

Abstract
PPARα, a member of the nuclear receptor superfamily, and thioredoxin, a critical redox-regulator in cells, were found to form a negative feedback loop, which autoregulates transcriptional activity of PPARα. Thioredoxin was identified as a target gene of PPARα. Activation of PPARα leads to increase of thioredoxin expression as well as its translocation from cytoplasm to nucleus, whereas ectopic overexpression of thioredoxin in the nucleus dramatically inhibited both constitutive and ligand-dependent PPARα activation. As PPARα-target genes, the expression of muscle carnitine palmitoyltransferase I, medium chain acyl CoA dehydrogenase, and apolipoprotein A-I were significantly down-regulated by nucleus-targeted thioredoxin at transcriptional or protein level. The suppression of PPARα transcriptional activity by Trx could be enhanced by overexpression of thioredoxin reductase or knockdown of thioredoxin-interacting protein, but abrogated by mutating the redox-active sites of thioredoxin. Mammalian one-hybrid assays showed that thioredoxin inhibited PPARα activity by modulating its AF-1 transactivation domain. It was also demonstrated by electrophoretic mobility-shift assay that thioredoxin inhibited the binding of PPARα to the PPAR-response element. Together, it is speculated that the reported negative-feedback loop may be essential for maintaining the homeostasis of PPARα activity.
INTRODUCTION
As the first identified genetic sensor for fats, PPARα is a ligand-activated transcription factor belonging to the nuclear receptor superfamily. The target genes of PPARα are relatively homogenous group of genes that participate in various aspects of lipid catabolism such as fatty acid uptake through membranes, fatty acid binding in cells, fatty acid oxidation in microsomes, peroxisomes and mitochondria, and lipoprotein assembly and transportation (Lemberger et al., 1996). The significance of PPARα in physiology and disease is evidenced by the fact that it and PPARγ, another subtype of PPARs, are molecular targets for the lipid-lowering fibrate drugs and insulin-sensitizing thiazolidinedione (TZD), respectively (Evans et al., 2004). Besides activation of PPARα by fatty acid and various exogenous ligands such as fibrates, the regulation of the PPARα transcription activity by coactivators (Dowell et al., 1997), corepressors (Dowell et al., 1999), other transcription factors (TFs; Zhou et al., 1999), and posttranslational modifications including phosphorylation (Juge-Aubry et al., 1999) and ubiquination (Blanquart et al., 2002) have been extensively studied. However, redox regulation of PPARα activity has not been reported so far.
Multicellular organisms have evolved complex homeostatic mechanisms to sense and respond to a diverse range of exogenous and endogenous signals. It may be reasonable to expect some mechanisms that rapidly sense and tightly control the activity of PPARα according to metabolic needs under normal physiological condition. Degradation of PPARα by the ubiquitin-proteasome system and decrease of the ubiquitination by the ligands of PPARα may represent one of such mechanisms (Blanquart et al., 2002). Because feedback regulation, by which a transcription factor is negatively regulated by its target gene product, has been found for several nuclear receptors such as thyroid hormone receptor and retinoid receptor (Thompson and Bottcher, 1997; Kerley et al., 2001), a question may be raised as if any PPARα-target gene product can also negatively regulate PPARα activity.
Thioredoxin (Trx), as one of the major components of the thiol-reducing system, is small, ubiquitous, and multifunctional protein that plays a variety of redox-related roles in organisms ranging from Escherichia coli to human (Holmgren, 1985). Trx contains a conserved redox-active site Cys-Gly-Pro-Cys that are essential for its redox regulatory function (Powis and Montfort, 2001). A bulk of evidences has shown that Trx regulates the transcriptional activities of quite a number of transcription factors. It has been reported that Trx enhances NF-κB transcriptional activities by enhancing its ability to bind DNA (Hirota et al., 1999) and mediates the interplay between cellular redox signaling and glucocorticoid receptor (GR) signaling via a direct association with its conserved DNA-binding domain (DBD; Makino et al., 1999). It was also reported that the function of estrogen receptor (ER), another nuclear receptor, was strongly influenced by its redox state and modulated by endogenous Trx (Hayashi et al., 1997). However, whether Trx modulates the activity of PPARα or other peroxisome proliferator-activated receptors has never been reported.
This study is aimed to know if PPARα activity is redox-regulated and if thioredoxin plays any role in such regulation. It is also attempted to find out if there is any regulatory mechanism to control the transcriptional activity of PPARα to a physiologically reasonable level.
MATERIALS AND METHODS
Reagents and Plasmid Constructs
WY14643,5,5′-dithiiobis-2-nitrobenzoic acid (DTNB), goldthioglucose, and other reagents, which are not specified, were purchased from Sigma (St. Louis, MO). Guanidine hydrochloride was purchased from Bebco (Kansas, AZ). The bovine wild-type thioredoxin reductase (TrxR), recombinant human Trx were kindly provided by Dr. A. Holmgren (Karolinska Institute, Sweden).
A 2.4-kb HindIII-BamHI fragment of the Trx promoter subcloned into pGL3 basic vector [Trx(2400)-Luc] is a kind gift from Dr. K. F. Tonissen (Griffith University, Australia; Bloomfield et al., 2003). The Trx(2210)-Luc, Trx(2085)-Luc, Trx(1898)-Luc, and Trx(841)-Luc vectors were constructed by inserting the PCR-amplified KpnI-BamHI fragments of the Trx(2400)-Luc vector into the KpnI-BglII sites of the pGL3 basic vector (Promega, Madison, WI). The (Trx-PPRE)3-SV40-Luc was constructed by cloning the oligonucleotide containing three direct tandem repeats of the PPAR response element (PPRE) sequence between -2204 and -2182 of the Trx promoter (TTACGGAGCTCACTGTTCATTAC) into the KpnI and XhoI restriction sites of pGL3 promoter vector (Promega). pcDNA3-Trx, pcDNA3-Trx(C32/35S), and pCMX-GAL4-Trx vectors were kindly provided by Dr. J. Yodoi (Kyoto University, Japan; Hirota et al., 1999). pcDNA3-Trx(C69/72/79S) was generated by site-directed mutagenesis of pcDNA3-Trx. pEYFP-Trx was constructed by subcloning the Trx cDNA into the EcoRI-BamHI sites of the pEYFP-C1 (Clontech, Palo Alto, CA) according to the literature (Hwang et al., 2004). pCMX-mPPARα, pCMX-VP16-mPPARα, and pCMX-mRXRα constructs were kindly provided by Dr. R. M. Evans (Howard Hughes Medical Institute). pEGFP-mPPARα, pECFP-mPPARα, and (PPRE)3-TK-Luc are generous gifts of Dr. F. Gonzalez (National Cancer Institute, Bethesda, MD; Akiyama et al., 2002), Dr. B. Desvergne (University of Lausanne, Switzerland; Feige et al., 2005), and Dr. B. Spiegelman (Harvard Medical School, Boston, MA; Drori et al., 2005), respectively. Vector VP16-ΔAF1 coding a fusion protein linking the VP16 to the upstream of the COOH-terminal region (amino acids 90-498) of PPARα was constructed by subcloning the corresponding PPARα cDNA fragment into the BamHI and KpnI sites of the pACT plasmid (Promega). The same fragment of PPARα with an additional ATG in the NH2-terminal was subcloned into the BamHI and Not I sites of the pcDNA3.0 to generate pcDNA3.0-ΔAF1. GAL4-PPARα (1-92), and GAL4-PPARα (LBD) were generous gifts from Dr. C. Meier (University Hospital Geneva, Switzerland; Juge-Aubry et al., 1999) and Dr. T. Goda (University of Shizuoka, Japan; Mochizuki et al., 2002), respectively. MCPT1.Luc.781 and MCAD.Luc.376 were kindly provided by Dr. D. P. Kelly (Washington University School of Medicine, St. Luis, MO). pCNX2-myc-TrxR is a kind gift from Dr. D. Gius (National Institutes of Health, Bethesda, MD; Karimpour et al., 2002).
Cell Culture and Transient Transfection
HepG2, HeLa, A549 (all obtained from ATCC, Rockville, MD), and EA.hy.926 cells (provided by C.J.S. Edgell, University of North Carolina, Chapel Hill, NC) were cultured in DMEM containing 10% fetal calf serum (FCS). The differentiated HepG2 cells were obtained by maintaining in culture without passage for 20 d as previously described (Stier et al., 1998). For transfection, cells were plated on 12- or 6-well plates at a density of 60-70% confluence and transfected in the same medium using Fugene 6 (Roche, Indianapolis, IN) for HepG2 and EA.hy 926 or jetPEI (Polyplus, Illkirch, France) for HeLa and A549. In each transfection, except for the quantity of reporter and other expression vectors specified in figure legends, 100 ng of the internal control plasmid CMV-β-galactosidase was always added and not specified again in the legends. Total DNA quantity was kept constant with pcDNA3 or relevant empty vectors. Twelve hours after transfection, the cells were washed and cultured in fresh medium containing WY14643 or vehicle for 24 h and then harvested. Both luciferase and β-galactosidase activities were measured with a homemade luminometer and a Bio-Rad plate Reader (Richmond, CA), respectively. The luciferase activity normalized against the β-galactosidase activity represents the transcription efficiency of the reporter gene. All transfections throughout the investigation were performed in triplicate, and each assay was repeated three times.
Mammalian One-Hybrid Analysis
Cells were cotransfected with pG5-Luc reporter vector (Promega) containing five GAL4 binding sites upstream of a minimal TATA box-linked luciferase gene [(UAS)5-TATA-Luc], GAL4-fusion protein expression vector, and Trx expression vectors in the presence or absence of WY14643 stimulation. The cells without cotransfected Trx were used as control. The β-galactosidase-normalized luciferase activities were assayed 36 h after transfection.
Thioredoxin-interacting Protein Gene Silencing by RNA Interference
A published sequence, ACAGACTTCGGAGTACCTG, for silencing thioredoxin-interacting protein (Txnip; Schulze et al., 2004) was cloned into pSilencer4.1-CMV neo RNA interference (RNAi) vector (Ambion, Austin, TX). The Txnip-silenced cells were obtained by transfection of Txnip siRNA vector and selection with 400 μg/ml G418 for 4 wk. The cells stably transfected with a scramble vector were also cloned and used as control.
Reverse Transcription-PCR
Total RNA was isolated from A549 cells with Trizol reagent (Invitrogen-BRL, Carlsbad, CA) and reverse-transcribed to cDNA using RNase-free Superscript reverse transcriptase (Invitrogen). PCR was performed using the following primers: 5′-TGGTGGATGTCAATACCCCT-3′/5′-ATTGGCAAGGTAAGTGTGGC-3′ for human Txnip, and 5′-GAAGCATTTGCGGTGGACCA-3′/5′-TCCTGTGGCATCCACCAAAC-3′ for β-actin. The PCR products were analyzed on a 1% agarose gel with ethidium bromide.
Western Blotting
To extract total proteins, cells were washed twice, collected, and lysed in lysis buffer (50 mM Tris, pH 7.4, 250 mM NaCl, 0.1% SDS, 2 mM DTT, 0.5% NP-40, 1 mM phenylmethylsulfonyl fluoride, and protease inhibitor cocktail) on ice for 30 min. Protein fraction was collected by centrifugation at 16,000 × g at 4°C for 20 min. The nuclear proteins were isolated by NE-PER Nuclear and Cytoplasmic Extraction Reagents (Pierce, Rockford, IL) according to the manufacturer's instruction. Equal amount of the protein fraction were mixed with sample buffer, separated on 10 or 15% SDS-polyacrylamide gel, and transferred to nitrocellulose membranes. Then, mouse anti-human Trx monoclonal antibody (BD PharMingen, San Diego, CA), rabbit polyclonal PPARα antibody (Santa Cruz Biotechnology, Santa Cruz, CA), rabbit polyclonal actin antibody (Santa Cruz), rabbit polyclonal anti-GAL4BD antibody (Clontech), and rabbit polyclonal anti-GFP antibody (Clontech) were used as primary antibodies to detect Trx, PPARα, actin, Gal4(BD)-AF1 fusion protein, and YFP-Trx fusion protein, respectively. The HRP-conjugated secondary antibody and enhanced chemiluminescence kit (Pierce) were used to visualize the separated proteins. Equal loading of proteins was monitored by the actin level.
Assay of Trx Activity in Cells
Trx activity was determined by insulin reduction-based assay as previously described (Zheng et al., 2005).
Fluorescence and Immunofluorescence Imaging of Cells
The cells transfected with GFP-PPARα were fixed with 3.7% paraformaldehyde in phosphate-buffered saline (PBS) for 30 min, permeabilized by 0.2% Triton X-100 in PBS for 10 min, and then blocked with PBS containing 5% BSA and 10% FCS for 30 min. The cells were subsequently incubated with Trx antibody overnight at 4°C and flooded by Texas Red-labeled secondary antibody (Molecular Probes, Eugene, OR) for 60 min. The cell nuclei were stained with Hoechst 33342. The fluorescence images of Hoechst 33342-stained nuclei and GFP-fused PPARα as well as the immunofluorescence image of Trx in the same cells were observed on an Olympus IX-71 inverted microscope (Tokyo, Japan) equipped with AquaCosmos Microscopic Image Acquisition and Analysis System (Hamamatsu Photonics K.K., Bridgewater, NJ) at the excitation of 360, 488, and 590 nm, respectively.
In Vitro Translation of PPARα and RXRα
The full-length PPARα and RXRα protein were obtained by in vitro transcription-coupled translation using the TNT Quick Coupled Transcription/Translation Systems (Promega) according to the manufacturer's instructions.
Electrophoretic Mobility-Shift Assay
Sense and antisense oligonucleotides for electrophoretic mobility-shift assay (EMSA) were synthesized, labeled with or without biotin at their 5′ end, and then annealed with each other to produce double-stranded oligonucleotide probes. The following sense strand sequences were used: 5′-CTTATGTTACGGAGCTCACTGTTCATTACTACTGTCT-3′ for Trx-PPRE, 5′-CTTAGAACTAGAAGGTCACTGGTCAAGCAGCCATTTG-3′ for HACOX-PPRE. EMSA assays were performed with in vitro-translated proteins or the nuclear extracts. The in vitro-translated proteins (3 μl) or nuclear extracts (3 μg) were added in EMSA-binding buffer (10 mM Tris, 50 mM KCl, 5 mM MgCl2, 1 mM EDTA, 1 mM DTT, 10% glycerol, and 1 μg of poly(dI/dC), pH 7.5) in a final volume of 20 μl and kept on ice for 20 min. In some cases, recombinant human Trx and/or rat TrxR/NADPH were also added in the binding system. Approximately 50 fmol of the biotin-labeled probe was then added and the incubation was continued on ice for a further 30 min. In competition experiments, molar excess of the unlabeled competitive oligonucleotides were added in the reaction mixture before addition of the biotin-labeled probe. Supershift assay was performed by adding anti-PPARα antibody in the reaction mixture during the last 15-min incubation. Protein-DNA complexes were separated by 5% nondenaturing PAGE and transferred onto nylon membrane. The DNA was then cross-linked to the membrane by UV illumination. The biotin-labeled DNA bands were visualized using LightShift Chemiluminescent EMSA Kit (Pierce).
Determination of apoA-I Secretion
Cells were cultured in serum-free DMEM medium for 12 h and then stimulated with 50 μM WY14643 or vehicle for 24 h. The medium was collected, and 20 μl of the medium was subjected to Western blot. The secreted protein was detected using rabbit polyclonal apoA-I antibody (Calbiochem, La Jolla, CA) and visualized as described in the section Western Blotting.
Decoy PPRE Approach
Cells were transfected with a double-stranded phosphorothioate oligonucleotide containing a human ACOX PPRE sequence. The sense sequence of the oligonucleotide is 5′-AACTAGAAGGTCACTGGTCAAGC-3′, in which the underlined part is the HACOX PPRE. The oligonucleotide with a mutated PPRE site, 5′-AACTAGAAGAACAAAGAACAAGC-3′, was used as control. The apoA-I secretion from the transfected cells was determined after WY14643-stimulation for 24 h.
RESULTS
Human Trx Is a PPARα-Target Gene
To know if activation of PPARα could affect the expression of Trx in cells, the Trx content in human hepatoma HepG2 cells stimulated by the selective PPARα agonist, WY14643, was assessed. It was found that the agonist did not cause any notable change of Trx under normal culture condition (unpublished data), possibly because of a rather low PPARα level in this type of cells (Palmer et al., 1998; Hsu et al., 2001). To evaluate the potential correlation between the protein level of PPARα and Trx expression, a PPARα expression vector, pCMX-mPPARα, was transiently transfected into HepG2 cells in order to increase PPARα. As Figure 1A shows, the PPARα level was obviously elevated and the cellular Trx level was correspondingly up-regulated in the transfected cells. In addition, it was found that WY14643 induced a rise of Trx expression in the differentiated HepG2 cells that express high level of PPARα (Stier et al., 1998). The protein level of Trx at various time within 48 h after WY14643-stimulation showed that the rise reached a plateau 24 h after stimulation (Figure 1B). In contrast, treatment of the cells with vehicle (0.1% dimethyl sulfoxide [DMSO]) alone did not induce any change of Trx expression (unpublished data).
Figure 1.

Identification of human Trx as a PPARα-target gene. (A) A typical immunoblotting analysis of Trx and PPARα in HepG2 cells 36 h after transfection with pCMX-mPPARα (mouse PPARα expression vector). The cells transfected with empty vector (pCMX) were used as control. Lanes 1 and 2, and 3 and 4 are shown as two parallel measurements. (B) Trx protein expression in the differentiated HepG2 cells at various times after stimulation by 100 μM WY14643. Fold of induction of Trx protein normalized with actin was quantified relative to that at zero time point. The Western blot showed here is representative of three independent assays. (C) Activation of Trx gene promoter by PPARα and RXRα in the HepG2 cells transfected with 1 μg Trx (2400)-Luc, 300 ng PPARα, and 600 ng RXRα or empty vector. (D) Promoter analysis of human Trx gene. HepG2 cells were transfected with 300 ng PPARα and 1 μg luciferase reporter constructs containing series deletions of the 5′ flanking region of human Trx promoter. (E) List of various identified PPRE sequences from human ACOX, rat Cu/Zn-SOD, rat catalase, and the sequence of the putative PPRE in the human Trx promoter. (F) EMSA analysis of the binding of PPARα/RXRα dimmer to Trx-PPRE in vitro. Either or both of in vitro translated PPARα and RXRα proteins were incubated with biotin-labeled Trx PPRE (lanes 2-6). Ten- and 50-fold excess unlabeled human ACOX PPRE was added in the reaction mixture to determine specificity of the binding (lanes 5 and 6). (G) EMSA analysis of the binding of the endogenous PPARα in nuclear extract of the differentiated HepG2 cells to the biotin-labeled Trx-PPRE under various conditions. Lane 1, the cells were stimulated by 50 μM WY14643 for 24 h, and anti-PPARα antibody was added during the last 15 min incubation of the extract with the Trx-PPRE probe; lanes 2 and 3, the cells were unstimulated and stimulated with 50 μM WY14643 for 24 h, respectively. (H) The activity of Trx-PPRE in driving exogenous core promoter (SV40)-linked luciferase transcription in response to PPARα activation. HepG2 cells were transfected with 1 μg (Trx-PPRE)3-SV40-Luc, 300 ng PPARα, and 600 ng RXRα or empty vector. In all experiments in C, D, and H, after a 12-h transfection, cells were treated with 50 μM WY14643 or DMSO (vehicle) for 24 h and then lysed. Normalized luciferase activities are shown as mean ± SE of three independent measurements.
To examine if Trx expression is regulated by PPARα at transcriptional level, a luciferase reporter vector, Trx (2400)-Luc, containing the 5′ flanking sequence from -2446 to -47 relative to the translation start site of human Trx gene and a PPARα expression vector were cotransfected into HepG2 cells, and transactivation of the Trx promoter was measured in the presence or absence of WY14643. As Figure 1C shows, the ligand did not induce any notable increase of the Trx promoter activity without transfected PPARα. However, cotransfection of PPARα resulted in 8- and 15-fold increases of the promoter activity in unstimulated and the WY14643-stimulated cells, respectively, in consistence with the above immunoblotting analysis of endogenous Trx level in HepG2 cells (Figure 1, A and B). Because all the natural PPREs described so far indeed consist of a direct repeat of two more or less conserved AGGTCA hexamers separated by a single base pair and are thus referred to as DR1 elements (Tugwood et al., 1992). PPAR, thyroid hormone receptor (TR), vitamin D receptor (VDR), and all-transretinoic acid receptor (RAR) form a subgroup within the superfamily that heterodimerize with the 9-cis-retinoic acid receptor (RXR) and bind to response elements composed of two AGGTCA half-sites predominantly organized in a direct repeat. Thus, RXRα was cotransfected with PPARα to examine if PPARα-mediated activation of the reporter would be further enhanced. It turned out that an obviously enhanced activation was observed (Figure 1C).
To identify if a putative PPRE exists in Trx promoter, a series of luciferase reporter plasmids containing various truncated form of the Trx promoter ranging from -2259, -2134, -1947, and -890 to -47 were cotransfected with PPARα vector, respectively. The luciferase activities were assayed in the presence or absence of WY14643. It was found that transactivation of the truncated Trx promoters in response to PPARα activation was nearly completely abolished when the region between -2259 and -2134 was missing (Figure 1D). The missing region contains a direct repeat-1-like sequence AGCTCACTGTTCA similar to the consensus PPRE for known PPAR target genes (Juge-Aubry et al., 1997; Figure 1E).
To confirm if the PPARα/RXRα heterodimer directly binds to the putative Trx PPRE, a double-stranded Trx PPRE oligonucleotide probe labeled with biotin was incubated with in vitro-translated PPARα and RXRα. The protein-DNA-binding activity was determined by EMSA. As Figure 1F shows, neither PPARα nor RXRα alone, but PPARα/RXRα heterodimers bound to the Trx PPRE probe (comparing lanes 3 and 4 with lane 2). Addition of 10- and 50-fold molar excess of the unlabeled human ACOX PPRE oligonucleotide (Varanasi et al., 1996) could abolish the binding in a dose-dependent manner, indicating the specificity of this binding (lanes 5 and 6). Furthermore, the binding of endogenous PPARα to Trx PPRE was confirmed by incubation of Trx PPRE probe with the nuclear extracts from the differentiated HepG2 cells. As shown in Figure 1G, the complexed Trx PPRE was obviously increased when the cells were stimulated with WY14643 for 24 h (compare lane 3 with lane 2) and supershifted by the PPARα antibody (lane 1). It suggests that higher activity of the endogenous PPARα in the agonist-stimulated cells results in more binding with Trx PPRE.
In addition, the function of the identified Trx PPRE was testified using a luciferase reporter, (Trx-PPRE)3-SV40-Luc, containing three copies of this element upstream a minimal SV40 promoter. As Figure 1H shows, cotransfection of PPARα resulted in sevenfold and 18-fold increase of the reporter activity in unstimulated and the WY14643-stimulated HepG2 cells, respectively. In addition, a further increase was achieved by cotransfection of RXRα, which is in consistent with the finding in Figure 1F.
All above results convincingly indicate that human Trx gene is a PPARα-targeted gene, and the identified PPRE in Trx promoter is required for PPARα-mediated transcription.
PPARα Induces the Translocation of Trx to Nucleus
In general, the stimuli that promote Trx expression are also capable of promoting its intracellular movement (Bai et al., 2003; Watson et al., 2003). We herein investigated if activation of PPARα could induce Trx translocation from cytosol to nucleus. To test this hypothesis, HeLa cells were transiently transfected with a functional GFP-tagged PPARα expression vector. Subcellular localization of the transfected GFP-PPARα and endogenous Trx were observed by microscope. As Figure 2 shows, GFP-PPARα only exists in nuclei (Figure 2B) consisting with a previous report (Akiyama et al., 2002). However, the subcellular distribution of endogenous Trx in the transfected cells differs dramatically from that in untransfected cells. In the formers, Trx was located predominantly in nuclei, though less Trx was still seen in cytosol (see the two cells in the lower left corner in Figure 2C), whereas Trx was predominantly located in cytosol of the wild-type cells (see the two top-right cells in Figure 2C). The study on Trx distribution in the cells transfected with GFP alone proved that PPARα rather than GFP is responsible for the nuclear translocation of Trx (unpublished data). These fluorescence and immunofluorescence images provide clear evidences for the PPARα-induced nuclear translocation of Trx.
Figure 2.

Translocation of Trx from cytoplasm to nucleus by overexpressing GFP-tagged PPARα. The HeLa cells transfected with EGFP-PPARα expression vector were stained with Hoechst 33342, fixed, and permeabilized. (A) Fluorescence image of nuclei in the cells. (B) Fluorescence image of GFP-tagged PPARα in the cells. (C) Immunofluorescence image of the endogenous Trx in the same cells using Trx antibody and Texas Red-labeled second antibody. Bar, 10 μm.
Down-regulation of PPARα Transcriptional Activity by Ectopic Overexpression of Trx in the Nucleus
Because Trx regulates many transcription factors (Watson et al., 2003), can Trx possibly regulate the transcriptional activity of PPARα? Trx is a cytosolic protein (Hirota et al., 1999) and shuttles between cytoplasm and nucleus under many circumstances (Bai et al., 2003; Watson et al., 2003). To study the potential intranuclear effect of Trx on regulation of PPARα transcriptional activity, a possible way is to deliberately increase nucleus-localized Trx. Because fluorescent proteins are able to diffuse passively into and out of the nucleus in mammalian cells (Chatterjee and Stochaj, 1998), it could be used to drive Trx into nucleus by fusion of the two proteins, as reported in a previous investigation (Hwang et al., 2004). Therefore, an EYFP-tagged Trx expression vector was transfected into HeLa cells, and its expression and redox function were confirmed by both immunoblotting and insulin-reduction assay (Figure 3, A and B). A marked increase of Trx activity was found in the cells transfected with YFP-Trx compared with that in the cells only transfected with YFP, indicating that the fused YFP did not affect the redox activity of Trx. Interestingly, unlike the predominant cytosolic localization of the endogenous Trx (bottom panel in Figure 3C), fusion with YFP resulted in a general cytoplasmic distribution with an increased nuclear localization of Trx (top panel, Figure 3C). Because PPARα was mainly localized in nucleus, the cotransfection of YFP-Trx with PPARα is a good approach to study the effect of nuclear Trx on the transcriptional activity of PPARα (Figure 3D). For this reason, HeLa cells were cotransfected with a luciferase reporter driven by three copies of an ACOX PPRE linked to a TK minimal promoter, (PPRE)3-TK-Luc, a PPARα expression vector, and increasing amount of YFP-Trx expression vector. Meanwhile, VP16-PPARα, a fusion protein expression vector linking a strong transactivation domain VP16 to the upstream of PPARα, was used as a positive control. As Figure 4A shows, the cotransfected PPARα led to 7- and 18-fold increase of the reporter activity in the absence and presence of WY14643, respectively. However, both constitutive and ligand-stimulated PPARα activities were significantly inhibited by cotransfection of YFP-Trx in a dose-dependent manner. A negative correlation was found between the nuclear level of YFP-Trx and the extent of inhibition of PPARα activity (see the Western blot analysis of YFP-Trx in the nuclear extracts of transfected HeLa cells shown on the bottom in Figure 4A). In addition, Western blot analysis (Figure 4B) showed no effect of the transfected YFP-Trx on expression of both endogenous (indicated as the hPPARα bands) and exogenous PPARα (indicated as mPPARα bands) in the cells, suggesting that the decrease of PPARα activity by the transfected Trx is not due to suppression of PPARα content.
Figure 3.
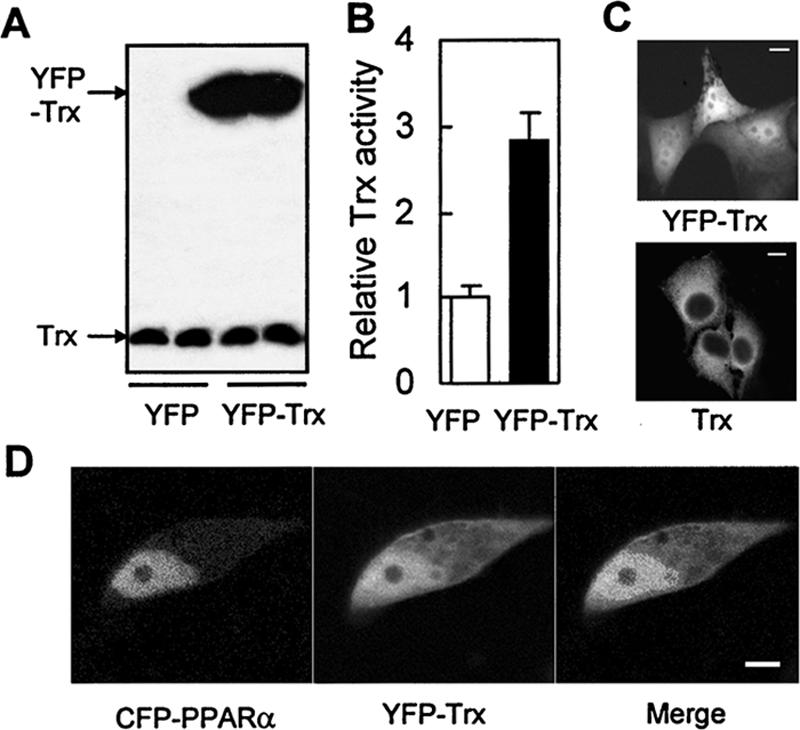
Intracellular distribution and redox activity of the YFP-fused Trx in the cells overexpressing YFP-Trx. (A) Immunoblotting analysis of the exogenous and endogenous Trx in HeLa cells 36 h after transfection of EYFP or EYFP-Trx using anti-Trx antibody (B) Trx activity in the same transfected cells as A. (C) Intracellular distribution of the endogenous Trx and the YFP-fused Trx in HeLa cells. Top, the fluorescence image of YFP-Trx in the transfected cells; bottom, the immunofluorescence image of the endogenous Trx in cells. (D) The fluorescence images of the CFP-fused PPARα and YFP-fused Trx in HeLa cells cotransfected with CFP-PPARα and YFP-Trx for 36 h. Bars, 10 μm.
Figure 4.

Inhibitory effect of YFP-Trx on the transcriptional activity of PPARα and expression of PPARα-target genes. (A) Inhibition of PPARα activity by YFP-Trx in HeLa cells transfected with 1 μg (PPRE)3-TK-Luc reporter construct, 300 ng PPARα vector, and increasing amounts of YFP-Trx vector (50-400 ng). The bottom Western blot shows the contents of the exogenous YFP-Trx in the nuclear extracts of the above-described transfected cells 36 h after transfection using anti-GFP(YFP) polyclonal antibody. (B) Immunoblotting analysis of the endogenous and exogenous PPARα protein in HeLa cells 36 h after cotransfection of pCMX-mPPARα (or VP16-mPPARα) vector with increasing amounts of YFP-Trx vector. hPPARα and mPPARα stands for the endogenous human PPARα and exogenous mouse PPARα respectively. (C) Inhibitory effect of YFP-Trx on transcriptional activation of the PPARα-target genes (MCPT1 and MCAD). HepG2 cells were cotransfected with 1 μg reporter constructs (MCPT1-Luc.781 or MCAD-Luc.376), 300 ng PPARα vector, and 200 ng YFP-Trx vector. In A and C, after a 12-h transfection, cells were treated with 50 μM WY14643 or DMSO (vehicle) for 24 h and then lysed. The data are shown as mean ± SE of three independent measurements. (D) Inhibitory effect of YFP-Trx on the secretion of apoA-I protein from the HepG2 cells transfected with YFP-Trx or decoy PPRE (1 μM) and cultured in serum-free medium containing 50 μM WY14643 or vehicle for 24 h. The secreted protein in the medium was analyzed by Western blotting using apoA-I antibody.
To investigate if overexpressed YFP-Trx inhibits transactivation of the promoters in PPARα-target genes, two well-identified PPARα-target genes involved in cellular fatty acid oxidation, muscle carnitine palmitoyltransferase I (MCPT1; Vega et al., 2000) and medium chain acyl CoA dehydrogenase (MCAD; Gulick et al., 1994), were examined. The reporter containing either the human MCPT1 gene promoter (MCPT1-Luc.781) or MCAD gene promoter (MCAD-Luc.376) was cotransfected with PPARα and YFP-Trx in HepG2 cells. Similar to the results obtained with (PPRE)3-TK-Luc, YFP-Trx significantly inhibited the MCPT1 or MCAD promoter-driven luciferase activity in the presence or absence of the PPARα ligand (Figure 4C). All above experiments with PPRE-driven heterologous promoter reporter, (PPRE)3-TK-Luc, or homologous promoter reporters (MCPT1-Luc.781 and MCAD-Luc.376) demonstrate well that the transfected YFP-Trx might inhibit transcription of the PPARα-target genes.
To verify if Trx can also inhibit the protein level of PPARα-target genes, the expression of apolipoprotein A-I (apoA-I), a PPARα-target gene that expresses the major structural component of the high-density lipoprotein (HDL) particle, was determined in HepG2 cells transfected with or without YFP-Trx. As shown in Figure 4D, secretion of apoA-I protein from HepG2 cells obviously increased under WY14643 stimulation, which is consistent with a previous report (Morishima et al., 2003). However, this increase was totally blocked by overexpression of YFP-Trx in the cells. As a positive control, the presence of the transfected PPRE decoy oligonucleotides also dramatically attenuated the WY14643-stimulated apoA-I expression in a manner similar to YFP-Trx. The result confirms that Trx suppresses apoA-I secretion through inhibiting PPARα activity.
To further confirm that nuclear Trx is responsible for down-regulation of the PPARα activity, a procytoplasmic and a pronuclear Trx expression vectors, pcDNA3-Trx and GAL4-Trx, were used to discriminate the roles of Trx in cytoplasm and in nucleus (Hirota et al., 1999). Because yeast GAL4 DBD (amino acids 1-147) has a strong nuclear localization signal amino acid sequence (Hirota et al., 1999), the GAL4(DBD)-tagged Trx preferably enters nucleus. With reporter (PPRE)3-TK-Luc, it was observed that the activity of PPARα was inhibited by cotransfection of either pcDNA3-Trx or pCMX-GAL4-Trx, but the latter had a much greater inhibitory effect (Figure 5A). The results suggest that Trx down-regulates PPARα activity in nucleus.
Figure 5.

Subcellular localization-dependent inhibitory effect of Trx on activity of exogenous and endogenous PPARα in HeLa (A and C), HepG2 (B and C), Ea.hy.926 (B and C), and A549 (B) cells. The activity of PPARα was measured as expression of reporter gene. (A) Cells were cotransfected with 1 μg (PPRE)3-TK-Luc reporter construct, 300 ng PPARα vector, and increasing amounts of the pronuclear (pCMX-GAL4-Trx) or the procytosolic (pcDNA3-Trx) Trx expression vector for 12 h and subsequently stimulated by 50 μM WY14643 or vehicle for 24 h. (B) Cells were cotransfected with 1 μg (PPRE)3-TK-Luc reporter construct, 300 ng PPARα vector, and 600 ng pcDNA3-Trx or pCMX-GAL4-Trx expression vector for 12 h and subsequently treated as A. (C) Cells were cotransfected with 1 μg (PPRE)3-TK-Luc reporter construct and 600 ng pcDNA3-Trx or pCMX-GAL4-Trx expression vector for 36 h and then the basal PPARα activity was measured. All data on the luciferase activities were normalized against activity of β-galactosidase, which is transfected as internal control, and are shown as mean ± SE of three independent measurements.
To exclude possible cell type specificity of the observed nucleus-localization-dependent inhibitory effect of Trx on PPARα activity, this effect was also examined in human HepG2 hepatoma cells, EA.hy.926 endothelial cells, and A549 lung epithelial cells. As shown in Figure 5B, overexpression of GAL4-linked Trx dramatically suppressed the PPARα activity in all examined cell lines. In contrast, cotransfection with the “nude Trx” (pcDNA3-Trx) resulted in less inhibition of the PPARα activity in these types of cells. The results suggest that the observed repression of PPARα activity by nucleus-localized Trx could be a common cellular phenomenon.
In all above experiments, the cells cotransfected with PPARα and Trx, which is either nude or tagged by YFP or GAL4, were used to investigate the inhibitory effect of overexpressed Trx on the overall activity of both endogenous and exogenous PPARα in the cells (see Figure 4, A and C, as well as Figure 5, A and B). To answer the question whether the endogenous PPARα would be also inhibited by the overexpressed Trx, the basal activity of PPARα was determined in HeLa, HepG2, and EA.hy.926 cells transfected with pcDNA-Trx or GAL4-Trx using (PPRE)3-TK-Luc as a reporter. It was reported that all of these cell types have endogenous PPARα (Rival et al., 2002). As shown in Figure 5C, the procytoplasmic Trx slightly, whereas the pronuclear Trx significantly inhibited the basal PPARα activity. The results suggest that the endogenous PPARα behaves the same as the exogenous one, and the observed inhibitory effect of Trx on overexpressed PPARα may reflect the real situation in the cells under normal physiological condition.
All above results clearly indicate that PPARα is the target for Trx-exerted inhibitory effect, whenever it is under basal condition, overexpressed or stimulated by its ligand.
Trx Inhibits PPARα Transcriptional Activity in a Redox-dependent Manner
Because most cellular functions of Trx depend on its reductive activity, it was examined whether the inhibition of PPARα by Trx depends on the redox activity of Trx. The A549 cells transiently cotransfected with the luciferase reporter [(PPRE)3-TK-Luc], PPARα vector, and pcDNA3-based Trx or its mutant were used as model system because A549 cells are more sensitive to “nude Trx” than other cell types (see Figure 5B) and lack endogenous PPARα (Pawliczak et al., 2002). As expected, cotransfection of the wild-type Trx markedly inhibited both constitutive and ligand-stimulated PPARα activity, whereas the redox-inactive mutant Trx(C32/35S), in which the Cys32 and Cys35 was mutated to serine, completely lost its inhibitory effect. However, the mutant Trx(C62/69/73S), in which three noncatalytic cysteines (Cys62, Cys69, and Cys73) were mutated to serines, still largely possessed its inhibitory activity (Figure 6A). These mutation experiments suggest that Cys32 and Cys35 are necessary for the effectiveness of Trx in inhibiting PPARα activity.
Figure 6.

Redox-dependent inhibitory effect of Trx on PPARα activity in A549 cells. (A) Basal and ligand-stimulated PPRE-driven transcription of luciferase in cells cotransfected with 1 μg (PPRE)3-TK-Luc reporter construct, 300 ng PPARα vector, and 600 ng of the vector expressing wild-type or mutated Trx (C32/35S or C62/69/72S). (B) Effect of TrxR on PPARα activity. Cells were cotransfected with 1 μg (PPRE)3-TK-Luc reporter construct, 300 ng PPARα vector, 600 ng pcDNA3-Trx(wt), or pcDNA3-Trx (C32/35S) vector, and 600 ng pCNX2-TrxR vector. (C) Effect of goldthioglucose on PPARα activity. Cells were cotransfected with 1 μg (PPRE)3-TK-Luc reporter construct, 300 ng PPARα vector, and 600 ng pcDNA3-Trx vector in the presence of goldthioglucose at various concentrations. (D) RT-PCR analysis of Txnip mRNA in the cells stably transfected with Txnip siRNA vector (refer as T1 and T2 cells) or scramble siRNA vector (refer as T0 cells). (E) Effect of Txnip silencing on PPARα activity in the presence of Trx. All three A549 clones (T0, T1, and T2) were cotransfected with 1 μg (PPRE)3-TK-Luc reporter construct, 300 ng PPARα vector, and 600 ng pcDNA3-Trx vector. Except for D, after a 12-h transfection, cells were treated with 50 μM WY14643 or DMSO (vehicle) for 24 h and then lysed. Normalized luciferase activities are shown as mean ± SE of three independent measurements.
Because inhibition of PPARα requires redox-active Trx, it is reasonable to expect that TrxR), the primary enzyme catalyzing the NADPH-dependent reduction of Trx, should play a role in the Trx-caused inhibition of PPARα. Therefore, a human TrxR expression vector was introduced in the assay system. As shown in Figure 6B, cotransfection of TrxR and Trx showed dramatically synergistic inhibitory effect on PPARα activity, though each of them alone exerted marked inhibition. However, TrxR lost its synergistic action when the mutant Trx (32S/35S) replaced the wild-type Trx. To further confirm the role of TrxR, its selective inhibitor, goldthioglucose (Sakurai et al., 2004), was used to inhibit the activity of the endogenous TrxR. It was found that the resulted PPARα activity increased as goldthioglucose concentration increased (Figure 6C).
Furthermore, it was also investigated if Txnip, the negative modulator of Trx (Junn et al., 2000), affects the Trx-regulated PPARα activity. Txnip siRNA was used to knockdown the endogenous Txnip. Two Txnip gene-silenced cell lines, T1 and T2, were cloned by stably transfecting the Txnip-siRNA vector in A549 cells. Reverse transcription (RT)-PCR analysis confirmed that the mRNA level of Txnip was substantially suppressed in the T1 and T2 cells in comparison with that in the cells transfected with a scrambled siRNA (referred as T0 cells; Figure 6D). It was found that PPARα-mediated transcription of the (PPRE)3-TK-Luc reporter was significantly suppressed in the Txnip-silenced cells. The suppression was further enhanced by cotransfection of Trx in the cells (Figure 6E). This study further demonstrates the role played by Trx in down-regulating PPARα activity.
AF-1 Is the Target Domain for Trx in the Inhibition of PPARα Transcriptional Activity
The inhibition of PPARα activity by Trx may imply a possible interaction between Trx and PPARα in the nucleus. Unfortunately, both a mammalian two-hybrid system using two fusion constructs linking the GAL4 DBD to Trx and the VP16 transactivation domain to PPARα, respectively, and a fluorescence resonance energy transfer (FRET) system containing CFP-fused PPARα and YFP-fused Trx demonstrated that there is no direct interaction between Trx and PPARα (unpublished data).
PPARα contains an NH2-terminal ligand-independent transactivation domain (AF-1), a central DBD and a COOH-terminal ligand-binding domain (LBD/AF-2) that displays a ligand-dependent transactivation activity. To identify which domain in PPARα is the target for Trx, the effect of Trx on transactivation function of AF-1 and AF-2 were investigated, respectively, by mammalian one-hybrid assay. A fusion construct linking GAL4(DBD) to either the N-terminal 92 amino acids of PPARα (GAL4-AF1) or the C-terminal 296 amino acids of PPARα (GAL4-LBD) was cotransfected with the (UAS)5-TATA-Luc reporter plasmid in A549 cells to establish two independent one-hybrid assay systems. The effect of Trx on AF-1 or LBD/AF2 transactivation function was evaluated by detecting the luciferase activities in the presence and absence of the cotransfected Trx. As shown in Figure 7A, stimulation with WY14643 markedly activated the GAL4-LBD chimera (4-fold increase), confirming the ligand-dependent nature of this domain in the transactivation function of PPARα. The overexpressed Trx had no effect on its function. In sharp contrast, the GAL4-AF1 chimera was constitutively activated without stimulation by the PPARα ligand (more than 30-fold increase over the basal activity of the reporter), whereas the cotransfected Trx(wt), but not its redox-inactive mutant, significantly inhibited the transactivation by the chimera (Figure 7B). However, the loss of GAL4-AF-1 activity was not due to any decrease in GAL4-AF-1 expression (Western blot in Figure 7B). Thus, the difference in affecting these two chimeric transcription factors suggests that AF-1 is the domain receiving Trx-modulation.
Figure 7.
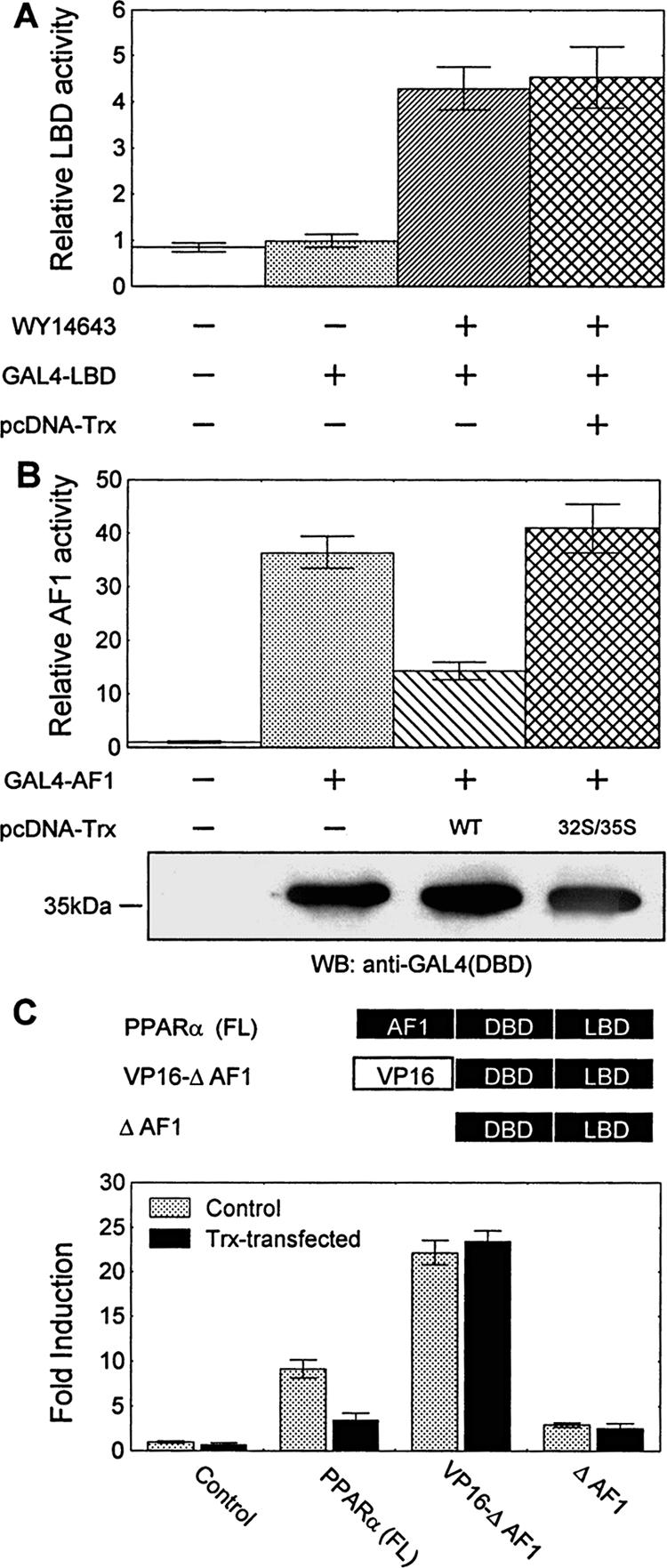
Trx inhibits PPARα activity by modulating the AF-1 domain of PPARα. (A) Effect of Trx on the ligand-dependent transactivation activity of LBD. A549 cells were cotransfected with 1 μg (UAS)5-TATA-Luc reporter construct, 300 ng GAL4-LBD vector, and 600 ng pcDNA3-Trx or its empty vector for 12 h and subsequently stimulated with 50 μM WY14643 or DMSO for 24 h. (B) Effect of Trx on the ligand-independent transactivation activity of AF-1 domain. A549 cells were cotransfected with 1 μg (UAS)5-TATA-Luc reporter construct, 300 ng GAL4-AF1 vector, and 600 ng of pcDNA3-Trx or pcDNA3-Trx (C32/35S) vector. The lower immunoblot shows the expression of GAL4-AF1 in each case using anti-GAL4 (BD) antibody. (C) Effect of Trx on the constitutive transcriptional activity of wild-type and modified PPARα. A549 cells were cotransfected with 1 μg (PPRE)3-TK-Luc reporter construct, 300 ng of the full-length or modified PPARα vector (ΔAF1 or VP16-ΔAF1), and 600 ng of pcDNA3-Trx or its empty vector. In all above experiments, after a 12-h transfection, cells were untreated (B and C) or treated with 50 μM WY14643 (A) for 24 h and then lysed. Normalized luciferase activities are shown as mean ± SE of three independent measurements.
To know if AF-1 is the only domain on PPARα modulated by Trx, two PPARα mutant vectors were constructed: one expresses an AF-1-deleted PPARα (ΔAF1) and the other expresses a modified PPARα with its AF-1 region replaced by the VP16 transactivation domain (VP16-ΔAF1). The transcriptional activities of the wild-type PPARα and its two mutants were then assayed with (PPRE)3-TK-Luc reporters in A549 cells in the presence or absence of cotransfected pcDNA3-Trx. As Figure 7C shows, overexpressed Trx only significantly inhibited the constitutive transcriptional activity of full-length PPARα, neither ΔAF1 nor VP16-ΔAF1 was influenced by the Trx. Because the both modified PPARα lack the AF-1 region, it could be concluded that AF-1 is the only domain modulated by Trx in PPARα.
Effects of Trx on PPARα-DNA-binding Activity
Binding to PPRE is a necessary step for PPARα to activate transcription of its target genes. To investigate whether the increased nuclear Trx affected PPARα-DNA-binding activity, EMSA were performed using the nuclear extracts (NE) from HepG2 cells and a biotin-labeled double-stranded oligonucleotide probe corresponding to ACOX PPRE. As Figure 8 shows, a prominent retarded complex was formed when NE was incubated with the probe (lane 1). The retarded complex further supershifted when anti-PPARα antibody was added (lanes 2) in NE and was completely eliminated by excessive unlabeled PPRE probe (lane 3), indicating a specific binding of the PPARα from NE to the PPRE probe. Incubation of NE with increasing concentrations of recombinant human Trx resulted in dose-dependent decrease of the PPARα-PPRE binding in the presence of TrxR and NADPH (lanes 6-9). It was noticed that removal of TrxR and NADPH markedly reduced the effect of Trx (lane 10), indicating that the reduced Trx is responsible for the inhibition of the PPARα-DNA binding.
Figure 8.

Effect of recombinant Trx on the DNA-binding activity of PPARα. EMSA was performed using biotin-labeled HACOX PPRE probe and nuclear extracts from HepG2 cells in the presence and absence of either anti-PPARα antibody or competitive unlabeled probe (lanes 1-3) and under various redox conditions as indicated, in which 200 nM TrxR, 500 μM NADPH, and various concentration of Trx were present or absent (lanes 4-11). The EMSA shown here is representative of two independent assays.
DISCUSSION
Redox regulation is critical for the functions of a number of TFs. Up to now, more than 60 redox-regulated TFs have been identified. Most of them have critical thiol moieties and can be regulated, at least in part, by Trx (Watson et al., 2003). Among them, Trx was found to up-regulate the activities of two members of the nuclear receptor superfamily, glucocorticoid receptor and estrogen receptor, seemingly only under oxidative conditions (Hayashi et al., 1997; Hirota et al., 1999). As regards PPARs, there have been quite a number of reports on their connection with redox. The majority of them are focused on the roles of PPARs in regulating generation of reactive oxygen species (ROS). For example, different agonists of PPARα, but not PPARγ, increase the production of ROS (H2O2 and 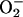 ) in macrophages due to the induction of NADPH oxidase (Teissier et al., 2004); PPARγ ligand pioglitazone modulates fibroblast growth and collagen type-I synthesis in cardiac fibroblasts exposed to anoxiareoxygenation through inhibition of ROS generation and induction of NF-κB (Chen et al., 2004); PPARα modulates catalase and superoxide dismutase (SOD) expression, and hence down-regulates the inflammatory response via scavenging ROS (Yoo et al., 1999; Girnun et al., 2002). Few of them showed the modulation of PPARs by ROS or nitric oxide (NO). It was reported that ROS generation and NF-κB activation down-regulating PPARα, resulting in intracellular lipid accumulation in skeletal muscle cells (Cabrero et al., 2002), and that NO switches monocyte/macrophage function from a proto an anti-inflammatory phenotype by activating PPARγ (Von Knethen and Bruene, 2002). However, no report on the regulation of PPAR activity by thioredoxin has been found. The present study revealed a novel function of thioredoxin in regulating the transcriptional activity of PPARα under normal physiological condition. We demonstrated that the increased nucleus-localized Trx by transfecting YFP-fused Trx or GAL4(DBD)-fused Trx markedly suppressed the transcriptional activity of PPARα. This inhibitory effect of nuclear Trx seems not specific for a particular cell line. Similar effect was actually seen in a variety of cell types, such as human hepatoma cells, endothelial cells, and cervical and lung epithelial cells. Besides the finding that Trx inhibits the transcriptional activity of PPARα, this study also identified Trx as the product of a PPARα-target gene. It was discovered that either overexpression of PPARα or activation of PPARα by its ligand up-regulated Trx expression. A functional PPRE (AGCTCACTGTTCA) was identified in the promoter region of human Trx gene. This PPRE sequence was confirmed by its abilities in binding to the PPARα/RXRα heterodimers and mediating PPARα-activated transcription. An even more interesting finding is that overexpression of PPARα induced a translocation of Trx from cytoplasm to nucleus. Together with the finding that increase of nucleus-localized Trx leads to a strong inhibition of PPARα transcriptional activity, the events: increase of PPARα activity, up-regulation of Trx expression, translocation of Trx into nucleus, and inhibition of PPARα activity, constitute a feedback loop shown as in Figure 9. The loop forms a negative feedback mechanism to maintain the homeostasis of the PPARα activity as well as cellular level of Trx. This “self-regulation” of PPARα activity through such a loop may therefore be defined as Trx-mediated negative autoregulation of PPARα activity. Thioredoxin, a PPARα-target gene product, negatively regulates the transcription activity of PPARα is the key feature of this negative autoregulation. So far, only positive regulation of PPARα activity by its target gene products, such as a mitochondrial protein (Meertens et al., 1998) and a peroxisomal protein (Juge-Aubry et al., 2001), has been reported. In this investigation, the human Trx gene was identified as the first target gene of PPARα that negatively regulates PPARα activity.
) in macrophages due to the induction of NADPH oxidase (Teissier et al., 2004); PPARγ ligand pioglitazone modulates fibroblast growth and collagen type-I synthesis in cardiac fibroblasts exposed to anoxiareoxygenation through inhibition of ROS generation and induction of NF-κB (Chen et al., 2004); PPARα modulates catalase and superoxide dismutase (SOD) expression, and hence down-regulates the inflammatory response via scavenging ROS (Yoo et al., 1999; Girnun et al., 2002). Few of them showed the modulation of PPARs by ROS or nitric oxide (NO). It was reported that ROS generation and NF-κB activation down-regulating PPARα, resulting in intracellular lipid accumulation in skeletal muscle cells (Cabrero et al., 2002), and that NO switches monocyte/macrophage function from a proto an anti-inflammatory phenotype by activating PPARγ (Von Knethen and Bruene, 2002). However, no report on the regulation of PPAR activity by thioredoxin has been found. The present study revealed a novel function of thioredoxin in regulating the transcriptional activity of PPARα under normal physiological condition. We demonstrated that the increased nucleus-localized Trx by transfecting YFP-fused Trx or GAL4(DBD)-fused Trx markedly suppressed the transcriptional activity of PPARα. This inhibitory effect of nuclear Trx seems not specific for a particular cell line. Similar effect was actually seen in a variety of cell types, such as human hepatoma cells, endothelial cells, and cervical and lung epithelial cells. Besides the finding that Trx inhibits the transcriptional activity of PPARα, this study also identified Trx as the product of a PPARα-target gene. It was discovered that either overexpression of PPARα or activation of PPARα by its ligand up-regulated Trx expression. A functional PPRE (AGCTCACTGTTCA) was identified in the promoter region of human Trx gene. This PPRE sequence was confirmed by its abilities in binding to the PPARα/RXRα heterodimers and mediating PPARα-activated transcription. An even more interesting finding is that overexpression of PPARα induced a translocation of Trx from cytoplasm to nucleus. Together with the finding that increase of nucleus-localized Trx leads to a strong inhibition of PPARα transcriptional activity, the events: increase of PPARα activity, up-regulation of Trx expression, translocation of Trx into nucleus, and inhibition of PPARα activity, constitute a feedback loop shown as in Figure 9. The loop forms a negative feedback mechanism to maintain the homeostasis of the PPARα activity as well as cellular level of Trx. This “self-regulation” of PPARα activity through such a loop may therefore be defined as Trx-mediated negative autoregulation of PPARα activity. Thioredoxin, a PPARα-target gene product, negatively regulates the transcription activity of PPARα is the key feature of this negative autoregulation. So far, only positive regulation of PPARα activity by its target gene products, such as a mitochondrial protein (Meertens et al., 1998) and a peroxisomal protein (Juge-Aubry et al., 2001), has been reported. In this investigation, the human Trx gene was identified as the first target gene of PPARα that negatively regulates PPARα activity.
Figure 9.

Proposed negative feedback loop for the Trx-mediated autoregulation of PPARα transcriptional activity.
Many efforts were devoted to understand how the redox-activity and subcellular localization of Trx influences the inhibitory effect of Trx on PPARα transcriptional activity. Inability of the overexpressed redox-inactive Trx in suppressing PPARα activity (Figure 6, A and B) suggests a critical role for the redox-active site (Cys32 and Cys35) of Trx. This conclusion was further supported by increased suppression of the binding activity of PPARα to PPRE in vitro in the presence of TrxR, the primary enzyme catalyzing Trx reduction (Figure 8), and an increased inhibition of PPARα transcriptional activity in the cells having Txnip, the endogenous redox inhibitor of Trx (Junn et al., 2000), knocked down by Txnip siRNA. Because TrxR and Txnip have been reported to be involved in promoting and blocking nuclear localization of Trx, respectively (Karimpour et al., 2002; Schulze et al., 2002), the observed enhanced inhibition PPARα activity by TrxR and Txnip siRNA may be also due to an alteration of subcellular distribution of Trx. In this study, YFP-fused Trx was found to inhibit PPARα activity more effectively than the native Trx because the YFP-Trx tends to be localized in the nucleus as compared with the latter. The reason for the altered localization of YFP-Trx may be attributed to the ability of fluorescent protein in diffusing passively into and out of the nucleus in mammalian cells (Chatterjee and Stochaj, 1998).
Concerning the regulation mechanism, we observed that Trx inhibited PPARα activity mainly by modulating the constitutive transactivation function of its AF-1 domain. In fact, the AF-1 transactivation domain of PPARα has already been demonstrated to be an effective target in transregulation of PPARα activity. It was reported that the signal transducer and activator of transcription (STAT5b) inhibited the transcription driven by the AF-1 transactivation domain of PPARα in a GAL4-linked chimera (Zhou and Waxman, 1999), and the peroxisomal bifunctional enzyme (BFE) bound and activated the AF-1 region of the PPARα (Juge-Aubry et al., 2001). The present study reveals that the AF-1 domain is essential for the inhibition of PPARα activity by Trx, because the transactivation ability of AF-1 is significantly inhibited by Trx whenever it is tethered to exogenous GAL4 DBD (Figure 7B) or included in the whole PPARα molecule (Figure 7C).
Despite the direct interaction between PPARα and Trx was not detected, it may still be expected that the modulation of AF-1 transactivity by Trx could be due to a transient thiol-redox modification of AF-1 domain or some unidentified AF-1-associated proteins such as steroid receptor coactivator 1 (SRC1; unpublished data). Nevertheless, it is also possible that modulation of AF-1 may alter other PPARα characters such as DNA-binding activity. In fact, the present study demonstrated well that the reduced form of recombinant human Trx inhibits the binding of the PPARα from HepG2 cell nuclear extract to the PPRE probe in vitro. The concentrations, at which Trx exerts inhibitory effect on the PPARα-DNA-binding activity in EMSA assay, are in the range of 4-12 μM, and are comparable to the physiological cellular concentration of Trx (2-12 μM) found in mammalian cells (Powis and Montfort, 2001). The slightly higher concentration used in the experiment is based on the following consideration: the nucleus is much smaller than the whole cell, and the concentration of Trx in nucleus could be largely increased when Trx is translocated from cytoplasm to nucleus in response to activation of PPARα or oxidative stress.
The present finding that Trx-mediated negative autoregulation of PPARα activity may have potential physiological, pathological, and pharmacological significances. To show cellular physiological evidence for the Trx-mediated negative regulation of PPARα activity, the secretion of apolipoprotein A-I, which is the major structural component of the HDL particle in human circulation system and regulated by PPARα, were determined from the HepG2 cells overexpressing YFP-Trx. The results clearly demonstrate that forced overexpression of Trx in the nucleus does suppress the expression of the PPARα-target gene at cellular level (see Figure 4D). Together with the strong inhibition of transcription from MCPT1 and MCAD promoter by nucleus-targeted Trx, our finding might reveal some physiologically detrimental aspects of thioredoxin. For example, on account of oxidative stress-caused increased expression and nucleus localization of Trx (Watson et al., 2003), our reported inhibitory role of nuclear Trx may mediate oxidative stress-induced metabolic dysfunction. In addition, the negative feedback mechanism probably renders body resistance to lipid-lowing drugs. Although the cytosolic Trx has been usually considered as an important antioxidant enzyme in cellular defense system and to be beneficial for human health, the detrimental effects of nuclear Trx should not be ignored. As a matter of fact, it has been reported that nuclear Trx are helpful for TNF-α-induced activation of NF-κB, a proinflammatory and procarcinogenic transcription factor (Hirota et al., 1999; Karin and Greten, 2005). In addition, the drug-resistance of cancer due to higher expression of thioredoxin (Sasada et al., 1996) and previously reported hyperlipidemic phenotype of the Txnip-deficient mice (Bodnar et al., 2002) are probably the two other examples of such detrimental effect of Trx system.
In summary, this study suggests that besides its antioxidant function in maintenance of cellular redox homeostasis, Trx may act as a regulator for timely quenching excessive PPARα signaling and controlling the expression levels of PPARα-regulated metabolic enzymes according to metabolic need. However, Trx-mediated negative autoregulation of PPARα probably causes a functional down-regulation of normal PPARα activities such as lipid and inflammatory regulation (Delerive et al., 1999). Thus, we may conclude that the autoregulation of PPARα activity by Trx may be a novel mechanism for controlling PPARα activities and PPARα-related physiological or pathological processes.
Acknowledgments
We particularly thank Drs. J. Yodoi, R. M Evans, C. Meier, D. P. Kelly, D. Gius, F. Gonzalez, K. F. Tonissen, F. Gonzalez, B. Desvergne, B. Spiegelman, and T. Goda for their kind help in providing some of the plasmids used in this study. We are also grateful to Dr. A. Holmgren for the gift of the recombinant thioredoxin and rat thioredoxin reductase. This work was supported by the National Natural Science Foundation of China (30570462 and 30330250) and in part by E-Institutes of Shanghai Municipal Education Commission (project E-04010).
This article was published online ahead of print in MBC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E05-10-0979) on February 21, 2006.
Abbreviations used: EMSA, electrophoretic mobility-shift assay; MCAD, medium chain acyl coenzyme A dehydrogenase; MCPT1, muscle carnitine palmitoyltransferase 1; PPARα, peroxisome proliferator-activated receptor α; PPRE, peroxisome proliferator response element; RXRα, retinoid X receptor-α; TF, transcription factor; Trx, thioredoxin; TrxR, thioredoxin reductase; Txnip, thioredoxin-interacting protein.
References
- Akiyama, T. E., Baumann, C. T., Sakai, S., Hager, G. L., and Gonzalez, F. J. (2002). Selective intranuclear redistribution of PPAR isoforms by RXR alpha. Mol. Endocrinol. 16, 707-721. [DOI] [PubMed] [Google Scholar]
- Bai, J., Nakamura, H., Kwon, Y. W., Hattori, I., Yamaguchi, Y., Kim, Y. C., Kondo, N., Oka, S., Ueda, S., Masutani, H., and Yodoi, J. (2003). Critical roles of thioredoxin in nerve growth factor-mediated signal transduction and neurite outgrowth in PC12 cells. J. Neurosci. 23, 503-509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanquart, C., Barbier, O., Fruchart, J. C., Staels, B., and Glineur, C. (2002). Peroxisome proliferator-activated receptor alpha (PPAR alpha) turnover by the ubiquitin-proteasome system controls the ligand-induced expression level of its target genes. J. Biol. Chem. 277, 37254-37259. [DOI] [PubMed] [Google Scholar]
- Bloomfield, K. L., Osborne, S. A., Kennedy, D. D., Clarke, F. M., and Tonissen, K. F. (2003). Thioredoxin-mediated redox control of the transcription factor Sp1 and regulation of the thioredoxin gene promoter. Gene 319, 107-116. [DOI] [PubMed] [Google Scholar]
- Bodnar, J. S. et al. (2002). Positional cloning of the combined hyperlipidemia gene Hyplip1. Nat. Genet. 30, 110-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabrero, A., Alegret, M., Sanchez, R. M., Adzet, T., Laguna, J. C., and Carrera, M. V. (2002). Increased reactive oxygen species production down-regulates peroxisome proliferator-activated α pathway in C2C12 skeletal muscle cells. J. Biol. Chem. 277, 10100-10107. [DOI] [PubMed] [Google Scholar]
- Chatterjee, S., and Stochaj, U. (1998). Diffusion of proteins across the nuclear envelope of HeLa cells. Biotechniques 24, 668-674. [DOI] [PubMed] [Google Scholar]
- Chen, K., Li, D., Zhang, X., Hermonat, P. L., and Mehta, J. L. (2004). Anoxiareoxygenation stimulates collagen type-I and MMP-1 expression in cardiac fibroblasts: modulation by the PPAR-gamma ligand pioglitazone. J. Cardiovasc. Pharmacol. 44, 682-687. [DOI] [PubMed] [Google Scholar]
- Delerive, P., De Bosscher, K., Besnard, S., Vanden Berghe, W., Peters, J. M., Gonzalez, F. J., Fruchart, J. C., Tedgui, A., Haegeman, G., and Staels, B. (1999). Peroxisome proliferator-activated receptor alpha negatively regulates the vascular inflammatory gene response by negative cross-talk with transcription factors NF-kappaB and AP-1. J. Biol. Chem. 274, 32048-32054. [DOI] [PubMed] [Google Scholar]
- Dowell, P., Ishmael, J. E., Avram, D., Peterson, V. J., Nevrivy, D. J., and Leid, M. (1997). p300 functions as a coactivator for the peroxisome proliferator-activated receptor alpha. J. Biol. Chem. 272, 33435-33443. [DOI] [PubMed] [Google Scholar]
- Dowell, P., Ishmael, J. E., Avram, D., Peterson, V. J., Nevrivy, D. J., and Leid, M. (1999). Identification of nuclear receptor corepressor as a peroxisome proliferator-activated receptor alpha interacting protein. J. Biol. Chem. 274, 15901-15907. [DOI] [PubMed] [Google Scholar]
- Drori, S., Girnun, G. D., Tou, L., Szwaya, J. D., Mueller, E., Xia, K., Shivdasani, R. A., and Spiegelman, B. M. (2005). Hic-5 regulates an epithelial program mediated by PPARgamma. Genes Dev. 19, 362-375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans, R. M., Barish, G. D., and Wang, Y. X. (2004). PPARs and the complex journey to obesity. Nat. Med. 10, 355-361. [DOI] [PubMed] [Google Scholar]
- Feige, J. N., Gelman, L., Tudor, C., Engelborghs, Y., Wahli, W., and Desvergne, B. (2005). Fluorescence imaging reveals the nuclear behavior of peroxisome proliferator-activated receptor/retinoid X receptor heterodimers in the absence and presence of ligand. J. Biol. Chem. 280, 17880-17890. [DOI] [PubMed] [Google Scholar]
- Girnun, G., Domann, F. E., Moore, S. A., and Robbins, M.E.C. (2002). Identification of a functional peroxisome proliferator-activated receptor response element in the rat catalase promoter. Mol. Endocrinol. 16, 2793-2801. [DOI] [PubMed] [Google Scholar]
- Gulick, T., Cresci, S., Caira, T., Moore, D. D., and Kelly, D. P. (1994). The peroxisome proliferator-activated receptor regulates mitochondrial fatty acid oxidative enzyme gene expression. Proc. Natl. Acad. Sci. USA 91, 11012-11016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi, S., Hajiro-Nakanishi, K., Makino, Y., Eguchi, H., Yodoi, J., and Tanaka, H. (1997). Functional modulation of estrogen receptor by redox state with reference to thioredoxin as a mediator. Nucleic Acids Res. 25, 4035-4040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirota, K., Murata, M., Sachi, Y., Nakamura, H., Takeuchi, J., Mori, K., and Yodoi, J. (1999). Distinct roles of thioredoxin in the cytoplasm and in the nucleus. A two-step mechanism of redox regulation of transcription factor NF-kappaB. J. Biol. Chem. 274, 27891-27897. [DOI] [PubMed] [Google Scholar]
- Holmgren, A. (1985). Thioredoxin. Annul. Rev. Biochem. 54, 237-271. [DOI] [PubMed] [Google Scholar]
- Hsu, M. H., Savas, U., Griffin, K. J., and Johnson, E. F. (2001). Identification of peroxisome proliferator-responsive human genes by elevated expression of the peroxisome proliferator-activated receptor alpha in HepG2 cells. J. Biol. Chem. 276, 27950-27958. [DOI] [PubMed] [Google Scholar]
- Hwang, C. Y., Ryu, Y. S., Chung, M. S., Kim, K. D., Park, S. S., Chae, S. K., Chae, H. Z., and Kwon, K. S. (2004). Thioredoxin modulates activator protein 1 (AP-1) activity and p27Kip1 degradation through direct interaction with Jab1. Oncogene 23, 8868-8875. [DOI] [PubMed] [Google Scholar]
- Juge-Aubry, C., Pernin, A., Favez, T., Burger, A. G., Wahli, W., Meier, C. A., and Desvergne, B. (1997). DNA binding properties of peroxisome proliferator-activated receptor subtypes on various natural peroxisome proliferator response elements. Importance of the 5′-flanking region. J. Biol. Chem. 272, 25252-25259. [DOI] [PubMed] [Google Scholar]
- Juge-Aubry, C. E., Hammar, E., Siegrist-Kaiser, C., Pernin, A., Takeshita, A., Chin, W. W., Burger, A. G., and Meier, C. A. (1999). Regulation of the transcriptional activity of the peroxisome proliferator-activated receptor alpha by phosphorylation of a ligand-independent trans-activating domain. J. Biol. Chem. 274, 10505-10510. [DOI] [PubMed] [Google Scholar]
- Juge-Aubry, C. E., Kuenzli, S., Sanchez, J. C., Hochstrasser, D., and Meier, C. A. (2001). Peroxisomal bifunctional enzyme binds and activates the activation function-1 region of the peroxisome proliferator-activated receptor alpha. Biochem. J. 353, 253-258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junn, E. et al. (2000). Vitamin D3 up-regulated protein 1 mediates oxidative stress via suppressing the thioredoxin function. J. Immunol. 164, 6287-6295. [DOI] [PubMed] [Google Scholar]
- Karimpour, S. et al. (2002). Thioredoxin reductase regulates AP-1 activity as well as thioredoxin nuclear localization via active cysteines in response to ionizing radiation. Oncogene 21, 6317-6327. [DOI] [PubMed] [Google Scholar]
- Karin, M., and Greten, F. R. (2005). NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat. Rev. Immunol. 5, 749-759. [DOI] [PubMed] [Google Scholar]
- Kerley, J. S., Olsen, S. L., Freemantle, S. J., and Spinella, M. J. (2001). Transcriptional activation of the nuclear receptor corepressor RIP140 by retinoic acid: a potential negative-feedback regulatory mechanism. Biochem. Biophys. Res. Commun. 285, 969-975. [DOI] [PubMed] [Google Scholar]
- Von Knethen, A., and Bruene, B. (2002). Activation of peroxisome proliferator-activated receptor α by nitric oxide in monocytes/macrophages down-regulates p47phox and attenuates the respiratory burst. J. Immunol. 169, 2619-2626. [DOI] [PubMed] [Google Scholar]
- Lemberger, T., Desvergne, B., and Wahli, W. (1996). Peroxisome proliferator-activated receptors: a nuclear receptor signaling pathway in lipid physiology. Annu. Rev. Cell. Dev. Biol. 12, 335-363. [DOI] [PubMed] [Google Scholar]
- Makino, Y., Yoshikawa, N., Okamoto, K., Hirota, K., Yodoi, J., Makino, I., and Tanaka, H. (1999). Direct association with thioredoxin allows redox regulation of glucocorticoid receptor function. J. Biol. Chem. 274, 3182-3188. [DOI] [PubMed] [Google Scholar]
- Meertens, L. M., Miyata, K. S., Cechetto, J. D., Rachubinski, R. A., and Capone, J. P. (1998). A mitochondrial ketogenic enzyme regulates its gene expression by association with the nuclear hormone receptor PPARalpha. EMBO J. 17, 6972-6978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochizuki, K., Suruga, K., Sakaguchi, N., Takase, S., and Goda, T. (2002). Major intestinal coactivator p300 strongly activates peroxisome proliferator-activated receptor in intestinal cell line, Caco-2. Gene 291, 271-277. [DOI] [PubMed] [Google Scholar]
- Morishima, A., Ohkubo, N., Maeda, N., Miki, T., and Mitsuda, N. (2003). NFκB regulates plasma apolipoprotein A-I and high density lipoprotein cholesterol through inhibition of peroxisome proliferator-activated receptor alpha. J. Biol. Chem. 278, 38188-38193. [DOI] [PubMed] [Google Scholar]
- Palmer, C. N., Hsu, M. H., Griffin, K. J., Raucy, J. L., and Johnson, E. F. (1998). Peroxisome proliferator activated receptor-alpha expression in human liver. Mol. Pharmacol. 53, 14-22. [PubMed] [Google Scholar]
- Pawliczak, R., Han, C., Huang, X. L., Demetris, A. J., Shelhamer, J. H., and Wu, T. (2002). 85-kDa cytosolic phospholipase A2 mediates peroxisome proliferator-activated receptor gamma activation in human lung epithelial cells. J. Biol. Chem. 277, 33153-33163. [DOI] [PubMed] [Google Scholar]
- Powis, G., and Montfort, W. R. (2001). Properties and biological activities of thioredoxins. Annu. Rev. Pharmacol. Toxicol. 41, 261-295. [DOI] [PubMed] [Google Scholar]
- Rival, Y., Beneteau, N., Taillandier, T., Pezet, M., Dupont-Passelaigue, E., Patoiseau, J. F., Junquero, D., Colpaert, F. C., and Delhon, A. (2002). PPARalpha and PPARdelta activators inhibit cytokine-induced nuclear translocation of NF-kappaB and expression of VCAM-1 in EAhy926 endothelial cells. Eur. J. Pharmacol. 435, 143-151. [DOI] [PubMed] [Google Scholar]
- Sakurai, A., Yuasa, K., Shoji, Y., Himeno, S., Tsujimoto, M., Kunimoto, M., Imura, N., and Hara, S. (2004). Overexpression of thioredoxin reductase 1 regulates NF-kappa B activation. J. Cell. Physiol. 198, 22-30. [DOI] [PubMed] [Google Scholar]
- Sasada, T., Iwata, S., Sato, N., Kitaoka, Y., Hirota, K., Nakamura, K., Nishiyama, A., Taniguchi, Y., Takabayashi, A., and Yodoi, J. (1996). Redox control of resistance to cis-diamminedichloroplatinum (II) (CDDP): protective effect of human thioredoxin against CDDP-induced cytotoxicity. J. Clin. Invest. 97, 2268-2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze, P. C., De Keulenaer, G. W., Yoshioka, J., Kassik, K. A., and Lee, R. T. (2002). Vitamin D3-upregulated protein-1 (VDUP-1) regulates redox-dependent vascular smooth muscle cell proliferation through interaction with thioredoxin. Circ. Res. 91, 689-695. [DOI] [PubMed] [Google Scholar]
- Schulze, P. C., Yoshioka, J., Takahashi, T., He, Z., King, G. L., and Lee, R. T. (2004). Hyperglycemia promotes oxidative stress through inhibition of thioredoxin function by thioredoxin-interacting protein. J. Biol. Chem. 279, 30369-30374. [DOI] [PubMed] [Google Scholar]
- Stier, H., Fahimi, H. D., Van Veldhoven, P. P., Mannaerts, G. P., Volkl, A., and Baumgart, E. (1998). Maturation of peroxisomes in differentiating human hepatoblastoma cells (HepG2): possible involvement of the peroxisome proliferator-activated receptor alpha (PPAR alpha). Differentiation 64, 55-66. [DOI] [PubMed] [Google Scholar]
- Teissier, E., Nohara, A., Chinetti, G., Paumelle, R., Cariou, B., Fruchart, J. C., Brandes, R. P., Shah, A., and Staels, B. (2004). Peroxisome proliferator-activated receptor α induces NADPH oxidase activity in macrophages, leading to the generation of LDL with PPAR-α activation properties. Circ. Res. 95, 1174-1182. [DOI] [PubMed] [Google Scholar]
- Thompson, C. C., and Bottcher, M. C. (1997). The product of a thyroid hormone-responsive gene interacts with thyroid hormone receptors. Proc. Natl. Acad. Sci. USA 94, 8527-8532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tugwood, J. D., Issemann, I., Anderson, R. G., Bundell, K. R., McPheat, W. L., and Green, S. (1992) The mouse peroxisome proliferator activated receptor recognizes a response element in the 5′ flanking sequence of the rat acyl CoA oxidase gene. EMBO J. 11, 433-439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varanasi, U., Chu, R., Huang, Q., Castellon, R., Yeldandi, A. V., and Reddy, J. K. (1996). Identification of a peroxisome proliferator-responsive element upstream of the human peroxisomal fatty acyl coenzyme A oxidase gene. J. Biol. Chem. 271, 2147-2155. [DOI] [PubMed] [Google Scholar]
- Vega, R. B., Huss, J. M., and Kelly, D. P. (2000). The coactivator PGC-1 cooperates with peroxisome proliferator-activated receptor alpha in transcriptional control of nuclear genes encoding mitochondrial fatty acid oxidation enzymes. Mol. Cell Biol. 20, 1868-1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Knethen, A., and Brune, B. (2002). Activation of peroxisome proliferator-activated receptor gamma by nitric oxide in monocytes/macrophages down-regulates p47phox and attenuates the respiratory burst. J. Immunol. 169, 2619-2626. [DOI] [PubMed] [Google Scholar]
- Watson, W. H., Yang, X., Choi, Y. E., Jones, D. P., and Kehrer, J. P. (2003). Thioredoxin and its role in toxicology. Toxicol. Sci. 78, 3-14. [DOI] [PubMed] [Google Scholar]
- Yoo, H. Y., Chang, M. S., and Rho, H. M. (1999). Induction of the rat Cu/Zn superoxide dismutase gene through the peroxisome proliferator-responsive element by arachidonic acid. Gene 234, 87-91. [DOI] [PubMed] [Google Scholar]
- Zheng, Y., Zhong, L., and Shen, X. (2005). Effect of selenium-supplement on the calcium signaling in human endothelial cells. J. Cell Physiol. 205, 97-106. [DOI] [PubMed] [Google Scholar]
- Zhou, Y. C., and Waxman, D. J. (1999). STAT5b down-regulates peroxisome proliferator-activated receptor alpha transcription by inhibition of ligand-independent activation function region-1 trans-activation domain. J. Biol. Chem. 274, 29874-29882. [DOI] [PubMed] [Google Scholar]