

Abstract
Interferons are cytokines with potent antiviral and antiproliferative activities. We report that although a transient exposure to β-interferon induces a reversible cell cycle arrest, a sustained treatment triggers a p53-dependent senescence program. β-Interferon switched on p53 in two steps. First, it induced the acetylation of p53 at lysine 320 and its dephosphorylation at serine 392 but not p53 activity. Later on, it triggered a DNA signaling pathway, the phosphorylation of p53 at serine 15 and its transcriptional activity. In agreement, β-interferon–treated cells accumulated γ-H2AX foci and phosphorylated forms of ATM and CHK2. The DNA damage signaling pathway was activated by an increase in reactive oxygen species (ROS) induced by interferon and was inhibited by the antioxidant N-acetyl cysteine. More important, RNA interference against ATM inhibited p53 phosphorylation at serine 15, p53 activity and senescence in response to β-interferon. β-Interferon–induced senescence was more efficient in cells expressing either, p53, or constitutive allele of ERK2 or RasV12. Hence, β-interferon–induced senescence targets preferentially cells with premalignant changes.
INTRODUCTION
Many cell types are able to enter a stable and viable post-mitotic state in response to oncogenic stresses such as DNA damage, short telomeres, and certain oncogenes. This condition, known as cellular senescence, is regulated by tumor suppressors such as p53, RB, p16INK4a, p19ARF, and PML and engages a specific gene expression program (Ferbeyre, 2002; Lowe et al., 2004; Shay and Roninson, 2004). By preventing the expansion of potentially malignant cells, senescence may act as a barrier to tumor formation. Several signaling pathways must connect different stressors to the senescence program. For example, short telomeres or DNA damage activate the senescence program through the checkpoint proteins ATM, ATR, CHK1, and CHK2 (d'Adda di Fagagna et al., 2003; Herbig et al., 2004; Zglinicki et al., 2005). On the other hand, oncogenic stresses do not regulate senescence through telomere shortening and they may signal to senescence regulators through the production of reactive oxygen species (Lee et al., 1999; Wu et al., 2004; Catalano et al., 2005). In addition, it is established that some senescence regulators (p53, PML, and IFI16) are also targets of interferon-stimulated transcription factors (Lavau et al., 1995; Stadler et al., 1995; Takaoka et al., 2003; Xin et al., 2003, 2004; de Stanchina et al., 2004). Accordingly, senescence prevented ras-transformation in wild-type fibroblasts, but not in fibroblasts from mice lacking the interferon regulated transcription factor IRF1 (Tanaka et al., 1994). Together, these studies suggest that signaling through the interferon pathway may play a role in senescence.
Interferons comprise a family of cytokines with antiviral and antiproliferative activity. They include the type I interferon family (mainly α and β-interferon) and type II or γ-interferon (Taniguchi and Takaoka, 2002). Type-I interferon, usually produced by cells infected with viruses, acts locally, at high concentrations and in a paracrine manner (Dianzani, 1992). β-Interferon can also act as an autocrine growth inhibitor during hematopoietic cell differentiation (Resnitzky et al., 1986) and is constitutively expressed by terminally differentiated skin keratinocytes (Bielenberg et al., 1999). These studies suggest that β-interferon can play a role in promoting a permanent withdrawal from the cell cycle.
We show here that prolonged treatment with β-interferon, but not α-interferon, induced senescence in normal cells. Interferon-induced senescence was associated with high levels of reactive oxygen species (ROS), activation of the ATM/CHK2 DNA damage signaling pathway and engagement of the tumor suppressors p53 and RB. Notably, inhibition of p53 activity in p53 null mouse fibroblasts, human cells expressing the viral oncoprotein E6, or an shRNA against ATM inhibited senescence in response to β-interferon. More important, β-interferon induced senescence more efficiently in cells expressing p53, an activated allele of ERK2 or RasV12. This suggests that the senescence program is preferentially activated in normal cells where viral or oncogenic stresses are persistent.
MATERIALS AND METHODS
Cells and Retroviruses
Normal human diploid fibroblasts IMR90 (ATCC, Manassas, VA) were cultured in DMEM (Invitrogen, Logan, Utah) supplemented with 10% fetal bovine serum (FBS; Invitrogen) and 1% penicillin G/streptomycin sulfate (Invitrogen). Retroviruses, pLPC and pLPCp53, were described in (Ferbeyre et al., 2000), MSCV and MSCVshRNA-ATM were described in Mukhopadhyay et al. (2005). TSP cells (p53 null fibroblasts expressing a temperature-sensitive p53) was previously described (Ferbeyre et al., 2002). Human β-interferon was kindly provided by Biogen; (Boston, MA) α-interferon A and mouse β-interferon were purchased from Biosource (Camarillo, CA).
Cell Proliferation and Senescence Determination
To determine cell proliferation rates we used a BrdU incorporation kit (BD Biosciences), a thymidine incorporation assay (Ferbeyre et al., 2000) and estimated cell counts at different times after plating using a crystal violet retention assay (Ferbeyre et al., 2000). Fluorescent-activated cell sorting (FACS) and cell cycle analysis were done according to Ferbeyre et al. (2000). Data were analyzed by the software CellQuest (version 3.3). Senescence-associated β-galactosidase (SA-β-gal) activity was assayed as described by (Dimri et al., 1995). Data were quantified from 200 cells counts in three independent experiments (n = 3).
Protein Expression
To prepare total cell protein, cells were collected by trypsinization, washed with phosphate-buffered saline (PBS), lysed in SDS sample buffer (60 mM TrisHCl. pH 6.8, 10% glycerol, 2% SDS. and 100 mM DTT) and boiled for 5 min. For Western blots, 30–50 μg of total cell protein was separated on SDS-PAGE gels and transferred to Immobilon-P membranes (Millipore. Bedford, MA). We measured the levels of α-tubulin to verify equal loading of the different samples. Primary antibodies used are listed in Supplementary Methods. Signals were revealed after incubation with anti-mouse or anti-rabbit secondary antibodies coupled to peroxidase (Amersham Pharmacia, Amersham, United Kingdom) by using enhanced chemiluminescence (ECL, Amersham Pharmacia) or Lumi-Light Plus (Roche).
Fluorescence Microscopy
For fluorescence microscopy, 2 × 104 IMR90 cells were plated on coverslips placed on six-well plates (Costar, Corning, NY). Twenty-four hours after plating, the cells were fixed with 4% paraformaldehyde for 15 min at room temperature. Subsequently, the cells were washed in PBS and permeabilized using ice-cold 0.2% Triton X-100 in PBS/3% BSA solution for 5 min. Then the cells were washed three times with PBS/BSA and incubated for 1 h at room temperature with anti-phospho ERK1/2 (9106, 1:300, Cell Signaling), anti-PML (PGM3, 1:200, Santa Cruz Biotechnology, Santa Cruz, CA), or anti-phospho-H2AX (JBW301, 1:200, Upstate Biotechnology, Lake Placid, NY) as primary antibodies. Images from independent field were captured and processed with the software Metamorph. Each experiment was repeated three times and representative fields are presented in each figure.
RT-PCR
Total cellular RNA was prepared from cells treated with 2000 IU of β-interferon or vehicle for 6 or 12 d using Ultraspec (Biotecx Laboratories, Houston, TX) reagents and instructions. The pool of first strand cDNAs was synthesized with RevertAid H Minus First Strand Synthesis Kit (Fermentas) using total RNA template and random hexameric primers. Primers used to amplify p21; PIG3, total PML, and PMLIV are listed in Supplementary Methods. We measured the levels of the β-actin mRNA to verify equal loading and amplification of the different samples.
Luciferase Assay
For luciferase assay, IMR90 cells were plated at 3 × 105 cells per 6-cm plate. Cells were transfected using FUGENE 6 (Roche) with 2.5 μg of the firefly luciferase reporter plasmid under the control of the HDM2 promoter, 0.5 μg of pLPC or its derivative expressing p53 and 0.1 μg of the renilla luciferase reporter plasmid under the control of the β-globin promoter. Then we added β-interferon (1000 U/ml), and cells were harvested 24 or 72 h later for Dual-luciferase assay (Promega, Madison, WI).
Determination of Mitochondrial Mass and ROS
To measure mitochondrial mass we used MitoFluor-Green (Molecular Probes. Eugene, OR) or MitoTracker-Red. Cells were incubated for 15 min with 100 nM MitoFluor-Green or MitoTracker-Red and then they were washed with PBS and the intensity of labeling was measured by FACS (MitoFluor-Green) or immunofluorescence (MitoTracker-Red). To measure ROS cells were incubated with dichlorodihydrofluorescein diacetate (H2DCFDA) from Molecular Probes. This compound emits fluorescence when oxidized by ROS. Fluorescence was monitored by immunofluorescence microscopy or FACS.
Statistics
Pairwise comparison between groups was performed using the Student's t test. A value of p ≤ 0.05 was considered significant.
RESULTS
Prolonged Treatment With β-Interferon Induces Senescence in Normal Human Fibroblasts
Expression of oncogenic ras in normal human fibroblasts IMR90 triggers a permanent cell cycle arrest with features of cellular senescence and expression of some interferon responsive genes (Serrano et al., 1997; Ferbeyre et al., 2000). To evaluate whether interferon was sufficient to activate the senescence program in normal fibroblasts, we treated these cells with 2000 U of β-interferon every 3 d. In these conditions, the cells proliferated very slowly during the first 9 d of treatment and most of them arrested with a typical senescent morphology around day 12 (Figure 1A). At this time, the ability of β-interferon–treated cells to synthesize DNA was lower than control growing cells and similar to that of cells arrested in low serum, as measured by incorporation of BrdU (p ≤ 0.014; Figure 1B) or labeled thymidine (p ≤ 0.01; Figure 1C). DNA content analysis by flow cytometry of propidium iodide–stained cells revealed that most of the cells were arrested in G1 (Figure 1D).
Figure 1.

Treatment with β-interferon induces a senescent cell cycle arrest in normal fibroblasts. (A) Growth curves. Cell numbers were followed for 12 d in IMR90 cells treated with β-interferon or vehicle. (B) Cells were treated with β-interferon or vehicle for 12 d and then they were pulsed with BrdU for 3 h and fixed, and BrdU incorporation was measured by indirect immunofluorescence. Cells on low serum (LS) were used as a control for cell cycle arrest. Data were quantified by counting 50 cells in different fields in three independent experiments and analyzed with the Student's t test. (C) Cells were treated with β-interferon or vehicle for 12 d; then they were pulsed with [3H] thymidine for 24 h, and thymidine incorporation was measured by scintillation counting and analyzed with the Student's t test. (D) Cell cycle analysis of cells treated with β-interferon. Cells were treated for 12 d with β-interferon or vehicle, stained with propidium iodide, and analyzed by flow cytometry. The data represent a typical profile that was reproduce in three independent experiments. (E) Senescence associated β-galactosidase at indicated times. (F and G) Reversibility of the senescence phenotype. Cells were treated with interferon for 18 d or hydroxyurea (HU) for 12 d and then washed and incubated in normal medium. Cells were fixed and stained for SA-β-Gal 0, 3, and 6 d after withdrawal of β-interferon or HU. The total amount of SA-β-Gal–positive cells per field in three independent fields of view was counted. Values represent the mean and SD of three independent measurements. In comparison to the control cells the number of SA-β-Gal–positive cells in samples treated with β-IFN of HU was statistically significant (p ≤ 0.001).
To confirm that β-interferon–treated cells entered a senescent cell cycle arrest, we measured several senescence markers along the treatment schedule. Cells did not significantly stain positive for the senescence associated β-galactosidase (SA-β-Gal) 6 d posttreatment. However, ∼75% of the cells were morphologically senescent and SA-β-Gal positive 12 d after treatment (Figure 1E). We also studied the status of the MAP kinases ERK1/2 and p38 in cells treated with β-interferon. β-Interferon induced a nuclear exclusion of phospho-ERK1/2 as early as 6 d after treatment, but most notably at later time points (Supplementary Figure 1, A and B) in correlation with an increase in the number of senescent cells. Of note, PEA-15, a protein capable of promoting the cytoplasmic localization of phospho-ERK1/2 (Formstecher et al., 2001; Gaumont-Leclerc et al., 2004), was increased in response to β-interferon (Supplementary Figure 1B). β-Interferon also induced the activation of the p38MAP kinase (Supplementary Figure 1B). Hence, β-interferon–treated cells display altered MAP kinase signaling pathways, as described for senescence in response to short telomeres or oncogenes (Iwase et al., 1997; Lim et al., 2000; Wang et al., 2002; Gaumont-Leclerc et al., 2004).
β-Interferon–treated cells also displayed an increased number of PML bodies and an increase in the levels of the PML protein (Supplementary Figure 1C), as reported for senescence induced by oncogenes or short telomeres (Ferbeyre et al., 2000). This increase in PML was also detected at the mRNA level and involved PML-IV (Supplementary Figure 1D), the only PML isoform known to induce senescence (Bischof et al., 2002). Finally, β-interferon–treated cells were strongly labeled by anti-K9-methyl histone 3, a marker of SAHFs (senescence-associated heterochromatin foci), which are typical of senescent cells (Narita et al., 2003; Supplementary Figure 1D).
We also treated normal fibroblasts with α-interferon but we could not induce senescence or cell cycle arrest with the same efficiency (Supplementary Figure 2A). α-Interferon was also less potent than β-interferon in inducing STAT1 phosphorylation (Supplementary Figure 2B), explaining its inability to induce senescence in normal human fibroblasts at doses similar to β-interferon. In agreement, it has been reported that treatment of epithelial cell lines with α- or γ-interferon induced a reversible cell cycle arrest in G1 (Harvat and Jetten, 1996; Akiyama et al., 1999).
Senescence is considered a stable cell phenotype. To determine whether β-interferon–treated cells remained senescent, we washed the cells and incubated them in medium without β-interferon. Then we counted the number of SA-β-Gal–positive cells in different fields of view under the microscope 3 or 6 d later. In this experiment, some cells resumed proliferation but the total number of SA-β-Gal–positive cells did not decrease with time (Figure 1, F and G). As a positive control for this experiment we induced senescence with hydroxyurea (HU; Yeo et al., 2000). The number of SA-β-Gal–positive cells after HU withdrawal also remained stable. Significantly, we reproducibly noticed an increase in the number of SA-β-Gal–positive cells in β-interferon–treated cells after we stopped the treatment. This suggested that some cells that were nonsenescent by the time of interferon withdrawal did carry out modifications that eventually triggered their senescence. Taken together, our data indicate that long-term treatment with β-interferon triggers a permanent cell cycle arrest with features of cellular senescence in a significant fraction of the cells.
β-Interferon Engages the p53- and RB-Tumor Suppressor Pathways
The p53 and the RB tumor suppressor pathways are central regulators of the senescence response (Ferbeyre, 2002; Lowe et al., 2004). It is then reasonable to expect that β-interferon activates specific signaling pathways to engage these tumor suppressors. To collect evidence for the activation of these two tumor suppressor pathways, we performed an expression analysis of several proteins regulated by RB or p53.
We first examined the status of several key cell cycle regulators controlled by the RB tumor suppressor pathway in cells treated with β-interferon. β-Interferon induced the accumulation of hypophosphorylated RB, a reduction in total RB levels and the repression of E2F genes such as cyclin A and Mcm6 (Figure 2A), as seen in other forms of senescence (Ferbeyre et al., 2000; Mallette et al., 2004). We conclude that there is a clear signature of RB engagement in β-interferon–treated cells.
Figure 2.

Status of several members of the RB and p53 pathways in cells treated with β-interferon. (A) Immunoblots for RB and various members of the RB pathway. (B) Immunoblots for p53, its modifications, and targets. C+, positive control. (C) RT-PCR for PIG-3, p21, and β-actin in cells treated for 6 or 12 d with β-interferon, or cells treated with adryamicin (Ad).
Next we measured the levels of several upstream regulators of the RB pathway to get insight into the mechanism by which β-interferon activates RB. We found no change in the levels of CDK2 (unpublished data), CDK4, and cyclin D. The levels of CDK6 were moderately reduced. Among the CDK inhibitors, only p27, but not p21 or p16INK4a, was found consistently increased (Figure 2A). The data suggest that β-interferon engages the RB tumor suppressor pathway by preventing RB phosphorylation due to low CDK6 activity and high p27 levels.
Then, we measured total p53 levels and observed that they were not significantly augmented in cells treated with β-interferon for 6 or 12 d (Figure 2B). Next we studied p53 modifications because they are a more reliable marker of p53 activity. For example, a decrease in serine 392 phosphorylation and an increase in serine 15 phosphorylation were reported in association with replicative senescence (Webley et al., 2000), Ras-induced senescence and PML-induced senescence (Ferbeyre et al., 2000, 2002). As expected, we found that β-interferon reduced phosphorylation at serine 392 and increased the phosphorylation at serine 15. It was reported that p53 is acetylated at lysine 382 during Ras-induced senescence (Pearson et al., 2000). However, we could not find p53 acetylation at this site in β-interferon–treated cells, even after treatment with trichostatin A, which inhibits endogenous deacetylases (unpublished data). One explanation for this result could be that interferon-stimulated transcription factors STAT1 or STAT2 titrate the acetylases CBP/p300 required for acetylation at lysine 382 (Hottiger et al., 1998). Remarkably, β-interferon–induced acetylation of p53 at lysine 320 (Figure 2B). This acetylation correlated with the induction of pCAF by interferon, both at the mRNA and protein level (Supplementary Figure 3, A and B). In agreement, the oncoprotein E7 that blocks pCAF, but not E7 mutants unable to bind pCAF, inhibited the ability of interferon to induce p53 acetylation at K320 (Supplementary Figure 3D). Taken together the posttranslational modifications of p53 induced by β-interferon suggest activation of the p53 pathway.
Next we measured the endogenous levels of two p53 target genes, HDM2 and p21. We found an accumulation of HDM2 but not p21 protein levels. This poor accumulation of p21 may be secondary to a decrease in p21 protein stability in β-interferon–treated cells, because we readily detected an induction of p21 by β-interferon after 12 d of treatment using quantitative RT-PCR (Figure 2C). β-Interferon also induced the p53 target gene PIG3 as early as 6 d after treatment (Figure 2C). Hence, β-interferon engages the p53 tumor suppressor pathway and promotes p53-dependent transcription.
β-Interferon Signals to p53 in Two Stages
To get more insight into the mechanisms of p53 activation by β-interferon, we first measured p53 levels and modifications at 24 h intervals after treatment with β-interferon. Interferon activity, as measured by PML induction, was detected 1 d after treatment and remained elevated during the following days. Also, acetylation at lysine 320 and dephosphorylation at serine 392 were both early events in response to β-interferon treatment (Figure 3A). In fact, acetylation at K320 appeared as early as 4 h after treatment with β-interferon (Figure 3B). In contrast, serine-15 phosphorylation appeared later, around 3 d after treatment, and coinciding with the induction of the p53 target gene HDM2. Thus, p53 activity and modifications in response to β-interferon evolved with time, probably reflecting different signaling pathways activated by β-interferon. In agreement with the immunoblots, transient reporter assays revealed that β-interferon was unable to stimulate p53 activity on the HDM2 promoter after 1 d of treatment. However, 3 d of β-interferon treatment were sufficient to stimulate the HDM2 promoter (p ≤ 0.0003; Figure 3C). The data suggest that the early signaling from β-interferon to p53 does not activate its transcriptional activity and that a second wave of signaling was required to do so.
Figure 3.
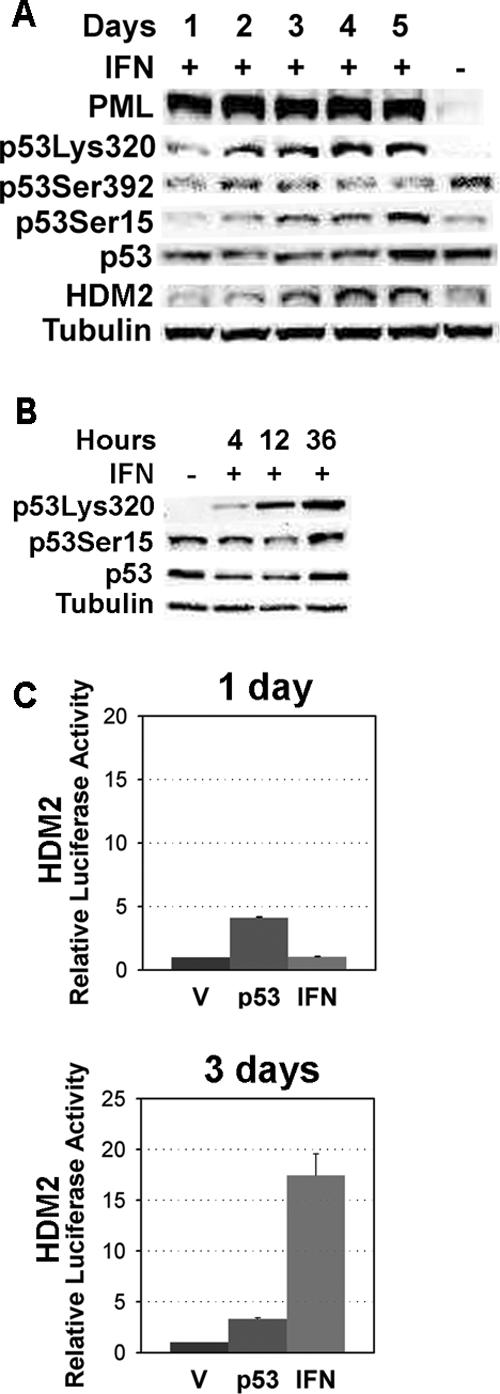
Two signaling stages from β-interferon to p53. (A) Immunoblots for several p53 modifications and HDM2 in cells treated with β-interferon or vehicle for the indicated amounts of time. (B) p53 and p53 modifications after 4, 12, or 36 h of β-interferon treatment. (C) Luciferase reporter assays with the HDM2 promoter in normal human fibroblasts transfected with p53 or treated with β-interferon. Data were analyzed with the Student's t test.
Role of p53 in β-Interferon–induced Senescence
Next we asked whether interferon would cooperate with enforced p53 expression to regulate senescence. We have shown that increasing p53 levels in normal human or mouse cells was not sufficient to trigger senescence. However, introduction of p53 in p53 null cells expressing oncogenic ras did induce senescence (Ferbeyre et al., 2000, 2002). We reasoned that β-interferon, like oncogenic ras, might also convey complementary signals to p53. To explore that possibility, we enforced p53 expression in normal human fibroblasts IMR90 using a retroviral vector. As shown before, p53 induced a cell cycle arrest from which the cells slowly escape 10 d after infection (unpublished data). However, treating p53-expressing cells with β-interferon induced a higher proportion of senescent cells than p53-expressing or interferon-treated cells alone (p ≤ 0.001). More important, β-interferon only required 6 d to induce senescence in p53-expressing cells, whereas it required 12 d in control cells (Figure 4A). Although β-interferon did not further stabilize exogenous p53, it did promote its phosphorylation at serine 15 (Figure 4B). These results suggest that stimuli that stabilize p53 might sensitize normal cells to β-interferon.
Figure 4.

Role of p53 in β-interferon–induced senescence. (A) SA-β-Gal: IMR90 cells infected with the retroviral vector pLPC or its derivative expressing p53 were treated with β-interferon or vehicle. Data were analyzed with the Student's t test. (B) Immunoblots from cells as above collected 6 d after retroviral infection. (C) SA-β-Gal: IMR90 cells were infected with the retroviral vector LXSN or its derivative expressing E6. Then the cells were treated with β-interferon or vehicle for 12 d and fixed for SA-β-Gal staining. (D) Immunoblots for cell cycle regulators and the p53 pathway in E6-expressing cells and control after treatment with β-interferon or vehicle for 12 d. (E) Colony formation assay at 39°C of TSP cells previously treated with α- or β-interferon for 8 d at 39°C or for 2, 4, or 8 d at 32°C. TSP cells expressing oncogenic ras were used as a positive control for induction of a permanent cell cycle arrest after activation of p53 at 32°C. Colonies were counted in three independent experiments, and the data were analyzed for significance with the Student's t test.
Then, we asked whether p53 was required for β-interferon–induced senescence. To do so, we prepared cells expressing the viral oncoprotein E6, widely used to interfere with p53 functions (Scheffner et al., 1990). In E6-expressing cells, β-interferon slowed cell growth and engaged the RB pathway, but p53 levels and activity were dramatically impaired and as a consequence most cells did not stain positive for SA-β-Gal (Figure 4, C and D and Supplementary Figure 4). Of note E6 did not interfere with interferon signaling to p53, because we could still detect modifications of p53 at serine 15 (Figure 4D) and lysine 320 (unpublished data).
E6 has been extensively used as a reagent to demonstrate the p53 requirements for a variety of processes in human cells. However, in principle E6 may have other targets relevant to senescence. To further demonstrate the requirement for p53 in β-interferon–induced senescence we used p53 null mouse embryonic fibroblasts engineered to express a temperature sensitive p53 (TSP cells; Ferbeyre et al., 2002). TSP cells are immortal as long as they are kept growing at 39°C. Activation of p53 in these cells can be achieved by incubating the cells at 32°C and results in a reversible cell cycle arrest. This is seen in Figure 4E where the mean number of colonies obtained from cells after incubating for 8 d at 32°C (170 ± 13) was slightly higher although not significantly different (p = 0.06) from that obtained from cells growing at 39°C (148 ± 22). We previously showed that expression of oncogenic ras in these cells turned the p53-mediated transient cell cycle arrest into a permanent senescent arrest (Ferbeyre et al., 2002). Treatment of TSP cells growing at 39°C with mouse β-interferon did not induce growth arrest (Figure 4E). However, treatment of cells at 32°C with β-interferon for 8 d reduced significantly (p ≤ 0.005) their colony formation ability in comparison with untreated cells arrested at 32°C for the same amount of time. These results indicate that β-interferon, like oncogenic ras, was able to signal to p53 to induce a permanent cell cycle arrest. Together, our data indicate that p53 is critical for the long-term cell cycle effects of β-interferon, raising the question about the mechanism by which β-interferon triggers these p53-activating signals.
Activation of the ATM/CHK2 Checkpoint Pathway by β-Interferon Is Required for p53 Activation and Senescence
β-Interferon induced phosphorylation of p53 at serine 15, a modification catalyzed by kinases of the DNA damage signaling pathway, such as ATR and ATM (Canman et al., 1998; Hammond et al., 2002). Intriguingly, β-interferon induced the phosphorylation of CHK2, but not CHK1 (Figure 5A), suggesting that it activates the ATM signaling pathway (Falck et al., 2001). To prove whether ATM acts downstream β-interferon to activate p53 and senescence, we used a previously validated anti-ATM shRNA (Mukhopadhyay et al., 2005). This shRNA was able to knockdown 90% of ATM expression (Figure 5B) and inhibited the ability of β-interferon to induce CHK2 phosphorylation (Figure 5A). In addition, the ATM shRNA reduced the stimulation of the HDM2-luciferase reporter (p = 0.005; Figure 5C) and senescence (p = 0.014; Figure 5D) in cells treated with β-interferon. This genetic study was supported by the presence of additional biochemical markers of ATM activation in β-interferon–treated cells. First, β-interferon–treated cells accumulated phospho-H2AX foci (Figure 5E), which are typical of DNA damage or dysfunctional telomeres (d'Adda di Fagagna et al., 2003; Takai et al., 2003). In addition, β-interferon–treated cells accumulated ATM protein phosphorylated at serine 1981, a modification that indicates ATM activation (Bakkenist and Kastan, 2003; Figure 5F). We concluded that β-interferon activates the DNA damage signaling pathway and requires the ATM kinase to signal to p53 and induce senescence after long-term stimulation. These results raised the important question about how interferon may activate the DNA damage signaling pathway.
Figure 5.

β-Interferon induces the DNA damage-signaling pathway. (A) Immunoblots of phospho-p53S15 and phospho-CHK2T68 in cells with an empty vector or its derivative expressing a shRNAs against ATM. (B) ATM levels in cells with an empty vector or its derivative expressing a shRNAs against ATM. (C) Luciferase reporter assays with the HDM2 promoter in normal human fibroblasts infected with a control vector or anti-ATM shRNA and treated with β-interferon or vehicle. (D) Relative percent of SA-β-Gal–positive cells bearing an empty vector or its derivative expressing shRNAs against ATM after treatment with β-interferon or vehicle for 12 d. Control cells treated with β-interferon were taken as 100%. Differences between each group and the control and between cells with the shRNA and control vector were statistically assessed with the Student's t test. (E) Representative fields of immunofluorescence for H2AX foci in cells treated with β-interferon for the indicated times or H2O2. (F) Immunoblots for phospho-ATMS1981.
To investigate the mechanism of activation of the DNA damage signaling pathway in β-interferon–treated cells, we look for the possibility of production of endogenous DNA-damaging agents. We first noticed that early after treatment, β-interferon–treated cells displayed an increase in mitochondrial mass (labeling with MitoFluor-Green; Figure 6A) or MitoTracker-Red (Figure 6B). Because mitochondria are an important source of ROS, we stained the cells with 2′,7′-dichlorofluorescein diacetate (H2DCFDA), a compound that fluoresces in the presence of high levels of ROS (Figure 6, C and D). We found that β-interferon induced ROS production, suggesting a mechanism for inducing DNA damage and activation of the DNA damage signaling pathway.
Figure 6.

Interferon increases mitochondrial mass and accumulation of reactive oxygen species. (A) FACS measurement of mitochondrial mass with MitoFluor-Green in cells treated for 2 d with β-interferon or vehicle. (B) Immunofluorescence of cells labeled with MitoTracker-Red. (C) Immunofluorescence detection of cells labeled by the ROS sensor H2DCFDA. (D) FACS detection of ROS with the H2DCFDA probe. Control cells or cells treated with interferon for 12 d were also treated with 4 mM N-acetyl l-cysteine for 12 d. (E) Immunoblots for phospho-CHK2Thr68 in cells as in C. (F) Immunofluorescence of H2AX foci in cells treated as in C. Cells with at least one foci were scored as positive and the mean and SD of three independent experiments is indicated at the bottom right of each panel. Cells with multiple foci were only seen in interferon-treated cells.
To prove the role of ROS in the DNA damage signaling pathway activated by β-interferon, we treated the cells with N-acetyl-cysteine (NAC). As expected, NAC reduced ROS accumulation in β-interferon–treated cells (Figure 6D). More important, NAC inhibited the phosphorylation of CHK2 and the accumulation of phospho-H2AX foci in β-interferon-treated cells (p = 0.02; Figure 6, E and F). However, we could not study whether NAC could block senescence in response to β-interferon, because long-term incubation with NAC was toxic for the cells. Together the data indicate that DNA damage, at least in part, as a consequence of ROS accumulation, engages the ATM-CHK2 pathway in normal cells treated for several days with β-interferon.
Constitutively Active ERK2 or RasV12 Sensitizes Normal Cells to Senescence Induced by β-Interferon
Normal cells would be rarely exposed to constitutively high levels of β-interferon unless they are experiencing viral infections, oncogenic stress, or specific cell differentiation programs (Resnitzky et al., 1986; Bielenberg et al., 1999). Therefore we wanted to know whether early oncogenic changes would sensitize normal cells to β-interferon. To investigate the effect of oncogenic threats on the response of normal cells to β-interferon, we prepared normal human fibroblasts IMR90 expressing a constitutively activated allele of ERK2 (Emrick et al., 2001; Gaumont-Leclerc et al., 2004). We also took advantage from the fact that cells expressing RasV12 from a weak promoter (PGK) do not enter senescence (Deng et al., 2004). We used these oncogenes because constitutive ERK signaling as a result of ras mutations or autocrine growth factors is a hallmark of many tumors (Johnson and Lapadat, 2002). As expected, more cells expressing activated ERK2 or RasV12 entered senescence in response to β-interferon in comparison with a control bearing an empty vector (Figure 7A). Also, growth arrest in response to β-interferon was more efficient in ERK2-expressing cells than in control vector–bearing cells (Figure 7B). Intriguingly, ERK2-expressing cells arrested their proliferation as small cells with few points of attachment to the plate. This morphology was originally reported in cells that entered senescence in response to a constitutively active allele of MEK, the kinase upstream ERK in the Ras/MAP kinase pathway (Lin et al., 1998).
Figure 7.

Constitutively active ERK2 sensitizes normal fibroblasts to β-interferon. (A) Cell expressing constitutively activated ERK2, RasV12, or vector control were treated with 1000 U of β-interferon for 12 d and then fixed and stained for of SA-β-Gal. Data were analyzed by the Student's t test: for (V; ERK2) p = 0.006 and for (V; RasV12) p = 0.003. (B) Growth curves. Cell numbers were followed for 12 d in IMR90 cells bearing a vector control or its derivative expressing constitutively active ERK2 and treated with β-interferon or vehicle. (C) Immunoblot for phospho-STAT1 in IMR90 cells with empty vector or its derivative expressing activated ERK2 and treated with β-interferon or vehicle for 12 d (Akiyama et al., 1999). (D) Luciferase reporter assays with the β-interferon promoter in normal human fibroblasts expressing constitutively active ERK2 or a vector control and treated with β-interferon or vehicle. Data were analyzed with the Student's t test.
To get insight into the mechanism by which the ERK1/2 MAP kinase pathway might sensitize normal cells to β-interferon, we studied STAT1 phosphorylation, which is a direct consequence of interferon binding to its receptor. We found that cells expressing the constitutively active ERK2 allele have more phospho-STAT1 than control cells. STAT1 in complex with STAT2 and IRF9 induce the expression of multiple interferon target genes, including STAT1, IRF1, and IRF7. The last two can activate the β-interferon promoter (Taniguchi and Takaoka, 2002), generating a powerful positive feedback mechanism that amplifies the interferon response and sensitizes cells to interferon stimulation. To check whether such a mechanism could operate in cells with constitutive activation of the MAP kinase pathway, we studied the activation of the β-interferon promoter reporter. As expected, β-interferon significantly activated transcription form its own promoter (p = 0.001). Interestingly, constitutive ERK dramatically increased the ability of β-interferon to stimulate the β-interferon promoter reporter (16 ± 0.9 vs. 2.3 ± 0.3; p = 0. 001; Figure 7D). Hence, constitutive ERK signaling sensitized normal fibroblasts to β-interferon and this effect correlates with higher STAT1 phosphorylation and increased β-interferon activity.
DISCUSSION
We report here that β-interferon can block cell cycle progression in normal human fibroblasts and trigger a cellular senescence program by engaging a DNA damage signaling pathway, ATM, and the tumor suppressor pathways controlled by RB and p53. The cellular response to β-interferon was time dependent. Early after treatment, β-interferon only induced a reversible growth arrest and was unable to activate p53 transcriptional activity or induce senescence. However, upon long-term treatment, β-interferon induced p53-dependent senescence in normal fibroblasts, a process that was potentiated in cells with a constitutive activation of the ERK1/2 MAP kinase pathway or enforced expression of p53. In agreement with our results, it was reported that β-interferon induced a “noncharacteristic senescent arrest” in a glioma cell line, but the mediators and mechanism of the senescence response were not described (Kaynor et al., 2002).
One critical question is what is the signal that converts a reversible cell cycle arrest into senescence after prolonged treatment with β-interferon. First, we found that p53 was required for senescence, suggesting that the signal that triggers senescence may interact with the p53 tumor suppressor pathway. Subsequently, we observed that early after interferon treatment p53 was acetylated at lysine 320, dephosphorylated at serine 392 and transcriptionally inactive. Serine 392 phosphorylation is catalyzed by casein kinase 2 in UV-treated cells (Blaydes and Hupp, 1998), and it may increase the DNA-binding ability of p53 in those conditions (Hupp et al., 1992). In agreement, hypoxia decreased phosphorylation at S392 and inhibited p53-dependent apoptosis (Achison and Hupp, 2003). Also, mice engineered to express a p53 allele that cannot be phosphorylated at serine 389, the equivalent to human Serine 392, are predisposed to UV-induced skin tumors and acetyl-aminofluorene–induced bladder tumors (Hoogervorst et al., 2005). Our observation that p53 is transcriptionally inactive and dephosphorylated at serine 392 early after interferon treatment is consistent with those reports. However, we did observe an increase in p53 activity in our cells, despite lack of phosphorylation at serine 392, after 3 d of treatment with β-interferon. Several models may account for this apparent contradiction. One idea is that, in some settings, phosphorylation at serine 392 may play a negative role on p53 because it was reported that this phosphorylation could promote p53 export from the nucleus (Kim et al., 2004). Another explanation is that the lack of phosphorylation at serine 392 is compensated by other p53 modifications that arise later in interferon-treated cells. This is consistent with the fact that p53.S389A mice are not tumor prone in normal conditions, perhaps because most oncogenic threats induce compensating p53 modifications (Hoogervorst et al., 2005). The other modification induced early by interferon on p53 is acetylation at lysine 320, a reaction catalyzed by pCAF. This modification may contribute to p53 activation by increasing the recruitment of histone acetyl transferases to p53-dependent promoters (Barlev et al., 2001). Hence, together acetylation at K320 and dephosphorylation at S392 may have induced a preactivation stage upon which further modifications could confer senescence-inducing abilities to p53. Although the constellation of p53 modifications required for p53-dependent senescence remains to be identified, our results suggest that serine 15 phosphorylation catalyzed by the ATM kinase is one of the critical modifications.
The mechanism of ATM activation in interferon-treated cells seems to depend on an increase in ROS production and subsequent DNA damage. However, the mechanism of ROS induction, in response to β-interferon, remains to be elucidated. Interferon-treated cells displayed a significant increase in mitochondrial mass as measured with MitoFluor-Green or MitoTracker-Red. An increase in mitochondrial mass is commonly associated to mitochondrial defects (Rossignol et al., 2003), which in general increase the rate of superoxide generation by the mitochondria (Raha and Robinson, 2000). Several groups have reported that interferon treatment reduces several components of the electron transport chain (ETC; Lou et al., 1994; Lewis et al., 1996; Inagaki et al., 1997; Le Roy et al., 2001). Taken together our data and previous reports suggest a model in which interferon first inhibits mitochondrial functions with the subsequent increase in the production of ROS and DNA damage. At the same time interferon signals directly to the p53 system, increasing its mRNA (Takaoka et al., 2003) and promoting modifications that can potentially enhance its transcriptional activity (this study). These two mechanisms cooperate to trigger a full p53 response in cells bearing oncogenes, high levels of p53 or exposed to sustained levels of β-interferon (Figure 8). Normal fibroblasts, for reasons not yet clear, enter into senescence in response to p53 activation. However, other cell types, or tumor cells where the senescence pathway is usually inactivated, die through apoptosis. In this context it is tempting to speculate that dephosphorylation at serine 392 may be involved in the regulation of the decision to regulate senescence versus apoptosis by p53. This idea is supported by studies showing that cells expressing p53.S389A have a reduced apoptotic response (Bruins et al., 2004) as seen in senescent cells (Marcotte et al., 2004), which also have p53 dephosphorylated at serine 392 (Webley et al., 2000).
Figure 8.
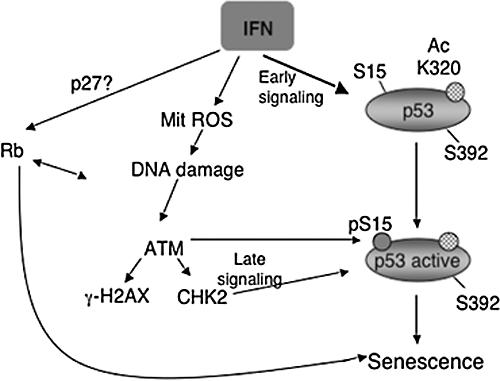
Proposed model for interferon-induced senescence. The immediate effects of interferon treatment are a reversible growth arrest, mitochondrial changes, and p53 modifications (acetylation at K320 and dephosphorylation at serine 392). These changes do not commit the cells to an irreversible withdrawal from the cycling pool. However, chronic stimulation results in an increase in ROS production and activation of the DNA damage signaling pathway. The ATM kinase is a central regulator of the DNA damage response induced by interferon and it activates p53 by promoting its phosphorylation at serine 15 and inducing its transactivation potential. Activation of p53 is then critical to trigger a senescent cell cycle arrest in normal fibroblasts.
We predict that stresses activating the DNA damage–signaling pathway will synergize with β-interferon to induce cellular senescence. For example, oncogenes may damage DNA by inducing the production of ROS (Lee et al., 1999; Benhar et al., 2002; Catalano et al., 2005) and therefore they may sensitize cells to β-interferon as shown here. In addition cells with defects in the repair of O6-methyl guanine are known to be more sensitive to the killing effects of interferon (Yarosh et al., 1985). Of note, viral replication accumulate replication intermediates that activate the DNA damage signaling pathway (Kudoh et al., 2005) and may sensitize them to interferon. Viruses in turn, have evolved proteins that interfere with the interferon pathway and with senescence regulators such as the tumor suppressors PML, p53 or RB (Whyte et al., 1988; Dyson et al., 1989; Scheffner et al., 1990; Gutch and Reich, 1991; Barnard and McMillan, 1999; Everett, 2001). Consequently, some of these viral proteins, alone or in combination, can block cellular senescence and promote transformation (Ferbeyre et al., 2000; Mallette et al., 2004). Together, the data suggest that the senescence response triggered by interferon may contribute both to tumor suppression and antiviral responses.
Concluding Remarks
The interferon response is known for its antiviral and antiproliferative activity and it has been proposed that it may also act as a tumor suppressor pathway (Taniguchi and Takaoka, 2002). Here we provide substantial evidence for placing p53 downstream the interferon pathway to trigger a cellular senescence program. β-Interferon triggered senescence in normal cells after long-term stimulation but the response was potentiated or accelerated in cells with high p53 levels or expressing a constitutively active allele of ERK2 or RasV12. These conditions may naturally occur in cells with DNA damage, viruses, or mutated oncogenes. Therefore, our results are relevant to understand both the tumor suppression and antiviral functions of interferon.
Supplementary Material
Acknowledgments
We thank Drs. D. Galloway, S. Goelz, M. Gilman, J. Hiscott, and P. Sun for plasmids and reagents and S. Sénéchal for assistance with flow cytometry. We also thank Dr. A. Koromilas and the members of the Ferbeyre's laboratory for useful suggestions. This work was supported by a grant from the Cancer Research Society to G. F.
This article was published online ahead of print in MBC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E05-09-0858) on January 25, 2006.
The online version of this article contains supplemental material at MBC Online (http://www.molbiolcell.org).
References
- Achison, M., and Hupp, T. R. (2003). Hypoxia attenuates the p53 response to cellular damage. Oncogene 22, 3431–3440. [DOI] [PubMed] [Google Scholar]
- Akiyama, M., Iwase, S., Horiguchi-Yamada, J., Saito, S., Furukawa, Y., Yamada, O., Mizoguchi, H., Ohno, T., and Yamada, H. (1999). Interferonalpha repressed telomerase along with G1-accumulation of Daudi cells. Cancer Lett. 142, 23–30. [DOI] [PubMed] [Google Scholar]
- Bakkenist, C. J., and Kastan, M. B. (2003). DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 421, 499–506. [DOI] [PubMed] [Google Scholar]
- Barlev, N. A., Liu, L., Chehab, N. H., Mansfield, K., Harris, K. G., Halazonetis, T. D., and Berger, S. L. (2001). Acetylation of p53 activates transcription through recruitment of coactivators/histone acetyltransferases. Mol. Cell 8, 1243–1254. [DOI] [PubMed] [Google Scholar]
- Barnard, P., and McMillan, N. A. (1999). The human papillomavirus E7 oncoprotein abrogates signaling mediated by interferon-alpha. Virology 259, 305–313. [DOI] [PubMed] [Google Scholar]
- Benhar, M., Engelberg, D., and Levitzki, A. (2002). ROS, stress-activated kinases and stress signaling in cancer. EMBO Rep. 3, 420–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielenberg, D. R., McCarty, M. F., Bucana, C. D., Yuspa, S. H., Morgan, D., Arbeit, J. M., Ellis, L. M., Cleary, K. R., and Fidler, I. J. (1999). Expression of interferon-beta is associated with growth arrest of murine and human epidermal cells. J. Invest. Dermatol. 112, 802–809. [DOI] [PubMed] [Google Scholar]
- Bischof, O., Kirsh, O., Pearson, M., Itahana, K., Pelicci, P. G., and Dejean, A. (2002). Deconstructing PML-induced premature senescence. EMBO J. 21, 3358–3369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaydes, J. P., and Hupp, T. R. (1998). DNA damage triggers DRB-resistant phosphorylation of human p53 at the CK2 site. Oncogene 17, 1045–1052. [DOI] [PubMed] [Google Scholar]
- Bruins, W., et al. (2004). Increased sensitivity to UV radiation in mice with a p53 point mutation at Ser389. Mol. Cell. Biol. 24, 8884–8894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canman, C. E., Lim, D. S., Cimprich, K. A., Taya, Y., Tamai, K., Sakaguchi, K., Appella, E., Kastan, M. B., and Siliciano, J. D. (1998). Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science 281, 1677–1679. [DOI] [PubMed] [Google Scholar]
- Catalano, A., Rodilossi, S., Caprari, P., Coppola, V., and Procopio, A. (2005). 5-Lipoxygenase regulates senescence-like growth arrest by promoting ROS-dependent p53 activation. EMBO J. 24, 170–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- d'Adda di Fagagna, F., Reaper, P. M., Clay-Farrace, L., Fiegler, H., Carr, P., Von Zglinicki, T., Saretzki, G., Carter, N. P., and Jackson, S. P. (2003). A DNA damage checkpoint response in telomere-initiated senescence. Nature 426, 194–198. [DOI] [PubMed] [Google Scholar]
- de Stanchina, E., Querido, E., Narita, M., Davuluri, R. V., Pandolfi, P. P., Ferbeyre, G., and Lowe, S. W. (2004). PML is a direct p53 target that modulates p53 effector functions. Mol. Cell 13, 523–535. [DOI] [PubMed] [Google Scholar]
- Deng, Q., Liao, R., Wu, B. L., and Sun, P. (2004). High intensity ras signaling induces premature senescence by activating p38 pathway in primary human fibroblasts. J. Biol. Chem. 279, 1050–1059. [DOI] [PubMed] [Google Scholar]
- Dianzani, F. (1992). Interferon treatments: how to use an endogenous system as a therapeutic agent. J. Interferon Res. Spec No, 109–118. [DOI] [PubMed]
- Dimri, G. P., et al. (1995). A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 92, 9363–9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyson, N., Howley, P. M., Munger, K., and Harlow, E. (1989). The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science 243, 934–937. [DOI] [PubMed] [Google Scholar]
- Emrick, M. A., Hoofnagle, A. N., Miller, A. S., Ten Eyck, L. F., and Ahn, N. G. (2001). Constitutive activation of extracellular signal-regulated kinase 2 by synergistic point mutations. J. Biol. Chem. 276, 46469–46479. [DOI] [PubMed] [Google Scholar]
- Everett, R. D. (2001). DNA viruses and viral proteins that interact with PML nuclear bodies. Oncogene 20, 7266–7273. [DOI] [PubMed] [Google Scholar]
- Falck, J., Mailand, N., Syljuasen, R. G., Bartek, J., and Lukas, J. (2001). The ATM-CHK2-Cdc25A checkpoint pathway guards against radioresistant DNA synthesis. Nature 410, 842–847. [DOI] [PubMed] [Google Scholar]
- Ferbeyre, G. (2002). PML a target of translocations in APL is a regulator of cellular senescence. Leukemia 16, 1918–1926. [DOI] [PubMed] [Google Scholar]
- Ferbeyre, G., de Stanchina, E., Lin, A. W., Querido, E., McCurrach, M. E., Hannon, G. J., and Lowe, S. W. (2002). Oncogenic ras and p53 cooperate to induce cellular senescence. Mol. Cell. Biol. 22, 3497–3508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferbeyre, G., de Stanchina, E., Querido, E., Baptiste, N., Prives, C., and Lowe, S. W. (2000). PML is induced by oncogenic ras and promotes premature senescence. Genes Dev. 14, 2015–2027. [PMC free article] [PubMed] [Google Scholar]
- Formstecher, E., et al. (2001). PEA-15 mediates cytoplasmic sequestration of ERK MAP kinase. Dev. Cell 1, 239–250. [DOI] [PubMed] [Google Scholar]
- Gaumont-Leclerc, M. F., Mukhopadhyay, U. K., Goumard, S., and Ferbeyre, G. (2004). PEA-15 is inhibited by adenovirus E1A and plays a role in ERK nuclear export and Ras-induced senescence. J. Biol. Chem. 279, 46802–46809. [DOI] [PubMed] [Google Scholar]
- Gutch, M. J., and Reich, N. C. (1991). Repression of the interferon signal transduction pathway by the adenovirus E1A oncogene. Proc. Natl. Acad. Sci. USA 88, 7913–7917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond, E. M., Denko, N. C., Dorie, M. J., Abraham, R. T., and Giaccia, A. J. (2002). Hypoxia links ATR and p53 through replication arrest. Mol. Cell. Biol. 22, 1834–1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvat, B. L., and Jetten, A. M. (1996). Gamma-interferon induces an irreversible growth arrest in mid-G1 in mammary epithelial cells which correlates with a block in hyperphosphorylation of retinoblastoma. Cell Growth Differ. 7, 289–300. [PubMed] [Google Scholar]
- Herbig, U., Jobling, W. A., Chen, B. P., Chen, D. J., and Sedivy, J. M. (2004). Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21(CIP1), but not p16(INK4a). Mol. Cell 14, 501–513. [DOI] [PubMed] [Google Scholar]
- Hoogervorst, E. M., Bruins, W., Zwart, E., van Oostrom, C. T., van den Aardweg, G. J., Beems, R. B., van den Berg, J., Jacks, T., van Steeg, H., and de Vries, A. (2005). Lack of p53 Ser389 phosphorylation predisposes mice to develop 2-acetylaminofluorene-induced bladder tumors but not ionizing radiation-induced lymphomas. Cancer Res. 65, 3610–3616. [DOI] [PubMed] [Google Scholar]
- Hottiger, M. O., Felzien, L. K., and Nabel, G. J. (1998). Modulation of cytokine-induced HIV gene expression by competitive binding of transcription factors to the coactivator p300. EMBO J. 17, 3124–3134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hupp, T. R., Meek, D. W., Midgley, C. A., and Lane, D. P. (1992). Regulation of the specific DNA binding function of p53. Cell 71, 875–886. [DOI] [PubMed] [Google Scholar]
- Inagaki, H., Matsushima, Y., Ohshima, M., and Kitagawa, Y. (1997). Interferons suppress mitochondrial gene transcription by depleting mitochondrial transcription factor A (mtTFA). J. Interferon Cytokine Res. 17, 263–269. [DOI] [PubMed] [Google Scholar]
- Iwase, S., Furukawa, Y., Kikuchi, J., Nagai, M., Terui, Y., Nakamura, M., and Yamada, H. (1997). Modulation of E2F activity is linked to interferon-induced growth suppression of hematopoietic cells. J. Biol. Chem. 272, 12406–12414. [DOI] [PubMed] [Google Scholar]
- Johnson, G. L., and Lapadat, R. (2002). Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science 298, 1911–1912. [DOI] [PubMed] [Google Scholar]
- Kaynor, C., Xin, M., Wakefield, J., Barsoum, J., and Qin, X. Q. (2002). Direct evidence that IFN-beta functions as a tumor-suppressor protein. J. Interferon Cytokine Res. 22, 1089–1098. [DOI] [PubMed] [Google Scholar]
- Kim, Y. Y., Park, B. J., Kim, D. J., Kim, W. H., Kim, S., Oh, K. S., Lim, J. Y., Kim, J., Park, C., and Park, S. I. (2004). Modification of serine 392 is a critical event in the regulation of p53 nuclear export and stability. FEBS Lett. 572, 92–98. [DOI] [PubMed] [Google Scholar]
- Kudoh, A., Fujita, M., Zhang, L., Shirata, N., Daikoku, T., Sugaya, Y., Isomura, H., Nishiyama, Y., and Tsurumi, T. (2005). Epstein-Barr virus lytic replication elicits ATM checkpoint signal transduction while providing an S-phase-like cellular environment. J. Biol. Chem. 280, 8156–8163. [DOI] [PubMed] [Google Scholar]
- Lavau, C., Marchio, A., Fagioli, M., Jansen, J., Falini, B., Lebon, P., Grosveld, F., Pandolfi, P. P., Pelicci, P. G., and Dejean, A. (1995). The acute promyelocytic leukaemia-associated PML gene is induced by interferon. Oncogene 11, 871–876. [PubMed] [Google Scholar]
- Le Roy, F., Bisbal, C., Silhol, M., Martinand, C., Lebleu, B., and Salehzada, T. (2001). The 2–5A/RNase L/RNase L inhibitor (RLI) [correction of (RNI)] pathway regulates mitochondrial mRNAs stability in interferon alpha-treated H9 cells. J. Biol. Chem. 276, 48473–48482. [DOI] [PubMed] [Google Scholar]
- Lee, A. C., Fenster, B. E., Ito, H., Takeda, K., Bae, N. S., Hirai, T., Yu, Z. X., Ferrans, V. J., Howard, B. H., and Finkel, T. (1999). Ras proteins induce senescence by altering the intracellular levels of reactive oxygen species. J. Biol. Chem. 274, 7936–7940. [DOI] [PubMed] [Google Scholar]
- Lewis, J. A., Huq, A., and Najarro, P. (1996). Inhibition of mitochondrial function by interferon. J. Biol. Chem. 271, 13184–13190. [DOI] [PubMed] [Google Scholar]
- Lim, I. K., Won Hong, K., Kwak, I. H., Yoon, G., and Park, S. C. (2000). Cytoplasmic retention of p-Erk1/2 and nuclear accumulation of actin proteins during cellular senescence in human diploid fibroblasts. Mech. Ageing Dev. 119, 113–130. [DOI] [PubMed] [Google Scholar]
- Lin, A. W., Barradas, M., Stone, J. C., van Aelst, L., Serrano, M., and Lowe, S. W. (1998). Premature senescence involving p53 and p16 is activated in response to constitutive MEK/MAPK mitogenic signaling. Genes Dev. 12, 2997–3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou, J., Anderson, S. L., Xing, L., and Rubin, B. Y. (1994). Suppression of mitochondrial mRNA levels and mitochondrial function in cells responding to the anticellular action of interferon. J. Interferon Res. 14, 33–40. [DOI] [PubMed] [Google Scholar]
- Lowe, S. W., Cepero, E., and Evan, G. (2004). Intrinsic tumour suppression. Nature 432, 307–315. [DOI] [PubMed] [Google Scholar]
- Mallette, F. A., Goumard, S., Gaumont-Leclerc, M. F., Moiseeva, O., and Ferbeyre, G. (2004). Human fibroblasts require the Rb family of tumor suppressors, but not p53, for PML-induced senescence. Oncogene 23, 91–99. [DOI] [PubMed] [Google Scholar]
- Marcotte, R., Lacelle, C., and Wang, E. (2004). Senescent fibroblasts resist apoptosis by downregulating caspase-3. Mech. Ageing Dev. 125, 777–783. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay, U. K., Senderowicz, A. M., and Ferbeyre, G. (2005). RNA silencing of checkpoint regulators sensitizes p53-defective prostate cancer cells to chemotherapy while sparing normal cells. Cancer Res. 65, 2872–2881. [DOI] [PubMed] [Google Scholar]
- Narita, M., Nunez, S., Heard, E., Lin, A. W., Hearn, S. A., Spector, D. L., Hannon, G. J., and Lowe, S. W. (2003). Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 113, 703–716. [DOI] [PubMed] [Google Scholar]
- Pearson, M., et al. (2000). PML regulates p53 acetylation and premature senescence induced by oncogenic Ras. Nature 406, 207–210. [DOI] [PubMed] [Google Scholar]
- Raha, S., and Robinson, B. H. (2000). Mitochondria, oxygen free radicals, disease and ageing. Trends Biochem. Sci. 25, 502–508. [DOI] [PubMed] [Google Scholar]
- Resnitzky, D., Yarden, A., Zipori, D., and Kimchi, A. (1986). Autocrine beta-related interferon controls c-myc suppression and growth arrest during hematopoietic cell differentiation. Cell 46, 31–40. [DOI] [PubMed] [Google Scholar]
- Rossignol, R., Faustin, B., Rocher, C., Malgat, M., Mazat, J. P., and Letellier, T. (2003). Mitochondrial threshold effects. Biochem. J. 370, 751–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheffner, M., Werness, B. A., Huibregtse, J. M., Levine, A. J., and Howley, P. M. (1990). The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 63, 1129–1136. [DOI] [PubMed] [Google Scholar]
- Serrano, M., Lin, A. W., McCurrach, M. E., Beach, D., and Lowe, S. W. (1997). Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 88, 593–602. [DOI] [PubMed] [Google Scholar]
- Shay, J. W., and Roninson, I. B. (2004). Hallmarks of senescence in carcinogenesis and cancer therapy. Oncogene 23, 2919–2933. [DOI] [PubMed] [Google Scholar]
- Stadler, M., Chelbi-Alix, M. K., Koken, M. H., Venturini, L., Lee, C., Saib, A., Quignon, F., Pelicano, L., Guillemin, M. C., Schindler, C., and de The, H. (1995). Transcriptional induction of the PML growth suppressor gene by interferons is mediated through an ISRE and a GAS element. Oncogene 11, 2565–2573. [PubMed] [Google Scholar]
- Takai, H., Smogorzewska, A., and de Lange, T. (2003). DNA damage foci at dysfunctional telomeres. Curr. Biol. 13, 1549–1556. [DOI] [PubMed] [Google Scholar]
- Takaoka, A., et al. (2003). Integration of interferon-alpha/beta signalling to p53 responses in tumour suppression and antiviral defence. Nature 424, 516–523. [DOI] [PubMed] [Google Scholar]
- Tanaka, N., Ishihara, M., Kitagawa, M., Harada, H., Kimura, T., Matsuyama, T., Lamphier, M. S., Aizawa, S., Mak, T. W., and Taniguchi, T. (1994). Cellular commitment to oncogene-induced transformation or apoptosis is dependent on the transcription factor IRF-1. Cell 77, 829–839. [DOI] [PubMed] [Google Scholar]
- Taniguchi, T., and Takaoka, A. (2002). The interferon-alpha/beta system in antiviral responses: a multimodal machinery of gene regulation by the IRF family of transcription factors. Curr. Opin. Immunol. 14, 111–116. [DOI] [PubMed] [Google Scholar]
- Wang, W., Chen, J. X., Liao, R., Deng, Q., Zhou, J. J., Huang, S., and Sun, P. (2002). Sequential activation of the MEK-extracellular signal-regulated kinase and MKK3/6-p38 mitogen-activated protein kinase pathways mediates oncogenic ras-induced premature senescence. Mol. Cell. Biol. 22, 3389–3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webley, K., Bond, J. A., Jones, C. J., Blaydes, J. P., Craig, A., Hupp, T., and Wynford-Thomas, D. (2000). Posttranslational modifications of p53 in replicative senescence overlapping but distinct from those induced by DNA damage. Mol. Cell. Biol. 20, 2803–2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte, P., Buchkovich, K. J., Horowitz, J. M., Friend, S. H., Raybuck, M., Weinberg, R. A., and Harlow, E. (1988). Association between an oncogene and an anti-oncogene: the adenovirus E1A proteins bind to the retinoblastoma gene product. Nature 334, 124–129. [DOI] [PubMed] [Google Scholar]
- Wu, C., Miloslavskaya, I., Demontis, S., Maestro, R., and Galaktionov, K. (2004). Regulation of cellular response to oncogenic and oxidative stress by Seladin-1. Nature 432, 640–645. [DOI] [PubMed] [Google Scholar]
- Xin, H., Curry, J., Johnstone, R. W., Nickoloff, B. J., and Choubey, D. (2003). Role of IFI 16, a member of the interferon-inducible p200-protein family, in prostate epithelial cellular senescence. Oncogene 22, 4831–4840. [DOI] [PubMed] [Google Scholar]
- Xin, H., Pereira-Smith, O. M., and Choubey, D. (2004). Role of IFI 16 in cellular senescence of human fibroblasts. Oncogene 23, 6209–6217. [DOI] [PubMed] [Google Scholar]
- Yarosh, D. B., Scudiero, D. A., Yagi, T., and Day, R. S., 3rd. (1985). Human tumor cell strains both unable to repair O6-methylguanine and hypersensitive to killing by human alpha and beta interferons. Carcinogenesis 6, 883–886. [DOI] [PubMed] [Google Scholar]
- Yeo, E. J., Hwang, Y. C., Kang, C. M., Kim, I. H., Kim, D. I., Parka, J. S., Choy, H. E., Park, W. Y., and Park, S. C. (2000). Senescence-like changes induced by hydroxyurea in human diploid fibroblasts. Exp. Gerontol. 35, 553–571. [DOI] [PubMed] [Google Scholar]
- Zglinicki, T., Saretzki, G., Ladhoff, J., Fagagna, F., and Jackson, S. P. (2005). Human cell senescence as a DNA damage response. Mech. Ageing Dev. 126, 111–117. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.