

Abstract
Among acetyltransferases, the MYST family enzyme Esa1p is distinguished for its essential function and contribution to transcriptional activation and DNA double-stranded break repair. Here we report that Esa1p also plays a key role in silencing RNA polymerase II (Pol II)-transcribed genes at telomeres and within the ribosomal DNA (rDNA) of the nucleolus. These effects are mediated through Esa1p's HAT activity and correlate with changes within the nucleolus. Esa1p is enriched within the rDNA, as is the NAD-dependent protein deacetylase Sir2p, and the acetylation levels of key Esa1p histone targets are reduced in the rDNA in esa1 mutants. Although mutants of both ESA1 and SIR2 have enhanced rates of rDNA recombination, esa1 effects are more modest yet result in distinct structural changes of rDNA chromatin. Surprisingly, increased expression of ESA1 can bypass the requirement for Sir2p in rDNA silencing, suggesting that these two enzymes with seemingly opposing activities both contribute to achieve optimal nucleolar chromatin structure and function.
INTRODUCTION
Histone modifications such as phosphorylation, methylation, acetylation, and deacetylation provide a mechanism by which chromatin structure is modulated to affect gene expression both positively and negatively (for reviews see Strahl and Allis, 2000; Iizuka and Smith, 2003; Kurdistani and Grunstein, 2003). The acetylation of lysine residues on histone N-terminal tails is classically linked to increases in gene expression and more directly to roles in transcriptional activation. By contrast, deacetylation of histones is most frequently correlated with transcriptionally silent chromatin. As more is learned about the enzymes catalyzing these modifications, the histone acetyltransferases (HATs) and histone deacetylases (HDACs), simple functional distinctions between acetylation and deacetylation of histones do not hold (Iizuka and Smith, 2003). For example, in several organisms, mutations in RPD3, a gene encoding a deacetylase, lead to increases in silencing rather than the expected decreases (DeRubertis et al., 1996; Rundlett et al., 1996). This brings forward the possibility that histone acetylation may play an important part in both silent and active chromatin. In addition, coactivator proteins, such as the histone acetyltransferases p300 and CBP and nuclear hormone receptors, appear to have roles in transcriptional repression through acetylation and association with particular binding partners (for examples, see Chen and Li, 1998; Waltzer and Bienz, 1998; Baluchamy et al., 2003; Girdwood et al., 2003; Rajabi et al., 2005). Thus, understanding of multiple roles for acetylation in controlling gene expression is expanding.
In Saccharomyces cerevisiae, there are three regions of the genome controlled by chromatin-mediated transcriptional silencing: telomeres, the silent mating-type loci HMR and HML, and the rDNA array. Many factors are important for transcriptional silencing at subsets of these loci including the four core histones, the repressor activator protein Rap1p, the Silent Information Regulator proteins, Sir1-4p, and other proteins with chromatin modifying activities (reviewed in Rusche et al., 2003; Fox and McConnell, 2005). Sir2p, an NAD-dependent histone deacetylase (Imai et al., 2000; Landry et al., 2000; Smith et al., 2000), is unique among the Sir proteins because it is important for silencing at all three loci (reviewed in Gottschling, 2000; Guarente, 2000; Shore, 2000; Moazed, 2001). Sir2p is part of the RENT complex and plays a role in rDNA silencing and rDNA chromatin structure that is mediated through its deacetylase activity (Bryk et al., 1997; Fritze et al., 1997; Smith and Boeke, 1997; Shou et al., 1999; Straight et al., 1999; Ghidelli et al., 2001). Sir2p also plays an important role in suppressing homologous recombination within the repetitive rDNA array, highlighting its contribution to chromatin structure at this locus (Gottlieb and Esposito, 1989). Studies of chromatin modification at silenced loci in yeast have been critical for the emerging understanding of how changes in chromatin structure lead to changes in gene expression.
Esa1p is an essential HAT in S. cerevisiae (Smith et al., 1998a; Clarke et al., 1999) that is a member of the MYST family that is distantly related to the GNAT family of HATs (reviewed in Neuwald and Landsman, 1997; Utley and Côté, 2003). There are two other nonessential MYST proteins in yeast, Sas2p and Sas3p. Sas2p is necessary for silencing at telomeres and together with Sir1p plays a role in silencing at the silent mating-type loci (Reifsnyder et al., 1996; Ehrenhofer-Murray et al., 1997), and it has been proposed that Sas2p and Sir2p act in opposition to one another (Kimura et al., 2002; Suka et al., 2002). Sas3p is the catalytic HAT component of the NuA3 complex, with roles in transcriptional activation through acetylation of histone H3 (Takechi and Nakayama, 1999; John et al., 2000).
Esa1p has a range of cellular functions distinct from those of other HATs. It primarily acetylates histone H4 and, to a limited extent H2A and H3, and is required for cell cycle progression (Smith et al., 1998a; Clarke et al., 1999). Several esa1 temperature-sensitive mutants display a conditional G2/M arrest, which is dependent upon the RAD9 DNA damage checkpoint, and these mutants fail to segregate their chromosomes properly. Under restrictive conditions, nucleolar structure is severely disrupted in esa1 mutants as revealed by electron microscopy and the overall level of histone H4 acetylation is reduced in the esa1 temperature-sensitive mutants under permissive and restrictive conditions (Clarke et al., 1999). Esa1p is the catalytic component of the 1.4-MDa NuA4 HAT complex that is important for transcriptional activation both in vitro and in vivo at specific target loci, including many ribosomal protein genes (reviewed in Doyon and Côté, 2004). Esa1p also functions in the highly active, smaller picNuA4 complex (Boudreault et al., 2003).
The NuA4 complex itself is broadly conserved (Doyon and Côté, 2004) and the human catalytic subunit Tip60 functions in nontranscriptional components of apoptosis and signaling processes (Ikura et al., 2000; Sheridan et al., 2001). Tip60 has been linked to DNA repair (Ikura et al., 2000) and a role for Esa1p and the Yng2p NuA4 subunit was found in replication-coupled DNA double-stranded break repair (Bird et al., 2002; Choy and Kron, 2002). Indeed, several subunits of NuA4 are shared with the Swr1 chromatin remodeling complex and are linked at additional levels to chromosomal stability (Kobor et al., 2004; Krogan et al., 2004; Mizuguchi et al., 2004).
Because Esa1p regulates global levels of acetylation in yeast and these modifications are not restricted to promoter-proximal regions (Clarke et al., 1999; Vogelauer et al., 2000), we considered that Esa1p may have additional roles in genomic regulation. We found that Esa1p contributes to locus-specific transcriptional silencing and appears to have a prominent role within the rDNA of the nucleolus. In particular, Esa1p is associated with the rDNA and affects its acetylation status. Our studies reveal a surprising connection between Esa1p and the NAD-dependent deacetylase Sir2p that also functions within the nucleolus. Sir2p is itself required for rDNA silencing and in all previous studies has been considered essential for this process. Yet, we find that modest increases in gene dosage of ESA1 can bypass the rDNA silencing defects of sir2 null mutants. This effect is reciprocal, although restricted to partially functional esa1 mutants. Together, these results demonstrate an increasingly universal role for Esa1p in genomic structure and function. In addition, these results suggest that seemingly opposing chromatin-modifying activities can both contribute to achieve optimal function.
MATERIALS AND METHODS
Structural Image Production
The Esa1p structural representation was generated using the RasMol program with Brookhaven file 1FY7 with the generous help of Dr. R. Dutnall.
Strain Construction and Genetic Methods
Strains used are listed in Table 1 and plasmids are listed in Table 2. The W303 esa1 mutant strains were generated by digesting pLP952 (esa1-414/URA3/integrating) with BstEII and digesting pLP949 (esa1-L254P/URA3/integrating) with SphI (New England Biolabs, Beverly, MA) and transforming these linearized plasmids into LPY5. These URA+ transformants were cultured on 5-FOA plates to identify cells that had excised the URA3 marker and either the ESA1 wild-type locus or the esa1 mutant locus. The 5-FOA–resistant, ura–colonies were tested for temperature sensitivity at 37°C to identify candidates in which the ESA1 locus had been excised. Strains LPY4679 and LPY4680 (esa1-414) and LPY5001 (esa1-L254P) were confirmed as such integrants. Subsequently, standard genetic analysis and molecular amplification were utilized to test for appropriate integration of the esa1 mutant alleles at the ESA1 locus. Molecular amplification was carried out with Taq polymerase (Promega, Madison, WI) following manufacturer instructions with primers ESAWAYUP 5′CTCACGTAACAGTTGGTG3′ and TAS1PST 5′TGCACTGCAGCGTGTGATATGTTCTCATC3′.
Table 1.
Strains used in this study
| Strain | Genotype | Source |
|---|---|---|
| LPY5 | MATa | This lab |
| LPY11 | MATasir2Δ::HIS3 | This lab |
| AMR53 | MATatop1-8::LEU2 | R. Sternglanz |
| LPY78 | MATα his4 | This lab |
| LPY2991 | MATahis3Δ200 leu2-3,112 trp1Δ1 ura3-52 esa1Δ::HIS3 + pLP795 | This lab |
| LPY3291 | MATahis3Δ200 leu2-3,112 trp1Δ1 ura3-52 esa1Δ::HIS3 + pLP863 | This lab |
| LPY3498 | MATahis3Δ200 leu2-3,112 trp1Δ1 ura3-52 | This lab |
| LPY3500 | MATahis3Δ200 leu2-3,112 trp1Δ1 ura3-52 esa1Δ::HIS3 esa1-L254P::URA3 | This lab |
| CCF101 | MATα hmr::TRP1 rDNA::ADE2-CAN1 TELVR::URA3 | K. Runge |
| LPY4679 | MATaesa1-414 | This study |
| LPY4680 | MATaesa1-414 | This study |
| LPY4801 | MATa/α +/esa1-414 +/hmr::TRP1 +/rDNA::ADE2-CAN1 +/TELVR::URA3 | This study |
| LPY4804 | MATa/α +/esa1-414 +/hmr::TRP1 +/rDNA::ADE2-CAN1 +/TELVR::URA3 | This study |
| LPY4819 | MATα hmr::TRP1 rDNA::ADE2-CAN1 TELVR::URA3 | This study |
| LPY4821 | MATα esa1-414 hmr::TRP1 rDNA::ADE2-CAN1 TELVR::URA3 | This study |
| LPY4848 | MATα esa1-414 hmr::TRP1 rDNA::ADE2-CAN1 TELVR::URA3 | This study |
| LPY4906 | MATa/α +/esa1-414 +/sir2Δ::HIS3 +/hmr::TRP1 +/rDNA::ADE2-CAN1+/TELVR::URA3 | This study |
| LPY4909 | MATα rDNA::ADE2-CAN1 | This study |
| LPY4911 | MATα esa1-414 rDNA::ADE2-CAN1 | This study |
| LPY4913 | MATα hmr::TRP1 | This study |
| LPY4915 | MATα esa1-414 hmr::TRP1 | This study |
| LPY4917 | MATα TELVR::URA3 | This study |
| LPY4919 | MATα esa1-414 TELVR::URA3 | This study |
| LPY4978 | MATα sir2Δ::HIS3 rDNA::ADE2-CAN1 | This study |
| LPY4980 | MATα sir2Δ::HIS3 hmr::TRP1 | This study |
| LPY5001 | MATaesa1-L254P | This study |
| LPY5017 | MATasir2Δ::TRP1 rDNA::ADE2-CAN1 TELVR::URA3 | This study |
| LPY5055 | MATα esa1-L254P rDNA::ADE2-CAN1 | This study |
| LPY5059 | MATα esa1-L254P hmr::TRP1 | This study |
| LPY5066 | MATα esa1-L254P hmr::TRP1 rDNA::ADE2-CAN1 TELVR::URA3 | This study |
| LPY5099 | MATa/α +/esa1-L254P +/hmr::TRP1 +/rDNA::ADE2-CAN1 +/TELVR::URA3 | This study |
| LPY5136 | MATα esa1-414 rDNA::ADE2-CAN1 + pLP1402 | This study |
| LPY5141 | MATα esa1-414 rDNA::ADE2-CAN1 + pLP796 | This study |
| LPY5143 | MATα esa1-414 rDNA::ADE2-CAN1 + pLP37 | This study |
| LPY5144 | MATα esa1-L254P rDNA::ADE2-CAN1 + pLP1402 | This study |
| LPY5150 | MATα esa1-L254P rDNA::ADE2-CAN1 + pLP796 | This study |
| LPY5151 | MATα esa1-L254P rDNA::ADE2-CAN1 + pLP37 | This study |
| LPY5159 | MATα sir2Δ::HIS3 rDNA::ADE2-CAN1 + pLP1402 | This study |
| LPY5164 | MATα sir2Δ::HIS3 rDNA::ADE2-CAN1 + pLP796 | This study |
| LPY5166 | MATα sir2Δ::HIS3 rDNA::ADE2-CAN1 + pLP37 | This study |
| LPY6181 | MATα TELVR::URA3 + pLP1347 | This study |
| LPY6182 | MATα TELVR::URA3 + pLP1348 | This study |
| LPY6184-7 | MATα TELVR::URA3 + pLP1349 | This study |
| LPY6189 | MATα esa1-414 TELVR::URA3 + pLP1347 | This study |
| LPY6190 | MATα esa1-414 TELVR::URA3 + pLP1348 | This study |
| LPY6192-5 | MATα esa1-414 TELVR::URA3 + pLP1349 | This study |
| LPY6217 | MATα rDNA::ADE2-CAN1 + pLP1347 | This study |
| LPY6219 | MATα rDNA::ADE2-CAN1 + pLP1348 | This study |
| LPY6220-3 | MATα rDNA::ADE2-CAN1 + pLP1349 | This study |
| LPY6225 | MATα esa1-414 rDNA::ADE2-CAN1 + pLP1347 | This study |
| LPY6227 | MATα esa1-414 rDNA::ADE2-CAN1 + pLP1348 | This study |
| LPY6228-31 | MATα esa1-414 rDNA::ADE2-CAN1 + pLP1349 | This study |
| LPY6244 | LPY5 with 3HA-ESA1 | This study |
All strains are in the W303 background with the genotype ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 ura3-1, and additional pertinent genotype information is noted. Exceptions are the mating tester strain LPY78 and the S288c background strains LPY2991, LPY3291, LPY3498, and LPY3500.
Table 2.
Plasmids used in this study
| Plasmid | Description | Source |
|---|---|---|
| pLP37 | SIR2/URA3/2-micron | This lab |
| pLP61 | TRP1/CEN vector | This lab |
| pLP690 | ESA1/TRP1/CEN | This lab |
| pLP796 | ESA1/URA3/2-micron | This lab |
| pLP948 | ESA1/URA3/integrating | This lab |
| pLP949 | esa1-L254P/URA3/integrating | This lab |
| pLP952 | esa1-414/URA3/integrating | This lab |
| pLP1070 | rDNA 5′-BglII fragment/PKC7 | S. Gasser |
| pLP1071 | rDNA 3′-BglII fragment/PKC7 | S. Gasser |
| pLP1237 | SIR2/LEU2/CEN | This lab |
| pLP1309 | PPR1/pUC8 | This lab |
| pLP1347 | TRP1/2-micron vector | S. Berger |
| pLP1348 | ESA1/TRP1/2-micron | S. Berger |
| pLP1402 | URA3/2-micron vector | This lab |
To generate strains marked at the three silenced loci in the W303 background, strains LPY4679, LPY4680, and LPY5001 were crossed with CCF101 (rDNA::ADE2-CAN1, TELVR::URA3, hmrΔRap1::TRP1), generating the diploids LPY4801, LPY4804, and LPY5099, respectively. LPY4801 was sporulated and dissected to yield strains LPY4819, LPY4821, LPY4909, LPY4911, LPY4913, and LPY4915. LPY4804 was sporulated and dissected to generate LPY4848. LPY5099 was sporulated and dissected to generate the strains LPY5055, LPY5059, and LPY5066. To generate sir2Δ::HIS3 mutant combinations with the three reporter genes, LPY4821 (esa1-414 triple-marked) was crossed with LPY11 (sir2Δ::HIS3) transformed with pLP1237 (SIR2/LEU2/CEN) to yield the diploid LPY4906. LPY4906 was sporulated and dissected to generate the strains LPY4978 and LPY4980. All sir2Δ strains generated no longer contain pLP1237.
Quantitative Mating and Silencing Assays
The MATa strains LPY3498 (ESA1), LPY3500 (esa1-L254P), LPY2991 (ESA1), and LPY3291 (esa1-414) were grown in YPD. Cultures in log phase were diluted 1:5000 in YPD, and 100 μl of each dilution was either plated on SC or mixed with 100 μl of a freshly growing MATα mating tester strain (LPY78) and spread onto minimal plates. Samples were plated in triplicate for each medium at 28°C. The ratio of the average number of colonies on the three minimal plates to the average number of total colonies on the three SC plates defines the mating efficiency of that culture. The wild-type mating efficiency was set to 1.00 and mutant mating efficiencies were normalized to the appropriate wild type. Mating efficiencies of all strains were measured in two independent experiments and are reported as their average.
Assays measuring telomeric silencing were carried out by incubating 2-ml YPD cultures for 4-5 d at 28°C and then plating the cultures in fivefold serial dilutions on synthetic complete medium (SC) and SC containing 5-FOA at 1 g/l. For transformants with TRP1 plasmids, tryptophan was left out of the medium to select for the plasmids. Plates were incubated at the indicated temperatures for 2-4 d, depending on media and temperature. Assays measuring HMR silencing were carried out by incubating 2-ml YPD cultures for 2-4 d at 28°C and plating these cultures on SC or SC without tryptophan at the indicated temperatures for 2-4 d, depending on the temperature.
Assays measuring rDNA silencing were carried out by incubating 2-ml cultures in liquid SC without adenine and arginine for 2-4 d at 28°C. For strains that were transformed with plasmids, cultures were grown in SC without adenine, arginine, and either tryptophan or uracil to select for the plasmids. Cultures were plated in fivefold serial dilutions on SC, SC without adenine, SC without adenine and arginine, and SC without adenine and arginine (except strains with plasmids, as above) but containing canavanine (Sigma, St. Louis, MO) at concentrations ranging from 8 to 60 μg/ml. Strains were tested for general canavanine sensitivity on SC containing 60 μg/ml canavanine without arginine.
Recombination Assays
Recombination assays were carried out by incubating cultures in medium lacking adenine and arginine to select for the ADE2 reporter gene in the rDNA array during logarithmic growth for several cell cycles (4-6 h). Cultures of different isolates were serially diluted 10-fold and then spread to 10 YPD plates with 500 cells each. Cell concentrations were estimated using the conversion of 2.0 × 107 cells per ml yields an A600 of 1.0. Plates were incubated at 28 or 33°C for 2 d, and colonies were counted. Approximately 3000-4000 total colonies for each isolate and experiment were counted. Plates were incubated at 4°C to allow red color development that occurs when cells become ade– due to loss of the reporter cassette. The number of half-sectored colonies was divided by the total number of colonies plated to measure the rate of recombination. This ratio provides the frequency of cells in a given population that had undergone a recombination event during the first cell division. Averages of 3-4 independent isolates for each temperature and strain are provided with standard deviations. Statistical analysis of these data (Supplementary Table S1) was performed using the Wilcoxon test (Wonnacott and Wonnacott, 1984) with the assistance of Dr. Vincent Carey (Harvard Medical School and Whitehead Institute).
Chromatin Immunoprecipitation
Immunoprecipitation of formaldehyde cross-linked chromatin was performed as described previously by Strahl-Bolsinger et al. (1997). Briefly, a 100-ml culture was grown to A600 = 1.0 and fixed with 0.86% formaldehyde for 30 min at room temperature. Harvested cells were washed twice with tris-buffered saline (20 mM Tris-HCl, pH 7.5, 200 mM NaCl) and lysed in 1.6 ml FA-lysis buffer (50 mM HEPES-KOH, pH 7.5, 140 mM NaCl, 1 mM EDTA, 0.5% Triton X-100, 0.1% sodium deoxycholate, and the protease inhibitors 1 mM phenylmethylsulfonyl fluoride, 1 mM benzamidine, 5 μg/ml TPCK, 2 μg/ml leupeptin and pepstatin) at 4°C by the glass bead method. The extract was sonicated to shear DNA and clarified by centrifugation at 16,000 × g for 5 min at 4°C. The clarified supernatant was incubated overnight with either α-HA (12CA5, BabCo), αH4AcLys5, αH4AcLys12, αH4AcLys16, or αH3Ac (Serotech) at 4°C. Immune complexes were immunoprecipitated with protein A Sepharose (Pharmacia Biotech, Piscataway, NJ). Precipitates were washed successively with 1 ml FA-lysis buffer, FA-lysis buffer with 0.5 M NaCl, LiCl solution (10 mM Tris-HCl, pH 8, 0.25 M LiCl, 0.5% NP-40, 0.5% sodium deoxycholate, 1 mM EDTA) and TE (10 mM Tris, pH 7.5, 1 mM EDTA) followed by RNase A and proteinase K treatment. After reversal of cross-linking, DNA was purified by phenol-chloroform extraction and recovered by ethanol precipitation. Precipitated DNA was analyzed by molecular amplification using the 25S-1, 25S-2, 5S, and NTS primers described in Gotta et al. (1997) and the POL1 primers described in Papamichos-Chronakis et al. (2002).
To generate the triple-HA–tagged ESA1 construct (ESA1-3HA), sequence immediately 3′ to the ESA1 ORF was amplified using Deep-Vent polymerase (New England Biolabs) and oligos T3 and D1-ESA1 (5′-CGCGTTTAAACGC-TATGTAGTTTCCTAAAC-3′). The amplification product was inserted into pFA6a-3HA-kanMX6 (Longtine et al., 1998) using PmeI and EcoRV (New England Biolabs) to construct pLP1452. The entire ESA1 coding region was amplified using oligos TAS-1059 (5′-AACATATCACACGAGGATG3′) and C-Bam-ESA1 (5′CTCGAGGATCCACCAGGCAAAGCGTAACT-3′). A PvuII PacI–digested fragment from pLP1455 was used to insert ESA1 in frame with 3HA in pLP1452 to generate pLP1453. To generate the ESA1-3HA strain, a SacII-SnaBI fragment of pLP1453 was transformed into LPY4819. Integrants that were G418 resistant were isolated and integration at the ESA1 locus was verified with oligos kanMX6-sense (5′-GATGACGAGCGTAATGGCTG-3′) and TAS1-2132 (5′-GGCTGTATATCTTAAGTAAG-3′) to generate LPY6934. Expression of Esa1-3HAp from its endogenous promoter was verified by SDS-PAGE and Western blotting analyses of whole-cell extracts.
Pulsed-Field Gel Electrophoresis and Southern Blotting
Sample preparation for pulsed-field gel electrophoresis was carried out, in part, as described (Birren and Lai, 1993). Briefly, the strains AMR53, LPY4909, and LPY4911 were grown in log phase in YPD for 24 h at 28 and 33°C and for 4 h at 37°C. Before sample preparation, portions of the cultures were reserved for flow cytometric analysis, viability determination, and standard genomic DNA preparation. The remainder was washed twice in 10 ml of CSE (20 mM citric acid, pH 5.6, 20 mM Na2HPO4, 50 mM EDTA, pH 8, 1.2 M sorbitol), resuspended in 10 ml CSE with 0.43 mg/ml zymolyase 70T (ICN), and incubated in a 37°C water bath for 1 h. Cells were pelleted and resuspended in TSE (10 mM Tris, pH 7.5, 45 mM EDTA, pH 8, 0.9 M sorbitol) at a concentration of 6 × 108 cells per ml. Cells were then prewarmed briefly at 37°C and mixed 1:1 with warmed (55°C) liquefied 1% Incert agarose (FMC) in TSE. Multiple 80-μl plugs were poured into a mold, and plugs were incubated in 5 ml of 0.25 M EDTA, pH 8, 50 mM Tris, pH 7.5, 1% SDS in sealed 50-ml conical tubes that were immersed in a 55°C water bath for 2 h. Plugs were transferred to fresh conical tubes and incubated in 5 ml of 1% lauryl sarcosine, 0.5 M EDTA, in a 55°C water bath for 72 h total. Fresh solutions of 5 ml of 1% lauryl sarcosine, 0.5 M EDTA, 1 mg/ml proteinase K were replaced every 24 h. Plugs were transferred to fresh 50-ml conical tubes and stored at 4°C in 5 ml of 1% lauryl sarcosine, 0.5 M EDTA.
Before electrophoresis, plugs were washed four times in TE over a 1-h period with gentle agitation. Gels containing 1% PFGE Seakem Gold agarose (FMC) in 0.5× TBE (89 mM Tris-borate, pH 8.3, 25 mM EDTA) were cast, and plugs were sealed in the wells with residual agarose. Gels were electrophoresed in chilled (4°C) 0.5× TBE for 72 h at 50 Volts (2 V/cm) with 30-min fixed pulse times. Gels were stained with ethidium bromide at a final concentration of 0.5 μg/ml for 30 min to visualize chromosomes.
To digest whole chromosomes with NotI, plugs were dialyzed in 50 ml TEN (10 mM Tris, pH 7.4, 50 mM NaCl) overnight with gentle agitation at room temperature. A single plug was then incubated in 250-μl final volume reactions with either no enzyme (mock) or 5 units of NotI (New England BioLabs) overnight at 37°C. After 8 h of digestion, 5 additional units of NotI were added to the reactions. BSA (bovine serum albumin) was added to NotI digests at a final concentration of 1 μg/ml.
To determine the relative number of rDNA repeats in a given sample, genomic DNA corresponding to a PFGE experiment was digested with Hind III or BglII, both of which liberate satellite genomic bands that were resolved on a standard 1% agarose gel stained with ethidium bromide. Satellite bands, corresponding to rDNA unit fragments, were normalized to nonsatellite resolved DNA and quantified using the Alpha Digital Imaging System 2000 software. This analysis yielded an estimate of the relative number of repeats between samples. In the HindIII-digested samples, the esa1-414 mutant possessed 98% of the number of repeats present in wild type, and in the BglII-digested samples the esa1-414 mutant possessed 84% of the number of repeats present in wild type, averaged together as 91% of the repeats in wild type. This small difference in repeat numbers cannot fully explain the mobility difference observed for the rDNA array in the esa1-414 mutant. For example, if there were 200 rDNA repeating units at 9.1 kb, the array size would be 1820 kb in wild-type cells and 1656 kb in esa1-414 cells, the difference of which is 164 kb. This size difference is not enough to explain the mobility differences observed for the entire chromosome XII nor for the NotI fragment containing the array between wild-type and esa1-414 mutants.
Pulsed-field gels were prepared for Southern blotting, in part, as described (Birren and Lai, 1993). Briefly, after gels were stained with ethidium bromide and photographed, each side of the gels was UV-irradiated in a Stratalinker apparatus (Stratagene, La Jolla, CA) at maximum output for 60 s. Gels were soaked in 0.4 M NaOH, 1.5 M NaCl for 15 min and transferred to Zetabind membrane (Cuno Laboratory Products) in 0.4 M NaOH, 1.5 M NaCl for 24-36 h. Blots were rinsed in 0.5 M Tris, pH 7.5, for 5 min, rinsed briefly in 2× SSC (333 mM NaCl, 30 mM sodium citrate), and dried, and each side of the blots were UV-irradiated in a Stratalinker (Stratagene) at 100 μJ. An rDNA/chromosome XII-specific probe for hybridization was prepared from pLP1070 by digestion with BglII to liberate a 4.5-kb fragment, which contains part of the rDNA repeat. pLP1309 was digested with BamHI and SalI to liberate a 2-kb fragment for use as a PPR1-specific probe for hybridization.
Indirect Immunofluorescence
Indirect immunofluorescence was carried out as described (Gotta et al., 1997; Stone et al., 2000). Briefly, cells were logarithmically grown in YPD for 4 h at either 28 or 37°C and fixed by adding paraformaldehyde to a final concentration of 3.3% at 30°C for 10 min. Cells were washed twice in YPD, resuspended at 1 ml per 0.1 g of cells in 0.1 M EDTA, KOH, pH 8.0, 10 mM DTT, and incubated at 30°C for 10 min with gentle agitation. Cells were resuspended at 1 ml per 0.1 g of cells in YPD containing 1.2 M sorbitol. Lyticase (ICN) was added to a 3000 U/ml final concentration, and samples were incubated at 30°C with gentle agitation for 30 min to form spheroplasts. Samples were diluted with 5 volumes of YPD, washed twice in 20 ml YPD containing 1.2 M sorbitol, resuspended in YPD at 3 ml per 0.1 g cells, and incubated on appropriate slides for 2 min. Slides were subsequently fixed in 100% methanol at –20°C for 6 min and in 100% acetone at –20°C for 1 min and blocked in PBST (phosphate-buffered saline [PBS], 0.1% Triton X-100) with 1% ovalbumin (Sigma) for 1 h. Slides were incubated with both the affinity-purified anti-Sir2p antibody (rabbit 2916, bleed 8, polyclonal; Smith et al., 1998b; Ersfeld and Stone, 2000) at 1:1 dilution and the anti-Nop1p antibody (mouse D77, monoclonal; Aris and Blobel, 1988) at 1:100 in PBST overnight at 37°C. Slides were washed five times in PBST. The secondary antibodies, FITC-conjugated goat anti-rabbit and Texas Red–conjugated goat anti-mouse (Jackson ImmunoResearch Laboratories, West Grove, PA) were diluted 1:50 in PBST and preincubated at 4°C for 30 min with cells from all samples, and the resulting supernatant was incubated on slides for 1 h at 37°C. Slides were then washed three times in PBST, stained with 0.25 mg/ml DAPI for 5 min, washed again, and mounted in 24 mg/ml DAPCO in PBS, 87% glycerol, pH 7.5. Staining was visualized with an Applied Precision Deltavision optical sectioning deconvolution microscope, Olympus IX70, with six deconvolved 0.2-μm sections. Deltavision version 2.10 software and a Princeton Instruments MicroMax camera were utilized to capture images.
RESULTS
A Structural Basis for esa1 Mutant Defects
To begin to investigate the function of Esa1p in transcriptional regulation, we focused on two temperature-sensitive esa1 mutants, esa1-L254P and esa1-414 (Clarke et al., 1999). From the crystal structure of Esa1p (Yan et al., 2000), the lesions in the two temperature-sensitive mutants are found to reside very near the highly conserved HAT domain (Figure 1B, purple). The leucine residue 254 (orange) is buried within helix α2, just four positions away from two residues that contact coenzyme-A (CoA). The esa1-414 lesion is a frameshift mutation resulting in 10 changed residues (amino acids 414-423, in blue, starting in helix α7) and the remaining C-terminal 22 residues deleted from 424 to 445 (black). The arginine residue 421 (blue spacefill), which is changed to glutamic acid in the esa1-414 mutant, normally contacts CoA. The highlighted glutamic acid residue 338 (red spacefill), between strand β10 and helix α4 is also an essential residue for catalysis (Yan et al., 2000) that by both structure and sequence is conserved among many HATs. A dominant mutant harboring a glutamine substitution at this position, ESA1-E338Q, interferes with growth in wild-type cells when overexpressed, mimicking the cell cycle arrest observed in esa1 mutants (Yan et al., 2000). Effects on transcriptional control, in either positive or negative directions, were investigated for the esa1-L254P and esa1-414 recessive temperature-sensitive mutants (below) and the ESA1-E338Q dominant HAT mutant (see Supplementary Figure S2).
Figure 1.

Mutants of ESA1 contain lesions in residues near and within the catalytic HAT domain. (A) A schematic box diagram of Esa1p highlights lesions (asterisks) residing near and within the highly conserved GNAT family HAT domain (purple) in three distinct esa1 mutants: esa1-L254P, esa1-E338Q, and esa1-414. The esa1-L254P and esa1-414 mutants are temperature-sensitive for growth above 34°C (Clarke et al., 1999) and the esa1-E338Q mutant is inviable and displays dominant negative properties (Yan et al., 2000). The chromodomain (aqua) is not part of the solved crystal structure. (B) The solved crystal structure of Esa1p (Yan et al., 2000) from residues 165-435 illustrates the location of esa1 mutant lesions with respect to CoA (green) and the HAT domain (purple) corresponding to motifs A and D (Yan et al., 2000). The serine residue 254 (orange spacefill), mutated in esa1-L254P, is located in helix α2, which precedes strand β7 of motif D. The esa1-414 frameshift mutation changes 10 amino acids (blue) beginning in helix α7 and deletes the remaining C-terminal 22 amino acids (black), 10 of which were not included in the original solved structure. The arginine residue 421 (blue spacefill), which contacts CoA, is replaced with glutamic acid in esa1-414. The critical catalytic glutamic acid residue 338 (red spacefill), changed to glutamine in the esa1-E338Q mutant, is within the pocket comprising the A motif where the acetyl group in acetyl-CoA would reside.
esa1 Mutants Are Defective in Transcriptional Silencing at Telomeres and the rDNA
Because Esa1p is an essential histone acetyltransferase with connections to transcriptional activation (Allard et al., 1999; Galarneau et al., 2000; Reid et al., 2000; Eisen et al., 2001), it might be expected that the esa1 temperature-sensitive mutants that have total decreases in histone H4 acetylation would have collateral increases in transcriptional silencing. To investigate whether Esa1p may function in both transcriptional activation and silencing, we asked if esa1 mutants displayed changes in silencing at three well-characterized loci. Expression of reporter genes placed at the silent mating-type locus HMR, within the rDNA array and at telomeres (reviewed in van Leeuwen and Gottschling, 2002) was examined in the two esa1 temperature-sensitive mutants to test for effects on transcriptional silencing.
Expression of a TRP1 reporter gene placed at a sensitized HMR locus was measured in the esa1-L254P and esa1-414 mutants by assessing growth on medium lacking tryptophan (Figure 2A). If HMR silencing is intact, as in wild-type cells, TRP1 expression is low and the strain will grow poorly on medium lacking tryptophan. However, if HMR silencing is disrupted, as in the sir2Δ control strain, TRP1 expression will be adequate to promote full growth. A modest defect in silencing at HMR was observed for the esa1-L254P mutant compared with a sir2Δ mutant at 28°C, and this defect was not influenced by temperature (unpublished data). No defect was detected for the esa1-414 mutant. We also tested silencing at HML by quantitative mating analysis, where only modest changes were observed (Table 3) by comparison with those of original sir mutants, (for example, see Rine and Herskowitz, 1987). In these assays, no significant increases in silencing were detected at either HMR or HML in the esa1 mutants, as might be expected for mutants with an overall loss in histone H4 acetylation. Instead, only slight disruptions of HMR and HML silencing were detected for either mutant. Therefore, it appears that ESA1 is neither essential for nor antagonizes transcriptional silencing of the HM loci.
Figure 2.

Temperature-sensitive esa1 mutants display transcriptional silencing defects within the rDNA array and at telomeres but only modest silencing defects at HMR. (A) The esa1-414 mutant (LPY4915) displayed no defect in silencing at HMR, whereas the esa1-L254P mutant (LPY5059) had a modest defect compared with wild-type (LPY4913) and to a sir2Δ mutant (LPY4980), which was completely disrupted for silencing at HMR. Silencing at HMR was assessed by measuring growth on plates lacking tryptophan (right) compared with a synthetic complete (SC) growth control plate (left) of strains containing the TRP1 reporter gene within the HMR locus. (B) The esa1-414 (LPY4821) mutant had an intermediate telomeric silencing defect compared with wild-type (LPY4819), yet not as severe as a sir2Δ mutant (LPY5017). Telomeric silencing was assessed for strains containing the telomere V-proximal URA3 reporter gene by measuring growth on plates containing 5-FOA (right) compared with an SC growth control plate (left). (C) The esa1-414 (LPY4821 and LPY4848) mutant displayed strong defects in rDNA silencing compared with wild-type (LPY4819), even more so than that of a sir2Δ mutant (LPY5017). Silencing in the rDNA array was assessed by measuring growth on 8 mg/ml canavanine plates lacking adenine (right) of strains containing the ADE2-CAN1 rDNA reporter cassette compared with a growth control plate (left). Note that the concentration of canavanine used in the rDNA silencing assay was eightfold lower than in earlier studies (Roy and Runge, 2000) to highlight the severity of the esa1 defect relative to that of sir2Δ. Plates in A were incubated at 28°C, those in B and C at 33°C.
Table 3.
The esa1 mutants silence HML
| MATa strain | Mating efficiency |
|---|---|
| ESA1 (LPY2991) | 1.00 |
| esa1-414 (LPY3291) | 0.42 |
| ESA1 (LPY3498) | 1.00 |
| esa1-L254P (LPY3500) | 1.15 |
Mating efficiencies were measured in two independent experiments at 28°C and are reported here as their average. See Materials and Methods for details.
In a second series of assays, telomeric silencing, also known as telomeric position effect (Gottschling et al., 1990) was evaluated. Here, expression of a URA3 reporter gene placed proximal to telomere V was measured by assessing growth on 5-FOA, a suicide substrate for the URA3 gene product. Surprisingly both esa1-414 (Figure 2B) and esa1-L254P (unpublished data) mutants had defects in telomeric silencing, as demonstrated by decreased growth on 5-FOA. This silencing defect is most severe at 33°C in the esa1 mutants but is also detectable at 28°C, both temperatures at which growth and viability are completely normal. Note that although the esa1 mutants are defective in telomeric silencing, and readily distinguishable from wild-type cells, their defect is not as severe as in a sir2Δ strain (Figure 2B).
Finally, the expression of an ADE2-CAN1 reporter cassette placed in the rDNA array within the 25S transcription unit (Fritze et al., 1997) was measured in the esa1 mutants by assessing growth on medium containing canavanine. Expression of CAN1 leads to production of the arginine permease that transports canavanine, a toxic arginine analog, into cells at the cell membrane. Thus, strains will grow on medium containing canavanine only if the CAN1 gene is silenced within the rDNA array. Both esa1-414 (Figure 2C) and esa1-L254P (unpublished data) mutants displayed severe rDNA silencing defects, as evidenced by lack of growth on the canavanine plate. At 33°C, this esa1 rDNA-silencing defect was more severe than that of a sir2Δ mutant (Figure 2C) and at 28°C was roughly equal to a sir2Δ (unpublished data). Viability was measured at 100% and importantly, cell growth rates are equivalent for wild-type and the esa1 temperature-sensitive mutants at both 28 and 33°C, so the defects observed were not due to nonspecific effects on growth. In addition, esa1 mutants are not generally sensitive to canavanine; therefore, the effects observed were due to aberrantly increased expression of the reporter gene.
ESA1 Function within the rDNA
The pronounced effects of loss of ESA1 function on rDNA silencing could be mediated directly or indirectly. For example, if SIR2 were a transcriptional target of ESA1, its expression might be down-regulated in the esa1 mutants, leading to defects at this Sir2p-dosage–sensitive locus (Smith et al., 1998b). This does not appear to be the case however because SIR2 is not a transcriptional target of ESA1 as assessed by either chromatin immunoprecipitation (ChIP) or expression-based microarray studies (Reid et al., 2000 and our unpublished observations). Further, the steady state levels of Sir2p appear normal in esa1 mutants (Supplementary Figure S1) and there are no gross defects in the Sir2p-dependent processes of mating (see above).
To test the alternative possibility that Esa1p might be acting directly within the rDNA, we took several independent approaches. In the first, we asked whether Esa1p was itself found within the rDNA. In previous ChIP experiments to evaluate promoter occupancy, Esa1p was found at many ribosomal protein gene promoters (Reid et al., 2000). It is also known that ESA1 influences total levels of H4 acetylation (Clarke et al., 1999) and that much of this global acetylation is not restricted to promoter regions (Vogelauer et al., 2000). We performed ChIP experiments using a fully functional Esa1-HA epitope-tagged strain and compared recovery of rDNA sequences to a control untagged strain (Figure 3, A and B). We observed Esa1-HA–dependent recovery of rDNA sequences from the 25S, 5S, and NTS regions of the repetitive rDNA array from chromosome XII compared with the untagged strain. That enrichment at these sites was specific was established by comparison to the control POL1 locus, where no differences were observed between tagged and untagged strains. Recovery of Esa1p from these sites in the rDNA thus demonstrates that Esa1p maps to the site of its effects on silencing, as observed for targeting of Sir2p (Huang and Moazed, 2003).
Figure 3.

Esa1p functions within the rDNA. (A) A diagram of a single rDNA transcription unit highlighting the features of each unit and orientations of transcription is shown. Dotted lines above show the position of primer sets. Replication fork barrier (RFB) and ARS1 (Abf1p binding site) are also indicated as black boxes within the nontranscribed spacer (NTS) regions. (B) Esa1p is enriched within the rDNA array. Formaldehyde cross-linked chromatin of untagged control ESA1 (LPY5) or 3HA-ESA1 (LPY6244) strains was immunoprecipitated using the α-HA antibody. Molecular amplification was carried out on total (Input) and immunoprecipitated chromatin (IP α-HA) using two primer sets specific to the 25S region (25S1 and 25S2), one set near the 5S region, an NTS primer set, and a control primer set for the POL1 locus. Two dilutions within the linear range of amplification are shown for each. The fold-enrichment values indicated are the ratio of IP/Input for tagged/untagged strains after normalization with POL1. In all cases 3HA-ESA1p is enriched within the rDNA array compared with the untagged strain. (C) Acetylation of Esa1p targets is reduced in the rDNA. Wild-type and esa1-414 cells were grown at a permissive temperature of 33°C with full viability. Chromatin was immunoprecipitated with antibodies against acetylated lysines H4 Lys5, H4 Lys12, H4 Lys16, and total AcH3. Levels of acetylation were compared in wild-type and esa1-414 strains in the rDNA using primer sets 25S1, 25S2, 5S, and NTS. The % wild-type value shown below each esa1 IP (gray boxes) is calculated by esa1-414/wild-type signals after input normalization. Acetylation of H4 Lys5 is reduced to the greatest extent throughout the rDNA at all sites tested, which correlates with the primary acetylation target of Esa1p. In addition, acetylation of H4 Lys12 and 16 are also somewhat reduced, as is total acetylated H3, again correlating with other minor acetylation targets of Esa1p.
Because esa1 mutants affect global levels of histone H4 acetylation (Clarke et al., 1999), we tested the effect of esa1 mutants on the levels of acetylation in the rDNA at specific lysine residues on histone tails. We performed ChIP analysis using modification specific antisera at the 25S, 5S, and NTS regions within the rDNA, comparing ESA1 and esa1-414 strains at 33°C where both strains have complete viability (Figure 3C). We observed multiple ESA1-dependent changes of acetylation within the rDNA that varied depending on both the location and the isoform examined. The overall acetylation level of H4 Lys5 was reduced to the greatest extent, varying from 25 to 42% to that of wild type. This correlates well with H4 Lys5 being the primary target of Esa1p in vitro and in in vivo analysis of esa1 total histones (Clarke et al., 1999). Acetylation of H4 Lys12 and 16 was also reduced. To a lesser extent, H3 was affected, correlating with minor targets of Esa1p. These results demonstrate that Esa1p both localizes to and functions directly within the rDNA.
In addition to being found directly in the nucleolus and rDNA (Gotta et al., 1997; Huang and Moazed, 2003), Sir2p functions to suppress recombination within the highly repetitive rDNA array (Gottlieb and Esposito, 1989). Indeed, many rDNA silencing defective mutants are correlated with increases in rDNA recombination (Smith et al., 1999). We tested this function for ESA1 at both 28 and 33°C using a sectoring assay based on colony color associated with the ADE2-CAN1 reporter. Recombination rates were measured as the number of events that excise the reporter cassette per cell division (Figure 4, A and B). In these experiments, both wild-type and sir2Δ mutant strains were included as controls. We observed that in the esa1-414 mutant, the mitotic rDNA recombination rate was slightly elevated relative to wild-type cells. In contrast, the sir2Δ strain had substantially elevated mitotic recombination, as previously reported. When the values for recombination were evaluated with the Wilcoxon ranking test (Wonnacott and Wonnacott, 1984; Supplementary Table S1), wild-type and esa1-414 mutant rates appeared different at 28°C, and both esa1-414 and wild type were significantly different from sir2Δ mutants. Thus, the esa1 mutant was modestly defective in suppressing mitotic recombination within the rDNA array under conditions in which Pol II silencing was disrupted, but viability was intact. Together, these effects are distinct from loss of Sir2p function.
Figure 4.
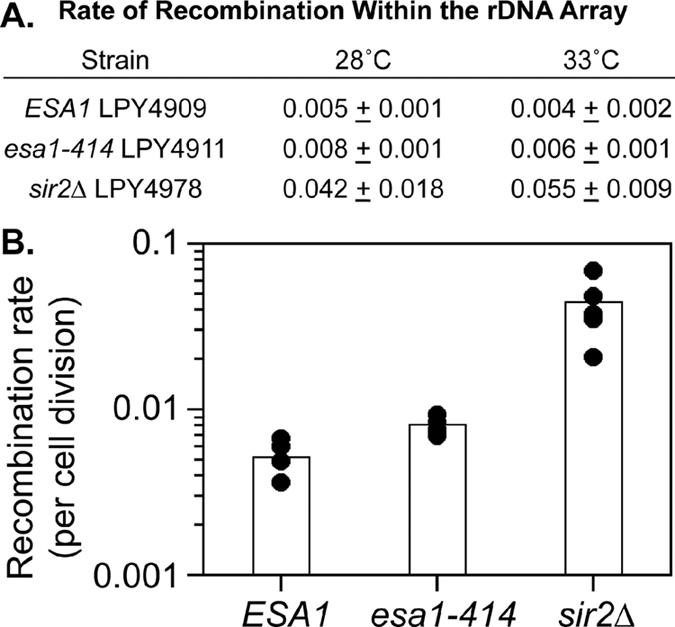
Mitotic recombination in the rDNA array. (A) Mitotic recombination within the rDNA array was evaluated using a marker loss assay. In the table, the average recombination rate of four independent isolates is shown with indicated standard deviations, except for the sir2Δ strain where the range of five (28°C) or two (33°C) isolates is presented. (B) Graphed are values for recombination assays performed at 28°C. Wilcoxon ranking tests indicated that esa1 and wild-type rates of loss are significantly different from sir2Δ rates and that esa1 rates are elevated relative to wild type (see Supplementary Table S1).
Alteration of the rDNA Array in esa1 Mutants
In S. cerevisiae, the rDNA resides as a repetitive array at a single locus on the right arm of chromosome XII (Figures 4A and 5A). The rDNA array consists of 100-200 repeating units on average, with each 9.1-kb unit containing the genes for the 35S and 5S transcription units. We observed previously that esa1 mutants have striking structural aberrations in the nucleolus, the site of the rDNA, when evaluated by electron microscopy (Clarke et al., 1999). Whereas the normal yeast nucleolus is a coalesced, hemispherical mass of electron-dense material in close apposition to the nuclear membrane, in esa1 mutants, this structure was not observed at elevated temperatures. Instead, there was very little electron-dense material and what was present appeared dispersed and scattered throughout the nucleus. These observations raised the possibility that chromatin structure itself was altered within the nucleolus. The strong rDNA silencing defects in the esa1 mutants also support the idea that chromatin structure might be changed within the rDNA array.
Figure 5.

The rDNA array is affected by esa1 mutants. (A) The 2.2+-Mb rDNA array as diagrammed resides on the right arm of chromosome XII, the largest S. cerevisiae chromosome (not drawn to scale). NotI restriction enzyme sites (N) flank the array and the region containing the PPR1 gene (gray box). A chromosomal NotI digest liberates a 2.2+-Mb fragment containing the rDNA array and a 1.4-Mb fragment containing the PPR1 gene. (B) The esa1-414 mutant exhibited altered chromatin structure of chromosome XII as evidenced by pulsed-field gel electrophoretic mobility. Whole chromosome preparations from ESA1 (LPY4909), esa1-414 (LPY4911), sir2Δ (LPY4978), and top1–8 (AMR53) strains incubated at the temperatures indicated (28, 33, and 37°C) were subjected to pulsed-field gel electrophoresis and analyzed by ethidium bromide staining (top) and Southern blotting (bottom) with a PPR1 probe, as diagrammed in A. In the esa1-414 mutant, chromosome XII (as identified in the Southern blot) displayed an altered electrophoretic mobility at all temperatures compared with wild-type (left arrows), which was distinct from the sir2Δ and top1-8 strains, where chromosome XII failed to enter the gel. (C) The altered electrophoretic mobility of chromosome XII in the esa1-414 mutant is due to the rDNA array. Whole chromosome preparations as in B were digested with NotI, subjected to pulsed-field gel electrophoresis, and analyzed by ethidium bromide staining (top) and Southern blotting (bottom) with the PPR1 probe. The rDNA array has an altered electrophoretic mobility in the esa1-414 mutant compared with wild-type, as evidenced by ethidium bromide staining (top arrows). However, the PPR1 NotI fragment (bottom arrow, Southern) migrated equivalently in all strains, including in the sir2Δ and top1-8 strains. In both B and C relevant marker sizes are indicated to the right. Asterisks indicate the marker chromosome XII and the wells are indicated. In B and C, the wells are detected by the PPR1 probe, because not all chromosomal material enters the gels.
To determine if the chromosomes of esa1 mutants were substantially different from wild-type, we examined them by pulsed-field gel electrophoresis (PFGE). Under conditions that maximized resolution of the entire chromosomal complement, we observed that wild-type and esa1 strains had similar chromosomal migration patterns, demonstrating that the esa1 mutants did not have massive, genome-wide defects (unpublished data). However, there appeared to be clear and reproducible differences in chromosome XII, the most slowly migrating chromosome in these gels. It has previously been observed that the electrophoretic mobility of chromosome XII and of the rDNA array can be greatly affected by their structure. In particular, mutations in the topoisomerase I gene (TOP1) lead to extreme chromatin alterations that prevent either chromosome XII or the rDNA array from entering pulsed-field gels and that influence chromosomal compaction (Christman et al., 1993; Castaño et al., 1996). In fact, top1 mutants are defective in the suppression of rDNA recombination as well as rDNA silencing (Christman et al., 1988; Smith et al., 1999). To investigate the electrophoretic mobility of chromosome XII in the esa1-414 mutant, whole chromosomes were prepared and analyzed by PFGE and Southern blotting, using wild-type, sir2Δ, and top1 mutants for comparison (Figure 5B).
We observed that the electrophoretic mobility of chromosome XII in the esa1-414 mutant was distinct from that of wild-type, irrespective of temperature (Figure 5B). This mobility difference was also clearly distinct from the behavior of either the sir2Δ or top1 strains. Chromosome XII entered the gel in the esa1-414 mutant, rather than being trapped in the wells as previously characterized for top1 mutants, or as shown here for sir2Δ mutants.
Because the rDNA array migrated more rapidly in the esa1 mutants compared with wild type, we considered the possibility that these strains had fewer repeat units, even though the recombination rate was significantly lower than that in sir2Δ or top1 mutants (Figure 4B and Christman et al., 1988; Gottlieb and Esposito, 1989). We quantified the rDNA repeats for ESA1 and esa1-414 strains during the experiment shown in Figure 4 and found that they had essentially equivalent numbers of repeats and that the small difference in number is unlikely to account for the PFGE migration differences (range 84-98%; see Materials and Methods). In addition, we independently measured the relative rDNA repeat number in the ESA1, esa1-414, and top1Δ strains by quantitative genomic dot blotting. The relative rDNA repeat hybridization signal was compared with a single copy control locus YBR071w on chromosome II for ESA1, esa1-414, and top1Δ strains under normal growth conditions. The results of this quantification also indicated that the esa1-414 strain had similar (although in this assay slightly more) amounts of rDNA as wild-type and top1Δ strains (Supplementary Table S2). Thus, data from two independent assays suggest that esa1-414 chromosome XII is not migrating faster in PFGE analysis due to fewer repeats.
To determine if the mobility difference observed in the esa1 mutant was due to the rDNA specifically or due to a more general property affecting all of chromosome XII, we took advantage of the fact that NotI sites fall outside, but not within, the rDNA repeats. A schematic representation of chromosome XII with relevant NotI sites surrounding the rDNA array (Figure 5A) shows that when chromosome XII is digested with NotI a ∼2.2-Mb fragment containing the rDNA array is liberated. A 1.4-Mb fragment containing the PPR1 gene is also liberated (Figure 5A), along with other additional fragments not indicated. Chromosomes from the same strains examined above were digested with NotI and analyzed by PFGE and Southern blotting. We observed that, again, the rDNA array migrated differently for the wild-type, esa1-414, sir2, and top1-8 strains (Figure 5C, top). However, the centromere-proximal 1.4-Mb fragment detected by the PPR1 probe migrated equivalently for all three strains (Figure 5C, bottom). Therefore, the electrophoretic mobility differences observed for chromosome XII in the esa1-414 appear due primarily to properties of the rDNA array. Because gross changes in rDNA repeat numbers are unlikely to explain the difference in mobility we observe, we hypothesize that the changes are due to alterations in chromatin structure that are sensitive to PFGE separation but that are distinct from severe recombination phenotypes such as those occurring in sir2 and top1 mutants.
Altered Localization of Two Nucleolar Proteins in esa1 Mutants
By both chromatin immunoprecipitation and fluorescence localization studies, Esa1p has been defined for its general nuclear roles and distribution (Galarneau et al., 2000; Reid et al., 2000; Vogelauer et al., 2000; Huh et al., 2003). Viewed collectively, the previously observed defects in nucleolar structure (Clarke et al., 1999), in addition to the rDNA silencing, recombination and PFGE analyses reported here all suggested that ESA1 also contributes to normal nucleolar structure and function. Many proteins are known to function in the nucleolus, so to investigate further the function of ESA1, we evaluated the effects of an esa1 mutation on two of these nucleolar markers. We used indirect-immunofluorescence to compare localization of the yeast fibrillarin homolog, Nop1p (Aris and Blobel, 1988), and Sir2p in wild-type and esa1 cells under permissive and restrictive conditions. Nop1p staining in wild-type cells at 28°C appeared as a characteristic discrete crescent shape apart from the bulk of the DAPI-staining chromatin (Figure 6A, top). Most of Sir2p colocalized with the Nop1p staining in the nucleolar crescent and some Sir2p localized to telomeric foci with the bulk of the chromatin, consistent with previous results (Gotta et al., 1997).
Figure 6.

Nop1p and Sir2p localization are affected in the esa1-414 mutant under both permissive and restrictive conditions. (A) At 28°C, Nop1p and Sir2p localization in the esa1-414 mutant was distinct from wild type by indirect-immunofluorescence. Nop1p staining in the esa1-414 mutant (bottom) was more punctate, although often still in a crescent shape (asterisk) though generally occupying a larger portion of the nucleus than in wild type. In some esa1-414 cells, the punctate staining was not crescent-shaped and was quite small or dispersed throughout the nucleus (arrowheads). Sir2p staining was also reduced in the nucleolus to very small punctate foci that coincided with the Nop1p staining (merge), yet often these foci were not crescent-shaped. Some telomeric Sir2p foci were still visible in the esa1-414 mutant. (B) After a 4-h shift to 37°C, the esa1-414 mutant displayed aberrant Nop1p and Sir2p localization by indirect-immunofluorescence. Nop1p staining in the esa1-414 mutant was distinct from wild-type with many cells displaying bilobed fragmented staining (arrowheads) or only a very small focus of staining (asterisk). Sir2p staining in the nucleolus was even more reduced and punctate in the esa1-414 mutant at 37 than at 28°C, with very few crescent shapes, although there was still partial overlap with the Nop1p staining (merge). In addition, Sir2p telomeric foci were difficult to identify in the esa1-414 mutant at 37°C.
In contrast, at 28°C in the esa1-414 mutant, the anti-Nop1p staining was more punctate, although usually still found in a crescent shape (Figure 6A, bottom, asterisk) and the staining appeared to occupy a larger portion of the nucleus than in wild-type. However, in some cells, no crescent was evident but Nop1p-staining instead appeared as a small focus or was dispersed throughout the nucleus (Figure 6A, bottom, arrowheads). Sir2p staining in the esa1 mutant was also aberrant at 28°C with more punctate nucleolar foci than in wild type. Often the Sir2p crescent shape was not visible and the staining in general appeared dispersed or diminished. Nonetheless, the Sir2p staining that was visible coincided with Nop1p and telomeric foci were visible indicating at least some Sir2p was localized properly to the nucleolus and telomeres.
After a 4-h shift to 37°C, the Nop1p and Sir2p staining in the esa1-414 mutant (Figure 6B, bottom) were again distinct from wild type (Figure 6B, top). Many cells displayed Nop1p staining that was either bilobed (arrowheads) or in very small foci (asterisk). Sir2p staining appeared even more abnormal and localized as small condensed foci that coincided with Nop1p staining. Together these results underscore that the substructure of the nucleolus is altered in esa1 mutants and that the effect is more severe at nonpermissive temperatures. The fact that at least part of the pool of Sir2p appears to still be localized to the nucleolus is consistent with the observation that some of its functions, such as suppression of recombination, remain at least partially intact.
Increased Gene Dosage of ESA1 and SIR2 Reciprocally Suppresses rDNA Silencing Defects
The data above and earlier observations establish that there are intersecting yet distinct functions for both Esa1p and Sir2p within the nucleolus. To explore this relationship further, we asked if changes in dosage of either gene affected the rDNA silencing mutant phenotype of the other. As an initial approach, we constructed the esa1-414 sir2Δ double mutant through standard genetic crosses. When rDNA silencing was assayed in this strain and compared with wild-type and the single mutant strains, we observed that silencing remained as defective as in an esa1 strain (unpublished data). Thus, these acetyltransferase and deacetylase mutants do not mutually suppress each other's transcriptional defects, as has been observed previously for other HAT/HDAC mutant pairs (Pérez-Martín and Johnson, 1998).
In addition to evaluating decreased expression with the double mutants, we also looked at effects of increased expression of the two genes. It had been noted earlier that silencing within the rDNA array is quite sensitive to the gene dosage of SIR2. For example, overexpression of SIR2 or deletion of SIR4, which presumably redirects a pool of Sir2p from the telomeres to the rDNA, strengthens rDNA silencing (Smith et al., 1998b). We first asked if increasing SIR2 gene dosage affected rDNA silencing in esa1 mutants. The esa1-414 strain was transformed with 2-μm plasmids to provide moderately increased copy number (Broach, 1983) of the URA3 vector alone, the ESA1 gene, or SIR2 (Figure 7A). As expected, ESA1 fully complemented the defect evident in the vector controls. The SIR2 transformants also had fully restored silencing, indicating that the esa1 defect could be suppressed by increasing SIR2. In companion experiments, a sir2Δ strain was transformed with the same plasmids (Figure 7B). Again, the wild-type SIR2 gene complemented the sir2Δ silencing defect. Strikingly, increased ESA1 gene dosage also restored silencing, indicating that ESA1 can fully bypass the requirement for Sir2p in rDNA silencing.
Figure 7.
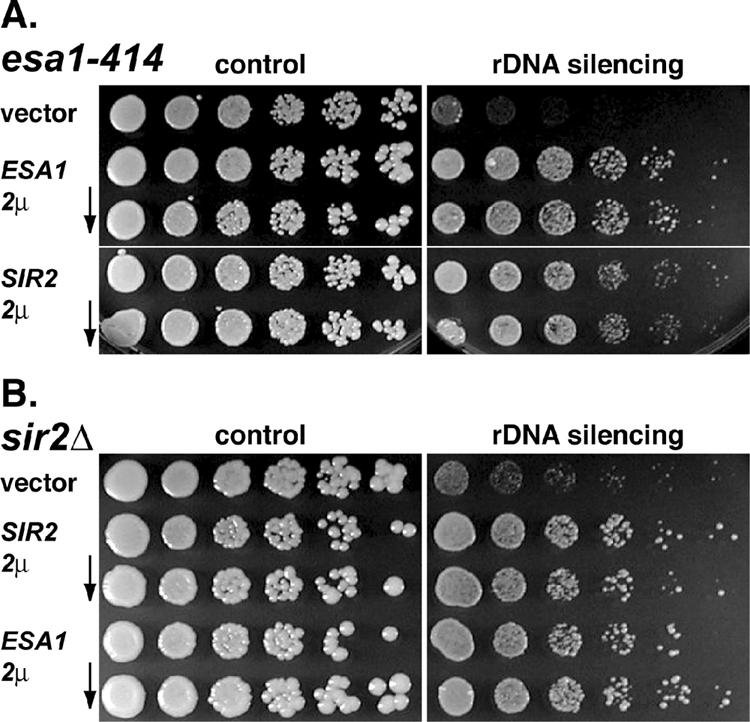
Increasing the gene dosage of SIR2 and ESA1 reciprocally suppresses the rDNA silencing defects associated with esa1-414 and sir2Δ mutants. (A) Increased gene dosage of SIR2 from a 2-micron plasmid suppressed the esa1-414 rDNA silencing defect. The esa1-414 strain transformed with ESA1 (LPY5141) or SIR2 (LPY5143) from URA3-marked 2-micron plasmids grew equivalently on plates assaying for rDNA silencing (right) compared with vector transformants (LPY5136) at 33°C. (B) Similarly, increasing the dosage of ESA1 suppressed the sir2Δ rDNA silencing defect. The sir2Δ strain transformed with SIR2 (LPY5166) or ESA1 (LPY5164) grew equivalently on plates assaying for rDNA silencing compared with vector transformants (LPY5159) at 28°C. The level of suppression was somewhat variable between isolates, perhaps reflecting a difference in expression levels of either ESA1 or SIR2 due to fluctuations in 2-micron plasmid copy number per isolate (Armstrong et al., 1989). The rDNA silencing assays were performed with 32 mg/ml canavanine in order to compare silencing effects in the sir2Δ and esa1 mutants directly.
Interestingly, the cross-suppression between ESA1 and SIR2 has restricted locus specificity because increased SIR2 could not bypass the essential requirements for ESA1 (unpublished data). Likewise, increased ESA1 did not restore mating to a sir2Δ strain (unpublished data), consistent with the lack of a central role for ESA1 at the silent mating-type loci (Figure 2 and Table 3). Furthermore, neither gene could suppress the telomeric silencing defect of the other mutant (unpublished data). Thus, whereas ESA1 and SIR2 clearly have independent roles within the genome, they have the previously unsuspected capacity to compensate for each other's defects in rDNA silencing of a Pol II reporter gene.
DISCUSSION
Starting from initial links to transcriptional activation of specific target genes, HATs have now been implicated in processes as diverse as DNA replication and double-stranded break repair and recombination. Acetylation is a readily reversible modification and long-standing correlations have associated HATs with activation and deacetylases with repression of transcription. In some cases, paired, opposing activities at a single promoter have been shown to either positively or negatively regulate transcription of the locus (reviewed in Carrozza et al., 2003).
We have found that ESA1's transcriptional functions are not limited to activation and repair. Instead, Esa1p also functions in transcriptional silencing, potentially through direct effects on chromatin structure, most evident within the rDNA and nucleolus. Perhaps even more surprisingly, we have uncovered a link between ESA1 and SIR2, a prominent nucleolar NAD-dependent HDAC. These two enzymes with seemingly opposing modifying activities both have the capacity to function in silencing within the rDNA.
Expanded Roles for an Essential MYST Family HAT
The MYST family of acetyltransferases was originally defined by the human MOZ leukemia translocation partner, the two yeast Sas proteins, and the human HIV-Tat interacting protein Tip60 (Borrow et al., 1996; Kamine et al., 1996; Reifsnyder et al., 1996). Family members have subsequently been characterized with diverse roles in DNA replication, apoptosis, sex dosage compensation, phospholipid signaling, development, neurogenesis, and androgen receptor function (reviewed in Utley and Côté, 2003). Because multiple MYST family proteins may be expressed simultaneously in the same cells and tissues, understanding the specificity of their function has been a key goal.
ESA1 encodes the essential HAT activity that is primarily directed toward histones H4 and H2A (Smith et al., 1998a; Clarke et al., 1999). Analysis of conditional esa1 mutants reveals that loss of activity results in changes in nuclear substructure and overall decreases in genomic H4 acetylation (Clarke et al., 1999). Biochemical studies demonstrate that Esa1p is the catalytic subunit of the NuA4 complex that includes other essential subunits such as Tra1p and Act3p and other key subunits with connections to growth control and epigenetic elements of transcriptional activation (Allard et al., 1999; Galarneau et al., 2000). Indeed, a number of distinct transcriptional targets have been identified for Esa1p, along with genome-wide validation of its role as a global H4 HAT (Galarneau et al., 2000; Reid et al., 2000; Vogelauer et al., 2000; Nourani et al., 2001). More extensive expression analysis suggests nearly equal numbers of positively and negatively affected transcriptional targets for Esa1p (Mnaimneh et al., 2004). Although some of these may be indirect, the data raise the strong possibility that Esa1p participates in either gene-specific or more general chromatin-mediated forms of repression.
Here, we report previously unanticipated roles for ESA1 in transcriptional silencing and genomic structure and function. In independent support of a role for Esa1p HAT activity in silencing, an ESA1-E338Q catalytically inactive dominant mutant (Yan et al., 2000) interferes with wild-type silencing at the rDNA and telomeres (Supplementary Figure S2). Nucleolar structure is also disrupted in esa1 mutants, as assessed by mislocalization of resident nucleolar proteins and alterations in PFGE and ultrastructural profiles (Clarke et al., 1999). The esa1 silencing defects are not globally communicated through all silenced loci, because regulation of mating type control at the HM loci is only modestly affected.
Esa1p maps to the rDNA locus by ChIP, and mutational analyses suggest a significant contribution to this region of the genome. Acetylation levels of key Esa1p targets are significantly reduced in mutants in the rDNA, supporting the idea that Esa1p functions there directly through its HAT activity. These data expand the map of genomic targets for Esa1p beyond previously identified Pol II-regulated sites (Vogelauer et al., 2000; Reid et al., 2000) to the territory ordinarily controlled by Pol I and Pol III.
A key question raised by the silencing phenotypes of esa1 mutants is whether these defects are mediated through loss of NuA4 activity, previously associated with activation. A second possibility is that Esa1p-silencing functions are mediated through multiple, functionally distinct complexes. Evidence for the latter possibility comes from the observation that the canonical yeast HAT Gcn5p works through alternative complexes with different target genes (Sterner and Berger, 2000) and the discovery of the Esa1p-containing Piccolo complex, picNuA4 (Boudreault et al., 2003). Considering alternative complexes, one might imagine discrete subunits that mediate activating versus silencing outputs to fine-tune locus-specific functions of Esa1p. In parallel, some Esa1p-interacting partners would function generically in both activating and silencing contexts. In this regard, it is noteworthy that like ESA1, mutants of EPL1, a component of both NuA4 and picNuA4 have defects in transcriptional activation and telomeric silencing (Boudreault et al., 2003). In contrast, other NuA4 subunits are themselves implicated in transcriptional activation but not silencing (Galarneau et al., 2000; Nourani et al., 2001). Future identification of Esa1p's interacting partners in nucleolar and telomeric silencing should refine understanding of its role in NuA4, picNuA4, or other complexes, but the results presented here demonstrate a clearly expanded role for Esa1p in genomic regulation.
Esa1p Is a Distinct rDNA Silencing Protein Important for Nucleolar Structure
A long-standing observation is that only about half of the ribosomal repeats in the rDNA cluster are transcriptionally activated under normal growth conditions. Many questions remain about the nature of the regulation of the transcriptional silencing of the remainder of the repeats (reviewed in Grummt and Pikaard, 2003). When it was discovered that Pol II-regulated genes that are engineered into the rDNA array are silenced in an epigenetic manner that is dependent on SIR2 (Bryk et al., 1997; Fritze et al., 1997; Smith and Boeke, 1997), the appealing possibility was raised that Pol I transcription itself might be at least partially regulated through heterochromatin-like SIR-dependent mechanisms. Indeed, it had previously been observed that a Pol III reporter gene could be silenced in a SIR-dependent manner (Schnell and Rine, 1986). However, in contrast to early indications (Smith and Boeke, 1997), subsequent more sensitive analysis indicates that SIR2 does not affect rDNA transcription (Sandmeier et al., 2002), but may instead affect firing of origins of rDNA replication (Pasero et al., 2002).
Because the discovery of nucleolar silencing of Pol II reporter genes by Sir2p, a growing number of factors have been shown to participate, including other chromatin modifying enzymes such as the Rpd3 deacetylase, the Set1 methyltransferase, and topoisomerase I (Smith et al., 1999; Bryk et al., 2002). Nucleolar proteins with no known catalytic activities, such as Net1p also influence rDNA silencing (Straight et al., 1999). In other mutants, such as those of the Set1p methyltransferase or the Swi/Snf remodeling complex, silencing defects and effects on chromatin modification are pronounced, yet no defects have been reported for Pol I transcription and rDNA recombination may be either unaffected or suppressed (Bryk et al., 2002; Dror and Winston, 2004). By comparison, Net1p directly binds Pol I and has the capacity to promote transcription (Shou et al., 2001), with no apparent effects on recombination. Intriguingly, transcription within the rDNA by Pol I itself is required for silencing and for the spread of silencing toward the centromere (Buck et al., 2002). In contrast, RPD3 is required for down-regulating rDNA transcription as cells enter the stationary phase (Sandmeier et al., 2002).
We have observed that esa1 mutants have extreme defects in rDNA silencing, but are distinguished from sir2Δ mutants because they have only modestly increased recombination rates between the homologous DNA repeats. The esa1 mutants are also distinct from sir2 mutants in that they have disrupted nucleolar structure as observed by electron microscopy (Clarke et al., 1999), by the position of nucleolar marker proteins and by PFGE. Among mutants with rDNA silencing defects, ESA1 is the only gene yet reported to also influence ribosomal protein gene transcription (Reid et al., 2000). Thus, Pol I transcription defects may be part of coordinate mechanisms to shut down ribosome production when either or both rRNA or ribosomal protein synthesis are defective. How these defects would be communicated is not yet fully known, but having at least one shared regulatory molecule or mechanism provides an attractive point for coordination.
HATs and HDACs Are Both Required for Functional Optimization
HATs function in concert with other types of chromatin-modifying enzymes, such as kinases, methyltransferases, and ubiquitin ligases. A key challenge is to understand how these activities and their substrates are specified to yield optimal biological regulation (reviewed in Strahl and Allis, 2000). In some cases, pairs of enzymes with distinct activities may function sequentially to potentiate function. Such is the case for the Snf1 histone kinase and the HAT Gcn5p (Lo et al., 2000, 2001). In other cases, two HATs with different substrate specificities may act on the same gene to establish a characteristic pattern of modification, as has been observed for Gcn5p and Esa1p (Vogelauer et al., 2000).
In contrast, opposing modifying activities have been observed as counterbalances to one another. In these situations, mutant phenotypes resulting from the loss of one activity, such as acetylation, are effectively suppressed by loss of a paired deacetylase. Such is the case for gcn5Δ and rpd3Δ deacetylase mutants when evaluating HO expression (Krebs et al., 1999). Whether such pairing is a general feature of chromatin modification is not yet known, but in some cases, direct interactions between HDACs and HATs have been observed (for example, see Yamagoe et al., 2003).
Our observation of the mutual suppression of sir2Δ or esa1 mutant rDNA silencing defects by overexpression of an opposing activity suggests the possibility for novel regulatory mechanisms. In this case, the ESA1-encoded HAT can actually bypass the requirement for Sir2p, which previously was a defining feature of rDNA silencing. Importantly, the suppression observed is locus-specific. That is, ESA1 cannot fully substitute for all of SIR2's functions, nor can SIR2 bypass the essential requirements for ESA1. Such specificity of suppression is intriguing and suggests that in discrete regions of the genome these two enzymes contribute to achieve optimal chromosomal structure and function. We cannot yet exclude the possibility that Esa1p and Sir2p function independently within the rDNA. For example, Sir2p may affect origin firing within the rDNA and Esa1p may affect transcriptional regulation, yet both functions may be required for appropriate chromatin structure. Likewise, it is known that some regions of the rDNA array are more sensitive to SIR2-silencing than others (Smith and Boeke, 1997; Buck et al., 2002). It should prove interesting to map regional effects of ESA1 control by comparison. Further investigation of the exact molecular consequences of Sir2p and Esa1p function in the rDNA is necessary to understand how these two enzymes with opposing activities contribute to silencing.
Mechanistic clues come from recent studies in which a cycle of acetylation and deacetylation was triggered at the site of an induced dsDNA lesion. The HATs Esa1p and Gcn5p were first recruited, followed by the Sir2p, Hst1p, and Rpd3p deacetylases. Histone acetylation and deacetylation patterns correlated with these ordered recruitments (Tamburini and Tyler, 2005). Deeper understanding of the in vivo substrates and dynamic activities of chromatin modifying enzymes such as Esa1p and Sir2p both within the rDNA and throughout the genome will establish how general the requirement for combinations of chromatin modifications at specific loci may be.
Supplementary Material
Acknowledgments
We thank K. Runge, S. Berger, D. Gottschling, R. Sternglanz, and J. Aris for providing strains and reagents; R. Dutnall for assistance with modeling; D. Liang for assistance with PFGE; and K. Gardner and C. Fox for ChIP advice and critique. V. Carey and E. Nalefski provided guidance with statistical analysis. We thank M. Nomura for discussion of independent studies on ESA1, P. Laurenson and S. Jacobson for critical comments and discussion, and E. Spedale and V. Le for technical assistance. This work was supported with funding from the National Institutes of Health.
This article was published online ahead of print in MBC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E05-07-0613) on January 25, 2006.
The online version of this article contains supplemental material at MBC Online (http://www.molbiolcell.org).
References
- Allard, S., Utley, R. T., Savard, J., Clarke, A., Grant, P., Brandl, C. J., Pillus, L., Workman, J. L., and Côté, J. (1999). NuA4, an essential transcription adaptor/histone H4 acetyltransferase complex containing Esa1p and the ATM-related cofactor Tra1p. EMBO J. 18, 5108–5119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aris, J. P., and Blobel, G. (1988). Identification and characterization of a yeast nucleolar protein that is similar to a rat liver nucleolar protein. J. Cell Biol. 107, 17–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong, K. A., Som, T., Volkert, F. C., Rose, A., and Broach, J. R. (1989). Propagation and expression of genes in yeast using 2-micron circle vectors. Biotechnology 13, 165–192. [PubMed] [Google Scholar]
- Baluchamy, S., Rajabi, H. N., Thimmapaya, R., Navaraj, A., and Thimmapaya, B. (2003). Repression of c-Myc and inhibition of G1 exit in cells conditionally overexpressing p300 that is not dependent on its histone acetyltransferase activity. Proc. Natl. Acad. Sci., USA 100, 9524–9529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird, A. W., Yu, D. Y., Pray-Grant, M. G., Qiu, Q., Harmon, K. E., Megee, P. C., Grant, P. A., Smith, M. M., and Christman, M. F. (2002). Acetylation of histone H4 by Esa1 is required for DNA double-strand break repair. Nature 419, 411–415. [DOI] [PubMed] [Google Scholar]
- Birren, B., and Lai, E. (1993). Pulsed Field Gel Electrophoresis: A Practical Guide, San Diego: Academic Press.
- Borrow, J., et al. (1996). The t(8;16)(p11;p13) translocation of acute myeloid leukemia fuses a putative acetyltransferase to the CREB-binding protein. Nat. Genet. 14, 33–41. [DOI] [PubMed] [Google Scholar]
- Boudreault, A. A., Cronier, D., Selleck, W., Lacoste, N., Utley, R. T., Allard, S., Savard, J., Lane, W. S., Tan, S., and Côté, J. (2003). Yeast enhancer of polycomb defines global Esa1-dependent acetylation of chromatin. Genes Dev. 17, 1415–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broach, J. R. (1983). Construction of high copy yeast vectors using 2-μm circle sequences. Methods Enzymol. 101C, 307–325. [DOI] [PubMed] [Google Scholar]
- Bryk, M., Banerjee, M., Murphy, M., Knudsen, K. E., Garfinkel, D. J., and Curcio, M. J. (1997). Transcriptional silencing of Ty1 elements in the RDN1 locus of yeast. Genes Dev. 11, 255–269. [DOI] [PubMed] [Google Scholar]
- Bryk, M., Briggs, S. D., Strahl, B. D., Curcio, M. J., Allis, C. D., and Winston, F. (2002). Evidence that Set1, a factor required for methylation of histone H3, regulates rDNA silencing in S. cerevisiae by a Sir2-independent mechanism. Curr. Biol. 12, 165–170. [DOI] [PubMed] [Google Scholar]
- Buck, S. W., Sandmeier, J. J., and Smith, J. S. (2002). RNA Polymerase I propagates unidirectional spreading of rDNA silent chromatin. Cell 111, 1003–1014. [DOI] [PubMed] [Google Scholar]
- Carrozza, M. J., Utley, R. T., Workman, J. L., and Côté, J. (2003). The diverse functions of histone acetyltransferase complexes. Trends Genet. 19, 321–329. [DOI] [PubMed] [Google Scholar]
- Castaño, I. B., Brzoska, P. B., Sadoff, B. U., Chen, H., and Christman, M. F. (1996). Mitotic chromosome condensation in the rDNA requires TRF4 and DNA topoisomerase I in Saccharomyces cerevisiae. Cell 10, 2564–2576. [DOI] [PubMed] [Google Scholar]
- Chen, J. D., and Li, H. (1998). Coactivation and corepression in transcriptional regulation by steroid/nuclear hormone receptors. Crit. Rev. Eukaryot. Gene Expr. 8, 169–190. [DOI] [PubMed] [Google Scholar]
- Choy, J. S., and Kron, S. J. (2002). NuA4 subunit Yng2 function in intra-S-phase DNA damage response. Mol. Cell Biol. 22, 8215–8225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christman, M. F., Dietrich, F. S., and Fink, G. R. (1988). Mitotic recombination is suppressed by the combined action of DNA topoisomerases I and II. Cell 55, 413–425. [DOI] [PubMed] [Google Scholar]
- Christman, M. F., Dietrich, F. S., Levin, N. A., Sadoff, B. U., and Fink, G. R. (1993). The rRNA-encoding DNA array has an altered structure in topoisomerase I mutants of Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 90, 7637–7641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke, A. S., Lowell, J. E., Jacobson, S. J., and Pillus, L. (1999). Esa1p is an essential histone acetyltransferase required for cell cycle progression. Mol. Cell. Biol. 19, 2515–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeRubertis, F., Kadosh, D., Henchoz, S., Pauli, D., Reuter, G., Struhl, K., and Spierer, P. (1996). The histone deacetylase RPD3 counteracts genomic silencing in Drosophila and yeast. Nature 384, 589–591. [DOI] [PubMed] [Google Scholar]
- Doyon, Y., and Côté, J. (2004). The highly conserved and multifunctional NuA4 HAT complex. Curr. Opin. Genet. Dev. 14, 147–154. [DOI] [PubMed] [Google Scholar]
- Dror, V., and Winston, F. (2004). The Swi/Snf chromatin remodeling complex is required for ribosomal DNA and telomeric silencing in Saccharomyces cerevisiae. Mol. Cell. Biol. 24, 8227–8235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrenhofer-Murray, A., Rivier, D., and Rine, J. (1997). The role of Sas2, an acetyltransferase homolog, in silencing and ORC function in Saccharomyces cerevisiae. Genetics 145, 923–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisen, A., Utley, R. T., Nourani, A., Allard, S., Schmidt, P., Lane, W. S., Lucchesi, J. C., and Côté, J. (2001). The yeast NuA4 and Drosophila MSL complexes contain homologous subunits important for transcription regulation. J. Biol. Chem. 276, 3484–3491. [DOI] [PubMed] [Google Scholar]
- Ersfeld, K., and Stone, E. M. (2000). Simultaneous in situ detection of DNA and proteins. In: Protein Localization by Fluorescence Microscopy, A Practical Approach, ed. V. J. Allan, Oxford: Oxford University Press, 51–66.
- Fox, C. A., and McConnell, K. H. (2005). Toward biochemical understanding of a transcriptionally silenced chromosomal domain in Saccharomyces cerevisiae. J. Biol. Chem. 280, 8629–8632. [DOI] [PubMed] [Google Scholar]
- Fritze, C. E., Verschueren, K., Strich, R., and Esposito, R. E. (1997). Direct evidence for SIR2 modulation of chromatin structure in yeast rDNA. EMBO J. 16, 6495–6509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galarneau, L., Nourani, A., Boudreault, A. A., Zhang, Y., Heliot, L., Allard, S., Savard, J., Lane, W. S., Stillman, D. J., and Côté, J. (2000). Multiple links between the NuA4 histone acetyltransferase complex and epigenetic control of transcription. Mol. Cell 5, 927–937. [DOI] [PubMed] [Google Scholar]
- Ghidelli, S., Donze, D., Dhillon, N., and Kamakaka, R. T. (2001). Sir2p exists in two nucleosome-binding complexes with distinct deacetylase activities. EMBO J. 20, 4522–4535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girdwood, D., Bumpass, D., Vaughan, O. A., Thain, A., Anderson, L. A., Snowden, A. W., Garcia-Wilson, E., Perkins, N. D., and Hay, R. T. (2003). P300 transcriptional repression is mediated by SUMO modification. Mol. Cell 11, 1043–1054. [DOI] [PubMed] [Google Scholar]
- Gotta, M., Strahl-Bolsinger, S., Renauld, H., Laroche, T., Kennedy, B. K., Grunstein, M., and Gasser, S. M. (1997). Localization of Sir2p: the nucleolus as a compartment for Silent Information Regulators. EMBO J. 16, 3243–3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlieb, S., and Esposito, R. E. (1989). A new role for a yeast transcriptional silencer gene, SIR2, in regulation of recombination in ribosomal DNA. Cell 56, 771–776. [DOI] [PubMed] [Google Scholar]
- Gottschling, D. E. (2000). Gene silencing: two faces of SIR2. Curr. Biol. 10, R708–R711. [DOI] [PubMed] [Google Scholar]
- Gottschling, D. E., Aparicio, O. M., Billington, B. L., and Zakian, V. A. (1990). Position effect at S. cerevisiae telomeres: reversible repression of Pol II transcription. Cell 63, 751–762. [DOI] [PubMed] [Google Scholar]
- Grummt, I., and Pikaard, C. S. (2003). Epigenetic silencing of RNA polymerase I transcription. Nat. Rev. Mol. Cell Biol. 4, 641–649. [DOI] [PubMed] [Google Scholar]
- Guarente, L. (2000). Sir2 links chromatin silencing, metabolism, and aging. Genes Dev. 14, 1021–1026. [PubMed] [Google Scholar]
- Huang, J., and Moazed, D. (2003). Association of the RENT complex with nontranscribed and coding regions of rDNA and a regional requirement for the replication fork block protein Fob1 in rDNA silencing. Genes Dev. 17, 2162–2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh, W. K., Falvo, J. V., Gerke, L. C., Carroll, A. S., Howson, R. W., Weissman, J. S., and O'Shea, E. K. (2003). Global analysis of protein localization in budding yeast. Nature 425, 686–691. [DOI] [PubMed] [Google Scholar]
- Iizuka, M., and Smith, M. M. (2003). Functional consequences of histone modifications. Curr. Opin. Genet. Dev. 13, 154–160. [DOI] [PubMed] [Google Scholar]
- Ikura, T., Ogryzko, V. V., Grigoriev, M., Groisman, R., Wang, J., Horikoshi, M., Scully, R., Qin, J., and Nakatani, Y. (2000). Involvement of the TIP60 histone acetylase complex in DNA repair and apoptosis. Cell 102, 463–473. [DOI] [PubMed] [Google Scholar]
- Imai, S., Armstrong, C. M., Kaeberlein, M., and Guarente, L. (2000). Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 403, 795–800. [DOI] [PubMed] [Google Scholar]
- John, S., Howe, L., Tafrov, S. T., Grant, P. A., Sternglanz, R., and Workman, J. L. (2000). The Something About Silencing protein, Sas3, is the catalytic subunit of NuA3, a yTAF (II) 30-containing HAT complex that interacts with the Spt16 subunit of the yeast CP (Cdc68/Pob3)-FACT complex. Genes Dev. 14, 1196–1208. [PMC free article] [PubMed] [Google Scholar]
- Kamine, J., Elangovan, B., Subramanian, T., Coleman, D., and Chinnadurai, G. (1996). Identification of a cellular protein that specifically interacts with the essential cysteine region of the HIV-1 Tat transactivator. Virology 216, 357–366. [DOI] [PubMed] [Google Scholar]
- Kimura, A., Umehara, T., and Horikoshi, M. (2002). Chromosomal gradient of histone acetylation established by Sas2p and Sir2p functions as a shield against gene silencing. Nat. Genet. 32, 370–377. [DOI] [PubMed] [Google Scholar]
- Kobor, M. S., Venkatasubrahmanyam, S., Meneghini, M. D., Gin, J. W., Jennings, J. L., Link, A. J., Madhani, H. D., and Rine, J. (2004). A protein complex containing the conserved Swi2/Snf2-related ATPase Swr1p deposits histone variant H2AZ into euchromatin. PLoS Biol. 2, E131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebs, J. E., Kuo, M. H., Allis, C. D., and Peterson, C. L. (1999). Cell cycle-regulated histone acetylation required for expression of the yeast HO gene. Genes Dev. 13, 1412–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogan, N. J., et al. (2004). Regulation of chromosome stability by the histone H2A variant Htz1, the Swr1 chromatin remodeling complex, and the histone acetyltransferase NuA4. Proc. Natl. Acad. Sci. USA 101, 13513–13518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurdistani, S. K., and Grunstein, M. (2003). Histone acetylation and deacetylation in yeast. Nat. Rev. Mol. Cell Biol. 4, 276–284. [DOI] [PubMed] [Google Scholar]
- Landry, J., Sutton, A., Tafrov, S. T., Heller, R. C., Stebbins, J., Pillus, L., and Sternglanz, R. (2000). The silencing protein SIR2 and its homologs are NAD-dependent protein deacetylases. Proc. Natl. Acad. Sci. USA 97, 5807–5811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo, W. S., Duggan, L., Emre, N. C., Belotserkovskya, R., Lane, W. S., Shiekhattar, R., and Berger, S. L. (2001). Snf1—a histone kinase that works in concert with the histone acetyltransferase Gcn5 to regulate transcription. Science 293, 1142–1146. [DOI] [PubMed] [Google Scholar]
- Lo, W. S., Trievel, R. C., Rojas, J. R., Duggan, L., Hsu, J. Y., Allis, C. D., Marmorstein, R., and Berger, S. L. (2000). Phosphorylation of serine 10 in histone H3 is functionally linked in vitro and in vivo to Gcn5-mediated acetylation at lysine 14. Mol. Cell 5, 917–926. [DOI] [PubMed] [Google Scholar]
- Longtine, M. S., McKenzie, A., 3rd, Demarini, D. J., Shah, N. G., Wach, A., Brachat, A., Philippsen, P., and Pringle, J. R. (1998). Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14, 953–961. [DOI] [PubMed] [Google Scholar]
- Mizuguchi, G., Shen, X., Landry, J., Wu, W. H., Sen, S., and Wu, C. (2004). ATP-driven exchange of histone H2AZ variant catalyzed by SWR1 chromatin remodeling complex. Science 303, 343–348. [DOI] [PubMed] [Google Scholar]
- Mnaimneh, S., et al. (2004). Exploration of essential gene functions via titratable promoter alleles. Cell 118, 31–44. [DOI] [PubMed] [Google Scholar]
- Moazed, D. (2001). Enzymatic activities of Sir2 and chromatin silencing. Curr. Opin. Cell Biol. 13, 232–238. [DOI] [PubMed] [Google Scholar]
- Neuwald, A. F., and Landsman, D. (1997). GCN5-related histone N-acetyltransferases belong to a diverse superfamily that includes the yeast SPT10 protein. Trends Biochem. Sci. 22, 154–155. [DOI] [PubMed] [Google Scholar]
- Nourani, A., Doyon, Y., Utley, R. T., Allard, S., Lane, W. S., and Côté, J. (2001). Role of an ING1 growth regulator in transcriptional activation and targeted histone acetylation by the NuA4 complex. Mol. Cell. Biol. 21, 7629–7640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papamichos-Chronakis, M., Petrakis T., Ktistaki, E. Topalidou, I., and Tzamarias. D. (2002). Cti6, a PHD domain protein, bridges the Cyc8-Tup1 corepressor and the SAGA coactivator to overcome repression at GAL1. Mol. Cell 9, 1297–1305. [DOI] [PubMed] [Google Scholar]
- Pasero, P., Bensimon, A., and Schwob, E. (2002). Single-molecule analysis reveals clustering and epigenetic regulation of replication origins at the yeast rDNA locus. Genes Dev. 16, 2479–2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Martín, J., and Johnson, A. D. (1998). Mutations in chromatin components suppress a defect of Gcn5 protein in Saccharomyces cerevisiae. Mol. Cell. Biol. 18, 1049–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajabi, H. N., Baluchamy, S., Kolli, S., Nag, A., Srinivas, R., Raychaudhuri, P., and Thimmapaya, B. (2005). Effects of depletion of CREB-binding protein on c-Myc regulation and cell cycle G1-S transition. J. Biol. Chem. 280, 361–374. [DOI] [PubMed] [Google Scholar]
- Reid, J. L., Iyer, V. R., Brown, P. O., and Struhl, K. (2000). Coordinate regulation of yeast ribosomal protein genes is associated with targeted recruitment of Esa1 histone acetylase. Mol. Cell 6, 1297–1307. [DOI] [PubMed] [Google Scholar]
- Reifsnyder, C., Lowell, J., Clarke, A., and Pillus, L. (1996). Yeast SAS silencing genes and human genes associated with AML and HIV-1 Tat interactions are homologous with acetyltransferases. Nat. Genet. 14, 42–49. [DOI] [PubMed] [Google Scholar]
- Rine, J., and Herskowitz, I. (1987). Four genes responsible for a position effect on expression from HML and HMR in Saccharomyces cerevisiae. Genetics 116, 9–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy, N., and Runge, K. W. (2000). Two paralogs that antagonistically control yeast life span. Curr. Biol. 10, 111–114. [DOI] [PubMed] [Google Scholar]
- Rundlett, S. E., Carmen, A. A., Kobayashi, R., Bavykin, S., Turner, B. M., and Grustein, M. (1996). HDA1 and RPD3 are members of distinct yeast histone deacetylase complexes. Proc. Natl. Acad. Sci. USA 93, 14503–14508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusche, L. N., Kirchmaier, A. L., and Rine, J. (2003). The establishment, inheritance, and function of silenced chromatin in Saccharomyces cerevisiae. Annu. Rev. Biochem. 72, 481–516. [DOI] [PubMed] [Google Scholar]
- Sandmeier, J. J., French, S., Osheim, Y., Cheung, W. L., Gallo, C. M., Beyer, A. L., and Smith, J. S. (2002). RPD3 is required for the inactivation of yeast ribosomal DNA genes in stationary phase. EMBO J. 21, 4959–4968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnell, R., and Rine, J. (1986). A position effect on the expression of a tRNA gene mediated by the SIR genes of Saccharomyces cerevisiae. Mol. Cell. Biol. 6, 494–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheridan, A. M., Force, T., Yoon, H. J., O'Leary, E., Choukroun, G., Taheri, M. R., and Bonventre, J. V. (2001). PLIP, a novel splice variant of Tip60, interacts with group IV cytosolic phospholipase A(2), induces apoptosis, and potentiates prostaglandin production. Mol. Cell. Biol. 21, 4470–4481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shore, D. (2000). The Sir2 protein family: a novel deacetylase for gene silencing and more. Proc. Natl. Acad. Sci. USA 97, 14030–14032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shou, W., et al. (2001). Net1 stimulates RNA polymerase I transcription and regulates nucleolar structure independently of controlling mitotic exit. Mol. Cell 8, 45–55. [DOI] [PubMed] [Google Scholar]
- Shou, W., Seol, J. H., Shevchenko, A., Baskerville, C., Moazed, D., Chen, Z. W., Jang, J., Charbonneau, H., and Deshaies, R. J. (1999). Exit from mitosis is triggered by Tem1-dependent release of the protein phosphatase Cdc14 from nucleolar RENT complex. Cell 97, 233–244. [DOI] [PubMed] [Google Scholar]
- Smith, E. R., Eisen, A., Gu, W., Sattah, M., Pannuti, A., Zhou, J., Cook, R. G., Lucchesi, J. C., and Allis, C. D. (1998a). ESA1 is a histone acetyltransferase that is essential for growth in yeast. Proc. Natl. Acad. Sci. USA 95, 3561–3565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, J. S., and Boeke, J. D. (1997). An unusual form of transcriptional silencing in yeast ribosomal DNA. Genes Dev. 11, 241–254. [DOI] [PubMed] [Google Scholar]
- Smith, J. S., et al. (2000). A phylogenetically conserved NAD+-dependent protein deacetylase activity in the Sir2 protein family. Proc. Natl. Acad. Sci. USA. 97, 6658–6663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, J. S., Brachmann, C. B., Pillus, L., and Boeke, J. D. (1998b). Distribution of a limited Sir2 protein pool regulates the strength of yeast rDNA silencing and is modulated by Sir4p. Genetics 149, 1205–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, J. S., Caputo, E., and Boeke, J. D. (1999). A genetic screen for ribosomal DNA silencing defects identifies multiple DNA replication and chromatin-modulating factors. Mol. Cell. Biol. 19, 3184–3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterner, D. E., and Berger, S. L. (2000). Acetylation of histones and transcription-related factors. Microbiol. Mol. Biol. Rev. 64, 435–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone, E. M., Heun, P., Laroche, T., Pillus, L., and Gasser, S. M. (2000). MAP kinase signaling induces nuclear reorganization in budding yeast. Curr. Biol. 10, 373–382. [DOI] [PubMed] [Google Scholar]
- Strahl, B. D., and Allis, C. D. (2000). The language of covalent histone modifications. Nature 403, 41–45. [DOI] [PubMed] [Google Scholar]
- Strahl-Bolsinger, S., Hecht, A., Luo, K., and Grunstein, M. (1997). SIR2 and SIR4 interactions differ in core and extended telomeric heterochromatin in yeast. Genes Dev. 11, 83–93. [DOI] [PubMed] [Google Scholar]
- Straight, A. F., Shou, W., Dowd, G. J., Turck, C. W., Deshaies, R. J., Johnson, A. D., and Moazed, D. (1999). Net1, a Sir2-associated nucleolar protein required for rDNA silencing and nucleolar integrity. Cell 97, 245–256. [DOI] [PubMed] [Google Scholar]
- Suka, N., Luo, K., and Grunstein, M. (2002). Sir2p and Sas2p opposingly regulate acetylation of yeast histone H4 lysine16 and spreading of heterochromatin. Nat. Genet. 32, 378–383. [DOI] [PubMed] [Google Scholar]
- Takechi, S., and Nakayama, T. (1999). Sas3 is a histone acetyltransferase and requires a zinc finger motif. Biochem. Biophys. Res. Commun. 266, 405–410. [DOI] [PubMed] [Google Scholar]
- Tamburini, B. A., and Tyler, J. K. (2005). Localized histone acetylation and deacetylation triggered by the homologous recombination pathway of double-strand DNA repair. Mol. Cell. Biol. 25, 4903–4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utley, R. T., and Côté, J. (2003). The MYST family of histone acetyltransferases. Curr. Top. Microbiol. Immunol. 274, 203–236. [DOI] [PubMed] [Google Scholar]
- van Leeuwen, F., and Gottschling, D. E. (2002). Assays for gene silencing in yeast. Methods Enzymol. 350, 165–186. [DOI] [PubMed] [Google Scholar]
- Vogelauer, M., Wu, J., Suka, N., and Grunstein, M. (2000). Global histone acetylation and deacetylation in yeast. Nature 408, 495–498. [DOI] [PubMed] [Google Scholar]
- Waltzer, L., and Bienz, M. (1998). Drosophila CBP represses the transcription factor TCF to antagonize Wingless signalling. Nature 395, 521–525. [DOI] [PubMed] [Google Scholar]
- Wonnacott, T. H., and Wonnacott, R. J. (1984). Introductory Statistics for Business and Economics, New York: John Wiley & Sons.
- Yamagoe, S., et al. (2003). Interaction of histone acetylases and deacetylases in vivo. Mol. Cell. Biol. 23, 1025–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan, Y., Barlev, N. A., Haley, R. H., Berger, S. L., and Marmorstein, R. (2000). Crystal structure of yeast Esa1 suggests a unified mechanism for catalysis and substrate binding by histone acetyltransferases. Mol. Cell 6, 1195–1205. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.