPAR (Sandelin et al. 2004). A gene may be regulated by a particular TF if its promoter contains the binding site of this TF, although pure searches for matching patterns can have many false positives.
Computational approaches for identifying the transcriptional regulators of a particular gene are greatly enhanced by large-scale expression-profiling experiments and sequence analysis of multiple genomes. Genome-wide mRNA-profiling experiments allow the identification of genes that have similar expression patterns. As coexpressed genes are likely to be regulated by the same TFs, it is thought that the analysis of noncoding sequences of coexpressed genes will be useful in identifying common cis-regulatory elements recognized by known or novel TFs. These methods have been successfully applied to simple organisms such as yeast and worm (Hughes et al. 2000; GuhaThakurta et al. 2002; Thijs et al. 2002; Ao et al. 2004), but have been largely unsuccessful in mammals because intergenic sequences in higher eukaryotes are very long and contain a large excess of nonregulatory sequences. To help solve this problem and reduce the false positive rate, the comparison of sequences of multiple genomes is crucial. This is based on a hypothesis termed “phylogenetic footprinting” (Tagle et al. 1988), which states that functional regulatory elements are more conserved in evolution than nonfunctional sequences. Therefore, methods have been developed to align noncoding DNA of evolutionarily mid-distant species, such as human and mouse, and to find TF-binding sites that are conserved in multiple species (Wasserman et al. 2000; Blanchette and Tompa 2002; Kellis et al. 2003; Wang and Stormo 2003). Although this process reduces the number of false positive sites, systematic identification of bona fide transcriptional regulators in mammals still remains a challenging problem.
Two important questions often encountered in biological studies regarding transcriptional regulation include the following: (1) Find the common transcriptional regulators of a set of genes that are involved in the same biological pathway, in the same cellular process, in response to the same stimulus, or in the same disease. (2) Find genes that are regulated by one or several TFs that have important roles in a particular biological function or a pathophysiological process. To answer the first question, previous studies have utilized statistical methods to test the enrichment of a TF's binding site in a set of coregulated genes against a “reference” set such as randomly selected genes in the genome (Aerts et al. 2003; Elkon et al. 2003; Qiu et al. 2003; Hu et al. 2004). Other methods took a database approach to store all of the predicted binding sites and use them to estimate the probability of observing a TF-binding site by chance (Karanam and Moreno 2004; Cole et al. 2005; Ho Sui et al. 2005). However, results obtained using this approach are highly variable because they are very dependent on several factors, including the choice of the reference set, the statistical method itself, the score cutoff to identify a site, and the length of the sequence being searched. Therefore, a more robust statistical model is needed.
While experimental methods such as chromatin immunoprecipitation, followed by promoter microarray (Lee et al. 2002) may identify transcriptional regulatory targets, computational methods have also been developed to identify genes regulated by one or several transcription factors. All of these methods first establish a model of regulatory sequences based on the binding sites of the TFs being studied. This model is then used to search genomic sequences to identify potential targets of the same TFs (Kel et al. 2001; Jin et al. 2004). Alternatively, regulatory models may also be trained by logistic regressions (Krivan and Wasserman 2001; Liu et al. 2003; Qiu et al. 2003). Although previous studies were able to generate reasonable results using these methods, establishment of the regulatory model requires a set of known gene targets. Thus, it is difficult to generalize these methods and apply them to large-scale analyses.
From the viewpoint of systems biology, the transcriptional regulatory network of an organism consists of all of the genes, including all of the TFs, and all network interactions between the genes and their transcriptional regulators. With the ever-increasing number of completely sequenced genomes and better annotation of transcription factors in the genome, it is now possible to take a systematic and statistical approach to establish an integrated model considering all of the genes and all characterized TFs in the genome. Such a model would allow one to make robust statistical inferences about transcriptional regulation. Specifically, this model would allow one to answer the two important questions mentioned above and would reliably assign the statistical significance of the findings.
In this study, we present such a model and demonstrate its utility to analyze the potential regulatory sequences of a set of coexpressed genes in mammalian genomes and to make predictions regarding their regulatory mechanisms. We implemented this proposed model in a Web-based workbench termed the Promoter Analysis Pipeline (PAP). PAP is suitable for predicting transcriptional regulators of a set of genes and for identifying the target genes of a set of transcription factors. Various tests, including the analysis of coregulated gene sets collected from the literature, comparison with the chromatin immunoprecipitation experiment data, and the analysis of a published time-course expression-profiling data set indicated the robustness and accuracy of PAP. Therefore, PAP is useful in making reliable predictions about the regulation of gene expression. PAP is available at http://bioinformatics.wustl.edu/PAP.
Results
PAP overview
The design of PAP includes two components (Fig. 1). The data-processing pipeline was assembled using a series of algorithms and data manipulation tools. This set of applications was used to carry out genome-wide promoter analysis, namely, orthologous sequence alignment, TF binding-site identification, and promoter score calculation. The calculated results were stored in a relational database termed the Promoter Analysis Pipeline Database (PAPdb). The graphical user interface of PAP includes a set of interactive Web pages. These pages allow the user to input a set of potentially coregulated genes, to identify a set of transcription factors that are most likely to regulate these genes, to browse binding sites of these TFs, and to predict other genes that might be regulated by the same set of TFs. This bipartite design of PAP uncouples the majority of the computation from the user interface. Therefore, PAP is able to return results of genome-wide promoter analyses in real time. Details of methods and algorithms used in PAP are described in the following sections.
Figure 1.

An overview of the Promoter Analysis Pipeline (PAP). PAP has two components. The data processing pipeline assembles a set of algorithms to generate the results of a genome-wide promoter analysis, whereas the user interface queries and processes the stored data according to the user's input. Promoters were acquired and repetitive elements in the promoters were masked. Promoters of orthologous genes were aligned and transcription factor (TF) binding sites were identified and mapped. Probability scores of each promoter and each transcription factor were calculated, and a distribution of probability scores was generated for each transcription factor. R-scores were then computed using these distributions. All of these results were stored in a database termed PAPdb, which was used to predict the TFs that are most likely to regulate a set of genes and the genes most likely regulated by a set of TFs.
Curation of potential regulatory sequences
To generate the data required for PAP, an interval of genomic sequence that contains putative regulatory signals was defined for each gene. In simple organisms such as yeast or worm, the intergenic sequences are usually very short. Therefore, previous studies were able to obtain meaningful results or make reliable predictions using 500–600 base pairs (bp) as the putative promoter length (Hughes et al. 2000; GuhaThakurta et al. 2002). In mammals, the intergenic sequences for some genes may be very long, and the regulatory sequences may be located distantly from the transcription start sites. In higher eukaryotes, regulatory elements have also been found in the first intron (Helledie et al. 2002; Wong et al. 2002; Mathew et al. 2004). In the current setup, the sequence range that is mostly likely to contain regulatory signals was defined as 10 kilobases (kb) of sequence upstream and 5 kb downstream of the transcription start site. This interval was truncated if another gene was encountered prior to 10 kb and if the translation start site was reached prior to 5 kb for the upstream and downstream sequence, respectively. Therefore, the maximum length of the sequence analyzed for each gene was 15 kb. Based on the annotated transcription start sites, most of the genes do not encounter another upstream gene within a distance of 10 kb, whereas only a portion of genes do not reach their translation start site within a distance of 5 kb downstream (Fig. 2A). These sequences, defined as described, will be referred to as a gene's “promoter” throughout this work.
Figure 2.

The sequence conservation of the promoter sequence defined in PAP. (A) The fraction of genes whose promoters extend to a particular upstream or downstream position from the transcription start site. Most of the genes do not encounter another upstream gene within a distance of 10 kb, whereas only a portion of genes do not reach their translation start sites within a distance of 5 kb downstream. (B) The fraction of promoters that are conserved at a particular upstream or downstream position from the transcription start site. This fraction was calculated using the total number of promoters at each position in A as the denominator. The most conserved and alignable region is around 2 kb upstream and downstream of the annotated transcription start sites.
Using this definition, promoter sequences were retrieved from the Genome Assembly Project of the National Center for Biotechnology Information (NCBI). Since some alternatively spliced transcripts might have the same promoter sequence according to our definition (e.g., alternatively spliced exons did not change the position of the transcription or translation start site), 22,276 and 21,089 distinct promoter candidates were collected for human and mouse, respectively (Table 1). In the current model, two transcripts of the same gene locus are treated separately if they have different promoter candidates. Therefore, they will have different statistical scores. Although TF-binding sites within interspersed repetitive sequences might be functional (Zhou et al. 2002), most of the regulatory elements discovered so far are located outside of the repeats. Therefore, repetitive elements in promoters were masked using the program Repeat-Masker.
Table 1.
Summary of the data stored in the Promoter Analysis Pipeline Database
|
Gene
|
||||||||||
| Species | Gene loci | Transcripts | Promoters | Gene loci with multiple transcripts | Gene loci with multiple promoters | |||||
| Human | 20,984 | 24,749 | 22,276 | 2202 | 951 | |||||
| Mouse | 20,830 | 21,172 | 21,089 | 287 | 227 | |||||
|
Promoter
|
Human—Mouse Homology
|
|||||||||
| Species | Average promoter length | Average percent of repeats | Species | Orthologous gene loci | Average percent promoter aligned | |||||
| Human | 10,252 | 45.0 | Human | 14,140 | 21.0 | |||||
| Mouse | 10,019 | 36.9 | Mouse | 14,224 | 20.2 | |||||
Identification of conserved sequences
The basic assumption of phylogenetic footprinting is that most functional regulatory elements or TF-binding sites are conserved through evolution. As such, although functional elements may indeed exist in nonconserved sequence, they are most likely to be found in regions of sequence conservation in promoters of multiple species. To identify such conserved regions, orthologous genes for each gene locus were identified using NCBI's HomoloGene database (see Methods). Although genes in some of these ortholog groups may not be true orthologs, aligning the promoters of these genes may be informative to identify functional elements. For each ortholog group, we then aligned promoters of human and mouse gene loci using the program TBA (Blanchette et al. 2004). TBA is a local alignment program that aligns multiple sequences in order of their pairwise distance in the phylogenetic tree. This program was chosen because it does not require a “reference sequence” and it identifies all of the multiple local alignments, including those alignments that only consist of a subset of input sequences. This allows PAP to identify TF-binding sites that are only conserved in a subset of organisms.
In the promoter regions being studied, the most conserved segment is around 2 kb upstream and downstream of the annotated transcription start sites (Fig. 2B), with 20% of the sequence alignable, on average. The average G/C content of all the human promoters at each position across the sequence range stored in PAP was calculated. The region between –570 bp and +730 bp was G/C rich, with the G/C content increasing nearer to the transcription start site from both directions (data not shown). These global analyses of alignable sequences and G/C content are comparable to other genome-wide promoter studies (Wasserman et al. 2000; Aerts et al. 2003; Louie et al. 2003).
Identification of conserved TF-binding sites
Using weight matrices from TRANSFAC and JASPAR databases and the software PATSER (Stormo et al. 1982), a list of putative binding sites in all of the promoters was generated. The default cutoff score calculated by PATSER is used as the threshold score to predict a TF-binding site. TF-binding sites were deemed conserved if they were aligned in the sequence alignments of the orthologous promoters. Only 12% of the predicted sites were conserved between human and mouse.
The probabilistic framework of PAP
Assuming that the scores calculated by PATSER using the weight matrix model are related to binding energies (Berg and von Hippel 1987; Stormo 1998; Stormo and Fields 1998), we can determine relative binding probabilities for each promoter based on the combined scores of all of the potential binding sites (see Methods). Actual binding probabilities will depend on a variety of other factors, including the cooperative binding of TFs and the concentration of the TFs within the nucleus. In the case where the true binding profile of a TF is accurately modeled by its weight matrix, we expect that the computed scores are highly correlated with binding probabilities, such that promoters with higher combined scores are more likely to be bound by the TF than promoters with lower scores. These relative probability scores need to be normalized to be compared between different TFs. Therefore, we rank each promoter in the genome by its computed binding probability score from 1 for the highest scoring promoter to N for the lowest scoring of the N promoter regions in the database. The rank is converted to an “R-score,” which is related to the fraction of promoters with a higher rank, by
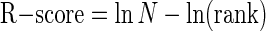 |
(1) |
which ranges from 0 to ln N for the lowest to highest ranking promoters. Promoters ranked in the top half have R-score ≥ ln 2, those in the top 10% have R-score ≥ ln 10, those in the top 1% have R-score ≥ ln 100, and so on. Furthermore, summing R-scores for several promoters is equivalent to multiplying the probabilities of their ranks, which provides a convenient means of determining the significance of the binding scores for sets of promoters or sets of TFs.
PAP's performance on experimentally verified TF-binding sites
To test the ability of PAP's model to analyze real biological data sets found in different experimental contexts, nine previously identified coregulated gene clusters were collected from the literature. These test cases covered the most common scenarios in which a set of correlated genes might be identified, including two tissue-specific gene clusters (muscle-specific and liver-specific genes), a coexpressed gene cluster (heat-shock response genes), a set of genes involved in a biological pathway (parathyroid hormone pathway genes), and known targets of the same transcription factor (NF-κB-regulated immune genes). For each set of genes we computed the average R-score (see Methods), denoted <R-score>, for every TF, and then determine the rank of each TF for each gene set. In 12 of 14 test cases, the known binding factor was ranked within the top six, and the only two exceptions were the ubiquitous TFs Sp1 and Ap1 (Table 2). This prediction accuracy was greatly improved compared with the case where human–mouse conservation was not applied as a filter (data not shown). These results showed PAP was able to predict the transcription regulators of coregulated genes identified in different experimental contexts.
Table 2.
Test results of PAP using coregulated gene sets collected from the literature
| Gene cluster | TF | Matrix | 〈R-score 〉 | Rank | P-value |
|---|---|---|---|---|---|
| Muscle specifica | SRF | M00810 | 4.605 | 1 | ∼0 |
| MEF | M00405 | 2.976 | 2 | ∼0 | |
| Myf | M00001 | 2.976 | 1 | ∼0 | |
| SP1 | MA0079 | 2.386 | 31 | 0.007 | |
| Liver specificb | HNF-1 | M00790 | 3.561 | 1 | ∼0 |
| HNF-3 | M00791 | 2.406 | 3 | 0.002 | |
| C/EBP | M00116 | 2.242 | 4 | 0.0095 | |
| HNF-4 | M00134 | 2.192 | 6 | 0.0036 | |
| Heat shock responsec | HSF2 | M00147 | 2.477 | 1 | 0.0033 |
| HSF1 | M00146 | 2.375 | 2 | 0.0048 | |
| PTH-regulatedd | CREB | M00801 | 3.73 | 2 | ∼0 |
| AML1 | M00751 | 3.27 | 6 | 0.0001 | |
| AP-1 | M00173 | 2.564 | 77 | 0.0069 | |
| NF-κB immune genese | NF-κB | M00774 | 2.882 | 2 | ∼0 |
Eight gene clusters that are regulated by common transcription factors were collected from the literature. For each TF, the 〈R-score 〉 was calculated as described in Results, and the rank of the true TF amongst all of the characterized TFs was determined. PAP predicted the true TF within the top six in 12 test cases. Low P-values indicate that the predictions are statistically significant.
Testing the statistical significance of PAP's findings
To evaluate the reliability of PAP's predictions, the statistical significance of PAP's findings was determined using randomly generated data sets. Genes were randomly selected from all 14,140 human genes that had a mouse ortholog and whose promoters were stored in PAP. The probability of observing a similar score or higher by chance was determined empirically from the distribution generated using these randomly selected gene sets:
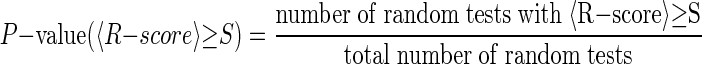 |
(2) |
For each transcription factor for the gene clusters collected from the literature (Table 2), 10,000 random gene clusters of the same size were generated and P-values were calculated. In all of the test cases, the P-value was <0.01, showing that PAP accurately predicted these true factors with high statistical significance.
Comparison of PAP's prediction with chromatin immunoprecipitation experiment data
To test PAP's performance of predicting regulatory targets of transcription factors, we compared PAP's predictions with the results of chromatin immunoprecipitation, followed by promoter microarray experiments. We collected lists of genes that are experimentally proven to be bound by transcription factors, including HSF1 (Trinklein et al. 2004), HNF1, HNF4, HNF6 (Odom et al. 2004), and E2F4 (Ren et al. 2002). For each of these transcription factors, PAP calculates a combined score for each gene according to all of the predicted binding sites in its promoter (see Methods). Therefore, a gene with an overrepresented binding site of a TF will have a high score for that TF. To see whether PAP's prediction is consistent with the experimental observations, the nonparametric Mann-Whitney U-test was applied to test whether the experimentally identified target genes have significantly higher scores, i.e., whether they have overrepresented sites of the true factor. For each of the five TFs, the Z score and the P-value were calculated (Fig. 3). In all of the test cases except HNF6, the P-values are very low (P < 0.0001), suggesting that PAP's prediction is highly consistent with the experimental results. The poor performance of predicting regulatory targets of HNF6 might result from the incompleteness of the HNF6-binding profile in TRANSFAC.
Figure 3.

PAP's prediction of target genes of transcription factors is consistent with chromatin immunoprecipitation experiment data. Target genes of transcription factors HSF1, HNF1a, HNF4a, HNF6, and E2F4 were determined by previous chromatin immunoprecipitation experiments and were collected from the literature. The nonparametric Mann-Whitney U test was used to test whether these validated genes have higher scores in PAP's predictions. In each case, the Z score and the P-value were calculated.
Prediction of genes regulated by a set of transcription factors
Starting with a set of coexpressed genes, PAP is able to predict the transcription factors that regulate these genes. Another interesting pursuit is the identification of other genes that might be regulated by the same set of transcription factors (Fig. 4A). This methodology may be applied to identify tissue-specific genes that are regulated by a well-defined regulatory module. To test the feasibility of this approach, a cluster of 14 liver-specific genes was used (Krivan and Wasserman 2001). These genes contain binding sites of several transcription factors, including HNF-1, HNF-3, HNF-4, and C/EBP, which are known to drive liver expression, and a “liver-specific regulatory module” was derived previously using this gene set. Leave-one-out cross validation was applied, such that one gene was chosen to be the verification gene each time. In every round, 13 genes were analyzed by PAP and high-scoring transcription factors were selected by a P-value cutoff of 0.001. The selected TFs were then used to calculate the joint probability scores (see Methods) of all of the human genes, and the rank of the verification gene is determined (Fig. 4B). In most cases, the verification gene was ranked very high, and the performance was compromised only when genes that are not regulated by all of the TFs in the liver-specific module (Krivan and Wasserman 2001), such as INS, DDC, or SLC2A2, were selected as the verification gene. Therefore, these results are consistent with established biological knowledge.
Figure 4.

The utility of PAP to identify additional genes regulated by the same set of factors. (A) Methodology of identifying additional similarly regulated genes. Starting from a set of coregulated genes, several transcription factors may be identified and hypothesized to be the common transcription regulator of the input genes. Additional genes that may be regulated by the same factors may be searched using these transcription factors. (B) Fourteen previously reported liver-specific genes were used to test this methodology by leave-one-out cross-validation. In each round, 13 genes were analyzed by PAP and putative common transcription factors were determined. High scoring matrices were then used to score all of the human genes, and the rank of the verification gene is reported.
Application of PAP to a published expression profiling experiment data
To demonstrate the usefulness of PAP to analyze multiple gene clusters identified by mRNA expression-profiling experiments, and to identify the underlying transcriptional regulatory events, we applied PAP to a published expression-profiling data set (Tomczak et al. 2004). In this study, gene-expression profiling in a 12-d time course was used to identify genes involved in myogenic differentiation. This time course was supposed to capture a sequence of events including cell proliferation, cell-cycle withdrawal, and maturation of myotubes. These processes are known to be transcriptionally regulated by MyoD family transcription factors and cell-cycle regulators. In this study, four groups of genes consisting of a total of 22 clusters with distinct expression patterns were identified by cluster analysis. When PAP was used to analyze these clusters and find potential transcription regulators, MyoD and E2F-1, a cell cycle regulator, were identified (Supplemental Table 1). Remarkably, E2F-1 was the top-ranking TF for the second and third cluster in the first group, where a majority of genes are cell cycle-regulating genes. And MyoD, as well as other muscle transcription factors such as TEF, SRF, and myogenin, were identified for many clusters in the third group, which contains many muscle-specific genes. This is consistent with the observation that genes in the first group were expressed in the early stage of the time course where proliferation is dominating, and that genes in the third group were expressed in the later stage of the time course where differentiation is dominating. This example demonstrated PAP's utility in analyzing multiple coregulated gene clusters and highlighting underlying transcriptional regulation.
Application of PAP to a novel cell proliferation-related gene cluster
The cell proliferation-related gene cluster was identified using previous mRNA expression-profiling experiments on two well-studied paradigms, prostate regeneration following castration and testosterone replacement (Magee et al. 2003) and peripheral nerve injury (Nagarajan et al. 2002). The first paradigm has been used to simulate and recapitulate the normal physiologic turnover of luminal prostatic epithelia, whereas in the second paradigm, Schwann cells are converted from a quiescent state to a proliferating state after nerve injury. In both paradigms, we found that the expression profile of Ki-67, a well-known marker of cell proliferation, is similar. Therefore, genes with expression profiles correlated to Ki-67 were collected from both expression experiments. By taking the intersection of the two sets of genes in which cell proliferation is involved, genes that were specific to each paradigm were filtered out and 32 common genes were identified. The most enriched GO term in the resulting gene set are cell cycle (GO:0007049, P = 7.34E-23) and cell proliferation (GO:0008283, P = 1.29E-18), which supports the hypothesis that these genes are involved in general cell proliferation and may be regulated by common transcription factors (Bluthgen et al. 2005). Among these 32 genes, six genes did not have identified human orthologs and were removed from the promoter analysis (Supplemental Table 2).
When PAP was used to analyze these genes, known transcriptional regulators of cell cycle regulatory genes including NF-Y (P = ∼0) and E2F (P = 0.0015) were identified (Table 3). Moreover, these factors have been proven to directly regulate several genes in our gene cluster (Supplemental Table 3), and previously characterized NF-Y and E2F-binding sites in these promoters were correctly identified by PAP (Fig. 5; data not shown). Gfi-1 is known to regulate cell proliferation in T cells and haematopoietic stem cells (Duan and Horwitz 2003; Hock et al. 2004). Human–mouse conserved Gfi-1 sites were present in 16 genes in the cell-proliferation gene cluster (Supplemental Table 4). ZF5-binding sites were previously shown to colocalize with NF-Y sites and E2F-1 sites in promoters of cell cycle regulated genes (Sharan et al. 2003). When the clustering of ZF5-binding sites with NF-Y or E2F sites was considered, 15 genes have a sequence window that is shorter than 500 bp and contains binding sites of each of the three TFs. These genes are more likely to be ZF5 targets (Supplemental Table 5).
Table 3.
Top ranking transcription factors predicted in cell proliferation-related genes
| Accession | Factor | Consensus | 〈R-score 〉 | P-value |
|---|---|---|---|---|
| M00185 | NF-Y | TRRCCAATSR | 2.975 | ∼0 |
| M00430 | E2F-1 | TTSGCGG | 1.911 | 0.0015 |
| MA0038 | Gfi-1 | AAATCACWGY | 1.82 | 0.002 |
| M00175 | AP-4 | RYCAGCTGYG | 1.65 | 0.0611 |
| M00007 | Elk-1 | AACMGGAAGT | 1.625 | 0.0935 |
| M00716 | ZF5 | GSGCGCGR | 1.551 | 0.1945 |
| M00773 | c-Myb | GNCAGTT | 1.546 | 0.1622 |
Primary cell cycle regulators. NF-Y, and E2F were accurately predicted as top ranking transcription factors. Other high scoring TFs predicted with higher P-values are likely to regulate a subset of these cell proliferation genes.
Figure 5.

PAP identified experimentally validated TF-binding sites in cell-proliferation related genes. Promoter regions that contain bona fide TF-binding sites are shown. Other TF-binding sites predicted by PAP are also shown. Numbers in the figure represent the sequence positions according to the transcription start site. Experimentally verified sites are designated by a star above the site.
As a complimentary study, we used PAP to analyze another cell-proliferation signature previously identified in a different study (Chang et al. 2004). This signature consists of 165 serum response genes thst are directly related to cell proliferation. Using this data set, PAP identified TFs that were also identified using our own proliferation genes, including NF-Y, E2F-1, ZF5, and Gfi-1 (Supplemental Table 6). This result confirmed the role of these TFs in cell proliferation.
Application of PAP to cholesterol biosynthesis pathway genes
The promoter analysis of genes encoding cholesterol biosynthetic enzymes was motivated by a previous study of Schwann cell expression profiling (Nagarajan et al. 2002). Cholesterol is an essential constituent of myelin. As expected, expression patterns of nine cholesterol synthetic enzymes were strictly correlated to myelination marker genes. Except for a few genes such as HMG-CoA synthase and HMG-CoA reductase, the transcriptional regulatory mechanisms of most cholesterol synthetic enzymes remain unknown. Egr2 is a key myelination transcriptional regulator, and expression profiles of many cholesterol synthetic enzymes have been shown to strictly correlate with those of myelination marker genes. Therefore, PAP was applied to determine whether Egr2 is a direct transcriptional regulator of cholesterol synthetic enzymes and if not, what transcription factors are potential downstream effectors of Egr2 activity.
When 11 cholesterol synthesis genes (Supplemental Table 7) with annotated human and mouse orthologs were analyzed using PAP, several known transcription regulators of cholesterol synthetic enzymes, including NF-Y, CREB, and YY1 (Supplemental Table 3) were identified with low P-values (Table 4). Although only three cholesterol synthetic enzymes have been shown to be directly regulated by CREB, PAP's result indicates that CREB may directly regulate other cholesterol synthesis genes as well.
Table 4.
Top ranking transcription factors predicted in cholesterol biosynthesis genes
| Accession | Factor | Consensus | 〈R-score 〉 | P-value |
|---|---|---|---|---|
| M00287 | NF-Y | RRCCAATSRG | 3.381 | ∼0 |
| M00801 | CREB | CGTCAN | 2.18 | 0.019 |
| M00413 | AREB6 | WCACCTGW | 2.104 | 0.0089 |
| M00059 | YY1 | CCATNTW | 1.808 | 0.0589 |
| M00716 | ZF5 | GSGCGCGR | 1.772 | 0.2331 |
| MA0089 | TCF11-MafG | NATGAC | 1.754 | 0.2127 |
| M00322 | c-Myc | GCCAYGYGS | 1.749 | 0.0665 |
| M00188 | AP-1 | RGTGACTMA | 1.72 | 0.1044 |
| M00217 | USF | CACGTG | 1.698 | 0.1733 |
Experimentally validated transcription regulators of cholesterol biosynthesis genes, such as NF-Y and CREB, were predicted as top ranking transcription factors. Other high scoring TFs predicted with higher P-values are likely to regulate a subset of these cholesterol biosynthesis genes.
Interestingly, PAP did not predict Egr2 as one of the top-ranking transcription factors (Egr matrix ranks the 38th). This implied that Egr2 may not be a direct transcriptional regulator of most of these enzymes. To investigate whether any of the high-scoring transcription factors predicted by PAP may be mediating the regulation of cholesterol synthetic genes by Egr2, the R-scores and P-values of genes encoding these factors were calculated using Egr2 as the transcription factor. Four of these transcription factor genes (NFYA, CREB1, YY1, and AP1) have overrepresented Egr-binding sites with low P-values (P < 0.05). Furthermore, when these four TF genes were collected as a gene cluster and analyzed by PAP, Egr2 matrix had a very low P-value of 0.0043, which indicated that NFYA, CREB1, YY1, and AP1 are likely to be regulated by Egr2. These results suggest a model of transcriptional regulation of cholesterol biosynthesis genes in Schwann cell, in which Egr2 does not directly regulate all of the cholesterol synthesis genes, but instead, coordinates cholesterol synthesis required for myelination through transcription factors, NF-Y, CREB, YY1, and/or AP1 (Fig. 6).
Figure 6.

Predicted transcriptional regulatory model of cholesterol biosynthesis genes in Schwann cells. In this model, Egr2 does not directly regulate all of the cholesterol synthetic enzymes in myelination. Instead, Egr2 coordinates the expression of these genes through other transcription factors, including NF-Y, CREB-1, YY1, and AP-1.
Discussion
In this study, a systematic and statistical approach was taken to establish a genome-wide promoter analysis model. The entire collection of promoters in the genome serves as a natural background for statistical analysis. Our model was tested using previously identified coregulated gene clusters, as well as many other data sets. In all of these tests, PAP performed robustly and was able to make reliable predictions about transcriptional regulatory mechanisms.
When tested using previously characterized coregulated gene clusters, PAP predicted the experimentally verified transcriptional regulators accurately. In addition, PAP also identified other TFs that may interact with them. For example, in the analysis of muscle-specific Myf target genes, E2A was also predicted as a high-scoring factor besides MyoD, and the hetero-oligomerization of E2A with MyoD is required for MyoD's function in muscle (Lassar et al. 1991). Another example was given in the NF-κB immune genes, where the top-ranking transcription factor, c-Rel, is known to interact with NF-κB (Miyamoto et al. 1994). These results confirmed that PAP's predictions are consistent with biological knowledge and previous experimental results.
While the current version of PAP has proven to be a useful tool for discovering and exploring regulatory networks, new data and enhanced analysis methods will provide further improvements. The two types of data that PAP utilizes, comparative genome sequences, and transcription-factor binding models are rapidly accumulating and will lead to improved analyses. In this study, only the mouse and human genomes were utilized, and it was shown that conservation was valuable for identifying true regulatory sites. The genomes of additional mammals and other vertebrates are now completed or in progress, and we expect that they will add useful information and allow for a more thorough investigation of distant regulatory regions. While TRANSFAC is the most comprehensive of transcription factor databases, it is far from complete, containing binding sites for only a fraction of the known and putative transcription factors. For many of the factors that are included, too few sites are known to build reliable models of their specificity and make accurate predictions of their binding sites throughout the genome. But new technologies, such as microarrays and ChIP-chip experiments (Lee et al. 2002), along with improved motif discovery algorithms, are rapidly increasing our knowledge of transcription factors and their binding sites. Those improvements in basic information can be rapidly imported into PAP to enhance its performance.
The analysis methods can also be improved to better take into account important correlations in the data. For example, currently, TF sets can be used to identify potentially coregulated genes. However, if two TFs have very similar binding profiles, they will have similar scores on any given promoters, which may confound the analysis. This issue may be resolved by considering constraints between the TFs, such as limited ranges of spacing or orientation, as well as other correlations that may indicate cooperative interactions. And when considering sets of genes, or sets of TFs, R-scores are tabulated and averaged over the entire set, which may miss important subsets with significant matches. Efficiently determining such significant subsets, and accurately assessing their P-values, is computationally challenging, and we are currently exploring techniques to accomplish the task. This will provide PAP with a much richer ability to discover important regulatory features in the genome sequences.
Methods
Promoter data preparation
Human and mouse chromosomal sequences and gene-annotation files were downloaded from the NCBI's Genome Assembly Project through their FTP site (ftp://ftp.ncbi.nih.gov/genomes/). Genome build 34 was used for human and genome build 32 was used for mouse. For each mRNA, the promoter sequence was obtained from the genomic sequence using the mRNA and coding start positions. Repetitive elements in promoter sequences were masked by the program RepeatMasker (http://www.repeatmasker.org/) using slow and sensitive search mode.
Ortholog groups' identification
The annotation of homolog groups was acquired from the NCBI's HomoloGene database. The information of each homolog group, including gene loci and the protein similarity between any two loci was available in an XML formatted file downloaded from ftp://ftp.ncbi.nih.gov/pub/HomoloGene/build36/. Since a homolog group in HomoloGene's annotation may contain two similar genes of the same organism, ortholog groups, which by definition have at most one gene for each organism, were identified for each gene in a homolog group.
TF-binding sites' identification
The program PATSER (Stormo et al. 1982) was used to search for TF-binding sites in the promoter sequences. For each characterized TF-binding matrix, PATSER scores each subsequence and calculates the P-value of observing a particular score or higher at that sequence position (Staden 1989). This score is assumed to be exponentially related to the probability of binding (see below). PATSER also calculates a P-value cutoff for each weight matrix using the information content (Staden 1989). This P-value cutoff is then used to eliminate low-scoring sites. Therefore, weight matrices that have low information contents will have more predicted sites and are less specific.
A total of 466 vertebrate matrices from TRANSFAC 7.2 and 79 vertebrate matrices from JASPAR were searched in the promoter sequences. The average G/C content of all human and mouse promoters, 46.5%, was used as the background base frequency of G/C. Promoters of orthologous genes were aligned using the program TBA (Blanchette et al. 2004). TF-binding sites in multiple sequences were defined as conserved if their first bases were aligned according to the sequence alignment.
Probability scores and R-scores
For each transcription factor and each promoter in the genome, the probability score of the TF binding to the promoter was computed by summing the exponential of the score of each individual site predicted in the promoter on either strand (GuhaThakurta et al. 2004). This score is set to a minimum value of 1 for a promoter with no sites exceeding the cutoff. A linear regression model was used to estimate the contribution of false positive sites to the probability scores, and this estimated contribution was then subtracted from the probability score.
Based on the probability score, the R-score of a promoter for a TF is computed by equation 1. For a set of n promoters, the average R-score, <R-score>, is calculated by
 |
(3) |
For a set of TFs, the R-score of a promoter is similarly computed by equation 1 considering all of the promoters in the genome, but the joint probability score of these TFs binding to a promoter is used. The joint probability score of a promoter is the product of the probability score of each individual TF binding to this promoter.
Acknowledgments
We thank Kai Tan and Jia-Jian Liu for useful discussions. We thank Deepak Kapur, Srikanth Adiga, Divyabhanu Singh, Aarti Sharma, and Sai Krishna Chitta for help in setting up the database and the Web server. L.C and G.D.S. are supported by the National Institutes of Health (NIH) grants HG00249 and GM63340. J.A.M. and J.M. are supported by the Prostate Cancer Foundation.
[Supplemental material is available online at www.genome.org.]
Article published online ahead of print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.4303406.
References
- Aerts, S., Thijs, G., Coessens, B., Staes, M., Moreau, Y., and De Moor, B. 2003. Toucan: Deciphering the cis-regulatory logic of coregulated genes. Nucleic Acids Res. 31 1753–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ao, W., Gaudet, J., Kent, W.J., Muttumu, S., and Mango, S.E. 2004. Environmentally induced foregut remodeling by PHA-4/FoxA and DAF-12/NHR. Science 305 1743–1746. [DOI] [PubMed] [Google Scholar]
- Baeuerle, P.A. and Baichwal, V.R. 1997. NF-κB as a frequent target for immunosuppressive and anti-inflammatory molecules. Adv. Immunol. 65 111–137. [PubMed] [Google Scholar]
- Berg, O.G. and von Hippel, P.H. 1987. Selection of DNA binding sites by regulatory proteins. Statistical-mechanical theory and application to operators and promoters. J. Mol. Biol. 193 723–750. [DOI] [PubMed] [Google Scholar]
- Blanchette, M. and Tompa, M. 2002. Discovery of regulatory elements by a computational method for phylogenetic footprinting. Genome Res. 12 739–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchette, M., Kent, W.J., Riemer, C., Elnitski, L., Smit, A.F., Roskin, K.M., Baertsch, R., Rosenbloom, K., Clawson, H., Green, E.D., et al. 2004. Aligning multiple genomic sequences with the threaded blockset aligner. Genome Res. 14 708–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bluthgen, N., Kielbasa, S.M., and Herzel, H. 2005. Inferring combinatorial regulation of transcription in silico. Nucleic Acids Res. 33 272–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, H.Y., Sneddon, J.B., Alizadeh, A.A., Sood, R., West, R.B., Montgomery, K., Chi, J.T., van de Rijn, M., Botstein, D., and Brown, P.O. 2004. Gene expression signature of fibroblast serum response predicts human cancer progression: Similarities between tumors and wounds. PLoS Biol. 2 E7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole, S.W., Yan, W., Galic, Z., Arevalo, J., and Zack, J.A. 2005. Expression-based monitoring of transcription factor activity: The TELiS database. Bioinformatics 21 803–810. [DOI] [PubMed] [Google Scholar]
- Covert, M.W., Knight, E.M., Reed, J.L., Herrgard, M.J., and Palsson, B.Ø. 2004. Integrating high-throughput and computational data elucidates bacterial networks. Nature 429 92–96. [DOI] [PubMed] [Google Scholar]
- Duan, Z. and Horwitz, M. 2003. Targets of the transcriptional repressor oncoprotein Gfi-1. Proc. Natl. Acad. Sci. 100 5932–5937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkon, R., Linhart, C., Sharan, R., Shamir, R., and Shiloh, Y. 2003. Genome-wide in silico identification of transcriptional regulators controlling the cell cycle in human cells. Genome Res. 13 773–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GuhaThakurta, D., Palomar, L., Stormo, G.D., Tedesco, P., Johnson, T.E., Walker, D.W., Lithgow, G., Kim, S., and Link, C.D. 2002. Identification of a novel cis-regulatory element involved in the heat shock response in Caenorhabditis elegans using microarray gene expression and computational methods. Genome Res. 12 701–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GuhaThakurta, D., Schriefer, L.A., Waterston, R.H., and Stormo, G.D. 2004. Novel transcription regulatory elements in Caenorhabditis elegans muscle genes. Genome Res. 14 2457–2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helledie, T., Grontved, L., Jensen, S.S., Kiilerich, P., Rietveld, L., Albrektsen, T., Boysen, M.S., Nohr, J., Larsen, L.K., Fleckner, J., et al. 2002. The gene encoding the Acyl-CoA-binding protein is activated by peroxisome proliferator-activated receptor γ through an intronic response element functionally conserved between humans and rodents. J. Biol. Chem. 277 26821–26830. [DOI] [PubMed] [Google Scholar]
- Ho Sui, S.J., Mortimer, J.R., Arenillas, D.J., Brumm, J., Walsh, C.J., Kennedy, B.P., and Wasserman, W.W. 2005. oPOSSUM: Identification of overrepresented transcription factor binding sites in co-expressed genes. Nucleic Acids Res. 33 3154–3164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hock, H., Hamblen, M.J., Rooke, H.M., Schindler, J.W., Saleque, S., Fujiwara, Y., and Orkin, S.H. 2004. Gfi-1 restricts proliferation and preserves functional integrity of haematopoietic stem cells. Nature 431 1002–1007. [DOI] [PubMed] [Google Scholar]
- Hu, Y., Wang, T., Stormo, G.D., and Gordon, J.I. 2004. RNA interference of achaete-scute homolog 1 in mouse prostate neuroendocrine cells reveals its gene targets and DNA binding sites. Proc. Natl. Acad. Sci. 101 5559–5564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes, J.D., Estep, P.W., Tavazoie, S., and Church, G.M. 2000. Computational identification of cis-regulatory elements associated with groups of functionally related genes in Saccharomyces cerevisiae. J. Mol. Biol. 296 1205–1214. [DOI] [PubMed] [Google Scholar]
- Jin, V.X., Leu, Y.W., Liyanarachchi, S., Sun, H., Fan, M., Nephew, K.P., Huang, T.H., and Davuluri, R.V. 2004. Identifying estrogen receptor α target genes using integrated computational genomics and chromatin immunoprecipitation microarray. Nucleic Acids Res. 32 6627–6635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karanam, S. and Moreno, C.S. 2004. CONFAC: Automated application of comparative genomic promoter analysis to DNA microarray data sets. Nucleic Acids Res. 32 W475–W484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kel, A.E., Kel-Margoulis, O.V., Farnham, P.J., Bartley, S.M., Wingender, E., and Zhang, M.Q. 2001. Computer-assisted identification of cell cycle-related genes: New targets for E2F transcription factors. J. Mol. Biol. 309 99–120. [DOI] [PubMed] [Google Scholar]
- Kellis, M., Patterson, N., Endrizzi, M., Birren, B., and Lander, E.S. 2003. Sequencing and comparison of yeast species to identify genes and regulatory elements. Nature 423 241–254. [DOI] [PubMed] [Google Scholar]
- Krivan, W. and Wasserman, W.W. 2001. A predictive model for regulatory sequences directing liver-specific transcription. Genome Res. 11 1559–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassar, A.B., Davis, R.L., Wright, W.E., Kadesch, T., Murre, C., Voronova, A. Baltimore, D., and Weintraub, H. 1991. Functional activity of myogenic HLH proteins requires hetero-oligomerization with E12/E47-like proteins in vivo. Cell 66 305–315. [DOI] [PubMed] [Google Scholar]
- Lee, T.I., Rinaldi, N.J., Robert, F., Odom, D.T., Bar-Joseph, Z., Gerber, G.K., Hannett, N.M., Harbison, C.T., Thompson, C.M., Simon, I., et al. 2002. Transcriptional regulatory networks in Saccharomyces cerevisiae. Science 298 799–804. [DOI] [PubMed] [Google Scholar]
- Liu, R., McEachin, R.C., and States, D.J. 2003. Computationally identifying novel NF-κB-regulated immune genes in the human genome. Genome Res. 13 654–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louie, E., Ott, J., and Majewski, J. 2003. Nucleotide frequency variation across human genes. Genome Res. 13 2594–2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee, J.A., Abdulkadir, S.A., and Milbrandt, J. 2003. Haploinsufficiency at the Nkx3.1 locus. A paradigm for stochastic, dosage-sensitive gene regulation during tumor initiation. Cancer Cell 3 273–283. [DOI] [PubMed] [Google Scholar]
- Mathew, S., Mascareno, E., and Siddiqui, M.A. 2004. A ternary complex of transcription factors, Nished and NFATc4, and co-activator p300 bound to an intronic sequence, intronic regulatory element, is pivotal for the up-regulation of myosin light chain-2v gene in cardiac hypertrophy. J. Biol. Chem. 279 41018–41027. [DOI] [PubMed] [Google Scholar]
- Matys, V., Fricke, E., Geffers, R., Gossling, E., Haubrock, M., Hehl, R., Hornischer, K., Karas, D., Kel, A.E., Kel-Margoulis, O.V., et al. 2003. TRANSFAC: Transcriptional regulation, from patterns to profiles. Nucleic Acids Res. 31 374–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto, S., Schmitt, M.J., and Verma, I.M. 1994. Qualitative changes in the subunit composition of κB-binding complexes during murine B-cell differentiation. Proc. Natl. Acad. Sci. 91 5056–5060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagarajan, R., Le, N., Mahoney, H., Araki, T., and Milbrandt, J. 2002. Deciphering peripheral nerve myelination by using Schwann cell expression profiling. Proc. Natl. Acad. Sci. 99 8998–9003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odom, D.T., Zizlsperger, N., Gordon, D.B., Bell, G.W., Rinaldi, N.J., Murray, H.L., Volkert, T.L., Schreiber, J., Rolfe, P.A., Gifford, D.K., et al. 2004. Control of pancreas and liver gene expression by HNF transcription factors. Science 303 1378–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu, P., Qin, L., Sorrentino, R.P., Greene, J.R., Wang, L., and Partridge, N.C. 2003. Comparative promoter analysis and its application in analysis of PTH-regulated gene expression. J. Mol. Biol. 326 1327–1336. [DOI] [PubMed] [Google Scholar]
- Ren, B., Cam, H., Takahashi, Y., Volkert, T., Terragni, J., Young, R.A., and Dynlacht, B.D. 2002. E2F integrates cell cycle progression with DNA repair, replication, and G(2)/M checkpoints. Genes & Dev. 16 245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandelin, A., Alkema, W., Engstrom, P., Wasserman, W.W., and Lenhard, B. 2004. JASPAR: An open-access database for eukaryotic transcription factor binding profiles. Nucleic Acids Res. 32 D91–D94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharan, R., Ovcharenko, I., Ben-Hur, A., and Karp, R.M. 2003. CREME: A framework for identifying cis-regulatory modules in human–mouse conserved segments. Bioinformatics 19 i283–i291. [DOI] [PubMed] [Google Scholar]
- Staden, R. 1989. Methods for discovering novel motifs in nucleic acid sequences. Comput. Appl. Biosci. 5 293–298. [DOI] [PubMed] [Google Scholar]
- Stormo, G.D. 1998. Information content and free energy in DNA—protein interactions. J. Theor. Biol. 195 135–137. [DOI] [PubMed] [Google Scholar]
- Stormo, G.D. and Fields, D.S. 1998. Specificity, free energy and information content in protein-DNA interactions. Trends Biochem. Sci. 23 109–113. [DOI] [PubMed] [Google Scholar]
- Stormo, G.D., Schneider, T.D., Gold, L., and Ehrenfeucht, A. 1982. Use of the `Perceptron' algorithm to distinguish translational initiation sites in E. coli. Nucleic Acids Res. 10 2997–3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagle, D.A., Koop, B.F., Goodman, M., Slightom, J.L., Hess, D.L., and Jones, R.T. 1988. Embryonic ε and γ globin genes of a prosimian primate (Galago crassicaudatus). Nucleotide and amino acid sequences, developmental regulation and phylogenetic footprints. J. Mol. Biol. 203 439–455. [DOI] [PubMed] [Google Scholar]
- Thijs, G., Marchal, K., Lescot, M., Rombauts, S., De Moor, B., Rouze, P., and Moreau, Y. 2002. A Gibbs sampling method to detect overrepresented motifs in the upstream regions of coexpressed genes. J. Comput. Biol. 9 447–464. [DOI] [PubMed] [Google Scholar]
- Tomczak, K.K., Marinescu, V.D., Ramoni, M.F., Sanoudou, D., Montanaro, F., Han, M., Kunkel, L.M., Kohane, I.S., and Beggs, A.H. 2004. Expression profiling and identification of novel genes involved in myogenic differentiation. FASEB J. 18 403–405. [DOI] [PubMed] [Google Scholar]
- Trinklein, N.D., Murray, J.I., Hartman, S.J., Botstein, D., and Myers, R.M. 2004. The role of heat shock transcription factor 1 in the genome-wide regulation of the mammalian heat shock response. Mol. Biol. Cell 15 1254–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visala Rao, D., Boyle, G.M., Parsons, P.G., Watson, K., and Jones, G.L. 2003. Influence of ageing, heat shock treatment and in vivo total antioxidant status on gene-expression profile and protein synthesis in human peripheral lymphocytes. Mech. Ageing Dev. 124 55–69. [DOI] [PubMed] [Google Scholar]
- Wang, T. and Stormo, G.D. 2003. Combining phylogenetic data with co-regulated genes to identify regulatory motifs. Bioinformatics 19 2369–2380. [DOI] [PubMed] [Google Scholar]
- Wasserman, W.W., Palumbo, M., Thompson, W., Fickett, J.W., and Lawrence, C.E. 2000. Human–mouse genome comparisons to locate regulatory sites. Nat. Genet. 26 225–228. [DOI] [PubMed] [Google Scholar]
- Wong, L.H., Sim, H., Chatterjee-Kishore, M., Hatzinisiriou, I., Devenish, R.J., Stark, G., and Ralph, S.J. 2002. Isolation and characterization of a human STAT1 gene regulatory element. Inducibility by interferon (IFN) types I and II and role of IFN regulatory factor-1. J. Biol. Chem. 277 19408–19417. [DOI] [PubMed] [Google Scholar]
- Zhou, Y.H., Zheng, J.B., Gu, X., Saunders, G.F., and Yung, W.K. 2002. Novel PAX6 binding sites in the human genome and the role of repetitive elements in the evolution of gene regulation. Genome Res. 12 1716–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]