

Abstract
Establishing transcriptional regulatory networks by analysis of gene expression data and promoter sequences shows great promise. We developed a novel promoter classification method using a Relevance Vector Machine (RVM) and Bayesian statistical principles to identify discriminatory features in the promoter sequences of genes that can correctly classify transcriptional responses. The method was applied to microarray data obtained from Arabidopsis seedlings treated with glucose or abscisic acid (ABA). Of those genes showing >2.5-fold changes in expression level, ∼70% were correctly predicted as being up- or down-regulated (under 10-fold cross-validation), based on the presence or absence of a small set of discriminative promoter motifs. Many of these motifs have known regulatory functions in sugar- and ABA-mediated gene expression. One promoter motif that was not known to be involved in glucose-responsive gene expression was identified as the strongest classifier of glucose-up-regulated gene expression. We show it confers glucose-responsive gene expression in conjunction with another promoter motif, thus validating the classification method. We were able to establish a detailed model of glucose and ABA transcriptional regulatory networks and their interactions, which will help us to understand the mechanisms linking metabolism with growth in Arabidopsis. This study shows that machine learning strategies coupled to Bayesian statistical methods hold significant promise for identifying functionally significant promoter sequences.
The identification and understanding of transcriptional regulatory networks and their interactions are a major challenge in biology, as transcriptional mechanisms contribute to the regulation of nearly all cellular processes. The time, location, and levels of gene transcripts are known to be specified by combinations of protein interactions with noncoding sequences surrounding genes, and significant progress is being made in defining protein interactions with regulatory motifs on a whole-genome scale. For example, experiments that localize transcription factor binding sites using chromatin immunoprecipitation to the yeast genome sequence have established pathways of gene regulation involving >100 of the 141 known yeast transcription factors (Lee et al. 2002). However, the multitude of transcription factors and the larger genomes of multicellular organisms make direct experimental approaches such as this daunting with current technology.
Computational methods that define relationships between gene expression levels and putative regulatory sequences in upstream regions of genes are increasingly used to establish genome-scale transcriptional regulatory networks (Smith et al. 2005). By correlating the frequency of occurrence of known promoter motifs in coregulated genes, it has been possible to relate promoter motifs with known functions to transcriptional pathways in yeast (Bussemaker et al. 2001). The clustering of genes that are coregulated during the yeast cell cycle according to their functions and alignment of promoter sequences of clustered genes identified promoter motifs with known regulatory functions and novel motifs with predicted functions (Tavazoie et al. 1999). This strategy was extended into a systematic approach analyzing a wide range of gene expression patterns in yeast and Caenorhabditis elegans with frequentist statistical methods for identifying promoter DNA elements and combinations of elements that optimally predict gene expression patterns. From this, the expression of a significant proportion of genes was accurately predicted according to promoter sequences (Beer and Tavazoie 2004). Regulatory modules have been defined in yeast based on coregulated gene expression patterns, and promoters in a significant number of these modules contained a promoter motif that was a known binding site for a coregulated transcription factor (Segal et al. 2003). Subsequent testing of these predictions defined the functions of several regulatory proteins and established the power of these approaches.
We are interested in elucidating the transcriptional regulatory mechanisms integrating carbohydrate availability and hormone action in the plant Arabidopsis thaliana (Arabidopsis). Widespread changes in cell function in response to carbohydrate status, such as reduced protein synthesis and the mobilization of alternative substrates for energy supply in response to carbohydrate starvation, have been predicted based on microarray analysis (Price et al. 2004; Thimm et al. 2004). These experiments also show that the expression of a wide range of genes is regulated by carbohydrates in Arabidopsis and ∼25% of the genes represented on the 8K Affymetrix chip also responded to both light and sugar treatments (Thum et al. 2004). Many of these genes encode enzymes of primary, secondary, and lipid metabolism, and a codependent interaction between light- and sugar-responsive gene expression was identified. These transcriptional responses were also interconnected with ABA- and ethylene-mediated gene expression and growth responses. Interactions between glucose- and ABA-response pathways have been established by the isolation of the ABA biosynthetic mutant aba2 and the ABA response mutant abi4 in screens for reduced responses of seedlings to high levels of glucose or sucrose (Arenas-Huertero et al. 2000; Huijser et al. 2000; Laby et al. 2000; Rook et al. 2001; Cheng et al. 2002).
Learning techniques are used in an increasingly wide variety of biological applications such as microarray analysis (Lavine et al. 2004), protein homology detection (Jaakkola et al. 1999), function prediction based on annotated sequence (Vinayagam et al. 2004), and functional predictions based on transcriptional coexpression (Zhang et al. 2004). Supervised learning methods construct a decision rule from a training set of known positive and negative examples and algorithms such as Support Vector Machines (SVM) (Boser et al. 1992) learn to discriminate between training examples from each category. SVMs have demonstrated both excellent performance in dealing with sparse and noisy data typically generated by biological experimentation and an ability to deal with high-dimensional data in a computationally efficient way (Scholkopf et al. 2004). Recently SVM applications have also been used to discriminate between promoter and nonpromoter regions of human DNA (Gangal and Sharma 2005), and to resolve promoter sequences and the positions of transcription initiation sites in plant DNA (Shahmuradov et al. 2005).
Here we describe the use of a Relevance Vector Machine (RVM) (Tipping 2000) to classify gene expression according to the composition of promoter sequences. The RVM was used with a Bayesian Automatic Relevance Determination (ARD) (MacKay 1994; Neal 1994) prior to select a small subset of promoter motifs for its discriminatory rule to optimally distinguish between regulated genes. Unlike correlation-based approaches, which consider the significance of individual features, the RVM considers the significance of a feature in the context of the features already selected, which may be useful in considering the effects of combinations of features on gene expression. This approach has been successfully used to find a small number of genes whose expression is diagnostic for certain cancer types (Li et al. 2002). The discriminatory features selected by the RVM classifier included promoter motifs that had known functions in both glucose- and ABA-activated gene expression and revealed that light-responsive promoter motifs were powerful features for classifying promoters controlling glucose down-regulated gene expression. One motif with no established function in glucose-responsive transcriptional responses that was the strongest classifier of glucose up-regulated gene expression was shown experimentally to confer glucose-activated gene expression in stable transgenic lines. The successful application of machine learning algorithms for promoter sequence analysis using Bayesian statistical principles established models of transcriptional pathways regulating glucose- and ABA-mediated gene expression and demonstrated that these methods hold promise for establishing transcriptional regulatory networks in Arabidopsis and other organisms.
Results
Transcript profiling reveals that glucose regulates genes with diverse functions
Affymetrix ATH1 Gene Chips were used to identify glucose- and ABA-regulated genes. Seedlings were grown in liquid culture for 7 d on low sugar concentrations (0.5% glucose) and constant light to abrogate diurnal responses. Treatments were designed to reveal transitions in gene expression from a sugar-restricted condition to a sugar-replete state. After 7 d of growth, the medium was replaced with glucose-free medium for 24 h, and then glucose or mannitol was added to 3% (w/v). Mannitol, a nontoxic nonmetabolized sugar, was used as an osmotic control in ABA experiments to define the interactions between ABA and 3% glucose. Seedlings that had developed the first pair of true leaves (stage 1.02) (Boyes et al. 2001) were sampled at 0, 2, 4, or 6 h after addition of glucose, mannitol, glucose + ABA, or mannitol + ABA. The time course was selected to detect proximal events, to minimize transcriptional changes due to accelerated growth and development in response to sugars, and to establish the dynamics of glucose- and ABA-mediated gene expression.
Scatterplots (Supplemental Fig. 1) show that >99% of the significantly expressed genes (Present) exhibit <2.5-fold variation in signal intensity between two independent chip hybridizations. Up- or down-regulated genes were defined independently for each time point as those with a statistically significant change in treatment/control pairs (Wilcoxon signed-rank test, P < 0.005) (Hubbell et al. 2002; Liu et al. 2002). Genes with expression ratios of glucose/mannitol and glucose/0 h of >2.5-fold or <2.5-fold at one time-course point or more were defined as glucose-inducible genes and glucose-repressible genes, respectively. The genes with expression ratios of ABA + mannitol/mannitol, ABA + mannitol/0 h, ABA + glucose/glucose and ABA + glucose/0 h of >2.5-fold or <2.5-fold at one time-course point or more were defined as ABA-inducible and ABA-repressible genes, respectively. The 0-h time point was common to all treatments, and time points for the glucose and mannitol treatments were replicated three times and hybridized independently to ATH1 arrays to measure changes over time. This scheme provided three experimental replicates of glucose treatment at each time point and nine experimental replicates for defining glucose-regulated genes. The ABA treatments provided a minimum of two experimental replicates for defining ABA-regulated genes. Accordingly, 983 genes were expressed by >2.5-fold in response to 3% glucose, 769 genes were expressed at 2.5-fold lower levels in response to 3% glucose (Supplemental Table 1), and 692 and 173 genes were identified as ABA inducible and ABA repressible with >2.5-fold change, respectively (Supplemental Table 2). To confirm the microarray expression profile analyses, semiquantitative RT-PCR analysis was performed on the RNA samples used for array analysis. Fifty genes exhibiting expression changes in response to glucose were selected and tested two times. Results from 15 of the selected genes are shown in Supplemental Figure 2. Gene expression patterns revealed by RT-PCR exhibited similar dynamics to those seen in array analysis, establishing the reliability of the microarray data.
We categorized glucose- and ABA-regulated genes according to their putative functions based on Arabidopsis Gene Ontology (GO) annotations in GeneSpring lists, the classification of the Munich Information Centre for Protein Sequencing (MIPS) database, pathway analysis defined by AraCyc (Mueller et al. 2003) and KEGG (Kanehisa 2002), and the literature. Table 1 shows the significance of finding glucose- and ABA-responsive genes in different functional categories calculated using the hypergeometric P-value (Tavazoie et al. 1999). ABA down-regulated genes were not considered because of the small number of genes in this category. The functional clusters enriched for glucose up-regulated genes include metabolic pathways and cellular processes associated with enhanced growth, such as amino acid and nucleotide synthesis, sulfur assimilation, and secondary metabolism. Genes involved in protein synthesis were significantly enriched in the glucose up-regulated set, as were protein targeting genes and abiotic stress proteins including chaperonins and heat-shock proteins, demonstrating that glucose-mediated transcriptional regulation mediates a coordinated increase in protein synthesis and processing. Glucose down-regulated genes were enriched in functional categories involved in metabolic responses such as amino acid degradation, gluconeogenesis, and glutaredoxins. The regulation of genes involved in trehalose metabolism was highly significant, consistent with the proposed role of trehalose 6 P levels in regulating carbon assimilation (Schluepmann et al. 2003). Many genes regulating light responses, such as transcription factors, light receptors and signaling proteins were also down-regulated in response to glucose, although in general this diverse functional group was not significantly down-regulated as a whole. The most significant categories of genes regulated by ABA in our conditions included abscisic acid metabolism, secondary metabolism, and carbohydrate degradation pathways. Our quantitative analysis is consistent with recent qualitative microarray analysis showing that glucose treatment regulates a broad range of gene functions (Price et al. 2004; Thimm et al. 2004).
Table 1.
Functional categorization of glucose- and ABA-responsive genes
| Category | Genes in category n | Regulated k | PvP — log10 |
|---|---|---|---|
| Glucose up-regulated | |||
| Sulfur assimilation | 62 | 16 | 8.2256* |
| Translation | 152 | 33 | 7.3065* |
| Abiotic stress | 454 | 47 | 6.3331* |
| Nucleotide synthesis | 186 | 25 | 6.1691* |
| Protein targeting | 524 | 48 | 5.2264* |
| Secondary metabolism methyl transferases | 19 | 5 | 3.6209* |
| Lipid transfer proteins | 15 | 4 | 3.2126 |
| Secondary metabolism flavonoid synthesis | 93 | 9 | 2.6203 |
| Nucleotide sugar transferases | 70 | 9 | 2.6128 |
| Ribosomal proteins | 258 | 30 | 2.5031 |
| Glucose down-regulated | |||
| Amino acid degradation | 142 | 21 | 6.9827* |
| Trehalose metabolism | 22 | 6 | 4.9561* |
| Glutaredoxins | 40 | 8 | 4.7538* |
| Gluconeogenesis | 6 | 3 | 4.5454* |
| Storage proteins | 28 | 5 | 3.2793 |
| Ethylene synthesis | 109 | 8 | 2.3114 |
| Pentose phosphate pathway | 19 | 3 | 2.30036 |
| Lipid degradation | 113 | 9 | 2.0985 |
| Ubiquitin conjugation | 37 | 4 | 1.9157 |
| Light-mediated signaling | 128 | 11 | 2.4742 |
| ABA up-regulated | |||
| Abscisic acid metabolism | 66 | 13 | 7.5126* |
| Secondary metabolism | 307 | 28 | 6.7421* |
| General carbohydrate degradation | 299 | 22 | 3.6926* |
| Storage and lipid transfer proteins | 88 | 9 | 3.1427 |
| Lipid metabolism | 485 | 28 | 2.9821 |
| Salicylic acid metabolism | 17 | 3 | 2.6772 |
| P450 and degradation enzymes | 739 | 37 | 2.4363 |
| Trehalose metabolism | 22 | 3 | 2.2472 |
P-values of <0.05 (corrected for multiple comparisons using the Bonferroni correction) in each regulated group are indicated with an asterisk:
- Glucose up-regulated, 143 functional categories, —log10 Pv = 3.445.
- Glucose down-regulated, 116 functional categories, —log10 Pv = 3.365.
- ABA up-regulated, 113 functional categories, —log10 Pv = 3.354.
Dynamics of glucose-responsive gene expression
Analysis of gene expression profiles during the first 6 h after addition of glucose or mannitol showed rapid and transient changes in the expression of many genes. A total of 469 genes were maximally expressed at the 2-h time point, and 719 and 628 genes were maximally expressed at the 4-h and 6-h time points, respectively (Fig. 1A). Nearly 42% of the induced genes exhibited overlapping expression at the 2-h and 4-h time points; 54% of the genes were maximally expressed at both the 4-h and 6-h time points, whereas only 32% of the glucose-induced genes had overlapping expression at the 2-h and 6-h time points (Fig. 1A). Furthermore, some genes were specifically induced or repressed by glucose at 2 h, 4 h, or 6 h, respectively (Fig. 1A,B). At these three time points, ∼25% of the induced genes had overlapping expression (Fig. 1A), whereas 45% of the repressed genes exhibited overlapping expression (Fig. 1B), suggesting that there are more dynamic changes in the expression of glucose-inducible genes compared to glucose-repressible genes.
Figure 1.

Expression dynamics of glucose-responsive genes. (A) Venn diagrams showing the number of genes up-regulated by glucose at 2-h, 4-h, and 6-h time course points determined by microarray analyses. Here, 248 genes were up-regulated by glucose at all time points; 353 genes were induced by glucose at both 2 h and 4 h; 475 genes were up-regulated by glucose at both 4 h and 6 h; and 263 genes were glucose inducible at both 2 h and 6 h. (B) Venn diagrams showing the number of genes down-regulated by glucose at 2-h, 4-h, and 6-h time points determined by microarray analyses. Here, 347 genes were down-regulated by glucose at all time-course points; 416 genes were down-regulated by glucose at both 2 h and 4 h; 449 genes were repressed by glucose at both 4 h and 6 h; and 368 genes were glucose repressible at both 2 h and 6 h. (C) The expression profiles of glucose-inducible genes according to Quality Threshold clustering. Cluster number and time course are indicated. (D) The expression profiles of glucose-repressible genes according to Quality Threshold clustering. Cluster number and time course are indicated. (E) The expression profiles of heat-shock genes, starch metabolism, phenylpropanoid biosynthesis, N-, P-, and S-assimilation genes, and amino acid biosynthesis genes are shown.
Quality Threshold (QT) clustering was used to divide glucose up-regulated genes into 10 clusters of 20 or more genes that shared similar expression dynamics (Fig. 1C; Supplemental Table 3). Cluster 11 (data not shown) contained the remaining genes. Cluster 1 comprises 281 transcripts that have similar expression levels at 2-h, 4-h, and 6-h time-course points and represents the main expression pattern of glucose-inducible genes. Clusters 2 and 7 exhibited similar profiles at the 4-h and 6-h time points, while clusters 3 and 8 are induced progressively. Genes in clusters 4 and 9 were induced maximally at the 2-h time point, and then the expression level decreased. Glucose down-regulated genes were classified into nine groups of 20 or more genes that shared a similar expression profile (Fig. 1D) and one (cluster 10) (data not shown) including the remaining genes (Supplemental Table 3).
Clusters 4 and 9 (Fig. 1C), which were maximally expressed 2 h after glucose treatment, contained a large proportion of heat-shock, peptidyl prolyltransferase, and transcription factor and protein kinase genes. Sixteen genes encoding heat-shock and DNAJ-like proteins (Fig. 1E; Supplemental Table 3) were maximally induced by glucose at 2 h; 10 heat-shock genes were maximally up-regulated by glucose at 4 h; and only one heat-shock gene was induced by glucose at 6 h, suggesting that expression of heat-shock proteins is rapidly modulated in response to glucose. This suggests that transiently increased levels of chaperonin activity are required to process newly synthesized proteins. Among the most rapidly glucose-repressed genes, found in clusters 1, 2, and 9 (Fig. 1D; Supplemental Table 3), were transcription factors regulating light responses. These included genes encoding the trihelix DNA-binding proteins GT1 and GT2, the GATA transcription factor 4, GBF1, and AT1g19000, encoding a 1-repeat MYB protein related to MYBST1, which interact with DNA sequences in many light-responsive gene promoters (Lam 1995; Puente et al. 1996; Chattopadhyay et al. 1998; Smalle et al. 1998). Genes encoding the blue-light photoreceptors CRY1 and CRY2 (Lin et al. 1995; Ahmad et al. 1998; Kleiner et al. 1999), the phytochrome A-specific light signaling component EID1 (Buche et al. 2000; Dieterle et al. 2001), phytochrome kinase substrate 1 (PKS1) (Fankhauser et al. 1999), and 6–4 photolyase (UVR3), which mediates light-dependent repair of UV-induced damage products (Jiang et al. 1997), were all rapidly and persistently repressed by glucose (Supplemental Table 3). Expression of TOC1, APRR5, and APRR7 genes belonging to the APRR1/TOC1 complex controlling circadian rhythms (Yamamoto et al. 2003) was also rapidly glucose-repressed (cluster 1) (Fig. 1D; Supplemental Table 3), suggesting that carbohydrate levels may influence the central oscillator controlling circadian rhythms.
Glucose treatment led to a rapid and progressive increase in the expression of genes involved in protein synthesis, including 32 ribosomal proteins, a putative ribosome recycling factor, and translation initiation and elongation factors, which were predominantly found in clusters 1, 2, and 3 (Fig. 1C; Supplemental Table 3). Genes in cluster 3, which are progressively expressed, tend to encode cell cycle and DNA-replication-related proteins such as a putative CDC21 protein (AT2G16440), the DNA-replication licensing factor MCM3 homolog, replication factor A (AT5G08020), and MCM5 and MCM7 (PROLIFERA) (Springer et al. 2000; Holding and Springer 2002; Moore et al. 2003), which ensure fidelity of DNA replication. Clusters 2 and 7 (Fig. 1C), which were maximally expressed at 4 and 6 h, were enriched for genes encoding metabolic enzymes, ribosomal proteins, and transporters (Supplemental Table 3). These included genes encoding a putative glucose-6-phosphate translocator (AT1G61800) and genes involved in starch biosynthesis enzymes, (AT1G32900), glucose-1-phosphate adenylyltransferase (AT2G21590), and ADPglucose pyrophosphorylase (AT2G21590)—were maximally induced at 4 h (Fig. 1E). Genes involved in secondary metabolism—such as 4-coumarate:CoA ligase 3 (AT1G65060), flavonol synthase (FLS), putative cinnamoyl CoA reductase (AT2G23910), flavonol 4-sulfotransferase (AT1G18590), flavanoid 3-hydroxylase (FH3), chalcone synthase (CHS), and cinnamyl-alcohol dehydrogenase (AT5G19440)—were also maximally induced at 4–6 h (Fig. 1E). Genes involved in sulfur and ammonium assimilation were up-regulated maximally by glucose between 2 h and 4 h, and genes involved in amino acid biosynthesis were also maximally induced by glucose at 4 h and 6 h (Fig. 1E).
Glucose- and ABA-responsive gene expression
Previous genetic analyses have shown that sugar- and ABA-mediated growth responses are closely interconnected in plants (Zhou et al. 1998; Rook et al. 2001). Array analysis revealed >14% of the ABA-inducible genes were also induced by glucose, indicating a substantial overlap between glucose- and ABA-regulated gene expression (Supplemental Table 4). Several transcriptional regulators of ABA responses were regulated by glucose. The homeodomain leucine zipper (HD-Zip) proteins in Arabidopsis are involved in ABA regulation (Himmelbach et al. 2002), and expression of ATHB6 is up-regulated by glucose (Supplemental Table 1), suggesting that sugars may participate in ABA signaling by regulating the expression of ABA-response regulators.
Ninety-five genes were up-regulated by both glucose and ABA. These genes are involved in stress, defense, and senescence responses, secondary metabolism and cell wall biosynthesis, amino acid metabolism, carbohydrate metabolism, fatty acid and lipid metabolism and transport, transcript regulation, and signal transduction (Fig. 2A). More than 12% of the genes induced by both glucose and ABA are involved in stress responses, indicating overlapping regulation by glucose and ABA. Expression of key regulators of abiotic stress responses such as CBF3, COR15A, and RD29A were induced by both glucose and ABA (Supplemental Table 4). Constitutive expression of CBF3 in transgenic Arabidopsis plants induces expression of target COR (cold-regulated) genes to enhance freezing tolerance in nonacclimated plants (Gilmour et al. 2000). Expression of COR15A and RD29A is regulated by CBF3, suggesting that both glucose and ABA may contribute to the regulation of cold stress tolerance. In addition, four genes encoding nonspecific lipid-transfer proteins were induced by both glucose and ABA (Fig. 2A), consistent with reports that nonspecific lipid-transfer proteins are induced by ABA, wounding, and cold stress (Yubero-Serrano et al. 2003). Four genes encoding heat-shock proteins were induced by both glucose and ABA (Supplemental Table 4). Finally, several genes involved in fatty acid and lipid metabolism are glucose and ABA inducible, revealing the roles of sugar and ABA in lipid metabolism (Fig. 2A; Supplemental Table 4).
Figure 2.

Glucose- and ABA-coregulated genes. (A) Functional classification of genes induced by both glucose and ABA. (B) Expression patterns of the set of 12 genes showing synergistic transcriptional responses to glucose and ABA. Expression patterns of the two genes encoding large subunits of AGPase (APL3 and AT2g21590) in response to glucose and ABA were indicated with a green line and black line, respectively. (C) Transcriptional responses of APL3:GUS promoter fusions to sugar and ABA in stable Arabidopsis transformants. Samples were taken from 7-d-old seedlings grown on the following media: 10 mM glucose + 90 mM mannitol (Mannitol), 100 mM glucose (Glucose), 10 mM glucose + 90 mM mannitol + 0.1 μM ABA (Mannitol + ABA), and 100 mM glucose + ABA (Glucose + ABA). The fold induction compared with the osmotic control (Mannitol) is given. Error bars represent the standard error from 10 independent transformants. (D) Response of the APL3 promoter to sugar and ABA in Arabidopsis protoplasts. Protoplasts were made from Col or isi3 7-d-old seedlings. Protoplasts were cultured in the following media: 400 mM mannitol, 400 mM glucose, 400 mM mannitol + 10 μM ABA and 400 mM glucose + 10 μM ABA. GUS activity was measured and normalized to Luciferase (Luc) activity expressed from the CaMV 35S promoter. The fold induction compared with the osmotic control of each genotype are given. Error bars represent the standard error of the mean from three samples.
Thirty-seven genes were identified as glucose- and ABA-corepressed genes, including protein kinases, transcription factors, transporters, and enzymes. Two genes (AT4G36670 and AT1G08930) encoding putative sugar transporters are down-regulated by both glucose and ABA. Two genes encoding 1-aminocyclopropane-1-carboxylate oxidase (AT1G77330) involved in ethylene biosynthesis and putative ethylene-responsive element binding factor (AT5G61590) are repressed by both glucose and ABA, revealing that aspects of ethylene biosynthesis and responses are modulated by both glucose and ABA. Genes regulated by glucose and ABA in opposed ways were also analyzed. Genes involved in ammonium assimilation, such as a putative ammonium transporter (AT1G64780), were glucose inducible and ABA repressible, and lysine-ketoglutarate reductase (AT4G33150) exhibited a decrease of expression level in glucose treatment and an increase of expression level in ABA treatment (Supplemental Table 4), suggesting that nitrogen metabolism may provide different compounds for stress and growth responses. Finally, the phosphate transporter gene ATPT2 (AT2G38940) is up-regulated by glucose and down-regulated by ABA (Supplemental Table 4), suggesting that the sugar-replete state may promote uptake and utilization of the phosphate required for carbon metabolism and ABA may repress this process.
Several examples of the synergistic effects of sugar and ABA on gene expression have been reported. For example, expression of the rice myo-inositol-1-phosphate synthase gene RINO1 was induced by both sucrose and ABA treatments, and the combination of both sucrose and ABA resulted in much higher expression levels (Yoshida et al. 2002). We defined synergistic interactions as those genes expressed at greater than twofold higher levels in response to glucose + ABA treatment compared to the sum of expression levels observed for glucose and ABA + mannitol treatments at two or more points in the time course. A set of 12 genes was in this class (Fig. 2B; Supplemental Table 5). These encoded proteins that are involved in lipid metabolism and transport, stress and senescence responses, and starch biosynthesis, such as CER1 involved in wax biosynthesis, lipid transfer protein gene 4 (LTP4), and two senescence-related genes (SAG29 and AT1G22160). Two of the genes encoding large subunits of ADP-glucose pyrophosphorylase, the first step in starch biosynthesis (Fig. 2B,C; Supplemental Table 5), were synergistically regulated, although the APL3 subunit was only synergistically regulated at one time point. The synergistic regulation defined by array analysis was confirmed by analysis of APL3::GUS promoter reporter gene expression in transgenic Arabidopsis seedlings (Fig. 2C,D). The APL3::GUS gene was 3.7-fold and 2.9-fold induced by sugar and ABA, respectively, and together they exerted a 15.6-fold induction (Fig. 2C). ABI4 has been implicated in regulation of the APL3 promoter (Rook et al. 2001). To test whether ABI4 contributed to the synergistic regulation of the APL3 promoter, the APL3 promoter::GUS reporter gene was analyzed by transient expression in Arabidopsis protoplasts. Similar synergistic regulation was seen in protoplasts and stable transformants (Fig. 2C,D). Expression of the APL3::GUS construct in isi3 protoplasts, which are defective in ABI4 activity (Rook et al. 2001), showed that ABA and glucose synergism was lost (Fig. 2D). This showed that ABI4 is involved in the synergistic responses of the APL3 promoter to glucose and ABA.
Regulatory gene expression
Glucose treatment led to rapid transient increases in the expression of diverse transcription factors including members of the MYB, bZIP, AP2, homeodomain, NAM-like, and heat-shock transcription factor protein families. Expression of MYB75/PAP1/AN2 (Borevitz et al. 2000; Stracke et al. 2001) and the flower pigmentation gene ATAN11 were rapidly induced by glucose (Supplemental Table 1). AN2 and AN11 have been well characterized and encode a MYB-domain transcriptional activator and a WD-repeat protein, respectively (de Vetten et al. 1997; Quattrocchio et al. 1999). In petunia flowers, AN2 and AN11 control flower pigmentation by stimulating the transcription of anthocyanin biosynthetic genes. Overexpression of PAP1 also leads to elevated expression of anthocyanin biosynthetic genes (Borevitz et al. 2000), suggesting that glucose may promote expression of phenylpropanoid biosynthetic genes by elevating expression of these MYB transcription factors and ATAN11. The MYB transcript factor gene ATR1, which activates tryptophan gene expression in Arabidopsis (Bender and Fink 1998), was also up-regulated by glucose, suggesting that glucose may increase expression of tryptophan biosynthetic genes by activating expression of ATR1. Expression of several MADS-box and WRKY-like family members was down-regulated by glucose. The expression of a WRKY class transcription factor (AT5g07100) encoding a protein related to sweet potato SPF1 (Kim et al. 1997) was reduced in response to glucose. SPF1 binds SP8a and SP8b promoter sequences of sporamin and beta-amylase genes expressed in storage roots of sweet potato, and reduced expression of SPF1 mRNA levels induced sporamin and beta-amylase expression (Ishiguro and Nakamura 1994). Our analysis suggests that AT5g07100 may modulate sugar-regulated gene expression in Arabidopsis by a similar mechanism.
Identification and analysis of promoter motifs
Promoter sequences comprising ∼1000 bp upstream of the predicted ATG initiation codon of all Arabidopsis genes predicted in the TIGR version 5 annotation (Haas et al. 2005) were assembled. Responsive genes were defined as those showing >2.5-fold changes at the 2-h, 4-h, and 6-h time points in response to glucose or ABA compared to control treatments. The set of 983 glucose up-regulated promoters was compared with 769 glucose down-regulated promoters and a set of 692 ABA up-regulated promoters was compared to a set of 647 promoters showing no responses to ABA. Matrices of (983 + 769) promoters regulated by glucose, and 381 experimentally defined plant transcriptional regulatory sequences established in the PLACE database (Higo et al. 1999) were assembled for feature extraction. Matrices of (692 + 647) ABA-regulated and nonregulated promoters and PLACE elements were also assembled, and features were extracted from both strands of the promoters. Similar matrices were also made with a set of all 1024 (45) possible 5-mers in an unbiased search for promoter motifs. 5-mers were chosen because 4-mers occurred too frequently to provide discriminatory power, while 6-mers may be too selective. These features served as input into a feature space by the RVM to construct classifiers of gene expression based on either PLACE elements or k-mer sequences.
These classifiers were tested in a 10-fold cross-validation procedure that partitioned the data into 10 disjoint subsets of approximately equal size. A model was then trained using nine segments as the training data and tested on the unused segment. This procedure was repeated 10 times, each time using a different combination of nine segments to form the training data, such that all 10 segments were used as test data for a different model. The average test set performance was reasonably stable after 10 trials; therefore, a 10-fold cross-validation provided a good estimate of model performance.
Classification accuracy was displayed in the Receiver-Operator Characteristic (ROC) curves shown in Figure 3, A and B. These show the sensitivity of classification compared to the specificity, or the true-positive rate versus the false-positive rate. The area under the ROC curve shows an optimum classification rate of ∼74% for both the k-mer and PLACE element features, indicating a robust performance. Only features that were selected in every fold of the cross-validation procedure were selected. These features were then ranked according to the magnitude of their weights over the 10-folds of the cross-validation procedure, and the top 75% are displayed in Tables 2 and 4. The top-ranked classifiers were seven PLACE elements for glucose up-regulated promoters, seven PLACE elements for glucose down-regulated promoters (Table 2), and nine PLACE elements for ABA-up regulated genes (Table 4). We identified 13 k-mers as top-ranking classifiers of glucose up-regulated genes and 13 k-mers as classifiers of glucose down-regulated genes (Table 3). Some of the k-mer motifs match PLACE elements identified as effective classifiers. Three of the highest-ranking k-mer motifs in glucose up-regulated genes had perfect matches to top-ranked PLACE elements: ACCCT matched the TELO-box PLACE element, TAGGT matched the MYB26S PLACE element, and CGGCA matched the E2FBNTRNR PLACE element. A single mismatch of the GGGAG 5-mer motif was found in the AMMORESIIUDCR NIA1 element. Among the k-mers associated with glucose down-regulated gene expression, GGATA perfectly matched the MYBST1 motif and the known sugar-repressible motif (TATCCA) and the OSRAMY3D motif (TATCCAY) (Hwang et al. 1998; Lu et al. 1998, 2002). The GATAA sequence is the IBOXCORE and the GATA factor binding site, TATCT is found in the EVENINGGAT element, and CGTGG is the core of G-box-type motifs such as LRENPCABE. Some k-mer features that were strong classifiers of glucose-regulated genes do not match functionally defined PLACE elements, suggesting that they may have novel functions in sugar regulation.
Figure 3.

ROC (Receiver Operating Characteristic) curves of RVM performance in classifying glucose- and ABA-regulated genes. (A) The ROC curves of glucose-regulated genes show the proportion of true positives selected by the RVM versus false positives. The performance is shown by the area under the ROC curve. PLACE element features (blue line) and k-mer features (pink line). A random selection is shown by the green line. (B) The ROC curves of ABA-up-regulated genes show the proportion of true positives selected by the RVM versus false positives. The performance is shown by the area under the ROC curve. PLACE element features (blue line) and k-mer features (pink line). A random selection is shown by the green line.
Table 2.
RVM selection of PLACE elements in glucose-regulated genes
| Element ID | Number of times picked by RVM | Average weight | Recognition sequence | Potential target genes (from Supplemental Table S6) |
|---|---|---|---|---|
| Up-regulated | ||||
| TELOBOXATEEF1AA1 | 10 | 2.9895 | aaaccctaa | Ribosomal proteins, protein synthesis |
| AMMORESIIUDCRNIA1 | 10 | 1.3346 | ggwagggt | Nucleotide metabolism |
| QARBNEXTA | 10 | 1.1033 | aacgtgt | No significant categories |
| BS1EGCCR | 10 | 0.9637 | agcggg | Carbohydrate metabolism enzymes |
| E2FBNTRNR | 10 | 0.9067 | gcggcaaa | Protein synthesis |
| MYB26PS | 10 | 0.8397 | gttaggtt | Carbohydrate metabolism, S uptake |
| DRECRTCOREAT | 10 | 0.8076 | rccgac | Abiotic stress |
| Down-regulated | ||||
| IBOXCORENT | 10 | -3.3202 | gataagr | Trehalose synthesis |
| IBOXCORE | 10 | -2.1431 | gataa | Abiotic stress |
| IBOX | 10 | -0.7107 | gataag | |
| MYBST1 | 10 | -3.2140 | ggata | Trehalose synthesis |
| Abiotic stress | ||||
| Amino acid degradation | ||||
| LRENPCABE | 10 | -1.1698 | acgtggca | Carbohydrate, lipid and amino acid metabolism |
| GARE2OSREP1 | 10 | -0.9167 | taacgta | Secondary metabolism |
| EVENINGAT | 10 | -0.8441 | aaaatatct | Jasmonate synthesis, abiotic stress |
Table 4.
RVM selection of place elements in ABA-regulated genes
| Element ID | Number of times picked by RVM | Average weight | Recognition sequence | Potential target genes (from Supplemental Table S6) |
|---|---|---|---|---|
| ABRELATERD1 | 10 | 4.4946 | acgtg | Abiotic stress, phenylpropanoid and ABA metabolism, carbohydrate breakdown |
| ABREATRD22 | 10 | 3.0406 | ryacgtggyr | Abiotic stress |
| ACGTABREMOTIFA2OSEM | 10 | 2.6213 | acgtgkc | Phenylpropanoid metabolism |
| DRE2COREZMRAB17 | 10 | 2.0471 | accgac | Abiotic stress, raffinose metabolism |
| ACGTATERD1 | 10 | 1.7750 | acgt | Phenylpropanoid metabolism |
| MYB1AT | 10 | 1.6179 | waacca | |
| DPBFCOREDCDC3 | 10 | 1.3682 | acacnng | |
| ABREMOTIFAOSOSEM | 10 | 1.3261 | tacgtgtc | Abiotic stress |
| SGBFGMGMAUX28 | 10 | 1.2677 | tccacgtgtc |
Table 3.
RVM selection of 5-mer motifs in glucose-regulated genes
| Recognition sequence | Number of times picked by RVM | Average weight |
|---|---|---|
| Up-regulated | ||
| accct | 10 | 2.4528 |
| gggag | 10 | 1.9367 |
| agtga | 10 | 1.6673 |
| gagaa | 10 | 1.3825 |
| attaa | 10 | 1.3428 |
| gaata | 10 | 1.1516 |
| gaatc | 10 | 1.0956 |
| taggt | 10 | 1.0248 |
| aatag | 10 | 0.927 |
| aatgt | 10 | 0.8825 |
| cggca | 10 | 0.8763 |
| accgt | 10 | 0.7836 |
| actct | 10 | 0.7546 |
| Down-regulated | ||
| ggata | 10 | -4.4913 |
| gataa | 10 | -3.0703 |
| tatct | 10 | -2.0260 |
| catcc | 10 | -1.2789 |
| aagat | 10 | -1.0549 |
| caatg | 10 | -1.014 |
| aatcc | 10 | -0.959 |
| gatta | 10 | -0.947 |
| gactc | 10 | -0.913 |
| catcg | 10 | -0.839 |
| cacac | 10 | -0.82 |
| cgtgg | 10 | -0.773 |
| gaccc | 10 | -0.721 |
The hypergeometric probability distribution function was used to assess the enrichment of these motifs in the promoters of genes in various functional categories. Supplemental Table 6 shows that many of the motifs were significantly enriched in the promoters of genes found in functional classes involved in glucose and ABA responses. These relationships were also consistent with the known functions of these promoter motifs in regulating different cellular functions.
The TELO motif, the top-ranked classifier of glucose-induced genes, was originally identified in promoters of genes encoding components of the translational machinery (Tremousaygue et al. 1999). Consistent with this, our analysis shows it is significantly enriched in the promoters of protein and nucleotide synthesis genes (Supplemental Table 6). The BS1EGCCR and MYB26PS motifs have been implicated in the regulation of phenylpropanoid biosynthesis genes (Uimari and Strommer 1997; Lacombe et al. 2000), and these were enriched in glucose-regulated carbohydrate metabolism and sulfate-uptake genes. The DRECRTCOREAT motif mediates stress responses (Dubouzet et al. 2003), and this motif was enriched in the promoters of abiotic stress-related genes. The PLACE elements that were top-ranking classifiers of glucose down-regulated gene expression, such as the I-box, the EVENINGAT, MYBST1, and the G-box-related motif, all have established functions in regulating light- and sugar-related gene expression. For example, the G-box-related element LRENPCABE was previously shown to repress gene expression by sugars (Hwang et al. 1998; Lu et al. 1998). The MYBST1 motif, TATCC, is very similar to the known sugar-repression motifs (TATCCA) and OSRAMY3D (TATCCAY) (Hwang et al. 1998; Lu et al. 1998, 2002), suggesting that TATCC is a core of motifs conferring sugar repression. Supplemental Table 6 shows these motifs are significantly enriched in the promoters of genes involved in catabolic responses, abiotic stress, and trehalose and jasmonate metabolism.
PLACE elements that were strong classifiers of ABA up-regulated promoters (Table 4) were also significantly enriched in classes of genes known to be regulated by ABA, such as stress responses, ABA biosynthesis, carbohydrate breakdown, and phenylpropanoid synthesis (Supplemental Table 6). Many of these PLACE elements have been shown to confer ABA- and stress-responsive gene expression, such as ABARELATERD1, AB AREATRD22, MYB1AT, and DRE2COREZMRAB17 (Busk and Pages 1998). Recently these ABRE motifs and the DRE element were also identified as overrepresented sequences in ABA-up-regulated genes (Leonhardt et al. 2004). Ten k-mer motifs were top-ranking classifiers of ABA up-regulated promoters (Table 5). ACGTG, the most significant motif, forms the core of ABRE LATERD1, ABREATRD22, and ACGTATBREMOTFA2OSEM; CGTGT is the core of ABREMOTIFAOSOSEM; CGTGG is the core of ABREATRD22; and CGTAC is the core of ABRE3HVA22.
Table 5.
RVM selection of 5-mer motifs in ABA-regulated genes
| Recognition sequence | Number of times picked by RVM | Average weight |
|---|---|---|
| acgtg | 10 | 4.5791 |
| cgtgt | 10 | 3.8186 |
| cgtgg | 10 | 1.9653 |
| cgtac | 10 | 1.8358 |
| ccgac | 10 | 1.7705 |
| cacac | 10 | 1.7430 |
| gaaca | 10 | 1.7009 |
| atatc | 10 | 1.4722 |
| gatac | 10 | 1.1721 |
| ccatc | 10 | 1.0937 |
The TELO motif was the best classifier of glucose up-regulated expression. It is required, together with other elements such as the TEF, trap40, and IIa/IIb elements, for high-level expression in actively dividing cells in root meristems (Tremousaygue et al. 1999, 2003; Manevski et al. 2000). Figure 4A shows that promoters containing the TELO motif are maximally expressed 4 h after glucose addition. Inspection of the 222 glucose up-regulated promoters containing the TELO motif revealed that all contained the motif CATAAT, which forms the core of the 16-bp TEF motif. Moreover, the performance of classifiers of glucose up-regulated expression that included both the TELO motif and all 5-mers was improved by 5-mer motifs AGGGG, GGGCA, CATAA, and ATAAT, which comprise 11 of the 16-nt TEF motifs (data not shown). We tested the function of the TELO motif in conferring glucose-responsive gene expression using stable transgenic lines. Oligonucleotide tetramers of TELO4 and TEF4 motifs and the combined motif TEF1TELO3, which included one TEF sequence and three TELO sequences, were inserted 5′ to a minimal –60 CaMV promoter (Fig. 4B,C). These promoters were fused upstream of the GUS reporter gene, inserted in a binary vector, and used to obtain transgenic Arabidopsis plants. For each construct, ∼100 independent transgenic plants were tested. We observed that the TEF1TELO3 promoter specifically conferred glucose-responsive expression of GUS activity in root meristems of transgenic plants (Fig. 4D,E,F). These results were consistent with previous studies showing that the TELO motif was required for GUS expression in root meristems and this activation required the TEF element (Tremousaygue et al. 1999). Quantitative analysis of GUS expression in TEF1TELO3::GUS transgenic plants showed 6.9-fold higher GUS activity in response to glucose compared to mannitol treatment (Fig. 4G). These results indicated that the TELO motif, the best classifier of glucose-up-regulated promoters, participates in the control of glucose-responsive gene expression in a cooperative manner with the TEF motif.
Figure 4.

The TELO motif confers glucose-mediated transcriptional regulation. (A) Expression patterns of the glucose-up-regulated genes with promoters containing the TELO motif. (B) Sequences of the TELO4, TEF4, and TEF1TELO3 motifs. (C) Constructs containing the TELO4, TEF4, and TEF1TELO3 motifs in a –60 CaMV::GUS reporter vector are shown. An oligonucleotide tetramer of TELO (TELO4) and TEF (TEF4) motifs and a combined motif (TEF1TELO3) containing one TEF sequence and three TELO sequences were inserted upstream of the –60 CaMV::GUS reporter construct. (D,E,F) Histochemical analysis of GUS activity of TEF1TELO3::GUS transgenic plants in response to 3% glucose (D), 3% mannitol (E), and water (F) for 12 h. GUS activities in lateral root primordia are shown. (G) GUS activity of TEF4::GUS, TELO4::GUS, and TEF1TELO3::GUS transgenic plants. Protoplasts made from 7-d-old TEF4::GUS, TELO4::GUS, and TEF1TELO3::GUS transgenic plants were cultured in 400 mM glucose or 400 mM mannitol for 48 h before GUS activity was measured. Error bars represent the standard error of the mean from five samples. These transgenic lines were assayed at least three times.
Discussion
Dynamic transcriptional responses to glucose
Glucose and ABA treatments lead to rapid dynamic changes in gene expression in Arabidopsis seedlings. Quantitative analysis of gene function and clustering of gene expression dynamics identified patterns of coregulation of classes of genes that revealed large-scale changes in cell function in response to glucose and ABA. Among the most rapid transient transcriptional responses to glucose involved the up-regulation of genes encoding heat-shock and DNAJ-like chaperonin proteins. Genes encoding components of protein synthesis were also rapidly induced, but their expression persisted, suggesting a temporal control the cellular machinery for protein synthesis that involves rapid initial synthesis of chaperonins for stabilizing newly synthesized proteins and longer-term expression of components involved in protein synthesis. Transcription factors and protein kinase genes were among the most rapidly modulated by glucose. Rapidly up-regulated genes in these classes included those encoding transcription factors regulating biosynthetic pathways such as MYB75/PAP1, ATR1, MYB28, and JAF13. This is consistent with these transcription factors mediating subsequent more persistent expression of many genes encoding enzymes, transporters, and other proteins involved in the reprogramming of biosynthetic and catabolic pathways. This is supported by the identification of cognate transcription-factor-binding sites as strong classifiers of glucose up-regulated expression of these classes of genes (see below). Among the rapidly induced and persistently expressed genes were those functioning in the cell cycle, cell division, DNA replication and recombination, and in growth. These rapid responses, which occur before any significant growth or development, suggest that glucose-mediated transcriptional responses directly orchestrate cell division and growth. One of the most striking responses to glucose was the rapid and persistent down-regulation of transcription factors regulating light responses and regulators of the circadian clock. Longer-term cellular responses to high sugar include suppression of photogene expression (Jang et al. 1997), and our analysis suggests a mechanism involving the rapid down-regulation of transcription factors conferring light-responsive expression of photogenes. This proposed mechanism is supported by the identification of cognate promoter elements that are strong classifiers of glucose down-regulated expression (see below). How these major changes in gene expression are regulated remains to be elucidated. A large number of genes were coregulated by glucose and ABA, including key regulators of ABA action such as ATHB6 (Himmelbach et al. 2002) and a diverse set of genes involved in signal transduction and transcription, stress responses, and metabolism. Furthermore, several genes involved in ethylene-mediated gene expression were also coregulated by ABA and glucose, identifying regulatory points for three-way interactions between these growth regulators (Yanagisawa et al. 2003; Price et al. 2004).
Regulatory mechanisms
Our application of machine learning methods for promoter classification linked known transcription factors and their cognate binding sites into a model of glucose- and ABA-mediated gene expression and revealed new glucose-mediated transcriptional control mechanisms. The TELO promoter motif was identified by the RVM as the strongest classifier of glucose up-regulated gene expression. It was found in >200 of the 983 glucose-up-regulated genes and was significantly enriched in the promoters of genes encoding components of protein and nucleotide synthesis pathways (Supplemental Table 6). The TELO motif and the associated TEF motif conferred increased gene expression in response to glucose, thus establishing a new role for this element and validating the feature extraction and classification strategy. The TELO motif, together with the adjacent TEF sequence in the eEF1A promoter, was previously shown to direct high-level expression in rapidly cycling primordia (Tremousaygue et al. 1999). Recently, the TELO motif was shown to be overrepresented in the promoters of genes up-regulated during axillary bud outgrowth in Arabidopsis, such as ribosomal protein and cell cycle genes (Tatematsu et al. 2005). Together these data demonstrate a key role for the TELO motif in regulating the expression of genes in response to growth stimuli such as glucose and decapitation.
The MYB26S and BS1EGCCR motifs, which are enriched in genes involved in carbohydrate metabolism and sulfur uptake (Supplemental Table 6), were previously shown to regulate genes in the phenylpropanoid pathway (Uimari and Strommer 1997; Lacombe et al. 2000). The E2FBNTRNR motif is enriched in protein synthesis genes, consistent with experimental evidence (Chaboute et al. 2000), and the AMMORESIIUDCRNIA1 motif involved in the transcriptional control of the nitrate reductase gene (Loppes and Radoux 2001) was enriched in nucleotide metabolism genes. This model proposes that glucose may either regulate the transcription of genes encoding transcription factors that then activate these classes of genes, or glucose promotes the activity of transcription factors by post-transcriptional mechanisms. The cycloheximide dependence of glucose up-regulated expression (Price et al. 2004) is consistent with the former mechanism.
Several examples of possible regulatory chains (Yu et al. 2003) involved in glucose-down-regulated gene expression were evident from the promoter features described in Tables 2 and 3. Four motifs involved in conferring light regulation (Puente et al. 1996), the I-box core motif, the GATA motif, light regulatory motifs related to the evening element, and a G-box-related element were all top-weighted classifiers of glucose-down-regulated gene expression (Table 2). GBF1 binds the G-box and confers light regulation, and the down-regulation of GBF1 in response to glucose suggests that glucose-down-regulates light-responsive gene expression by reducing expression of GBF1 (Supplemental Table 1). Glucose down-regulates the expression of GATA4 expression (Supplemental Table 1), which encodes a GATA transcription factor. This binds the sequences GGATA and GATAA (Puente et al. 1996), the top-weighted k-mer motifs for classifying glucose-down-regulated expression and establishes another putative regulatory chain. Glucose also down-regulates the expression of AT1G19000 (Supplemental Table 1) encoding a 1 repeat MYB protein related to MYBST1. This transcription factor binds to the GGATA motif and I-box-related sequences (Lu et al. 2002), which are also top-weighted classifiers of glucose down-regulated expression. This suggests another transcriptional regulatory chain contributing to glucose-mediated transcriptional repression of light-regulated genes. Expression of genes encoding the trihelix proteins GT1 and GT2, which confer light activation (Lam 1995), was also reduced by glucose treatment (Supplemental Table 1), but their cognate GT promoter elements were not selected as classifiers by the RVM. This analysis provides potential mechanisms linking glucose- and light-mediated gene expression suggested by earlier analyses (Thum et al. 2004).
The promoter of the Amy3D α-amylase gene contains a TATCCA- and a G-box-related motif required for repression by sugars or induction by sugar starvation (Hwang et al. 1998; Lu et al. 1998, 2002; Toyofuku et al. 1998). Three rice MYB proteins (OsMybS1, OsMybS2, and OsMybS3) bind to the TATCCA element and mediate these sugar responses. The expression of two Arabidopsis genes (AT1G19000 and AT5G47390) encoding MYB proteins with high overall similarity to OsMybS2 and OsMybS3 is glucose repressible (Supplemental Table 1), and the TATCCA-related motif (TATCC) is a strong classifier of glucose down-regulated gene expression. This suggests a third regulatory chain in which these Arabidopsis MYB proteins mediate glucose down-regulated transcription through the TATCC element.
Several cis-acting promoter elements confer ABA-responsive gene expression. These include the ABA-responsive element (ABRE) (Marcotte Jr. et al. 1989), coupling elements (Shen et al. 1996), and recognition sites for MYB and MYC classes of transcription factors (Iwasaki et al. 1995; Abe et al. 1997). Our RVM analyses of ABA-responsive promoters identified ABRE-like motifs, recognition sequences for the ATMYB2 transcription factor, a G-box-related motif and DRE-related motifs as top-weighted classifiers of ABA-induced genes. These motifs were enriched in the promoters of genes encoding proteins involved in stress responses, secondary metabolism, and hormone metabolism (Table 4; Supplemental Table 6). Our RVM classification is consistent with recently reported analysis of motif frequencies in ABA-regulated genes, which identified ABRE and DRE motifs as overrepresented (Leonhardt et al. 2004). The expression of genes encoding ABF3, DREB1A, DREB1B, DREB1C, and DREB2A transcription factors, which mediate ABA-responsive gene expression through ABRE- and DRE-related motifs, respectively, was induced by ABA, suggesting a regulator chain model in which these transcription factors mediate ABA responsiveness through the motifs identified as strong classifiers of ABA-regulated expression. Similarly, expression of ATMYB2 is up-regulated by ABA (Supplemental Table 2). It has been shown to function as a transcriptional activator in ABA-inducible gene expression under drought stress in plants (Abe et al. 2003) and its recognition motif (WAACCA) was a strong classifier of ABA-up-regulated promoters (Table 4). The DRE-related motif (ACCGAC) conferred glucose-, ABA-, drought-, high salt-, and cold-responsive gene expression (Busk et al. 1997; Kizis and Pages 2002; Dubouzet et al. 2003). Its cognate transcription factor DREB1A/CBF3 was also transcriptionally up-regulated by both glucose and ABA, suggesting a regulator chain model for glucose and ABA regulation of stress-responsive and other target genes.
Promoter analysis
A variety of approaches have been taken to establish regulatory networks based on whole-genome analysis of gene expression levels. Many of these use frequentist probabilistic methods to identify overrepresented sequence motifs associated with expression profiles (Beer and Tavazoie 2004), which can then be used to infer relationships between motifs and gene expression patterns. Our analysis of promoter sequences uses an RVM classifier to give an estimate of the probability that a gene is up- or down-regulated based on promoter sequence features. The advantage of the RVM (Tipping 2001) with a Bayesian Automatic Relevance Determination (MacKay 1994; Neal 1994) prior is that it selects a small subset of promoter motifs for its discriminatory rule that optimally distinguish between regulated genes. The RVM also has the useful property that no parameters are set, such as the threshold of significance of a feature, since the entire model is generated automatically from the data. It also considers the significance of a feature in the context of the features already selected. This makes the application especially suitable for biological problems with many variables of unknown significance that may influence each other. The RVM correctly predicted the up- or down-regulation of ∼70% of the 1752 promoters in the glucose regulon and 692 promoters in the ABA-up regulon. This success is similar to that achieved in a recent study (Beer and Tavazoie 2004), which correctly predicted the expression patterns of 73% of 2587 yeast genes in 255 conditions using probabilistic methods. Our analysis also shows that there are other features affecting gene expression that are not captured by PLACE elements or 5-mer sequences within 1 kb of the initiation codon of Arabidopsis genes. These “missing” features probably include combinatorial effects and protein–protein interactions.
The promoter sequences selected by the RVM strategy were validated by demonstrating that the TELO motif, which was the top-weighted classifier of glucose-up-regulated gene expression, conferred glucose-mediated expression in conjunction with the TEF motif. Furthermore, other promoter motifs selected as top-weighted classifiers had established functions in glucose- and ABA-mediated gene regulation. The transcriptional coregulation of transcription factors and promoters containing cognate promoter elements selected by the RVM provides further validation of the classification strategy and permitted regulatory networks to be established.
The sparse feature selection of our RVM provides a computationally efficient way of dealing with the wide range of variables commonly encountered in biology and is suitable for biologists to apply, as the classification rule is built automatically without any statistical assumptions. Bayesian statistical methods such as we have used also provide more realistic probability models based on these large data sets (Eddy 2004). Our work reveals that these approaches have significant promise in classifying promoter functions according to their sequence and establishing transcriptional regulatory networks.
Methods
Plant material, growth condition, and time course
Arabidopsis thaliana seedlings (ecotype Columbia-0) were grown in liquid culture for 7 d on MS medium containing 0.5% glucose in constant light. After 7 d of growth, the medium was replaced with glucose-free medium for 24 h, and then seedlings were treated with 3% glucose, 3% mannitol, 3% glucose + 10 μM ABA or 3% mannitol + 10 μM ABA, and sampled at 0, 2, 4, or 6 h after treatment. Three independent sets of cultures grown in 3% glucose and 3% mannitol were sampled for RNA isolation.
RNA preparation, cRNA synthesis, and microarray hybridization
Total RNA was extracted from the treated Arabidopsis seedlings using an RNeasy Plant Mini Kit (Qiagen) according to the kit manual. Affymetrix Gene Chip array expression profiling was carried out at the John Innes Genome Lab (http://www.jicgenomelab.co.uk) according to Affymetrix Expression Analysis Technical Manual II (Affymetrix Manual II; http://www.affymetrix.com/support/technical/manuals.affx). Further information on processing microarray data and clustering is provided in the Supplemental material.
Machine learning methods
The Relevance Vector Machine (RVM) (Tipping 2001) was selected as the most appropriate technique for learning to distinguish between up- and down-regulated genes according to the sequence composition of their promoter regions. A MATLAB implementation of the RVM is available from http://www.relevancevector.com.
Assume that our data set, D, is comprised of ℓ coregulated genes
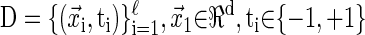 |
where 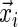 represents a set of features describing the i-th training pattern, in this case k-mers representing putative promoter protein-binding sites, and ti indicates whether the i-th gene is up-regulated (ti = +1) or down-regulated or nonregulated (ti = –1). The Relevance Vector Machine, in a statistical pattern recognition setting, essentially implements a familiar logistic regression model,
represents a set of features describing the i-th training pattern, in this case k-mers representing putative promoter protein-binding sites, and ti indicates whether the i-th gene is up-regulated (ti = +1) or down-regulated or nonregulated (ti = –1). The Relevance Vector Machine, in a statistical pattern recognition setting, essentially implements a familiar logistic regression model,
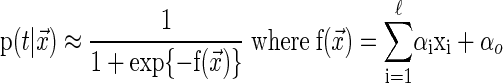 |
However, a Bayesian training algorithm was used, with an Automatic Relevance Determination (ARD) (MacKay 1994; Neal 1994) prior over the vector of model parameters, 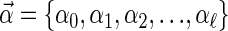 . The advantage of this approach was that the model was able to determine a small set of the most discriminatory features to form its decision rule. In this application it chooses, from a large set of arbitrary motifs, a small number of motifs that “optimally” distinguish between differentially regulated genes. A more extensive explanation is provided in the Supplemental material, and the method is available as a Web service for Arabidopsis promoter analysis from http://theoval.cmp.uea.ac.uk/~gcc/cbl/bred/, using the TIGR version 5 annotation (Haas et al. 2005).
. The advantage of this approach was that the model was able to determine a small set of the most discriminatory features to form its decision rule. In this application it chooses, from a large set of arbitrary motifs, a small number of motifs that “optimally” distinguish between differentially regulated genes. A more extensive explanation is provided in the Supplemental material, and the method is available as a Web service for Arabidopsis promoter analysis from http://theoval.cmp.uea.ac.uk/~gcc/cbl/bred/, using the TIGR version 5 annotation (Haas et al. 2005).
Calculating enrichment in functional categories
To ascribe functions to genes represented on the ATH1 chip, Gene Ontology (GO) annotations were integrated within GeneSpring 6.1 (Silicon Genetics, Redwood City, CA) as “GeneLists.” This was achieved by converting the Gene Ontology graph structures as exported from DAG-Edit (GO flat-file format, http://www.geneontology.org/) into a file-system-based data structure, where vertices are represented by directories. A list of Arabidopsis genes annotated to each GO term was prepared from the TIGR version 5 XML files (ftp://ftp.tigr.org/pub/data/a_thaliana/ath1/PSEUDOCHROMOSOMES/), and each list was stored in GeneSpring XML format within the appropriate directory. We classified sugar-regulated genes according to their putative functions based on Arabidopsis Gene Ontology (GO) annotations in GeneSpring lists, the classification of the Munich Information Centre for Protein Sequencing (MIPS) database, pathway analysis defined by AraCyc (Mueller et al. 2003) and KEGG (Kanehisa 2002), and the literature.
We calculated the P-value of the enrichment of regulated genes and promoter elements in functional categories using the hypergeometric cumulative distribution function (Tavazoie et al. 1999). Values were expressed as –log10 of P, where at least x genes in category of size k were regulated. k was determined from gene annotations as described above. The total number of genes on the array (M) was 21,000, and the total numbers of regulated genes were glucose up-regulated genes (N = 983), glucose down-regulated (N = 769), and ABA up-regulated (N = 692). The Bonferroni Correction was used to establish the significance of multiple comparisons of functional categories. Functional categories containing fewer than five genes were not considered for statistical reasons, and larger and heterogeneous functional groups were also not included in the analysis.
Construction of synthetic promoter motifs, Arabidopsis transformation, and β-glucuronidase (GUS) assays
Promoter motifs were synthesized, annealed into double-stranded DNA oligomers, cloned into a minimal promoter-reporter cassette, and transformed into Arabidopsis as described in the Supplemental material. Transformants were selected and assayed as described in the Supplemental material.
Acknowledgments
We thank Georg Harberer and Klaus Mayer (MIPS, GSF, Munich) for an initial version of the promoter database, James Hadfield of the John Innes Genome Laboratory for advice on RNA isolation and Affymetrix array processing, and members of the Bevan group for advice. This work was supported by BBSRC Exploiting Genomics Grants EGM16126 and EGM16128 to M.W.B. and G.C., respectively, and EC grant QLRT-1999-00351 (PlaNET) to M.W.B.
[Supplemental material is available online at www.genome.org. The microarray data from this study have been submitted to ArrayExpress under accession no. E-MEXP-475.]
Article published online ahead of print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.4237406.
References
- Abe, H., Yamaguchi-Shinozaki, K., Urao, T., Iwasaki, T., Hosokawa, D., and Shinozaki, K. 1997. Role of Arabidopsis MYC and MYB homologs in drought- and abscisic acid-regulated gene expression. Plant Cell 9 1859–1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abe, H., Urao, T., Ito, T., Seki, M., Shinozaki, K., and Yamaguchi-Shinozaki, K. 2003. Arabidopsis AtMYC2 (bHLH) and AtMYB2 (MYB) function as transcriptional activators in abscisic acid signaling. Plant Cell 15 63–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad, M., Jarillo, J.A., and Cashmore, A.R. 1998. Chimeric proteins between cry1 and cry2 Arabidopsis blue light photoreceptors indicate overlapping functions and varying protein stability. Plant Cell 10 197–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arenas-Huertero, F., Arroyo, A., Zhou, L., Sheen, J., and Leon, P. 2000. Analysis of Arabidopsis glucose insensitive mutants, gin5 and gin6, reveals a central role of the plant hormone ABA in the regulation of plant vegetative development by sugar. Genes & Dev. 14 2085–2096. [PMC free article] [PubMed] [Google Scholar]
- Beer, M.A. and Tavazoie, S. 2004. Predicting gene expression from sequence. Cell 117 185–198. [DOI] [PubMed] [Google Scholar]
- Bender, J. and Fink, G.R. 1998. A Myb homologue, ATR1, activates tryptophan gene expression in Arabidopsis. Proc. Natl. Acad. Sci. 95 5655–5660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borevitz, J.O., Xia, Y., Blount, J., Dixon, R.A., and Lamb, C. 2000. Activation tagging identifies a conserved MYB regulator of phenylpropanoid biosynthesis. Plant Cell 12 2383–2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boser, B.E., Guyon, I.M., and Vapnik, V.N. 1992. A training algorithm for optimal margin classifiers. In Proceedings of the Fifth Annual ACM Workshop on Computational Learning Theory, (ed. D. Haussler), pp. 144–152. ACM Press, Pittsburgh.
- Boyes, D.C., Zayed, A.M., Ascenzi, R., McCaskill, A.J., Hoffman, N.E., Davis, K.R., and Gorlach, J. 2001. Growth stage-based phenotypic analysis of Arabidopsis: A model for high throughput functional genomics in plants. Plant Cell 13 1499–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buche, C., Poppe, C., Schafer, E., and Kretsch, T. 2000. eid1: A new Arabidopsis mutant hypersensitive in phytochrome A-dependent high-irradiance responses. Plant Cell 12 547–558. [PMC free article] [PubMed] [Google Scholar]
- Busk, P.K. and Pages, M. 1998. Regulation of abscisic acid-induced transcription. Plant Mol. Biol. 37 425–435. [DOI] [PubMed] [Google Scholar]
- Busk, P.K., Jensen, A.B., and Pages, M. 1997. Regulatory elements in vivo in the promoter of the abscisic acid responsive gene rab17 from maize. Plant J. 11 1285–1295. [DOI] [PubMed] [Google Scholar]
- Bussemaker, H.J., Li, H., and Siggia, E.D. 2001. Regulatory element detection using correlation with expression. Nat. Genet. 27 167–171. [DOI] [PubMed] [Google Scholar]
- Chaboute, M.E., Clement, B., Sekine, M., Philipps, G., and Chaubet-Gigot, N. 2000. Cell cycle regulation of the tobacco ribonucleotide reductase small subunit gene is mediated by E2F-like elements. Plant Cell 12 1987–2000. [PMC free article] [PubMed] [Google Scholar]
- Chattopadhyay, S., Ang, L.H., Puente, P., Deng, X.W., and Wei, N. 1998. Arabidopsis bZIP protein HY5 directly interacts with light-responsive promoters in mediating light control of gene expression. Plant Cell 10 673–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng, W.H., Endo, A., Zhou, L., Penney, J., Chen, H.C., Arroyo, A., Leon, P., Nambara, E., Asami, T., Seo, M., et al. 2002. A unique short-chain dehydrogenase/reductase in Arabidopsis glucose signaling and abscisic acid biosynthesis and functions. Plant Cell 14 2723–2743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vetten, N., Quattrocchio, F., Mol, J., and Koes, R. 1997. The an11 locus controlling flower pigmentation in petunia encodes a novel WD-repeat protein conserved in yeast, plants, and animals. Genes & Dev. 11 1422–1434. [DOI] [PubMed] [Google Scholar]
- Dieterle, M., Zhou, Y.C., Schafer, E., Funk, M., and Kretsch, T. 2001. EID1, an F-box protein involved in phytochrome A-specific light signaling. Genes & Dev. 15 939–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubouzet, J.G., Sakuma, Y., Ito, Y., Kasuga, M., Dubouzet, E.G., Miura, S., Seki, M., Shinozaki, K., and Yamaguchi-Shinozaki, K. 2003. OsDREB genes in rice, Oryza sativa L., encode transcription activators that function in drought-, high-salt- and cold-responsive gene expression. Plant J. 33 751–763. [DOI] [PubMed] [Google Scholar]
- Eddy, S.R. 2004. What is Bayesian statistics? Nat. Biotechnol. 22 1177–1178. [DOI] [PubMed] [Google Scholar]
- Fankhauser, C., Yeh, K.C., Lagarias, J.C., Zhang, H., Elich, T.D., and Chory, J. 1999. PKS1, a substrate phosphorylated by phytochrome that modulates light signaling in Arabidopsis. Science 284 1539–1541. [DOI] [PubMed] [Google Scholar]
- Gangal, R. and Sharma, P. 2005. Human Pol II promoter prediction: Time series descriptors and machine learning. Nucleic Acids Res. 33 1332–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmour, S.J., Sebolt, A.M., Salazar, M.P., Everard, J.D., and Thomashow, M.F. 2000. Overexpression of the Arabidopsis CBF3 transcriptional activator mimics multiple biochemical changes associated with cold acclimation. Plant Physiol. 124 1854–1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas, B.J., Wortman, J.R., Ronning, C.M., Hannick, L.I., Smith Jr., R.K., Maiti, R., Chan, A.P., Yu, C., Farzad, M., Wu, D., et al. 2005. Complete reannotation of the Arabidopsis genome: Methods, tools, protocols and the final release. BMC Biol. 3 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higo, K., Ugawa, Y., Iwamoto, M., and Korenaga, T. 1999. Plant cis-acting regulatory DNA elements (PLACE) database: 1999. Nucleic Acids Res. 27 297–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Himmelbach, A., Hoffmann, T., Leube, M., Hohener, B., and Grill, E. 2002. Homeodomain protein ATHB6 is a target of the protein phosphatase ABI1 and regulates hormone responses in Arabidopsis. EMBO J. 21 3029–3038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holding, D.R. and Springer, P.S. 2002. The Arabidopsis gene PROLIFERA is required for proper cytokinesis during seed development. Planta 214 373–382. [DOI] [PubMed] [Google Scholar]
- Hubbell, E., Liu, W.M., and Mei, R. 2002. Robust estimators for expression analysis. Bioinformatics 18 1585–1592. [DOI] [PubMed] [Google Scholar]
- Huijser, C., Kortstee, A., Pego, J., Weisbeek, P., Wisman, E., and Smeekens, S. 2000. The Arabidopsis SUCROSE UNCOUPLED-6 gene is identical to ABSCISIC ACID INSENSITIVE-4: Involvement of abscisic acid in sugar responses. Plant J. 23 577–585. [DOI] [PubMed] [Google Scholar]
- Hwang, Y.S., Karrer, E.E., Thomas, B.R., Chen, L., and Rodriguez, R.L. 1998. Three cis-elements required for rice α-amylase Amy3D expression during sugar starvation. Plant Mol. Biol. 36 331–341. [DOI] [PubMed] [Google Scholar]
- Ishiguro, S. and Nakamura, K. 1994. Characterization of a cDNA encoding a novel DNA-binding protein, SPF1, that recognizes SP8 sequences in the 5′ upstream regions of genes coding for sporamin and β-amylase from sweet potato. Mol. Gen. Genet. 244 563–571. [DOI] [PubMed] [Google Scholar]
- Iwasaki, T., Yamaguchi-Shinozaki, K., and Shinozaki, K. 1995. Identification of a cis-regulatory region of a gene in Arabidopsis thaliana whose induction by dehydration is mediated by abscisic acid and requires protein synthesis. Mol. Gen. Genet. 247 391–398. [DOI] [PubMed] [Google Scholar]
- Jaakkola, T., Diekhans, M., and Haussler, D. 1999. ISMB99. AAAI Press, Menlo Park, CA.
- Jang, J.C., Leon, P., Zhou, L., and Sheen, J. 1997. Hexokinase as a sugar sensor in higher plants. Plant Cell 9 5–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, C.Z., Yee, J., Mitchell, D.L., and Britt, A.B. 1997. Photorepair mutants of Arabidopsis. Proc. Natl. Acad. Sci. 94 7441–7445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa, M. 2002. The KEGG database. Novartis Found. Symp. 247 91–101; discussion 101–103, 119–128, 244–252. [PubMed] [Google Scholar]
- Kim, D.J., Smith, S.M., and Leaver, C.J. 1997. A cDNA encoding a putative SPF1-type DNA-binding protein from cucumber. Gene 185 265–269. [DOI] [PubMed] [Google Scholar]
- Kizis, D. and Pages, M. 2002. Maize DRE-binding proteins DBF1 and DBF2 are involved in rab17 regulation through the drought-responsive element in an ABA-dependent pathway. Plant J. 30 679–689. [DOI] [PubMed] [Google Scholar]
- Kleiner, O., Kircher, S., Harter, K., and Batschauer, A. 1999. Nuclear localization of the Arabidopsis blue light receptor cryptochrome 2. Plant J. 19 289–296. [DOI] [PubMed] [Google Scholar]
- Laby, R.J., Kincaid, M.S., Kim, D., and Gibson, S.I. 2000. The Arabidopsis sugar-insensitive mutants sis4 and sis5 are defective in abscisic acid synthesis and response. Plant J. 23 587–596. [DOI] [PubMed] [Google Scholar]
- Lacombe, E., Van Doorsselaere, J., Boerjan, W., Boudet, A.M., and Grima-Pettenati, J. 2000. Characterization of cis-elements required for vascular expression of the cinnamoyl CoA reductase gene and for protein–DNA complex formation. Plant J. 23 663–676. [DOI] [PubMed] [Google Scholar]
- Lam, E. 1995. Domain analysis of the plant DNA-binding protein GT1a: Requirement of four putative α-helices for DNA binding and identification of a novel oligomerization region. Mol. Cell. Biol. 15 1014–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavine, B.K., Davidson, C.E., and Rayens, W.S. 2004. Machine learning based pattern recognition applied to microarray data. Comb. Chem. High Throughput Screen. 7 115–131. [DOI] [PubMed] [Google Scholar]
- Lee, T.I., Rinaldi, N.J., Robert, F., Odom, D.T., Bar-Joseph, Z., Gerber, G.K., Hannett, N.M., Harbison, C.T., Thompson, C.M., Simon, I., et al. 2002. Transcriptional regulatory networks in Saccharomyces cerevisiae. Science 298 799–804. [DOI] [PubMed] [Google Scholar]
- Leonhardt, N., Kwak, J.M., Robert, N., Waner, D., Leonhardt, G., and Schroeder, J.I. 2004. Microarray expression analyses of Arabidopsis guard cells and isolation of a recessive abscisic acid hypersensitive protein phosphatase 2C mutant. Plant Cell 16 596–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Y., Campbell, C., and Tipping, M. 2002. Bayesian automatic relevance determination algorithms for classifying gene expression data. Bioinformatics 18 1332–1339. [DOI] [PubMed] [Google Scholar]
- Lin, C., Robertson, D.E., Ahmad, M., Raibekas, A.A., Jorns, M.S., Dutton, P.L., and Cashmore, A.R. 1995. Association of flavin adenine dinucleotide with the Arabidopsis blue light receptor CRY1. Science 269 968–970. [DOI] [PubMed] [Google Scholar]
- Liu, W.M., Mei, R., Di, X., Ryder, T.B., Hubbell, E., Dee, S., Webster, T.A., Harrington, C.A., Ho, M.H., Baid, J., et al. 2002. Analysis of high density expression microarrays with signed-rank call algorithms. Bioinformatics 18 1593–1599. [DOI] [PubMed] [Google Scholar]
- Loppes, R. and Radoux, M. 2001. Identification of short promoter regions involved in the transcriptional expression of the nitrate reductase gene in Chlamydomonas reinhardtii. Plant Mol. Biol. 45 215–227. [DOI] [PubMed] [Google Scholar]
- Lu, C.A., Lim, E.K., and Yu, S.M. 1998. Sugar response sequence in the promoter of a rice α-amylase gene serves as a transcriptional enhancer. J. Biol. Chem. 273 10120–10131. [DOI] [PubMed] [Google Scholar]
- Lu, C.A., Ho, T.H., Ho, S.L., and Yu, S.M. 2002. Three novel MYB proteins with one DNA binding repeat mediate sugar and hormone regulation of α-amylase gene expression. Plant Cell 14 1963–1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKay, D.J.C. 1994. Bayesian methods for back-propagation networks. Springer, New York.
- Manevski, A., Bertoni, G., Bardet, C., Tremousaygue, D., and Lescure, B. 2000. In synergy with various cis-acting elements, plant insterstitial telomere motifs regulate gene expression in Arabidopsis root meristems. FEBS Lett. 483 43–46. [DOI] [PubMed] [Google Scholar]
- Marcotte Jr., W.R., Russell, S.H., and Quatrano, R.S. 1989. Abscisic acid-responsive sequences from the em gene of wheat. Plant Cell 1 969–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore, B., Zhou, L., Rolland, F., Hall, Q., Cheng, W.H., Liu, Y.X., Hwang, I., Jones, T., and Sheen, J. 2003. Role of the Arabidopsis glucose sensor HXK1 in nutrient, light, and hormonal signaling. Science 300 332–336. [DOI] [PubMed] [Google Scholar]
- Mueller, L.A., Zhang, P., and Rhee, S.Y. 2003. AraCyc: A biochemical pathway database for Arabidopsis. Plant Physiol. 132 453–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neal, R. 1994. Bayesian learning for neural networks. University of Toronto, Toronto.
- Price, J., Laxmi, A., St Martin, S.K., and Jang, J.C. 2004. Global transcription profiling reveals multiple sugar signal transduction mechanisms in Arabidopsis. Plant Cell 16 2128–2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puente, P., Wei, N., and Deng, X.W. 1996. Combinatorial interplay of promoter elements constitutes the minimal determinants for light and developmental control of gene expression in Arabidopsis. EMBO J. 15 3732–3743. [PMC free article] [PubMed] [Google Scholar]
- Quattrocchio, F., Wing, J., van der Woude, K., Souer, E., de Vetten, N., Mol, J., and Koes, R. 1999. Molecular analysis of the anthocyanin2 gene of petunia and its role in the evolution of flower color. Plant Cell 11 1433–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rook, F., Corke, F., Card, R., Munz, G., Smith, C., and Bevan, M.W. 2001. Impaired sucrose-induction mutants reveal the modulation of sugar-induced starch biosynthetic gene expression by abscisic acid signalling. Plant J. 26 421–433. [DOI] [PubMed] [Google Scholar]
- Schluepmann, H., Pellny, T., van Dijken, A., Smeekens, S., and Paul, M. 2003. Trehalose 6-phosphate is indispensable for carbohydrate utilization and growth in Arabidopsis thaliana. Proc. Natl. Acad. Sci. 100 6849–6854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholkopf, B., Tsuda, K., and Ver, J.P. 2004. Kernel methods in computational biology. MIT Press, Cambridge, MA.
- Segal, E., Yelensky, R., and Koller, D. 2003. Genome-wide discovery of transcriptional modules from DNA sequence and gene expression. Bioinformatics 19 Suppl 1: i273–i282. [DOI] [PubMed] [Google Scholar]
- Shahmuradov, I.A., Solovyev, V.V., and Gammerman, A.J. 2005. Plant promoter prediction with confidence estimation. Nucleic Acids Res. 33 1069–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen, Q., Zhang, P., and Ho, T.H. 1996. Modular nature of abscisic acid (ABA) response complexes: Composite promoter units that are necessary and sufficient for ABA induction of gene expression in barley. Plant Cell 8 1107–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smalle, J., Kurepa, J., Haegman, M., Gielen, J., Van Montagu, M., and Straeten, D.V. 1998. The trihelix DNA-binding motif in higher plants is not restricted to the transcription factors GT-1 and GT-2. Proc. Natl. Acad. Sci. 95 3318–3322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, A.D., Sumazin, P., and Zhang, M.Q. 2005. Identifying tissue-selective transcription factor binding sites in vertebrate promoters. Proc. Natl. Acad. Sci. 102 1560–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Springer, P.S., Holding, D.R., Groover, A., Yordan, C., and Martienssen, R.A. 2000. The essential Mcm7 protein PROLIFERA is localized to the nucleus of dividing cells during the G1 phase and is required maternally for early Arabidopsis development. Development 127 1815–1822. [DOI] [PubMed] [Google Scholar]
- Stracke, R., Werber, M., and Weisshaar, B. 2001. The R2R3-MYB gene family in Arabidopsis thaliana. Curr. Opin. Plant Biol. 4 447–456. [DOI] [PubMed] [Google Scholar]
- Tatematsu, K., Ward, S., Leyser, O., Kamiya, Y., and Nambara, E. 2005. Identification of cis-elements that regulate gene expression during initiation of axillary bud outgrowth in Arabidopsis. Plant Physiol. 138 757–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavazoie, S., Hughes, J.D., Campbell, M.J., Cho, R.J., and Church, G.M. 1999. Systematic determination of genetic network architecture. Nat. Genet. 22 281–285. [DOI] [PubMed] [Google Scholar]
- Thimm, O., Blasing, O., Gibon, Y., Nagel, A., Meyer, S., Kruger, P., Selbig, J., Muller, L.A., Rhee, S.Y., and Stitt, M. 2004. MAPMAN: A user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J. 37 914–939. [DOI] [PubMed] [Google Scholar]
- Thum, K.E., Shin, M.J., Palenchar, P.M., Kouranov, A., and Coruzzi, G.M. 2004. Genome-wide investigation of light and carbon signaling interactions in Arabidopsis. Genome Biol. 5 R10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tipping, M.E. 2000. The Relevance Vector Machine. Adv. Neural Inf. Process. Syst. 12 652–658. [Google Scholar]
- Tipping, M.E. 2001. Sparse Bayesian learning and the Relevance Vector Machine. J. Mach. Learn. Res. 1 211–244. [Google Scholar]
- Toyofuku, K., Umemura, T., and Yamaguchi, J. 1998. Promoter elements required for sugar-repression of the RAmy3D gene for α-amylase in rice. FEBS Lett. 428 275–280. [DOI] [PubMed] [Google Scholar]
- Tremousaygue, D., Manevski, A., Bardet, C., Lescure, N., and Lescure, B. 1999. Plant interstitial telomere motifs participate in the control of gene expression in root meristems. Plant J. 20 553–561. [DOI] [PubMed] [Google Scholar]
- Tremousaygue, D., Garnier, L., Bardet, C., Dabos, P., Herve, C., and Lescure, B. 2003. Internal telomeric repeats and `TCP domain' protein-binding sites co-operate to regulate gene expression in Arabidopsis thaliana cycling cells. Plant J. 33 957–966. [DOI] [PubMed] [Google Scholar]
- Uimari, A. and Strommer, J. 1997. Myb26: A MYB-like protein of pea flowers with affinity for promoters of phenylpropanoid genes. Plant J. 12 1273–1284. [DOI] [PubMed] [Google Scholar]
- Vinayagam, A., Konig, R., Moormann, J., Schubert, F., Eils, R., Glatting, K.H., and Suhai, S. 2004. Applying Support Vector Machines for Gene Ontology based gene function prediction. BMC Bioinformatics 5 116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto, Y., Sato, E., Shimizu, T., Nakamich, N., Sato, S., Kato, T., Tabata, S., Nagatani, A., Yamashino, T., and Mizuno, T. 2003. Comparative genetic studies on the APRR5 and APRR7 genes belonging to the APRR1/TOC1 quintet implicated in circadian rhythm, control of flowering time, and early photomorphogenesis. Plant Cell Physiol. 44 1119–1130. [DOI] [PubMed] [Google Scholar]
- Yanagisawa, S., Yoo, S.D., and Sheen, J. 2003. Differential regulation of EIN3 stability by glucose and ethylene signalling in plants. Nature 425 521–525. [DOI] [PubMed] [Google Scholar]
- Yoshida, S., Ito, M., Nishida, I., and Watanabe, A. 2002. Identification of a novel gene HYS1/CPR5 that has a repressive role in the induction of leaf senescence and pathogen-defence responses in Arabidopsis thaliana. Plant J. 29 427–437. [DOI] [PubMed] [Google Scholar]
- Yu, H., Luscombe, N.M., Qian, J., and Gerstein, M. 2003. Genomic analysis of gene expression relationships in transcriptional regulatory networks. Trends Genet. 19 422–427. [DOI] [PubMed] [Google Scholar]
- Yubero-Serrano, E.M., Moyano, E., Medina-Escobar, N., Munoz-Blanco, J., and Caballero, J.L. 2003. Identification of a strawberry gene encoding a non-specific lipid transfer protein that responds to ABA, wounding and cold stress. J. Exp. Bot. 54 1865–1877. [DOI] [PubMed] [Google Scholar]
- Zhang, W., Morris, Q.D., Chang, R., Shai, O., Bakowski, M.A., Mitsakakis, N., Mohammad, N., Robinson, M.D., Zirngibl, R., Somogyi, E., et al. 2004. The functional landscape of mouse gene expression. J. Biol. 3 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, L., Jang, J.C., Jones, T.L., and Sheen, J. 1998. Glucose and ethylene signal transduction crosstalk revealed by an Arabidopsis glucose-insensitive mutant. Proc. Natl. Acad. Sci. 95 10294–10299. [DOI] [PMC free article] [PubMed] [Google Scholar]