

Abstract
Mast cells are not only major effector cells in allergy and host defense against parasites and bacteria but also important cellular components in other immune responses. Recent studies on the effects of monomeric IgE on mast cell survival and activation have made an impact on our view of the IgE binding to its high-affinity receptors, Fc∊RI. Traditionally, IgE binding to Fc∊RI has been considered as a passive action of “sensitization” before receptor aggregation by Ag. However, recent studies indicate that at high concentrations some monoclonal IgEs have effects on mast cells similar to or identical to those induced by IgE+Ag stimulation. These effects may be due to induction of Fc∊RI aggregation by these IgEs in the absence of Ag. This review will synthesize recent findings of the heterogeneity of IgEs in their ability to induce survival and activation events, their mechanisms, the potential in vivo significance of IgE−Fc∊RI interactions, and the implications of the mouse studies to human diseases.
Mast cells reside close to the mucosal and epithelial interfaces with the environment and around blood vessels. A vast majority of mast cell studies have dealt with their predominant role in acute allergic reactions (immediate hypersensitivity) and more recently their roles in late-phase allergic reactions (1) and in the host defense against certain bacteria (2, 3) and parasites (4, 5). Beyond this traditional understanding, our knowledge of mast cell functions has recently been substantially expanded (6, 7) and now includes those in autoimmunity such as experimental allergic encephalomyelopathy and rheumatoid arthritis (8, 9), delayed-type hypersensitivity (10), angiogenesis (11), and congestive heart failure (12).
Fc∊RI expressed on murine mast cells consists of four subunits (αβγ2): an IgE-binding α subunit, a signal-amplifying, receptor-stabilizing β subunit, and two disulfide-bonded γ subunits that are the main signal transducer (13). Stimulation of IgE-sensitized mast cells with Ag (this mode of stimulation hereafter referred to as IgE+Ag) or anti-IgE Ab (referred to as IgE+anti-IgE) induces receptor aggregation or cross-linking. Aggregation of Fc∊RI leads to the activation of β subunit-associated Lyn, a Src family protein tyrosine kinase (PTK).4 Activated Lyn phosphorylates tyrosine residues in the ITAMs in the cytoplasmic regions of β and γ subunits. Phosphorylated β and γ ITAMs recruit Lyn and Syk, respectively. Another Src family PTK, Fyn, was also shown to associate with Fc∊RI and to play a complementary role, particularly by activating PI3K (14). These PTKs phosphorylate numerous targets and activate several signaling pathways, including the PI3K, phospholipase C/Ca2+, and several MAPK pathways (15, 16). These signaling events lead to degranulation and cytokine and chemokine production. Chemicals (e.g., histamine and serotonin), lipids (leukotrienes and PGs), nucleotides (e.g., adenosine), and polypeptides (e.g., proteases, cytokines, and chemokines) released from activated mast cells are effector molecules that induce allergic or inflammatory reactions or modulate innate and adaptive immune responses (7, 17).
IgE-induced mast cell survival and activation in the absence of Ag
One of the traditional views that have been widely accepted is on effects of IgE binding to the Fc∊RI. IgE binding was once thought of as a passive step of sensitization, which would keep mast cells at a “resting” state, before receptor aggregation with multivalent Ag (allergen) or anti-IgE. In stark contrast to this traditional view, it was shown that IgE binding to Fc∊RI in the absence of specific Ag (the stimulation mode referred to as IgE(−Ag)) engenders several biological outcomes in mast cells: IgE(−Ag) can induce up-regulation of cell surface expression of the receptor (i.e., Fc∊RI) (18-20), survival (21, 22), increase in histamine content (23), histamine release, leukotriene release, receptor internalization, DNA synthesis (24), increased responses to compound 48/80 and substance P (25), increase in F-actin content (26), membrane ruffling (27), mast cell adhesion to fibronectin (28), and migration (29). These events are almost all inducible by IgE+Ag or IgE+anti-IgE (Fig. 1). Importantly, these IgE(−Ag) effects are induced at high concentrations of IgE 2–3 logs more than required to sensitize a mast cell for Ag-dependent activation. Parenthetically, IgE was shown to augment IL-3-induced mast cell proliferation (30), but unfortunately, this finding in 1986 received little attention.
FIGURE 1.
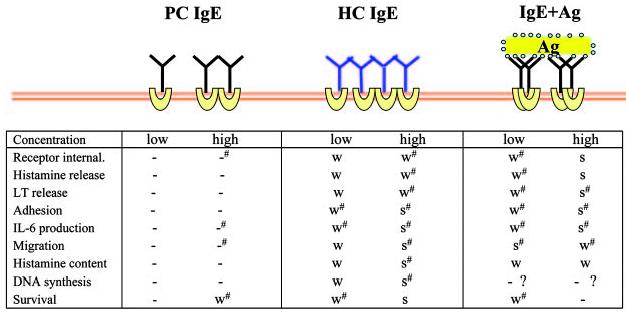
Biological effects by various modes of stimulation via the Fc∊RI. Experiments using mouse BMMCs are summarized. Concentrations of stimuli are as follows: PC IgE (H1 DNP-∊-206) and HC IgE (SPE-7), 0.5 μg/ml (low) and 5 μg/ml (high); and IgE+Ag (DNP21-BSA), 1 ng/ml (low) and 100 ng/ml (high). Notice that biological events are listed in a rough order of occurrences and that receptor aggregation presumably occurs in all modes of stimulation, except for low PC IgE concentrations. -, not detected, -#, very weak; W, weak; W#, weak∼moderate; S#, moderate∼strong; and S, strong. References: receptor internalization (24, 44, 50); histamine release (14, 21, 22, 24, 26, 27, 43, 44, 50); leukotriene release (21, 22, 24); adhesion (28, 56); IL-6 production (21, 22, 24, 43, 44, 50, 51); migration (29); histamine content (23, 24, 57); DNA synthesis (21, 22, 24, 86); and survival (21, 22, 24, 43, 44, 50, 51, 58-62). In Ref. 60, MC9 mouse mast cells were used. In Ref. 62, BMMCs were stimulated with anti-TNP IgE (hybridoma supernatants) and TNP4.8-BSA (1 μg/ml). However, it is not clear whether the used Ag concentration corresponds to weak or strong stimulus.
Earlier studies established that receptor aggregation induced by IgE+Ag or IgE+anti-IgE is required for mast cell activation (31), which is in line with receptor dimerization or oligomerization as the common mechanism for receptor-mediated cell activation, e.g., that by peptide growth factors (32, 33). A biophysical method termed time-resolved phosphorescence anisotropy measurement (that quantifies motions of phosphorescent dye-labeled membrane proteins in the nano- to microsecond range, thus assessing the size and rigidity of the membrane structures containing the labeled proteins) suggests that IgE(−Ag) also induces receptor aggregation (24). The enhancement in the affinity of binding an Ag-combining site of an IgE molecule to an epitope on another IgE was estimated by calculating the probability of finding as nearest neighbor a distinct receptor at distances between r0 and r0 +Δr on a cell surface (34). This simulation (r0 = 75Å and Δr = 30Å) predicted a significant probability (>0.4 or ∼0.1, respectively, for cells with 3 × 105 (RBL-2H3 rat mast cells) or 5 × 104 (mouse bone marrow-derived mast cell (BMMC)) receptors per cell), with which IgE-IgE interactions (presumed Fc∊RI aggregations) take place on the cell surface at IgE concentrations > 1 μg/ml in the absence of Ag. These calculations match the experimental data obtained in different laboratories in that IgE effects on survival and activation are seen at ≥0.5 μg/ml IgE concentrations, and they are consistent with IgE(Ag)-mediated Fc∊RI aggregation suggested by the phosphorescence anisotropy data. By contrast, the same simulation predicts a ∼104-fold lower probability of nearest neighbor (IgE-IgE) interactions in solution. Several studies suggested that survival and other activation effects were due to binding to Fc∊RI by monomeric, but not aggregate, IgE molecules in the IgE preparations. However, IgE(−Ag)-induced Fc∊RI aggregation will need to be confirmed by other methodologies.
Despite the rapid progress in this area, there are many issues to be resolved (Table I). It has never been absolutely shown if the very high amounts of IgE added under the culture conditions used do not self-Aggregate. IgE effects, interpreted as due to monomeric IgE, would be in fact due to aggregated IgE within the culture or IgE sticking to plastic plates. Also, the cultured mast cells exposed to these high levels of IgE have not seen IgE during maturation. This is not what happens in vivo, where an individual IgE receptor expressed on the developing mast cell is immediately engaged by IgE. Furthermore, how would such a system with no Ag specificity mechanistically function in tissues?
Table I.
Outstanding questions to be addressed in future
| 1. | Why high concentrations of IgE are required for mast cell survival and activation? |
| 2. | More definitive evidence for receptor aggregation by monomeric IgE should be obtained. |
| 3. | What is the mechanism for receptor aggregation? Is the same mechanism used by monomeric IgE and IgE+Ag to induce receptor aggregation? Does p28 play any role? |
| 4. | What receptor components and parts thereof are required for monomeric IgE-induced receptor up-regulation, survival, cytokine production, and other activation events? Are coreceptor or costimulatory receptors present? |
| 5. | Are the same sets of signaling pathways used by monomeric IgE and IgE+Ag to cause survival, cytokine production, and other activation events? |
| 6. | What makes the difference between HC and PC IgEs? Is there a wide spectrum in the ability of human IgEs similar to that of mouse monoclonal IgEs from HC to PC IgEs. If so, do such differences in human IgEs have pathophysiological significance? Do allergic patients tend to have HC IgEs than nonallergic individuals? |
Heterogeneity of IgE molecules
Not all monoclonal IgE molecules can induce all the activation events listed above in the absence of Ag: IgEs display a wide spectrum in their ability to induce the production and secretion of IL-6 and TNF-α with highly cytokinergic (HC) IgEs at one extreme end and poorly cytokinergic (PC) IgEs at the other (24). Anisotropy data suggest that more extensive receptor aggregations take place with HC IgEs than with PC IgEs. Consistent with such differences in the extent of receptor aggregation, a strong HC IgE could induce all the tested activation events, whereas PC IgEs cause only a limited set of activation events and even for those to a lesser extent. Importantly, a mixture of seven different monoclonal IgEs exhibited survival effects (24), suggesting that polyclonal IgE should also have survival effects.
Models for IgE-induced receptor aggregation
Despite this remarkable heterogeneity among IgE molecules, no information is available about differences among IgEs at molecular levels. Affinity of IgEs to Fc∊RI does not appear to be a determining factor of the differences between HC and PC IgEs, as a typical HC IgE, SPE-7, and a much weaker HC IgE, H1 DNP-∊-26, have similar reported affinities (KD = ∼15 nM) (35, 36). The variable (V) regions of IgE molecules seem important for receptor aggregation and consequent survival and cytokine production, as monovalent hapten (for which the used IgE is specific) can inhibit IgE(−Ag)-induced receptor aggregation, survival, and cytokine production (24). Potentially related to the receptor aggregation mechanism and as a likely factor that determines the potency to induce receptor aggregation, a recent study showed that the SPE-7 Ab expressed as a recombinant single-chain Fv (VH-VL) molecule can adopt different Ag binding site conformations before Ag binding and that binding of different Ags can induce isomerization of the binding site, leading to high-affinity complexes with a deep or narrow binding site (37). Therefore, it is conceivable that IgEs can induce Fc∊RI aggregation in the absence of specific Ag when the Fab portion of a Fc∊RI-bound IgE molecule interacts with a neighboring Fc∊RI-bound IgE directly or indirectly via a third molecule whose epitopes are recognized by neighboring IgEs. Similar aggregation models via Ig domain-Ig domain interactions or Ig-X-Ig (“X” represents an unknown bridging molecule) interactions were proposed for pre-BCR (38).
Interestingly, a recent study described a molecule termed p28 in RBL-2H3 cells (39). This molecule, which cross-reacts with a mAb against phospholipid scramblase 1, is associated with ∼50% of Fc∊RI in RBL-2H3 cells in the absence of IgE and dissociates from the receptor when the cells are incubated with a high concentration (20 μM) of IgEs. Therefore, p28 might be involved in sensing IgE binding. However, the identity and function of p28 remain to be revealed. Both IgE and Fc∊RI α subunit are glycoproteins. However, potential involvement of the carbohydrate moiety vs polypeptide backbone of IgE in receptor aggregation remains to be studied in future.
Another interesting feature of IgE(−Ag) effects is that at least some of them require the continuous, but not transient, presence of IgE, hence prolonged receptor aggregation: survival promotion by both HC and PC IgEs and proliferation by HC IgEs could be seen only in the presence of IgE but not after washing off IgE even after surface expression of Fc∊RI was dramatically increased by preincubation with IgE (21). Prolonged Fc∊RI aggregation was also suspected to be required for augmented proliferation of peritoneal mast cells, although this proliferation was induced in a semisolid culture by IgE+Ag in the presence of IL-3 (40).
Fc∊RI components and membrane microenvironments required for IgE(−Ag) effects
IgE+Ag stimulation induces localization of the aggregated receptors to cholesterol/sphingolipid-rich plasma membrane domains termed lipid rafts (41, 42). Experimental disruption of lipid rafts suggests that localization of the receptor in lipid rafts is required for the activation of ERK induced by IgE(−Ag) stimulation and therefore for survival as well (see Strength and duration of Fc∊RI stimulation determine mast cell survival) (22). Among the obligate subunits (α, β, and γ) of the murine Fc∊RI, both α and γ subunits were shown to be required for IgE(−Ag)-mediated survival (21, 43). Interestingly, the ITAM of γ subunit, which is critical for IgE+Ag-induced mast cell degranulation and cytokine production and IgE(−Ag)-mediated survival, is not essential for IgE(−Ag)-mediated receptor up-regulation (43). Consistent with this but unlike degranulation or other activation events, receptor up-regulation induced by IgE(−Ag) does not require Lyn, Syk, or other PTKs (44). This phenomenon is due to the increased lifetime of the surface-resident receptors rather than to its increased synthesis and/or transport to the plasma membrane (18, 45, 46). Thus, this effect of IgE binding to the Fc∊RI is of a nature totally different from the other effects.
Strength and duration of Fc∊RI stimulation determine mast cell survival
Mast cells die by apoptosis in the paucity of growth factors in vitro and in vivo. In vivo apoptosis was reported in mast cells that had been proliferated by repetitive injections of stem cell factor, following the cessation of the injections (47) and splenic mast cells during parasite infection (48). Survival-enhancing stimuli of Fc∊RI include IgE(−Ag), IgE+anti-IgE, and IgE+Ag (low Ag concentrations) (44). Importantly, a slow on-rate of IgE interaction with Fc∊RI together with prolonged activation of ERK matches with the required pattern of ERK activity for survival of T cells and other cell types (49). Consistent with this, relatively weak signals emanated from the γ subunit were required for the survival of mast cells expressing CD8/γ chimera in the genetic background of γ−/−, compared with the signals required for degranulation (50). However, the duration of the γ signals seems more essential for survival because sustained ERK activation, even strong enough to induce degranulation, could induce survival.
Autocrine and paracrine mechanisms for mast cell survival and activation
Survival mechanisms elicited by IgE(−Ag) and IgE+Ag are not completely understood. At cellular levels, HC IgEs use an autocrine or paracrine mechanism at least in part by secreting a cytokine(s) that supports mast cell survival (22, 24). Kohno et al. (51) recently identified IL-3 as the predominant cytokine induced by HC IgEs that is responsible for the survival effects; survival effects were drastically reduced (but not abrogated) by neutralization of IL-3 or IL-3 deficiency. By contrast, PC IgEs do not support Fc∊RIα−/− mast cells in coculture with wild-type cells (24), indicating that PC IgEs do not induce secretion of survival-mediating cytokines. These data suggest that there are at least two different survival mechanisms evoked by IgE(−Ag) at cellular levels.
In addition to the predominant role of autocrine IL-3 for mast cell survival, recent data indicate that the mediators released from activated mast cells affect not only other cell types (e.g., neutrophil recruitment by mast cell-derived TNF-α in late-phase allergic responses (52)) but also themselves and neighboring mast cells in terms of scale and duration of several aspects of activation. Thus, recent studies demonstrated the importance of adenosine and sphingosine 1-phosphate acutely released by IgE+Ag in Ca2+ influx, degranulation, and chemotaxis to Ag (53, 54). Unlike degranulation that is completed within several minutes after the initiation of IgE+Ag stimulation, migration induced by IgE(−Ag), as well as IgE+Ag, is affected by many factors; in addition to sphingosine 1-phosphate, adenosine, leukotriene B4, and several chemokines (MIP-1α/CCL3, MIP-1β/CCL4, MIP-2/CXCL1–3, MCP-1/CCL2, and RANTES/CCL5) contribute to the full migratory activity using both directional (chemotaxis) and nondirectional (chemokinesis) mechanisms; as these factors all use G protein-coupled receptors, this form of migration is dependent on PI3Kγ (29). Therefore, the PTK-based signaling pathways and G protein-coupled receptor-based signaling pathways seem to converge or cross-talk to enhance degranulation and migratory responses (and probably other activation events as well).
Signal transduction for IgE(−Ag)- and IgE+Ag-induced survival and activation
Because both IgE(−Ag) and IgE+Ag engage the Fc∊RI, it would not be irrational to assume that similar, if not identical, signaling events are elicited by the two modes of Fc∊RI stimulations. Indeed, recent studies using pharmacological agents and mast cells derived from gene-targeted mice showed very similar signaling requirements for IgE(−Ag)- and IgE+Ag-induced biological outcomes (refer to Refs. 15, 16, 55 for detailed reviews of IgE+Ag-induced signaling): degranulation induced by these stimuli depends on Syk; production of IL-6 and TNF-α also requires Syk; Fc∊RI internalization requires Lyn but not other Src family PTKs, Syk, or Btk; optimal adhesion via fibronectin requires Lyn and Syk but not Fyn; and migration toward HC IgEs or Ag depends heavily on Lyn and Syk and Fyn, PKCβ, PKC∊, and PKCθ to a lesser extent (24, 28, 29, 44, 56). However, quantitative differences were observed for the required signaling molecules in the IgE(−Ag)- and IgE+Ag-induced biological consequences: for instance, the Lyn requirement for HC IgE-induced adhesion is almost absolute, but the role of Lyn for IgE+Ag-induced adhesion is at best contributory (56). Qualitative differences were also observed between the two stimulation modes: concentrations of intracellular Ca2+ are increased transiently by HC IgEs (26-28). A recent study showed that Ca2+ responses induced by SPE-7 IgE are suppressed by SK&F 96365, a specific inhibitor of store-operated Ca2+ channel but not by broad-spectrum Ca2+ channel inhibitors, La3+ or Gd3+, whereas Ca2+ responses induced by IgE+Ag are suppressed by these three inhibitors (57). These results suggest that IgE(−Ag) and IgE+Ag induce Ca2+ influx by different mechanisms (Fig. 2).
FIGURE 2.
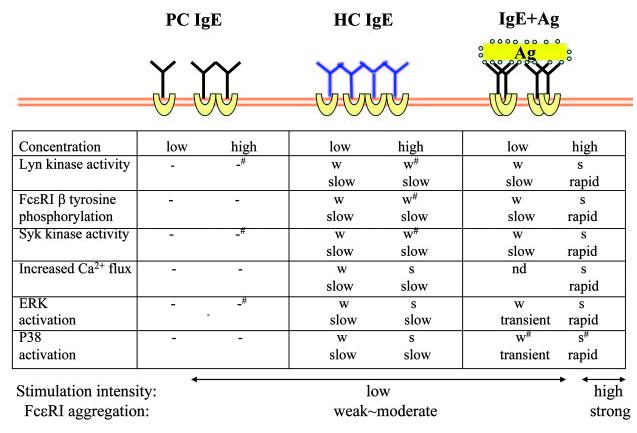
Comparison of signaling events induced by different modes of Fc∊RI stimulation. Data with mouse BMMCs are summarized. Concentrations of stimuli are as follows: PC IgE (H1 DNP-∊-206) and HC IgE (SPE-7), 0.5 μg/ml (low) and 5 μg/ml (high); IgE+Ag (DNP21-BSA), 1 ng/ml (low) and 100 ng/ml (high). -, not detected; -#, very weak; W, weak; W#, weak∼moderate; S#, moderate∼ strong; and S, strong. References: Lyn kinase activity (14, 21-24, 44); Fc∊RI β tyrosine phosphorylation (14, 21, 22, 24); Syk kinase activity (14, 21, 22, 24, 43, 44, 50); increased Ca2+ flux (14, 21, 23, 26, 27, 57); ERK activity (14, 21, 22, 24, 43, 50, 51); and p38 activity (14, 21, 22, 24).
A recent study showed that Bcl-2 and Bim, anti- and proapoptotic members of the Bcl-2 family proteins, respectively, antagonizes and promotes mast cell apoptosis induced by growth factor withdrawal (58). The major survival pathway induced by IgE(−Ag) appears to be dissected into two consecutive events: the Fc∊RI-activated signaling pathway leading to autocrine secretion of IL-3 and then activation of IL-3R signaling pathways leading to survival (51). Although details still remain to be worked out, the initial signaling events seem to include the activation of Lyn and Syk (24) leading to the activation of ERK. Interestingly, however, Fyn, Gab2, PI3K p85, or Akt are not required for IgE(−Ag)-mediated survival (51). Downstream of IL-3R signaling, Stat5 was shown to be important for mast cell survival (59). Expression of Bcl-xL and Bcl-2 is induced in an IL-3-dependent manner. Importantly, Bcl-xL can support mast cell survival in IL-3−/− mast cells (51).
IgE+Ag stimulation can also enhance mast cell survival under certain conditions (44): IgE+Ag prevents apoptosis of MC9 mast cells by an autocrine mechanism, producing IL-3, IL-4, and GM-CSF (60). Although secretion of endogenous IL-3 and GM-CSF is not sufficient for MC9 survival, IL- 4 renders the cells reactive to these cytokines. IgE+Ag can induce enhanced expression of FLIP, a caspase-8 inhibitor, and consequently a resistance to Fas-induced apoptosis (61). An anti-apoptotic member of the Bcl-2 family, A1, was shown to be important for the IgE+Ag-induced survival effects (62). IgE+Ag stimulation induces the expression of not only anti-apoptotic proteins such as A1 and Bcl-xL but also proapoptotic proteins such as BimEL and BimL (58). However, it remains to be examined whether these proteins with opposing functions contribute to IgE(−Ag)-induced mast cell survival. An IL-3-independent pathway used by IgE(−Ag) remains enigmatic, but it requires Syk because no survival effects of mIgEs were seen in syk−/− cells (24).
In vivo effects of IgE(−Ag) stimulation and their implications in diseases
IgE(−Ag) effects on survival and activation may not represent mere in vitro artifacts in mouse mast cells, although the definitive in vivo evidence for these phenomena has yet to come. However, there are several in vivo observations that support roles for IgE in mast cell survival and activation. Increased serum IgE levels in mice transplanted with IgE-producing hybridomas were correlated with increased mast cell numbers in some mucosal tissues (24). However, the presence of IgE is not required for the development of mast cells (63), and no increase in mast cell numbers was reported in IgE-transgenic mice (64). These results do not contradict the IgE(−Ag) effects on mast cell survival because relatively high concentrations of IgE are required for survival effects. These observations suggest that IgE levels do not determine the homeostasis of mast cell populations in vivo under physiological conditions. By contrast, IgE might play a critical role in maintaining mastocytosis during parasite infection. Infection with the parasitic helminth, Trichinella spiralis, elicits a vigorous IgE response and pronounced intestinal and splenic mastocytosis in wild-type mice. However, splenic mastocytosis was diminished in IgE−/− mice while similar levels of eosinophilia and jejunal mastocytosis occurred in wild-type and mutant mice (48). Mast cells in T. spiralis-infected mice migrate from the gut to the spleen and die there by apoptosis. Therefore, high-level IgEs seem essential to protect mast cells from apoptosis in the infected spleens. Furthermore, mucosal mastocytosis is impaired in IL-3−/− mice during nematode (Strongyloides venezuelensis) infection (5). These observations fit well with the IL-3 autocrine mechanism for IgE(−Ag)-induced mast cell survival. Therefore, under conditions where IgE may reach very high levels, e.g., parasite infection (53.3 μg/ml serum IgE in T. spiralis-infected mice (48)) and atopy (sometimes >100 μg/ml in NC/Nga mice (65)), tissue density of mast cells could be regulated in part by the survival (and proliferative) effects of IgE. However, the potential involvement of Ag, for which IgEs are specific, in the role for IgEs in enhancing mast cell survival has not been addressed in parasite infections, although much of the IgE that is produced in response to nematode infections is found not to be specific to the parasite (66). Additionally, contributions to mast cell survival by other factors, e.g., cytokines and chemokines, have not been known in the above parasite experiments.
A recent study showed that mast cell accumulation can be induced by epicutaneous application of HC IgEs and IgE+Ag (29). These results suggest that HC IgEs (in the absence of specific Ag) and IgE+Ag can induce accumulation of mast cells without prior inflammation. However, it is conceivable that, in allergic individuals, some inflammatory reactions such as the infiltration of helper T cells have occurred when IgE synthesis in B cells takes place at mucosal sites in response to Ag exposure (67-72). Given the vast variety of proinflammatory mediators secreted from activated mast cells (73), IgE(−Ag)- and IgE+Ag-induced mast cell accumulation would amplify inflammatory reactions by recruiting other immune cells such as T cells, eosinophils, monocytes, and neutrophils: for instance, histamine plays an important role in the pathogenesis of atopic asthma by enhancing the secretion of Th2 cytokines and inhibiting the production of Th1 cytokines (74). Leukotriene B4 recruits T cells and myeloid cells (75-77), and mast cell-produced cytokines and chemokines can recruit T cells, eosinophils, monocytes, and neutrophils. CC chemokine transcripts coding for I-309/CCL1, MIP-1α/CCL3 MIP-1β/CCL4, and MCP-3/CCL7 are among the most dramatically enhanced in IgE+Ag-stimulated mast cells (78). The ability of HC IgEs to attract mast cells suggests that this amplification of inflammation can last as long as local IgE synthesis continues even after the elimination of Ag. Therefore, IgEs in the presence as well as absence of allergen are implicated in mast cell accumulation at allergic tissue sites with local high IgE levels.
Importantly, IgE was shown to be essential for optimal sensitization in contact sensitivity but not for the elicitation phase, unlike mast cells, which are required for both the sensitization and elicitation phases (79). Contact sensitivity was markedly impaired in IgE−/− mice but was restored by either transfer of sensitized cells from wild-type mice or administration of hapten-irrelevant IgEs (including two HC IgEs) before sensitization. IgE was required for generating an appropriate cytokine milieu for immunization. IL-6, IL-1β, and MCP-1/CCL2 (and weakly TNF-α) can replace IgE in sensitization. However, it is not clear in this study whether or how IgE-mast cell interactions are required for the oxazolone sensitization, whether specific endogenous Ag interacts with IgE, or whether PC IgEs can restore responses in IgE−/− mice.
Several studies indicate the ability of IgE to enhance mast cell and basophil function in both mouse and human cells (20, 80, 81). Together with the enhanced cell survival, a positive feedback-loop hypothesis was proposed to explain inflammatory situations often found in atopic diseases associated with high-serum IgE levels: ↑ IgE → ↑ Fc∊RI, ↑ mast cells → ↑ Ag-, IgE-, and Fc∊RI-dependent release of IL-4, IL-13, MIP-1α/CCL3, and so on → ↑ IgE. Now, studies have been extended to human monoclonal IgEs: human umbilical-cord-blood-derived mast cells incubated with IgE synthesize and release I-309/CCL1 (a ligand for CCR8 responsible for chemoattraction of Th2 cells), GM-CSF, and MIP-1α/CCL3 (82).
The distinction between HC and PC IgEs is reminiscent of the dichotomy found in human IgE molecules in their ability to prime basophils for the stimulation with histamine-releasing factor, a cytokine produced by macrophages and platelets: basophils bound by IgEs (termed IgE+) derived from atopic patients, but not those (termed IgE−) from normal subjects, can release histamine and cytokines such as IL-4 and IL-13 in response to histamine-releasing factor (83, 84). Structural basis for the difference between IgE+ and IgE− is not known yet. Interestingly, unusual usages of VH regions have been noted in allergic individuals: VH5, one of the smallest gene families containing two members out of the total of 52 functional VH genes, is overrepresented in the IgE from allergic patients (85). These observations together indicate the structural and functional heterogeneity among human IgE molecules. Therefore, it is interesting to study the human IgE heterogeneity and its relevance to human atopic and other diseases.
Conclusions
Although numerous unanswered issues still remain (Table I), IgE(−Ag) effects on mast cell survival and activation have been well established. Several reports summarized in the previous section also presage future directions. Particularly, translational research together with basic mechanistic studies should be encouraged. Such studies will lead to a better understanding and, hopefully, better treatment of asthma, other allergic diseases, and host responses to parasite infections. Additionally, we hope this review stimulate studies in related fields, e.g., studies on effects of IgG-FcγRs.
Acknowledgments
We thank Ralph Kubo and two anonymous reviewers for critical reading of the manuscript.
Footnotes
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
This study was supported by Public Health Service Grants AI50209 from the National Institutes of Health (to T.K.).
Abbreviations used in this paper: PTK, protein tyrosine kinase; BMMC, bone marrow-derived mast cell; HC, highly cytokinergic; PC, poorly cytokinergic.
References
- 1.Galli SJ, Maurer M, Lantz CS. Mast cells as sentinels of innate immunity. Curr. Opin. Immunol. 1999;11:53–59. doi: 10.1016/s0952-7915(99)80010-7. [DOI] [PubMed] [Google Scholar]
- 2.Malaviya R, Ikeda T, Ross E, Abraham SN. Mast cell modulation of neutrophil influx and bacterial clearance at sites of infection through TNF-α. Nature. 1996;381:77–80. doi: 10.1038/381077a0. [DOI] [PubMed] [Google Scholar]
- 3.Echtenacher B, Mannel DN, Hultner L. Critical protective role of mast cells in a model of acute septic peritonitis. Nature. 1996;381:75–77. doi: 10.1038/381075a0. [DOI] [PubMed] [Google Scholar]
- 4.Matsuda H, Watanabe N, Kiso Y, Hirota S, Ushio H, Kannan Y, Azuma M, Koyama H, Kitamura Y. Necessity of IgE antibodies and mast cells for manifestation of resistance against larval Haemaphysalis longicornis ticks in mice. J. Immunol. 1990;144:259–262. [PubMed] [Google Scholar]
- 5.Lantz CS, Boesiger J, Song CH, Mach N, Kobayashi T, Mulligan RC, Nawa Y, Dranoff G, Galli SJ. Role for interleukin-3 in mast-cell and basophil development and in immunity to parasites. Nature. 1998;392:90–93. doi: 10.1038/32190. [DOI] [PubMed] [Google Scholar]
- 6.Gurish MF, Austen KF. The diverse roles of mast cells. J. Exp. Med. 2001;194:F1–F5. doi: 10.1084/jem.194.1.f1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Galli SJ, Nakae S, Tsai M. Mast cells in the development of adaptive immune responses. Nat. Immunol. 2005;6:135–142. doi: 10.1038/ni1158. [DOI] [PubMed] [Google Scholar]
- 8.Secor VH, Secor WE, Gutekunst CA, Brown MA. Mast cells are essential for early onset and severe disease in a murine model of multiple sclerosis. J. Exp. Med. 2000;191:813–822. doi: 10.1084/jem.191.5.813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee DM, Friend DS, Gurish MF, Benoist C, Mathis D, Brenner MB. Mast cells: a cellular link between autoantibodies and inflammatory arthritis. Science. 2002;297:1689–1692. doi: 10.1126/science.1073176. [DOI] [PubMed] [Google Scholar]
- 10.Biedermann T, Kneilling M, Mailhammer R, Maier K, Sander CA, Kollias G, Kunkel SL, Hultner L, Rocken M. Mast cells control neutrophil recruitment during T cell-mediated delayed-type hypersensitivity reactions through tumor necrosis factor and macrophage inflammatory protein 2. J. Exp. Med. 2000;192:1441–1452. doi: 10.1084/jem.192.10.1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Coussens LM, Raymond WW, Bergers G, Laig-Webster M, Behrendtsen O, Werb Z, Caughey GH, Hanahan D. Inflammatory mast cells up-regulate angiogenesis during squamous epithelial carcinogenesis. Genes Dev. 1999;13:1382–1397. doi: 10.1101/gad.13.11.1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hara M, Ono K, Hwang MW, Iwasaki A, Okada M, Nakatani K, Sasayama S, Matsumori A. Evidence for a role of mast cells in the evolution to congestive heart failure. J. Exp. Med. 2002;195:375–381. doi: 10.1084/jem.20002036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kinet JP. The high-affinity IgE receptor (Fc∊RI): from physiology to pathology. Annu. Rev. Immunol. 1999;17:931–972. doi: 10.1146/annurev.immunol.17.1.931. [DOI] [PubMed] [Google Scholar]
- 14.Parravicini V, Gadina M, Kovarova M, Odom S, Gonzalez-Espinosa C, Furumoto Y, Saitoh S, Samelson LE, O'Shea JJ, Rivera J. Fyn kinase initiates complementary signals required for IgE-dependent mast cell degranulation. Nat. Immunol. 2002;3:741–748. doi: 10.1038/ni817. [DOI] [PubMed] [Google Scholar]
- 15.Turner H, Kinet JP. Signalling through the high-affinity IgE receptor Fc∊RI. Nature. 1999;402:B24–B30. doi: 10.1038/35037021. [DOI] [PubMed] [Google Scholar]
- 16.Kawakami T, Galli SJ. Regulation of mast-cell and basophil function and survival by IgE. Nat. Rev. Immunol. 2002;2:773–786. doi: 10.1038/nri914. [DOI] [PubMed] [Google Scholar]
- 17.Wedemeyer J, Tsai M, Galli SJ. Roles of mast cells and basophils in innate and acquired immunity. Curr. Opin. Immunol. 2000;12:624–631. doi: 10.1016/s0952-7915(00)00154-0. [DOI] [PubMed] [Google Scholar]
- 18.Furuichi K, Rivera J, Isersky C. The receptor for immunoglobulin E on rat basophilic leukemia cells: effect of ligand binding on receptor expression. Proc. Natl. Acad. Sci. USA. 1985;82:1522–1525. doi: 10.1073/pnas.82.5.1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hsu C, MacGlashan D., Jr. IgE antibody up-regulates high affinity IgE binding on murine bone marrow-derived mast cells. Immunol. Lett. 1996;52:129–134. doi: 10.1016/0165-2478(96)02599-0. [DOI] [PubMed] [Google Scholar]
- 20.Yamaguchi M, Lantz CS, Oettgen HC, Katona IM, Fleming T, Miyajima I, Kinet JP, Galli SJ. IgE enhances mouse mast cell Fc∊RI expression in vitro and in vivo: evidence for a novel amplification mechanism in IgE-dependent reactions. J. Exp. Med. 1997;185:663–672. doi: 10.1084/jem.185.4.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Asai K, Kitaura J, Kawakami Y, Yamagata N, Tsai M, Carbone DP, Liu FT, Galli SJ, Kawakami T. Regulation of mast cell survival by IgE. Immunity. 2001;14:791–800. doi: 10.1016/s1074-7613(01)00157-1. [DOI] [PubMed] [Google Scholar]
- 22.Kalesnikoff J, Huber M, Lam V, Damen JE, Zhang J, Siraganian RP, Krystal G. Monomeric IgE stimulates signaling pathways in mast cells that lead to cytokine production and cell survival. Immunity. 2001;14:801–811. doi: 10.1016/s1074-7613(01)00159-5. [DOI] [PubMed] [Google Scholar]
- 23.Tanaka S, Takasu Y, Mikura S, Satoh N, Ichikawa A. Antigen-independent induction of histamine synthesis by immunoglobulin E in mouse bone marrow-derived mast cells. J. Exp. Med. 2002;196:229–235. doi: 10.1084/jem.20012037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kitaura J, Song J, Tsai M, Asai K, Maeda-Yamamoto M, Mocsai A, Kawakami Y, Liu FT, Lowell CA, Barisas BG, Galli SJ, Kawakami T. Evidence that IgE molecules mediate a spectrum of effects on mast cell survival and activation via aggregation of the Fc∊RI. Proc. Natl. Acad. Sci. USA. 2003;100:12911–12916. doi: 10.1073/pnas.1735525100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yamada N, Matsushima H, Tagaya Y, Shimada S, Katz SI. Generation of a large number of connective tissue type mast cells by culture of murine fetal skin cells. J. Invest. Dermatol. 2003;121:1425–1432. doi: 10.1046/j.1523-1747.2003.12613.x. [DOI] [PubMed] [Google Scholar]
- 26.Oka T, Hori M, Tanaka A, Matsuda H, Karaki H, Ozaki H. IgE alone-induced actin assembly modifies calcium signaling and degranulation in RBL-2H3 mast cells. Am. J. Physiol. 2004;286:C256–C263. doi: 10.1152/ajpcell.00197.2003. [DOI] [PubMed] [Google Scholar]
- 27.Pandey V, Mihara S, Fensome-Green A, Bolsover S, Cockcroft S. Monomeric IgE stimulates NFAT translocation into the nucleus, a rise in cytosol Ca2+, degranulation, and membrane ruffling in the cultured rat basophilic leukemia-2H3 mast cell line. J. Immunol. 2004;172:4048–4058. doi: 10.4049/jimmunol.172.7.4048. [DOI] [PubMed] [Google Scholar]
- 28.Lam V, Kalesnikoff J, Lee CW, Hernandez-Hansen V, Wilson BS, Oliver JM, Krystal G. IgE alone stimulates mast cell adhesion to fibronectin via pathways similar to those used by IgE + antigen but distinct from those used by Steel factor. Blood. 2003;102:1405–1413. doi: 10.1182/blood-2002-10-3176. [DOI] [PubMed] [Google Scholar]
- 29.Kitaura J, Kinoshita T, Matsumoto M, Chung S, Kawakami Y, Leitges M, Wu D, Lowell CA, Kawakami T. IgE- and IgE+Ag-mediated mast cell migration in an autocrine/paracrine fashion. Blood. 2005;105:3222–3229. doi: 10.1182/blood-2004-11-4205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kawanishi H. Role of IgE as a mast cell development co-factor in the differentiation of murine gut-associated mast cells in vitro. Eur. J. Immunol. 1986;16:689–692. doi: 10.1002/eji.1830160617. [DOI] [PubMed] [Google Scholar]
- 31.Beaven MA, Metzger H. Signal transduction by Fc receptors: the Fc∊RI case. Immunol. Today. 1993;14:222–226. doi: 10.1016/0167-5699(93)90167-j. [DOI] [PubMed] [Google Scholar]
- 32.Ullrich A, Schlessinger J. Signal transduction by receptors with tyrosine kinase activity. Cell. 1990;61:203–212. doi: 10.1016/0092-8674(90)90801-k. [DOI] [PubMed] [Google Scholar]
- 33.Heldin CH. Dimerization of cell surface receptors in signal transduction. Cell. 1995;80:213–223. doi: 10.1016/0092-8674(95)90404-2. [DOI] [PubMed] [Google Scholar]
- 34.Schweitzer-Stenner R, Pecht I. Cutting edge: death of a dogma or enforcing the artificial: monomeric IgE binding may initiate mast cell response by inducing its receptor aggregation. J. Immunol. 2005;174:4461–4464. doi: 10.4049/jimmunol.174.8.4461. [DOI] [PubMed] [Google Scholar]
- 35.Liu FT, Bohn JW, Ferry EL, Yamamoto H, Molinaro CA, Sherman LA, Klinman NR, Katz DH. Monoclonal dinitrophenyl-specific murine IgE antibody: preparation, isolation, and characterization. J. Immunol. 1980;124:2728–2737. [PubMed] [Google Scholar]
- 36.Eshhar Z, Ofarim M, Waks T. Generation of hybridomas secreting murine reaginic antibodies of anti-DNP specificity. J. Immunol. 1980;124:775–780. [PubMed] [Google Scholar]
- 37.James LC, Roversi P, Tawfik DS. Antibody multispecificity mediated by conformational diversity. Science. 2003;299:1362–1367. doi: 10.1126/science.1079731. [DOI] [PubMed] [Google Scholar]
- 38.Ohnishi K, Melchers F. The nonimmunoglobulin portion of λ5 mediates cell-autonomous pre-B cell receptor signaling. Nat. Immunol. 2003;4:849–856. doi: 10.1038/ni959. [DOI] [PubMed] [Google Scholar]
- 39.Charles N, Monteiro RC, Benhamou M. p28, a novel IgE receptor-associated protein, is a sensor of receptor occupation by its ligand in mast cells. J. Biol. Chem. 2004;279:12312–12318. doi: 10.1074/jbc.M309456200. [DOI] [PubMed] [Google Scholar]
- 40.Takagi M, Nakahata T, Koike K, Kobayashi T, Tsuji K, Kojima S, Hirano T, Miyajima A, Arai K, Akabane T. Stimulation of connective tissue-type mast cell proliferation by cross-linking of cell-bound IgE. J. Exp. Med. 1989;170:233–244-244. doi: 10.1084/jem.170.1.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Field KA, Holowka D, Baird B. Fc∊RI-mediated recruitment of p53/56lyn to detergent-resistant membrane domains accompanies cellular signaling. Proc. Natl. Acad. Sci. USA. 1995;92:9201–9205. doi: 10.1073/pnas.92.20.9201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Field KA, Holowka D, Baird B. Compartmentalized activation of the high affinity immunoglobulin E receptor within membrane domains. J. Biol. Chem. 1997;272:4276–4280. doi: 10.1074/jbc.272.7.4276. [DOI] [PubMed] [Google Scholar]
- 43.Sakurai D, Yamasaki S, Arase K, Park SY, Arase H, Konno A, Saito T. Fc∊RIγ-ITAM is differentially required for mast cell function in vivo. J. Immunol. 2004;172:2374–2381. doi: 10.4049/jimmunol.172.4.2374. [DOI] [PubMed] [Google Scholar]
- 44.Kitaura J, Xiao W, Maeda-Yamamoto M, Kawakami Y, Lowell CA, Kawakami T. Early divergence of Fc∊ receptor I signals for receptor up-regulation and internalization from degranulation, cytokine production, and survival. J. Immunol. 2004;173:4317–4323. doi: 10.4049/jimmunol.173.7.4317. [DOI] [PubMed] [Google Scholar]
- 45.Borkowski TA, Jouvin MH, Lin SY, Kinet JP. Minimal requirements for IgE-mediated regulation of surface Fc∊RI. J. Immunol. 2001;167:1290–1296. doi: 10.4049/jimmunol.167.3.1290. [DOI] [PubMed] [Google Scholar]
- 46.Kubo S, Matsuoka K, Taya C, Kitamura F, Takai T, Yonekawa H, Karasuyama H. Drastic up-regulation of Fc∊ri on mast cells is induced by IgE binding through stabilization and accumulation of Fc∊ri on the cell surface. J. Immunol. 2001;167:3427–3434. doi: 10.4049/jimmunol.167.6.3427. [DOI] [PubMed] [Google Scholar]
- 47.Iemura A, Tsai M, Ando A, Wershil BK, Galli SJ. The c -kit ligand, stem cell factor, promotes mast cell survival by suppressing apoptosis. Am. J. Pathol. 1994;144:321–328. [PMC free article] [PubMed] [Google Scholar]
- 48.Gurish MF, Bryce PJ, Tao H, Kisselgof AB, Thornton EM, Miller HR, Friend DS, Oettgen HC. IgE enhances parasite clearance and regulates mast cell responses in mice infected with Trichinella spiralis. J. Immunol. 2004;172:1139–1145. doi: 10.4049/jimmunol.172.2.1139. [DOI] [PubMed] [Google Scholar]
- 49.Werlen G, Hausmann B, Naeher D, Palmer E. Signaling life and death in the thymus: timing is everything. Science. 2003;299:1859–1863. doi: 10.1126/science.1067833. [DOI] [PubMed] [Google Scholar]
- 50.Yamasaki S, Ishikawa E, Kohno M, Saito T. The quantity and duration of FcRγ signals determine mast cell degranulation and survival. Blood. 2004;103:3093–3101. doi: 10.1182/blood-2003-08-2944. [DOI] [PubMed] [Google Scholar]
- 51.Kohno M, Yamasaki S, Tybulewicz VL, Saito T. Rapid and large amount of autocrine IL-3 production is responsible for mast cell survival by IgE in the absence of antigen. Blood. 2005;105:2059–2065. doi: 10.1182/blood-2004-07-2639. [DOI] [PubMed] [Google Scholar]
- 52.Wershil BK, Wang ZS, Gordon JR, Galli SJ. Recruitment of neutrophils during IgE-dependent cutaneous late phase reactions in the mouse is mast cell-dependent: partial inhibition of the reaction with antiserum against tumor necrosis factor α. J. Clin. Invest. 1991;87:446–453. doi: 10.1172/JCI115016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Laffargue M, Calvez R, Finan P, Trifilieff A, Barbier M, Altruda F, Hirsch E, Wymann MP. Phosphoinositide 3-kinase γ is an essential amplifier of mast cell function. Immunity. 2002;16:441–451. doi: 10.1016/s1074-7613(02)00282-0. [DOI] [PubMed] [Google Scholar]
- 54.Jolly PS, Bektas M, Olivera A, Gonzalez-Espinosa C, Proia RL, Rivera J, Milstien S, Spiegel S. Transactivation of sphingosine-1-phosphate receptors by Fcvar∊RI triggering is required for normal mast cell degranulation and chemotaxis. J. Exp. Med. 2004;199:959–970. doi: 10.1084/jem.20030680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Siraganian RP. Mast cell signal transduction from the high-affinity IgE receptor. Curr. Opin. Immunol. 2003;15:639–646. doi: 10.1016/j.coi.2003.09.010. [DOI] [PubMed] [Google Scholar]
- 56.Kitaura J, Eto K, Kinoshita T, Kawakami Y, Leitges M, Lowell CA, Kawakami T. Regulation of highly cytokinergic IgE-induced mast cell adhesion by Src, Syk, Tec, and protein kinase C family kinases. J. Immunol. 2005;174:4495–4504. doi: 10.4049/jimmunol.174.8.4495. [DOI] [PubMed] [Google Scholar]
- 57.Tanaka S, Mikura S, Hashimoto E, Sugimoto Y, Ichikawa A. Ca2+ influx-mediated histamine synthesis and IL-6 release in mast cells activated by monomeric IgE. Eur. J. Immunol. 2005;35:460–468. doi: 10.1002/eji.200425622. [DOI] [PubMed] [Google Scholar]
- 58.Alfredsson J, Puthalakath H, Martin H, Strasser A, Nilsson G. Proapoptotic Bcl-2 family member Bim is involved in the control of mast cell survival and is induced together with Bcl-xL upon IgE-receptor activation. Cell Death Differ. 2005;12:136–144. doi: 10.1038/sj.cdd.4401537. [DOI] [PubMed] [Google Scholar]
- 59.Shelburne CP, McCoy ME, Piekorz R, Sexl V, Roh KH, Jacobs-Helber SM, Gillespie SR, Bailey DP, Mirmonsef P, Mann MN, et al. Stat5 expression is critical for mast cell development and survival. Blood. 2003;102:1290–1297. doi: 10.1182/blood-2002-11-3490. [DOI] [PubMed] [Google Scholar]
- 60.Yoshikawa H, Nakajima Y, Tasaka K. Glucocorticoid suppresses autocrine survival of mast cells by inhibiting IL-4 production and ICAM-1 expression. J. Immunol. 1999;162:6162–6170. [PubMed] [Google Scholar]
- 61.Yoshikawa H, Nakajima Y, Tasaka K. Enhanced expression of Fas-associated death domain-like IL-1-converting enzyme (FLICE)-inhibitory protein induces resistance to Fas-mediated apoptosis in activated mast cells. J. Immunol. 2000;165:6262–6269. doi: 10.4049/jimmunol.165.11.6262. [DOI] [PubMed] [Google Scholar]
- 62.Xiang Z, Ahmed AA, Moller C, Nakayama K, Hatakeyama S, Nilsson G. Essential role of the prosurvival bcl-2 homologue A1 in mast cell survival after allergic activation. J. Exp. Med. 2001;194:1561–1569. doi: 10.1084/jem.194.11.1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Oettgen HC, Martin TR, Wynshaw-Boris A, Deng C, Drazen JM, Leder P. Active anaphylaxis in IgE-deficient mice. Nature. 1994;370:367–370. doi: 10.1038/370367a0. [DOI] [PubMed] [Google Scholar]
- 64.Matsuoka K, Taya C, Kubo S, Toyama-Sorimachi N, Kitamura F, Ra C, Yonekawa H, Karasuyama H. Establishment of antigen-specific IgE transgenic mice to study pathological and immunobiological roles of IgE in vivo. Int. Immunol. 1999;11:987–994. doi: 10.1093/intimm/11.6.987. [DOI] [PubMed] [Google Scholar]
- 65.Matsuda H, Watanabe N, Geba GP, Sperl J, Tsudzuki M, Hiroi J, Matsumoto M, Ushio H, Saito S, Askenase PW, Ra C. Development of atopic dermatitis-like skin lesion with IgE hyperproduction in NC/Nga mice. Int. Immunol. 1997;9:461–466. doi: 10.1093/intimm/9.3.461. [DOI] [PubMed] [Google Scholar]
- 66.Pritchard DI, Hewitt C, Moqbel R. The relationship between immunological responsiveness controlled by T helper 2 lymphocytes and infections with parasitic helminths. Parasitology. 1997;115(Suppl):S33–S44. doi: 10.1017/s0031182097001996. [DOI] [PubMed] [Google Scholar]
- 67.Cameron LA, Durham SR, Jacobson MR, Masuyama K, Juliusson S, Gould HJ, Lowhagen O, Minshall EM, Hamid QA. Expression of IL-4, C∊ RNA, and I∊ RNA in the nasal mucosa of patients with seasonal rhinitis: effect of topical corticosteroids. J. Allergy Clin. Immunol. 1998;101:330–336. doi: 10.1016/s0091-6749(98)70244-1. [DOI] [PubMed] [Google Scholar]
- 68.Coker HA, Durham SR, Gould HJ. Local somatic hypermutation and class switch recombination in the nasal mucosa of allergic rhinitis patients. J. Immunol. 2003;171:5602–5610. doi: 10.4049/jimmunol.171.10.5602. [DOI] [PubMed] [Google Scholar]
- 69.Durham SR, Gould HJ, Thienes CP, Jacobson MR, Masuyama K, Rak S, Lowhagen O, Schotman E, Cameron L, Hamid QA. Expression of epsilon germ-line gene transcripts and mRNA for the epsilon heavy chain of IgE in nasal B cells and the effects of topical corticosteroid. Eur. J. Immunol. 1997;27:2899–2906. doi: 10.1002/eji.1830271123. [DOI] [PubMed] [Google Scholar]
- 70.Smurthwaite L, Walker SN, Wilson DR, Birch DS, Merrett TG, Durham SR, Gould HJ. Persistent IgE synthesis in the nasal mucosa of hay fever patients. Eur. J. Immunol. 2001;31:3422–3431. doi: 10.1002/1521-4141(200112)31:12<3422::aid-immu3422>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 71.Snow RE, Djukanovic R, Stevenson FK. Analysis of immunoglobulin E VH transcripts in a bronchial biopsy of an asthmatic patient confirms bias towards VH5, and indicates local clonal expansion, somatic mutation and isotype switch events. Immunology. 1999;98:646–651. doi: 10.1046/j.1365-2567.1999.00910.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ying S, Humbert M, Meng Q, Pfister R, Menz G, Gould HJ, Kay AB, Durham SR. Local expression of epsilon germline gene transcripts and RNA for the epsilon heavy chain of IgE in the bronchial mucosa in atopic and nonatopic asthma. J. Allergy Clin. Immunol. 2001;107:686–692. doi: 10.1067/mai.2001.114339. [DOI] [PubMed] [Google Scholar]
- 73.Galli SJ, Costa JJ. Mast-cell-leukocyte cytokine cascades in allergic inflammation. Allergy. 1995;50:851–862. doi: 10.1111/j.1398-9995.1995.tb02490.x. [DOI] [PubMed] [Google Scholar]
- 74.Packard KA, Khan MM. Effects of histamine on Th1/Th2 cytokine balance. Int. Immunopharmacol. 2003;3:909–920. doi: 10.1016/S1567-5769(02)00235-7. [DOI] [PubMed] [Google Scholar]
- 75.Goodarzi K, Goodarzi M, Tager AM, Luster AD, von Andrian UH. Leukotriene B4 and BLT1 control cytotoxic effector T cell recruitment to inflamed tissues. Nat. Immunol. 2003;4:965–973. doi: 10.1038/ni972. [DOI] [PubMed] [Google Scholar]
- 76.Ott VL, Cambier JC, Kappler J, Marrack P, Swanson BJ. Mast cell-dependent migration of effector CD8+ T cells through production of leukotriene B4. Nat. Immunol. 2003;4:974–981. doi: 10.1038/ni971. [DOI] [PubMed] [Google Scholar]
- 77.Tager AM, Bromley SK, Medoff BD, Islam SA, Bercury SD, Friedrich EB, Carafone AD, Gerszten RE, Luster AD. Leukotriene B4 receptor BLT1 mediates early effector T cell recruitment. Nat. Immunol. 2003;4:982–990. doi: 10.1038/ni970. [DOI] [PubMed] [Google Scholar]
- 78.Nakajima T, Inagaki N, Tanaka H, Tanaka A, Yoshikawa M, Tamari M, Hasegawa K, Matsumoto K, Tachimoto H, Ebisawa M, et al. Marked increase in CC chemokine gene expression in both human and mouse mast cell transcriptomes following Fc∊ receptor I cross-linking: an interspecies comparison. Blood. 2002;100:3861–3868. doi: 10.1182/blood-2002-02-0602. [DOI] [PubMed] [Google Scholar]
- 79.Bryce PJ, Miller ML, Miyajima I, Tsai M, Galli SJ, Oettgen HC. Immune sensitization in the skin is enhanced by antigen-independent effects of IgE. Immunity. 2004;20:381–392. doi: 10.1016/s1074-7613(04)00080-9. [DOI] [PubMed] [Google Scholar]
- 80.Yano K, Yamaguchi M, de Mora F, Lantz CS, Butterfield JH, Costa JJ, Galli SJ. Production of macrophage inflammatory protein-1α by human mast cells: increased anti-IgE-dependent secretion after IgE-dependent enhancement of mast cell IgE-binding ability. Lab. Invest. 1997;77:185–193. [PubMed] [Google Scholar]
- 81.Yamaguchi M, Sayama K, Yano K, Lantz CS, Noben-Trauth N, Ra C, Costa JJ, Galli SJ. IgE enhances Fc∊ receptor I expression and IgE-dependent release of histamine and lipid mediators from human umbilical cord blood-derived mast cells: synergistic effect of IL-4 and IgE on human mast cell Fc∊ receptor I expression and mediator release. J. Immunol. 1999;162:5455–5465. [PubMed] [Google Scholar]
- 82.Gilchrest H, Cheewatrakoolpong B, Billah M, Egan RW, Anthes JC, Greenfeder S. Human cord blood-derived mast cells synthesize and release I-309 in response to IgE. Life Sci. 2003;73:2571–2581. doi: 10.1016/s0024-3205(03)00607-6. [DOI] [PubMed] [Google Scholar]
- 83.MacDonald SM, Lichtenstein LM, Proud D, Plaut M, Naclerio RM, MacGlashan DW, Kagey-Sobotka A. Studies of IgE-dependent histamine releasing factors: heterogeneity of IgE. J. Immunol. 1987;139:506–512. [PubMed] [Google Scholar]
- 84.Schroeder JT, Lichtenstein LM, MacDonald SM. An immunoglobulin E-dependent recombinant histamine-releasing factor induces interleukin-4 secretion from human basophils. J. Exp. Med. 1996;183:1265–1270. doi: 10.1084/jem.183.3.1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gould HJ, Sutton BJ, Beavil AJ, Beavil RL, McCloskey N, Coker HA, Fear D, Smurthwaite L. The biology of IGE and the basis of allergic disease. Annu. Rev. Immunol. 2003;21:579–628. doi: 10.1146/annurev.immunol.21.120601.141103. [DOI] [PubMed] [Google Scholar]
- 86.Plaut M, Pierce JH, Watson CJ, Hanley-Hyde J, Nordan RP, Paul WE. Mast cell lines produce lymphokines in response to cross-linkage of Fc∊RI or to calcium ionophores. Nature. 1989;339:64–67. doi: 10.1038/339064a0. [DOI] [PubMed] [Google Scholar]