

Abstract
We have previously found that hypoxia stimulates proliferation of vascular fibroblasts through Gαi-mediated activation of ERK1/2. Here, we demonstrate that hypoxia also activates the atypical protein kinase Cζ (PKCζ) isozyme and stimulates the expression of ERK1/2-specific phosphatase, MAP kinase phosphatase-1 (MKP-1), which attenuates ERK1/2-mediated proliferative signals. Replication repressor activity is unique to PKCζ because the blockade of classical and novel PKC isozymes does not affect fibroblast proliferation. PKCζ is phosphorylated upon prolonged (24 h) exposure to hypoxia, whereas ERK1/2, the downstream kinases, are maximally activated in fibroblasts exposed to acute (10 min) hypoxia. However, PKCζ blockade results in persistent ERK1/2 phosphorylation and marked increase in hypoxia-induced replication. Similarly prolonged ERK1/2 phosphorylation and increase in hypoxia-stimulated proliferation are also observed upon blockade of MKP-1 activation. Because of the parallel suppressive actions of PKCζ and MKP-1 on ERK1/2 phosphorylation and proliferation, the role of PKCζ in the regulation of MKP-1 expression was evaluated. PKCζ attenuation reduces MKP-1 expression, whereas PKCζ overexpression increases MKP-1 levels. In conclusion, our results indicate for the first time that hypoxia activates PKCζ, which acts as a terminator of ERK1/2 activation through the regulation of downstream target, MKP-1 expression and thus serves to limit hypoxia-induced proliferation of fibroblasts.
INTRODUCTION
Fibroblast proliferation is associated with various forms of vascular diseases (Sartore et al., 2001), different fibrotic conditions (Atamas, 2002) and cancer (Bhowmick et al., 2004). Hypoxia is the critical contributor to the pathophysiological conditions of these diseases. We have found that cultured vascular adventitial fibroblasts have the distinct capability to proliferate directly in response to hypoxia in the absence of any exogenous growth factors (Das et al., 2001). Intracellular signaling intermediates, e.g., protein kinase C (PKC) and MAP kinase families are the major mediators of hypoxic signal stimulating replication of cells (Das et al., 2000, 2001; Sodhi et al., 2000). However, cellular proliferation is tightly regulated by proper exit from the cell cycle to maintain normal physiological conditions. The molecular pathways that direct attenuation of hypoxia-induced proliferative signals in fibroblasts remain unknown.
PKC, a family of serine/threonine kinases, have been divided into three distinct groups: the conventional: calcium-, phospholipid-, and diacylglycerol-dependent PKC isozymes (cPKCα, βι, βιι, γ); the novel: calcium-independent PKC isozymes (nPKCδ, ε, η, θ); and the atypical PKC isozymes (aPKCζ, ι, λ), which are calcium-, phospholipid-, and diacylglycerol-independent (Nishizuka, 1992; Hug and Sarre, 1993). PKCζ can be activated directly or indirectly by a variety of important signaling molecules including ceramide (Powell et al., 2004), phosphatidic acid (Le Good et al., 1998), phosphoinositide 3-kinase lipid products and activation of the p21Ras pathway (Pal et al., 2001). PKCζ has emerged as a critical regulator of a number of cellular functions including proliferation, differentiation, and apoptosis (Hirai and Chida, 2003). This isozyme mediates proliferation in NIH3T3 cells (Berra et al., 1993; Kim et al., 1997), endothelial cells (Kent et al., 1995), and smooth muscle cells (Yano et al., 1999). In contrast, cytokine- and ceramide-induced activation of PKCζ leads to inhibition of proliferation and growth arrest in vascular smooth muscle cells, respectively (Bourbon et al., 2002; Hussain et al., 2002). Therefore, the biological functions of PKCζ in cellular responses are cell-type and stimulus specific. The mechanisms responsible for diverse physiological functions of PKCζ at the cellular level are not known.
A recent report has demonstrated that phosphorylation of the Na,K-ATPase α1 subunit in lung alveolar epithelial cells under hypoxic conditions is mediated through PKCζ (Dada et al., 2003). Datta et al. (2004) have found that PKCζ participates in the activation of hypoxia-inducible factor-1α (HIF-1α) by inhibiting the expression of asparagine hydroxylase (enzyme inhibitor of HIF-1), thereby promoting the transcription of hypoxia-inducible genes such as vascular permeability factor and vascular endothelial growth factor. Despite the importance of PKCζ in cellular signaling under hypoxic conditions, it is unknown whether PKCζ is a proliferative stimulator or suppressor in fibroblasts under hypoxic conditions.
Another set of protein kinases that plays an important role in transducing signal from intracellular PKC isozymes to the cell nucleus is MAP kinase family (Kim et al., 1997; Corbit et al., 2000; Mas et al., 2003). Previously, we have demonstrated that hypoxia induces transient activation of ERK1/2, one member of the MAP kinase family, and that ERK1/2 activation mediates replication of hypoxic fibroblasts (Das et al., 2001). PKCζ acts as an upstream regulator of ERK1/2 activation in response to various stimuli in different cell types (Hirai and Chida, 2003). However, the functional role of PKCζ in the regulation of hypoxia-induced activation of ERK1/2 in fibroblasts is not known.
Once activated, ERK1/2 can be rapidly inactivated through dephosphorylation by phosphatases known as dual specificity MAP kinase phosphatases (MKPs; Keyse and Emslie, 1992). The existence of at least eleven MKPs in mammals implies a considerable complexity in the regulation of MAP kinase signaling by these enzymes. Among these phosphatases, MKP-1 is encoded by an immediate early gene (Noguchi et al., 1993). Though MKP-1 is identified as a hypoxia-responsive gene (Laderoute et al., 1999; Seta et al., 2001; Liu et al., 2003), the role of this phosphatase in cellular responses under hypoxic conditions, is poorly understood. It is important to understand the mechanisms regulating MKP expression because the physiological functions of MKPs are largely determined by their expression patterns. Multiple pathways, e.g., ERK1/2, c-Jun N-terminal kinase (JNK), p38 MAP kinase and Ca2+-dependent pathways regulate MKP-1 expression (Reffas and Schlegel, 2000; Slack et al., 2001). PKC is also implicated as an important regulator in the expression of these phosphatases in various cell types (Stawowy et al., 2003). However, the signaling mechanisms that control hypoxia-stimulated MKP-1 expression in fibroblasts remain unknown.
The current study was undertaken to determine whether PKCζ activation and MKP-1 expression are required for the attenuation of ERK1/2 activation and proliferation in response to hypoxia in cultured vascular fibroblasts and whether PKCζ plays a role in the expression of MKP-1. Collectively, we hypothesized that PKCζ-induced up-regulation of MKP-1 mediates replication repressor activity of PKCζ to suppress ERK1/2 activation in fibroblasts upon hypoxic exposure.
MATERIALS AND METHODS
Materials
Eagle's MEM, bromodeoxyuridine (BrdU), monoclonal antibody (mAb) against β-tubulin and protease inhibitor cocktail were purchased from Sigma-Aldrich Chemical Company (St. Louis, MO). mAb against BrdU was obtained from Becton Dickinson Immunocytometry Systems (San Jose, CA). Fetal bovine serum (FBS) and porcine serum were from Gemini Bio-Products (Woodland, CA). Other antibodies were purchased from the following companies: polyclonal and monoclonal antibodies against PKCζ and PKCι from Santa Cruz Biotechnology (Santa Cruz, CA), biotinylated anti-mouse IgG, streptavidin-conjugated Alexa 488- and Alexa 594-conjugated anti-rabbit IgG from Molecular Probes (Eugene, OR), phosphoERK1/2 and phosphoPKCζ/λ (Thr410) from Cell Signaling (Beverly, MA). Polyclonal antibody against MKP-1, goat anti-rabbit and goat anti-mouse IgG conjugated with alkaline phosphatase were obtained from Santa Cruz and Upstate Biotechnology (Waltham, MA). Mounting medium containing 4′,6-diamino-2-phenylindole dihydrochloride (DAPI) was obtained from Vector Laboratories (Burlingame, CA). Myristoylated pseudosubstrate peptide inhibitors of PKCζ and PKC classical and novel isozymes (PKC inhibitor) were purchased from Biomol (Plymouth, PA). Sodium vanadate was obtained from Fisher Scientific (Pittsburgh, PA). Additionally, PKCζ antisense phosphorothioate oligonucleotides were obtained from Integrated DNA Technologies (Skokie, IL) and Oligos Etc. (Wilsonville, OR). The rabbit PKCζ sequences used are as follows: rabbit PKCζ antisense (5′-GCTGCGCCGGCCTCACACG-3′); PKCζ scrambled antisense (5′-ACCCCGTCGGCCCATGGCG-3′). Plasmids containing phosphatase inactive mutant (Cys258 to Ser, pSG5-MKP-1-CS) of MKP-1 (MKP-1PI) was kindly provided by Dr. N. K. Tonks (Cold Spring Harbor Laboratory, Cold Spring Harbor, NY). Vector (pCMV6) containing constitutively active PKCζ (MyrPKCζ) was a gift of Dr. A. Toker (Harvard Medical School, Boston, MA).
Cell Culture
Main pulmonary artery adventitia was harvested from the 15-d-old neonatal control calves, carefully dissected free of blood vessels and fat under a dissecting microscope and then cut into small pieces. Fibroblasts from the tissue were isolated, characterized, and maintained according to previously described methods (Das et al., 2002). Cells were maintained in MEM, pH 7.4, supplemented with 10% FBS, 100 U/ml penicillin, 0.1 mg/ml streptomycin, l-glutamine and incubated in a humidified atmosphere with 5% CO2 at 37°C. Medium was changed twice per week and cells were harvested with trypsin (0.2 g/L) containing EDTA (0.5 g/L). Passages ranging from 3 to 12 were used for all experiments. The growth characteristics and the appearance of the cells, examined by light microscopy, did not change for the passages studied.
Cell Proliferation Assay
Cells were plated at a density of 20,000/well in a 24-well plate in 10% FBS-containing medium and growth arrested for 72 h according to the previously described method (Das et al., 2001). Quiescent cells were transiently transfected with Optimem medium (Invitrogen, Carlsbad, CA) by combining 2 μg/ml of lipofectin (Invitrogen) with DNA for 5-6 h at 37°C. We used MyrPKCζ, MKP-1PI, PKCζ antisense and scrambled oligonucleotides for different transfection experiments. The cells were allowed to recover from transfection either overnight (PKCζ antisense experiments) or 48 h (myrPKCζ and MKP-1PI experiments) and then exposed to either normoxia (21% O2) or hypoxia (1% O2) in the presence of BrdU (10 μM) for 24 h. For the hypoxic experiments, cells were placed either in sealed humidified gas chambers as previously described (Das et al., 2002) or Bactron X (Sheldon Manufacturing, Cornelius, OR) environmental hood. At the end of the treatment, cells were fixed with cold 0.3% H2O2 in methanol for 15 min, treated with 2 N HCl for 20 min, and then incubated with anti-BrdU antibody. VectaStain DAB kit (Vector Laboratories, Burlingame, CA) was used to visualize nuclei. The BrdU-positive and -negative nuclei were counted in five randomly chosen areas in each well and reported as a percent of control.
For the studies with PKCζ inhibitor and the inhibitor of PKC classical and novel isozymes, quiescent cells were preincubated with the pseudosubstrate peptide inhibitors for 1 h at 37°C and then exposed to either normoxia or hypoxia for 24 h in the presence of BrdU. Cells were processed for BrdU immunostaining as described above.
PKCζ Activation Assay
Cells were plated at a density of ∼0.5-1 × 106 cells/100-mm Petri dish in medium containing 10% FBS, and growth was arrested for 72 h. Quiescent cells were exposed to hypoxia (1% O2) for either 10 min or 24 h. Calyculin A (100 nM) was added to the cells to block the serine/threonine phosphatases. Fibroblasts were exposed to calyculin A for 10 min. For 24-h hypoxic exposure, cells were treated with calyculin A for last 10 min of the experimental period. At the end of the exposure, cells were harvested with lysis buffer (Cell Signaling) containing protease inhibitor cocktail (1:100), 0.1% SDS, and 1% sodium deoxycholate, freeze-thawed to disrupt cell membranes, and centrifuged at 10,000 × g. Lysates were collected and protein concentrations were estimated using the Bradford method (BIORAD, Hercules, CA). To evaluate PKCζ activation, 500-700 μg protein was incubated with anti-PKCζ antibody (1:200, monoclonal) overnight at 4°C and then with AG agarose beads (Santa Cruz Biotechnology and Upstate Biotechnology) for 2 h at 4°C with agitation. The antigen-antibody complex was isolated by centrifugation at 10,000 × g for 1 min. The supernatant was discarded and the beads were washed multiple times with lysis buffer. The recovered protein was separated by gel electrophoresis, transferred to polyvinylidene difluoride membranes (PVDF; Amersham Pharmacia Biotech, Piscataway, NJ), and probed with an antibody against phosphoThr-PKCζ (1:1000). Immunoreactivity was detected on x-ray film using chemiluminescent reagents (Amersham Pharmacia Biotech). Immunoprecipitation followed by immunoblotting using the anti-PKCζ antibody (1:1000) served as a control to monitor any changes in total PKCζ protein levels. Because the mAb against PKCζ may cross-react with PKCι/λ isozyme, immunoprecipitates were also immunoblotted against PKCι using PKCι-specific antibody (1:1000). Images of Western blots were scanned using EPSON TWAIN software with EPSON PERFECTION 2450 PHOTO scanner. Densitometric quantitation of the scanned bands was performed using the National Institutes of Health (NIH) Image 1.58 program. The area under the curve (AUC) for each band was determined and represents the band intensity in arbitrary units. The AUC at 0-h time was considered as 100% and percent increases in hypoxic fibroblasts were calculated with respect to 0 h.
Immunoblotting
For the examination of PKCζ and PKCι expression in the presence of either MyrPKCζ or PKCζ-specific antisense oligonucleotides, vascular fibroblasts were plated at the density of 50,000-100,000 cells/well in a six-well plate in 10% FBS-containing medium and growth was arrested for 72 h. Quiescent cells were transiently transfected with the corresponding DNA according to the method described above for BrdU incorporation. Transfected cells were exposed to either normoxia or hypoxia for 24 h. Cells were harvested and processed for the evaluation of PKCζ and PKCι expression at the end of the hypoxic exposure.
For the evaluation of ERK1/2 activation, cells were plated and growth-arrested according to the abovementioned method. In one set of experiments, the cells were transiently transfected with PKCζ scrambled and antisense oligonucleotides and exposed to either normoxia or hypoxia for 10 min and 24 h. In the other set of experiments, the cells were pretreated with PKCζ pseudosubstrate peptide inhibitor for 1 h and then exposed to either normoxia or hypoxia for 24 h. In both instances, the cells were harvested with lysis buffer containing protease inhibitor/deoxycholate/SDS and freeze-thawed, and lysates were recovered by centrifugation at 10,000 × g. Proteins were separated by gel electrophoresis, transferred to a PVDF membrane, and probed with anti-phosphoERK1/2 antibody (1:1000).
MKP-1 expression was also evaluated by immunoblotting techniques. Antibody against MKP-1 was used at a concentration of 1:200-1:1000. The time course of MKP-1 expression was determined at 0, 24, 48, and 72 h of hypoxic exposure. β-Tubulin expression, used as an internal control, was detected by incubating the PVDF membrane with a mAb against β-tubulin (1:1000).
Immunofluorescence Staining
Fibroblasts were plated in medium containing 10% FBS at a density of ∼20,000/well in eight-well glass chamber slides (Life Sciences, Denver, CO). After allowing to attach overnight, cells were growth-arrested with medium containing 0.1% FBS for 72 h. Quiescent cells were then exposed to either normoxia or hypoxia for 24 h. After treatment, cells were fixed with 4% paraformaldehyde at 4°C, permeabilized with 0.5% Triton X-100, blocked with porcine serum, and incubated with either an anti-PKCζ antibody (1:25 dilution) or anti-MKP-1 antibody (1:50 dilution). Immunofluorescent staining of PKCζ was completed by incubating cells with biotin-conjugated anti-mouse IgG (1:300) and streptavidin-conjugated Alexa 488 (1:2000). For MKP-1, antigen-antibody complexes were visualized by incubating the cells with Alexa 594-conjugated anti-rabbit secondary antibody (1:500). Nuclei were stained with Hoechst dye (1:1000). Slides were mounted with H1000 mounting medium. All steps were done at room temperature unless otherwise stated. Images were captured using an Olympus Infinity microscope (Melville, NY) coupled to a Photometrics Quantix cooled CCD camera (Tucson, AZ) and Deltavision digital deconvolution software.
Data Analysis
All data are expressed as arithmetic means ± SE; n equals the number of replicate wells per test condition in representative experiments. One- and two-way analyses of variance, followed by the Student-Newman-Keuls multiple-comparisons tests, were conducted within and between groups of data points. Data are considered significantly different if p ≤ 0.05.
RESULTS
PKCζ Attenuation Greatly Up-regulates Hypoxia-induced Proliferation of Fibroblasts
We previously demonstrated that hypoxia stimulates an increase in DNA synthesis, which results in cell division in vascular fibroblasts (Das et al., 2001). PKC is an important intracellular regulator of hypoxia-induced proliferation of fibroblasts (Das et al., 2000). To evaluate the role of atypical PKCζ isozyme in these proliferative responses, we first used the isozyme-specific myristoylated peptide inhibitor derived from the pseudosubstrate region. The peptide inhibitor mimics the substrate and maintains PKCζ in its inactive form (Hirai and Chida, 2003). Figure 1 depicts concentration-dependent effects of peptide inhibitor on the proliferative responses of fibroblasts. The inhibitor at low dose (1 μM) did not affect the DNA synthesis (Figure 1). However, BrdU incorporation in the quiescent cells was significantly increased in the presence of 10 μM inhibitor (Figure 1A). Hypoxia alone stimulated a twofold increase in DNA synthesis, whereas BrdU incorporation in hypoxia-exposed fibroblasts which were treated with 10 μM inhibitor was up-regulated by sevenfold (Figure 1A).
Figure 1.

Myristoylated PKCζ pseudosubstrate peptide inhibitor stimulates DNA synthesis in vascular fibroblasts. (A) BrdU incorporation in hypoxic fibroblasts is greatly augmented by PKCζ inhibitor. For all the experiments where BrdU incorporation was evaluated, cells were plated at the density of 20 × 103/well in 24-well plates and growth-arrested with 0.1% FBS/MEM for 72 h. Quiescent fibroblasts were preincubated with different concentrations of PKCζ inhibitor at 37°C for 1 h and then exposed to either normoxia (21% O2) or hypoxia (1% O2) in the presence of BrdU (10 μM) for 24 h. At the end of the treatment, cells were fixed with cold 0.3% H2O2/methanol and processed for immunoperoxidase staining with anti-BrdU antibody. BrdU-positive and total nuclei were counted at five different randomly selected areas in individual well. n = 4 replicate wells. * p < 0.05 compared with normoxic control data. ** p < 0.05 compared with the hypoxic data. Similar results were obtained in three independent experiments with cell populations isolated from three independent animals. (B) PKCζ peptide inhibitor also up-regulates bFGF-stimulated DNA synthesis. Quiescent fibroblasts were treated with PKCζ inhibitor as mentioned above and then stimulated with bFGF (30 ng/ml) for 24 h in the presence of BrdU. n = 4 replicate wells. * p < 0.05 compared with the unstimulated control value. ** p < 0.05 compared with bFGF-stimulated data. Similar results were reproduced with at least two other cell populations.
The role of PKCζ in hypoxia-induced proliferation was compared with that of basic FGF (bFGF) stimulation, a wellknown fibroblast mitogen (Figure 1B). bFGF-induced DNA synthesis in the presence of the PKCζ inhibitor (10 μM) was again significantly greater than that induced by bFGF or inhibitor alone (Figure 1B).
To confirm the selectivity of PKCζ inhibitor-mediated stimulatory effects on BrdU incorporation, we also used a myristoylated pseudosubstrate peptide inhibitor, which was designed against classical and novel isozymes of PKC (PKC inhibitor). Neither hypoxia-induced nor bFGF-stimulated DNA synthesis was affected by PKC inhibitor (Figure 2, A and B). In contrast, and as shown independently above (Figure 1, A and B), PKCζ inhibitor selectively augmented hypoxia-stimulated and bFGF-induced BrdU incorporation (Figure 2, A and B). However, one can interpret, by contrasting hypoxia-induced and bFGF-stimulated BrdU incorporation in the presence of PKCζ inhibitor, that PKCζ attenuation unmasked proliferative responses in greater extent in hypoxic cells (Figure 1, 3-fold, and Figure 2, 7.6-fold, over hypoxia-induced DNA synthesis) than bFGF-stimulated fibroblasts (Figure 1, 1.3-fold, and Figure 2, 4-fold, compared with bFGF-stimulated proliferation). Therefore, functional role of PKCζ might be more critical during hypoxic exposure than bFGF-stimulated fibroblasts. Taken together, our data suggest that replicative up-regulation by myristoylated PKCζ pseudosubstrate peptide inhibitor is specific to this peptide and that PKCζ might act as a proliferation suppressing kinase in fibroblasts.
Figure 2.

PKCζ inhibitor, but not PKC inhibitor of classical and novel isozymes, stimulates proliferation of vascular fibroblasts. (A) Hypoxia-induced DNA synthesis is selectively up-regulated by PKCζ inhibitor. Quiescent fibroblasts were preincubated with peptide inhibitors of PKC classical and novel isozymes (10 μM) and PKCζ isozyme (10 μM) at 37°C for 1 h. BrdU incorporation in both normoxic and hypoxic fibroblasts was evaluated. n = 4 replicate wells. * p < 0.05 compared with normoxic control results. ** p < 0.05 compared with hypoxic data. (B) PKC inhibitor of classical and novel isozymes does not affect bFGF-stimulated DNA synthesis. Cells were preincubated with inhibitors as mentioned above and then stimulated with bFGF (30 ng/ml) in the presence of BrdU (10 μM) for 24 h. n = 4 replicate wells. * p < 0.05 compared with nonstimulated control results. ** p < 0.05 compared with bFGF-induced data. Similar results were obtained from three independent experiments using fibroblasts isolated from three different animals.
To confirm the role of PKCζ as a replication repressor, we then used antisense oligonucleotides specific for the message encoding PKCζ. Transient transfection of the cells with PKCζ antisense oligonucleotides selectively reduced the PKCζ expression, but not the PKCι levels (Figure 3, A and B). For detection of the PKCι level in our cells by immunoblotting techniques, we used 4-5 times higher protein concentration than that used for the evaluation of the PKCζ levels. In addition, x-ray film was exposed up to 30 min for the detection of the PKCι signal. Collectively, these observations suggest that a very low level of PKCι is present in vascular fibroblasts. A recent report (Cogolludo et al., 2005) also supports the notion that PKCζ, but not PKCι, is the major atypical isozyme of pulmonary vasculature.
Figure 3.
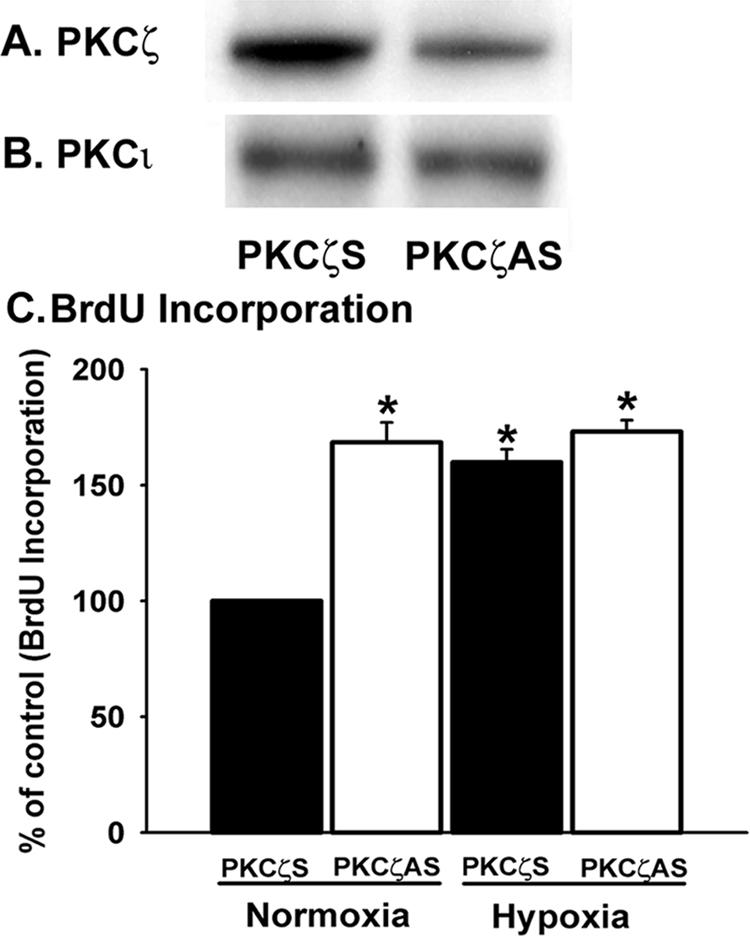
PKCζ attenuation with antisense oligonucleotides stimulates BrdU incorporation in vascular fibroblasts. (A) PKCζ expression is blocked by antisense oligonucleotides in fibroblasts. For all the transfection experiments, cells were plated at a density of 50-100 × 103/well in six-well plates in 10% FBS/MEM and growth-arrested with 0.1% FBS/MEM for 72 h. Quiescent cells were transiently transfected with PKCζ scramble and antisense oligonucleotides. After overnight recovery from transfection, cells were harvested with the lysis buffer. Whole cell lysates were processed for immunoblot analysis with anti-PKCζ mAb. (B) PKCζ antisense oligonucleotides have no effects on PKCι expression. The abovementioned cell lysates were processed for Western blot analysis for PKCι detection using mAb against PKCι. (C) BrdU incorporation in fibroblasts is greatly up-regulated by PKCζ antisense oligonucleotides. Quiescent cells were transiently transfected with PKCζ scramble and antisense oligonucleotides. Transfected cells were incubated at 37°C for overnight. Then BrdU (10 μM) was added to the cells and exposed to either normoxia or hypoxia for 24 h at 37°C. n = 4 replicate wells. * p < 0.05 compared with the data in the presence of scramble oligonucleotides under normoxic conditions. Similar results were reproduced in at least two different cell populations isolated from two different animals.
Proliferative responses of fibroblasts in the presence of PKCζ antisense oligonucleotides were then evaluated by measuring the BrdU incorporation. Marked increase in DNA synthesis was observed upon attenuation of PKCζ expression with antisense oligonucleotides in normoxic fibroblasts (Figure 3C). Hypoxia stimulated BrdU incorporation in fibroblasts both in the presence of scrambled and PKCζ-specific antisense oligonucleotides (Figure 3C). Equal magnitude in the up-regulation of DNA synthesis upon blockade of PKCζ expression with antisense oligonucleotides under both normoxic and hypoxic conditions suggests that proliferative responses are primarily regulated by PKCζ-mediated signaling pathways in fibroblasts in response to hypoxic exposure. Collectively, these results further support the role of PKCζ as a proliferative repressor for vascular fibroblasts.
PKCζ Overexpression Attenuates Proliferation of Hypoxic Fibroblasts
The myristoylated pseudosubstrate peptide inhibitor can block the activation of both PKCζ and PKCι because sequence homology exists in the pseudosubstrate region between these two atypical isozymes (Toker, 1998). Therefore, to confirm that the proliferation stimulatory effects of the inhibitor (Figures 1 and 2) are due to PKCζ inhibition and not attenuation of PKCι activity, PKCζ was overexpressed in fibroblasts by transient transfection with constitutively active PKCζ (MyrPKCζ). Increased expression of PKCζ, but not PKCι, in the presence of MyrPKCζ was confirmed by Western blot analysis (Figure 4, A and B).
Figure 4.
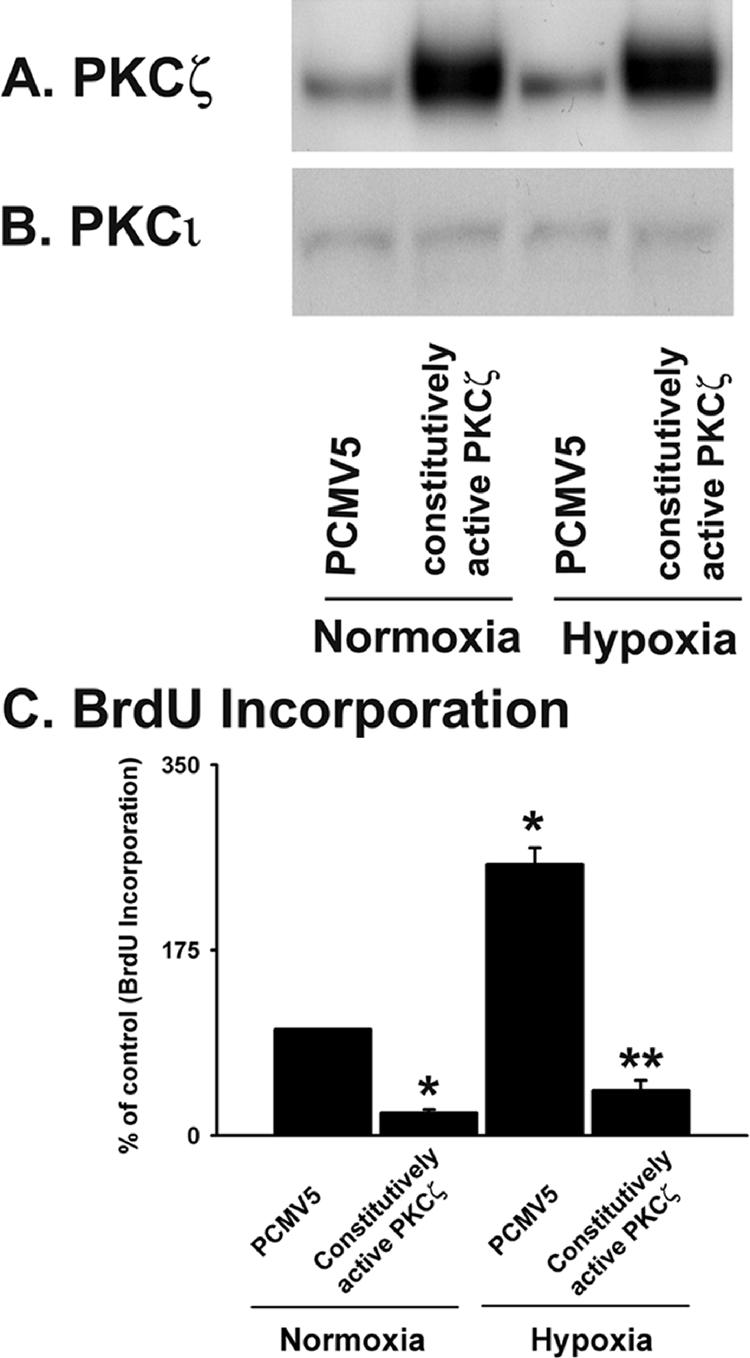
PKCζ overexpression attenuates DNA synthesis in vascular fibroblasts. (A) PKCζ expression is augmented in fibroblasts transfected with constitutively active PKCζ (MyrPKCζ). Quiescent cells were transiently transfected with vector containing MyrPKCζ and empty vector (PCMV5). After 48 h of transfection, cells were exposed to either normoxia or hypoxia for 24 h and harvested with lysis buffer. Cell lysates were processed for PKCζ detection by immunoblot analysis. (B) MyrPKCζ does not affect PKCι expression in fibroblasts. In the abovementioned cell lysates, PKCι expression was evaluated by Western blot analysis using PKCι-specific mAb. (C) BrdU incorporation in hypoxic fibroblasts is attenuated by MyrPKCζ. Transfected fibroblasts were exposed to either normoxia or hypoxia in the presence of BrdU for 24 h. n = 4 replicate wells. * p < 0.05 compared with PCMV5-transfected normoxic results. ** p < 0.05 compared with the results of PCMV5-transfected hypoxic cells. Similar results were obtained from three independent experiments using fibroblasts isolated from three different animals.
We then evaluated BrdU incorporation in the presence of MyrPKCζ. Hypoxia induced a 2.5-fold up-regulation of BrdU incorporation in vector-transfected cells (Figure 4C). However, marked down-regulation (6-fold) in DNA synthesis was observed in PKCζ-overexpressing cells compared with the vector-transfected fibroblasts under hypoxic conditions (Figure 4C). These data strongly suggest that PKCζ acts as a proliferative repressor in fibroblasts under hypoxic conditions.
Hypoxia Up-regulates PKCζ Phosphorylation
PKCζ phosphorylation at the Thr410 residue in the activation loop is required for its activity in response to a stimulus (Hirai and Chida, 2003). To evaluate hypoxia-induced PKCζ phosphorylation at the Thr410 site, PKCζ was immunoprecipitated from the lysates of control and hypoxia-exposed fibroblasts and immunoblotted with an anti-phosphoThr410 antibody. PKCζ phosphorylation levels were significantly higher in prolonged (24 h) hypoxia-exposed cells compared with that of the control fibroblasts (Figure 5A). Equal amounts of PKCζ precipitation from all the lysates were confirmed by immunoblotting the immunoprecipitates against PKCζ (Figure 5B). Quantitative data of the hypoxia-induced increase in PKCζ phosphorylation are presented in Figure 5C.
Figure 5.

Hypoxia induces PKCζ phosphorylation in vascular fibroblasts. (A and B) Phosphorylation of the Thr410 residue of PKCζ is increased by hypoxic exposure. Growth-arrested cells were exposed to hypoxia for 10 min and 24 h. Whole cell lysates (0.5-0.7 mg total protein) were immunoprecipitated with monoclonal anti-PKCζ antibody and the immunocomplexes were analyzed by immunoblot analysis with antibodies specific for either phosphoPKCζ (A) or total PKCζ (polyclonal; B). Representative immunoblot of three independent experiments is shown. (C) Quantitative data of hypoxia-induced PKCζ phosphorylation in vascular fibroblasts. * p < 0.05 compared with the data of normoxic condition.
We used an mAb directed toward the C-terminus of PKCζ to immunoprecipitate the isozyme. This antibody may cross-react with the other atypical isozyme, i.e., PKCι. To rule out that possibility, PKCζ immunoprecipitates were immunoblotted for PKCι. PKCι signal was not detected in the PKCζ precipitate (our unpublished observation), suggesting the antibody against PKCζ does not cross-react with PKCι in vascular fibroblasts. Therefore, our data suggest that hypoxia stimulates phosphorylation of PKCζ (Thr410) in vascular fibroblasts and is consistent with our previous observation of hypoxia-induced increase in PKCζ-specific activity in fibroblasts (Das et al., 2000).
Hypoxia Stimulates ERK1/2 Activation
PKCζ has been demonstrated to regulate the downstream ERK1/2 activation in response to various stimuli in different cell types (Berra et al., 1993). We, therefore, evaluated the activation of ERK1/2 as a possible downstream target of PKCζ in hypoxic fibroblasts. In contrast to the abovementioned up-regulation of PKCζ phosphorylation in fibroblasts exposed to prolonged hypoxia (Figure 5A), increase in ERK1/2 phosphorylation was evident in both acute (10 min) and prolonged (24 h) hypoxia-exposed fibroblasts. Greatest magnitude of hypoxia-stimulated ERK1/2 phosphorylation was observed in acute (10 min) hypoxia-exposed fibroblasts (Figure 6A), which is consistent with our previous observations (Das et al., 2001). In that report, we have also demonstrated that the activation of ERK1/2 mediates hypoxia-induced proliferation of fibroblasts. With prolonged (24 h) hypoxic exposure, the increase in phosphorylated ERK1/2 was significantly reduced compared with that of acute (10 min) hypoxic exposure (Figure 6C). Western blot analysis of these lysates for total ERK1/2 confirmed equal protein loading among all samples (Figure 6B). Quantitative data of hypoxia-induced increase in phosphoERK1/2 levels are presented in Figure 6C. These results suggest that hypoxia stimulates ERK1/2 phosphorylation in vascular fibroblasts.
Figure 6.

Hypoxia stimulates ERK1/2 phosphorylation in vascular fibroblasts. (A and B) ERK1/2 phosphorylation is greatest in acute (10 min) hypoxia-exposed fibroblasts. Quiescent cells were exposed to hypoxia for 10 min and 24 h and harvested with lysis buffer. Whole cell lysates were processed for immunoblot analysis. PhosphoERK1/2 (A) and total ERK1/2 (B) were evaluated using phosphoERK1/2- and total ERK1/2-specific antibodies. Representative blots of four independent experiments are shown here. (C) Quantitative data of hypoxia-stimulated ERK1/2 phosphorylation in vascular fibroblasts. * p < 0.001 compared with normoxic results. ** p < 0.001 compared with data of normoxic and acute (10 min) hypoxia-exposed conditions.
PKCζ Inhibition Up-regulates ERK1/2 Phosphorylation
To evaluate a possible role of PKCζ in hypoxia-induced ERK1/2 activation, ERK1/2 phosphorylation was examined upon blockade of PKCζ expression. First, attenuation of PKCζ level with antisense oligonucleotides was confirmed by Western blot analysis (Figure 7A). Acute (10 min) hypoxia-induced ERK1/2 phosphorylation was observed in the presence of scrambled oligonucleotides (Figure 7B). To our surprise, ERK1/2 were also significantly phosphorylated in both normoxic and hypoxic fibroblasts upon down-regulation of PKCζ expression with antisense oligonucleotides, implicating that PKCζ might be a suppressor of hypoxia-induced ERK1/2 phosphorylation (Figure 7B). The quantitative data of acute hypoxia-induced ERK1/2 activation in the presence of PKCζ antisense and scrambled oligonucleotides are presented in Figure 7D.
Figure 7.

PKCζ attenuation induces persistent ERK1/2 activation in vascular fibroblasts. (A) Transient transfection of quiescent fibroblasts with PKCζ scramble and antisense oligonucleotides induced selective blockade of PKCζ expression, but did not alter PKCι levels. (B) ERK1/2 phosphorylation is increased by PKCζ antisense oligonucleotides in acute (10 min) hypoxia-exposed cells. Transfected cells were exposed to either normoxia or hypoxia for 10 min. Total protein was extracted with lysis buffer. Western blots were performed on the extracts using an anti-phosphoERK1/2 antibody and anti-ERK1/2 antibody to examine the levels of phosphorylated and total ERK1/2. Similar results were obtained from three independent experiments. Representative immunoblots of phosphoERK1/2 and total ERK1/2. (C) In prolonged (24 h) hypoxia-exposed fibroblasts, PKCζ blockade results in persistent ERK1/2 phosphorylation. Transfected fibroblasts were exposed to either normoxia or hypoxia for 24 h. At the end of the experimental period, whole cell lysates were processed for immunoblot analysis with anti-phosphoERK1/2 and total ERK1/2 antibodies. (D) Quantitative measurement of acute (10 min) hypoxia-induced ERK1/2 phosphorylation in the presence of PKCζ scramble and antisense oligonucleotides. * p < 0.05 compared with the results of normoxic cells transfected with scramble oligonucleotides. (E) Quantitative measurement of ERK1/2 phosphorylation upon PKCζ attenuation with antisense oligonucleotides in prolonged (24 h) hypoxia-stimulated fibroblasts. * p < 0.05 compared with data of the normoxic cells that were transfected with scramble oligonucleotides. ** p < 0.05 compared with the results of hypoxic fibroblasts transfected with scramble oligonucleotides.
With prolonged (24 h) hypoxic exposure, phosphoERK1/2 levels were significantly decreased compared with the acute (10 min) hypoxia-exposed fibroblasts in the presence of scrambled oligonucleotides (Figure 7C), which is consistent with the time course of hypoxia-induced ERK1/2 activation data (Figure 6). However, ERK1/2 phosphorylation was markedly up-regulated upon PKCζ blockade with antisense oligonucleotides in both normoxic and hypoxic fibroblasts (Figure 7, C and E). Persistent ERK1/2 phosphorylation upon PKCζ attenuation suggests that PKCζ is the terminator of hypoxia-induced ERK1/2 activation in fibroblasts.
The role of PKCζ on ERK1/2 dephosphorylation was further investigated using myristoylated PKCζ pseudosubstrate peptide inhibitor. Significant up-regulation of ERK1/2 phosphorylation has occurred in the presence of PKCζ inhibitor (Figure 8A). We then used PKCζ inhibitor to evaluate phopshoERK1/2 localization in response to hypoxic stimulation. We were not able to detect any signal for activated ERK1/2 in quiescent fibroblasts by immunofluorescent staining (Figure 8C). In cells exposed to prolonged hypoxia (24 h), phosphoERK1/2 was expressed as distinct spots outside the nucleus (Figure 8C). However, in the presence of the PKCζ inhibitor, the intensity of phosphoERK1/2 immunofluorescent staining was strikingly enhanced (Figure 8C). Also, phosphoERK1/2 was compartmentalized in the nucleus as well as in the cytoplasm of hypoxic fibroblasts upon PKCζ inhibition (Figure 8C). PhosphoERK1/2 must be inside the nuclear compartment to initiate cellular proliferative responses (Pouyssegur et al., 2002). Therefore, our data suggest that PKCζ inhibition initiates exuberant replication of hypoxic fibroblasts (Figures 1 and 2) by permitting persistent nuclear localization of phosphoERK1/2 in hypoxic cells. Taken together the data, we conclude that PKCζ is the regulatory switch of ERK1/2 dephosphorylation status in fibroblasts in response to hypoxic exposure.
Figure 8.

PKCζ pseudosubstrate peptide inhibitor stimulates prolonged ERK1/2 phosphorylation in the nuclear compartment of vascular fibroblasts. (A) Western blot analysis of phosphoERK1/2 in the presence of PKCζ inhibitor. Quiescent fibroblasts were preincubated with PKCζ inhibitor (10 μM) for 1 h at 37°C, exposed to either normoxia or hypoxia for 24 h, and then harvested with lysis buffer. Whole cell lysates were separated by Western blot analysis and probed with anti-phosphoERK1/2 and total ERK1/2 antibodies. Similar results were obtained in three different experiments. Cells used for the three experiments were isolated from three different animals. A representative blot of phosphoERK1/2 is shown. (B) Representative blot of total ERK1/2. (C) PKCζ inhibition induces nuclear localization of activated ERK1/2 in fibroblasts. Representative photographs of phosphoERK1/2 compartmentalization in hypoxic fibroblasts. Cells were plated at the density of 20 × 103/well/0.5 ml of 10% FBS/MEM in eight-well glass chamber slides, allowed to attach overnight, and then growth-arrested with 0.1% FBS/MEM for 72 h. Fibroblasts were incubated with PKCζ inhibitor and then exposed to either normoxia or hypoxia according to the abovementioned method. At the end of the treatment, cells were fixed with cold 4% paraformaldehyde and indirect immunofluorescent staining of phosphoERK1/2 was performed. Nuclei were stained with Hoechst dye. Magnification, ×100. Similar results were obtained in two other experiments using different cell populations.
MKP-1 Regulates ERK1/2 Dephosphorylation
A reduction in ERK1/2 phosphorylation can be achieved by up-regulation of protein phosphatases, which dephosphorylate activated ERK1/2 (Keyse and Emslie, 1992). To explore the role of phosphatases in ERK1/2 dephosphorylation in these primary fibroblasts, phosphoERK1/2 levels were evaluated in the presence of sodium vanadate, an antagonist of phosphotyrosine phosphatases. There was marked increase in phosophorylated ERK1/2 levels (Figure 9, A and B) in fibroblasts, implicating the role of tyrosine phosphatases in ERK1/2 dephosphorylation events.
Figure 9.

MKP-1 regulates ERK1/2 dephosphorylation in vascular fibroblasts. (A and B) Significant up-regulation in phosphoERK1/2 levels is observed in the presence of sodium vanadate, an antagonist of tyrosine phosphatases. Growth-arrested cells were preincubated with sodium vanadate (200 μM) for 24 h. Whole cell lysates were separated by Western blot analysis and probed with anti-phosphoERK1/2 and total ERK1/2 antibodies. Similar results were obtained in another independent experiment using different cell populations. (C and D) MKP-1 is overexpressed in fibroblasts transfected with phosphatase inactive MKP-1 (MKP-1PI). Quiescent fibroblasts were transiently transfected with MKP-1PI or empty vector. Cells were harvested with lysis buffer after 48 h of transfection. Whole cell lysates were separated by immunoblot analysis for the evaluation of MKP-1 and tubulin expression. (E and F) PhosphoERK1/2 levels are greatly higher in the cells transfected with MKP-1PI than in those transfected with empty vector. MKP-1PI- and empty vector-transfected cells were exposed to either normoxia or hypoxia (10 and 60 min). Proteins from the cell lysates were separated by Western blot analysis and probed with anti-phosphoERK1/2 and total ERK1/2 antibodies. Similar results were obtained in three different experiments using three different cell populations.
Among potential candidates for such phosphatases, MKP-1 was chosen for investigation because this hypoxia-responsive phosphatase dephosphorylates ERK1/2 in other cell types (Keyse and Emslie, 1992). We utilized phosphatase inactive MKP-1 (MKP-1PI), which acts as a dominant negative MKP-1, and examined the effects of MKP-1 blockade on ERK1/2 phosphorylation. Overexpression of MKP-1 in cells transfected with MKP-1PI was confirmed by Western blot analysis (Figure 9C). Persistent phosphorylation of ERK1/2 was observed in both normoxic and hypoxic fibroblasts upon expression of MKP-1PI (Figure 9E). However, ERK1/2 phosphorylation in the vector-transfected cells was up-regulated only upon acute (10 min) hypoxic exposure (Figure 9E), which is consistent with the time course of ERK activation in hypoxic cells as demonstrated in Figure 6. Total ERK1/2 levels were not affected by MKP-1PI in fibroblasts (Figure 9F). These results suggest that MKP-1 regulates ERK1/2 dephosphorylation in vascular fibroblasts.
Hypoxia-induced Increase in MKP-1 Levels Represses Fibroblast Proliferation
MKP-1 is a hypoxia-responsive gene (Laderoute et al., 1999; Seta et al., 2001; Liu et al., 2003). To evaluate the effects of hypoxia on MKP-1 expression in vascular fibroblasts, quiescent cells were exposed to hypoxia (1% O2) for different lengths of time and MKP-1 expression was examined by immunoblot analysis. Growth-arrested cells expressed a significant amount of constitutive MKP-1 protein (Figure 10A). After 24 h of hypoxic exposure, MKP-1 levels were greatly increased in the cells (Figure 10A). MKP-1 is an early response tumor suppressor gene and has a rapid turnover (Noguchi et al., 1993). MKP-1 might accumulate over time in both normoxic as well as hypoxic fibroblasts (Figure 10A). In spite of the high levels of MKP-1 under normoxic conditions, hypoxic cells always had greater MKP-1 levels (Figure 10A). Equal protein loading among the samples was verified by immunoblotting with an antibody against tubulin (Figure 10). Therefore, these data suggest that hypoxia up-regulates MKP-1 expression in fibroblasts.
Figure 10.

Hypoxia-induced increase in MKP-1 levels terminates proliferation of hypoxic fibroblasts. (A) Hypoxia up-regulates MKP-1 expression. Quiescent fibroblasts were exposed to either normoxia or hypoxia for 24-72 h and harvested with lysis buffer. Western blot analysis of the cell lysates was performed using anti-MKP-1 and tubulin antibodies. Representative blot of the three independent experiments is shown. (B) Transfection of fibroblasts with MKP-1PI stimulates BrdU incorporation in the cells. Growth-arrested cells were transiently transfected with empty vector and MKP-1PI. After 48 h of transfection, cells were exposed to either normoxia or hypoxia in the presence of BrdU for 24 h. n = 4 replicate wells. * p < 0.05 compared with vector transfected normoxic results. ** p < 0.05 compared with the vector-transfected hypoxic data. Similar results were obtained from three independent experiments using fibroblasts from three different animals.
We then explored the role of MKP-1 in hypoxia-induced proliferative responses using MKP-1PI. Marked up-regulation (5-fold) in BrdU incorporation was observed in the presence of MKP-1PI under normal conditions (Figure 10B). Hypoxia induced DNA synthesis of the vector-transfected cells by twofold (Figure 10B). However, MKP-1PI induced an up-regulation of hypoxia-induced BrdU incorporation in fibroblasts by 5.5-fold (Figure 10B). Similar magnitude in the up-regulation of DNA synthesis between the normoxic and hypoxic fibroblasts by MKP-1PI, suggests that hypoxia-induced proliferation of fibroblasts is primarily regulated through the MKP-1 pathway (Figure 10B). Collectively, these data imply that hypoxia-induced increase in MKP-1 expression represses hypoxia-induced proliferation of fibroblasts.
PKCζ Regulates MKP-1 Expression
The strong parallel between proliferative up-regulation and persistent ERK1/2 phosphorylation with PKCζ attenuation (Figures 1, 2, 7, and 8) and MKP-1 blockade (Figures 9 and 10) suggest that PKCζ might direct ERK1/2 dephosphorylation events by regulating MKP-1. Therefore, the role of PKCζ in MKP-1 expression was first evaluated using PKCζ antisense oligonucleotides. In the presence of scrambled oligonucleotides, MKP-1 levels were up-regulated in hypoxic fibroblasts (Figure 11A). PKCζ blockade with antisense oligonucleotides induced marked reduction in MKP-1 expression (Figure 11A). Preincubation of fibroblasts with myristoylated PKCζ pseudosubstrate peptide inhibitor also resulted in complete blockade of MKP-1 expression under both normoxic and hypoxic conditions (our unpublished observation).
Figure 11.
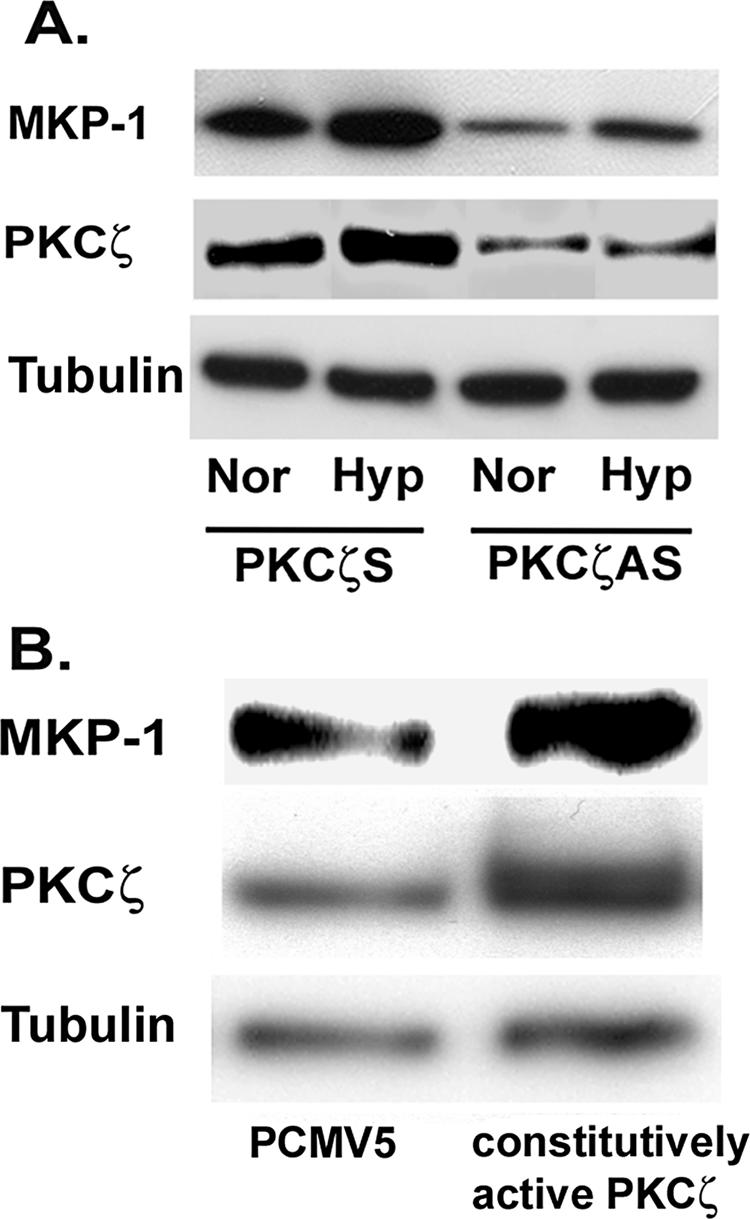
PKCζ regulates MKP-1 expression in vascular fibroblasts. (A) PKCζ blockade attenuates MKP-1 expression. Quiescent fibroblasts were transiently transfected with PKCζ scramble and antisense oligonucleotides. After allowing overnight recovery from transfection, cells were exposed to either normoxia or hypoxia for 24 h. Whole cell lysates were separated by gel electrophoresis and probed with anti-MKP-1, PKCζ, and tubulin antibodies. (B) MyrPKCζ stimulates MKP-1 expression in fibroblasts. Growth-arrested fibroblasts were transiently transfected with PCMV5 and MyrPKCζ. Cells were harvested with lysis buffer after 48 h of transfection. MKP-1, PKCζ and tubulin were detected in the cell lysates by Western blot analysis. Similar results were obtained in three independent experiments using three different fibroblast populations isolated from three different animals.
MKP-1 regulation by PKCζ was further confirmed by using MyrPKCζ. Growth-arrested fibroblasts were transiently transfected with empty vector (PCMV5) and MyrPKCζ. PKCζ overexpression stimulated an increase in MKP-1 levels in fibroblasts (Figure 11B). Collectively, these data suggest that PKCζ regulates MKP-1 expression in vascular fibroblasts.
PKCζ and MKP-1 Colocalize in the Nucleus of Vascular Fibroblasts
To evaluate the localization of PKCζ and MKP-1, both normoxic and hypoxia-exposed (24 h) cells were subjected to double immunofluorescent staining for PKCζ and MKP-1. Under normoxic conditions, punctate staining pattern of both MKP-1 and PKCζ was detected in the nuclear compartment of fibroblasts (Figure 12A). Intensity of the immunoreactivity of both PKCζ and MKP-1 was enhanced in the nuclear compartment of hypoxic fibroblasts (Figure 12B).
Figure 12.

MKP-1 and PKCζ colocalize in the nuclear compartment of vascular fibroblasts. (A) Representative photographs of MKP-1 and PKCζ in normoxic fibroblasts. Quiescent cells were fixed with cold 4% paraformaldehyde and permeabilized with 0.5% Triton X-100. Double immunofluorescent staining was performed with anti-MKP-1 and PKCζ antibodies. Magnification, ×100. (B) Representative photographs of nuclear localization of MKP-1 and PKCζ in hypoxic fibroblasts. Growth-arrested cells were exposed to hypoxia for 24 h. At the end of hypoxic exposure, MKP-1 and PKCζ were visualized by double immunofluorescent staining. (C) Nuclear staining of PKCζ is abolished in the presence of PKCζ peptide and anti-PKCζ antibody. To confirm the specificity of nuclear PKCζ staining, the PKCζ peptide was preincubated with the anti-PKCζ antibody overnight at 4°C. Immunofluorescent staining of PKCζ was performed with this antigen-antibody complex. (D) Myristoylated PKCζ pseudosubstrate peptide inhibitor induces disorganization of defined nuclear localization of MKP-1 in fibroblasts. Growth-arrested fibroblasts were preincubated with PKCζ-specific peptide inhibitor (10 μM) for 1 h at 37°C and then exposed to either normoxia or hypoxia for 24 h. At the end of the treatment, cells were fixed and processed for indirect immunofluorescent staining of MKP-1. Similar results were reproduced in two other fibroblast populations. Magnification, ×100. n = 4 replicate wells. A representative micrograph of the three independent experiments is shown in each case.
Nuclear localization of MKP-1 in vascular fibroblasts is consistent with other reports (Reffas and Schlegel, 2000; Plows et al., 2002; Pouyssegur et al., 2002). However, subcellular localization of PKCζ is cell-type dependent (Fields et al., 1989; Cho and Ziboh, 1995). Specificity of nuclear PKCζ immunofluorescent staining in fibroblasts was therefore confirmed with the peptide against which PKCζ-specific antibody was raised. Anti-PKCζ antibody was preincubated with the peptide and the mixture then was used for PKCζ detection. Punctate staining in the nuclear compartment was completely abolished in the presence of this antigen-antibody complex, (Figure 12C) confirming that PKCζ resides in well-defined foci and colocalizes with MKP-1 in the nucleus of vascular fibroblasts.
Fibroblasts were then preincubated with PKCζ-specific peptide inhibitor and exposed to either normoxia or hypoxia for 24 h. MKP-1 immunofluorescent staining in the nucleus was disrupted upon PKCζ inhibition (Figure 12D). Nuclear effects of myristoylated PKCζ pseudosubstrate peptide inhibitor are neither surprising nor unique for this cell type. Theodore et al. (1995) have reported that myristoylated PKC pseudosubstrate peptide inhibitor is capable of translocating across biological membranes to accumulate in all compartments of neuronal cells. Therefore, our results show that MKP-1 expression is tightly regulated by PKCζ in the nucleus of vascular fibroblasts.
DISCUSSION
Despite our previous studies on signaling pathways that mediate hypoxia-induced proliferation of fibroblasts (Das et al., 2000 and 2001), it remains unclear how replication suppressors, which simultaneously coexist with proliferative stimulators, work collaboratively to attenuate normal proliferation of hypoxic fibroblasts. In the present study, we report for the first time that PKCζ is the master regulator of hypoxia-induced ERK1/2 dephosphorylation events through the regulation of MKP-1 expression and thereby limits proliferation of hypoxic fibroblasts (Figure 13). PKCζ attenuation leads to striking up-regulation in proliferation as well as ERK1/2 phosphorylation in hypoxic fibroblasts. In contrast, PKCζ overexpression induces a significant down-regulation of replication in hypoxic cells. PKCζ regulates hypoxia-induced MKP-1 expression in fibroblasts. MKP-1 blockade mimics the results of PKCζ attenuation on hypoxia-stimulated ERK1/2 phosphorylation and proliferation. These results strongly support the idea that PKCζ acts as a replication repressor through its regulation of MKP-1 expression in hypoxic fibroblasts.
Figure 13.

Schematic diagram: role of PKCζ in the termination of proliferation of hypoxic fibroblasts through the regulation of ERK1/2 dephosphorylation and MKP-1 expression.
Our data in vascular fibroblasts contrast with the majority of published reports where PKCζ is described as a proliferative mediator in a variety of cells (Hirai and Chida, 2003). Recently, Braun and Mochly-Rosen (2003) have demonstrated that PKCζ is required for TGFβ1-induced proliferation of neonatal primary cardiac fibroblasts. PKCζ attenuation also inhibits platelet-derived growth factor (PDGF)-induced proliferation of human airway smooth muscle cells (Carlin et al., 2000). However, a recent report also demonstrates that PKCζ blockade does not inhibit rabbit vascular smooth muscle cell proliferation (Hussain et al., 2002). In fact, PKCζ inhibition increases growth factor- and cytokine-induced proliferation, which supports the notion that PKCζ is an antiproliferative kinase under specific circumstances and is in agreement with our results describing the role of PKCζ as proliferative repressor of hypoxic fibroblasts. Therefore, the role of PKCζ in proliferation is not only cell type specific, but also species specific and hence, the function of this particular atypical isozyme in cellular proliferative responses requires more rigorous characterization.
PKCζ plays an important functional role in mitogenic signaling by initiating the activation of the downstream MAP kinases such as MEK and ERK family proteins (Berra et al., 1993). Activation of the ERK cascade is known to be associated with cellular proliferation (Chang et al., 2003). Previously, we have also reported that proliferation of hypoxic fibroblasts is regulated by hypoxia-induced ERK1/2 activation (Short et al., 2004). However, in the present study, PKCζ attenuation results in persistent ERK1/2 phosphorylation, which contributes to the exuberant proliferation of hypoxic fibroblasts. Another important point is that PKCζ blockade induces accumulation of activated ERK1/2 in the nucleus, which is consistent with the fact that presence of activated ERK1/2 in the nucleus is necessary for the induction of cell proliferation (Pouyssegur et al., 2002). PKCζ-induced termination of ERK phosphorylation observed in the present studies is in contrast to the previously published reports demonstrating that PKCζ is the upstream kinase of MEK and ERK in other cell types (Berra et al., 1993). Also, in spite of the similar increases in proliferation of our cells and rabbit vascular smooth muscle cells (Hussain et al., 2002) upon PKCζ inhibition, the role of PKCζ in ERK1/2 activation is different between the two cell types. In the vascular fibroblasts, PKCζ is the master regulatory switch of ERK1/2 dephosphorylation events, whereas in rabbit smooth muscle cells ERK1/2 activation is independent of PKCζ activation (Hussain et al., 2002). Therefore, the functional role of PKCζ in the regulation of ERK1/2 activation upon a stimulus is a highly cell- and condition-specific event.
Because of the critical importance of ERK1/2 in cellular signaling, the activities of ERKs must be tightly regulated and this can be achieved by ERK-specific phosphatases, e.g., MKP family members. There are at least 11 MKPs in mammals, which imply the existence of a complex regulatory network for MAP kinase signaling. MKPs differ by properties such as tissue-specific expression, differential regulation in response to various stimuli, distinct subcellular localization and substrate specificity. MKP-1 has also been identified as a hypoxia-responsive gene in a variety of cell types (Keyse and Emslie, 1992; Noguchi et al., 1993; Laderoute et al., 1999) and is an immediate-early gene product, characterized by rapid transcriptional induction after MAP kinase family activation, short half-life, and rapid destruction by the 26S proteasome (Keyse and Emslie, 1992; Keyse, 1995). MKP-1 participates in the determination of the kinetics and thus the cellular outcome of MAP kinase signaling (e.g., proliferation vs. differentiation; Charles et al., 1993; Nishida and Gotoh, 1993; and survival vs. apoptosis; Xaus et al., 2001). The interaction between MKP-1/MAP kinase signaling also exhibits cell type specificity (Zhang et al., 2003).
In the present study, hypoxia-induced up-regulation of MKP-1 provides a novel mechanism, to account for the inhibitory effects of PKCζ on ERK1/2 phosphorylation and proliferation in hypoxic fibroblasts. Marked prolongation of ERK1/2 phosphorylation in cells treated with either PKCζ antisense oligonucleotides or myristoylated PKCζ peptide inhibitor correlates with the inhibition of MKP-1 expression upon PKCζ attenuation. Consequently, our results allow us to conclude that PKCζ is the suppressor of ERK1/2 phosphorylation by regulating MKP-1 expression. Prolonged activation of PKCζ in hypoxic fibroblasts (Figure 5) might be required by the system to maintain the level of MKP-1 upon hypoxic exposure. MKP-1 overexpression inhibits ERK-regulated reporter gene expression, Ras-induced DNA synthesis, and growth-factor-stimulated entry into the S phase in fibroblasts (Brunet et al., 1995). As a result, MKP-1 expression constitutes a control mechanism for attenuation of mitogenic signaling pathways. Our data further suggest that PKCζ-mediated MKP-1 expression plays a crucial role in the proper termination of ERK1/2 activation during the hypoxic exposure and contributes to “switching off” the signals directing the proliferation of hypoxic fibroblasts.
Evidence is accumulating to indicate that PKC is associated with nuclear events both in resting cells as well as in actively dividing cells (Capitani et al., 1987; Chiarugi et al., 1990; Buchner et al., 1992; Neri et al., 1994). In the present study, PKCζ localization in the nuclear compartment is consistent with the report demonstrating its expression in the nuclei of unstimulated adipocytes (Lacasa et al., 1995). Stimulation of the adipocytes with insulin or serum caused a rapid increase in nuclear PKCζ activity, suggesting that PKCζ could directly phosphorylate structural and/or regulatory nuclear proteins. Nucleolin and heterogeneous nuclear ribonucleoprotein are the substrates of PKCζ, suggesting that PKCζ may play an important role in nuclear signal transduction (Tuteja and Tuteja, 1998). Nuclear PKCζ might be the critical mediator of the hypoxic responses in fibroblasts because PKCζ transactivates HIF-1α by blocking the expression of a factor inhibiting HIF-1 in renal cancer cells (Datta et al., 2004). Interestingly, MKP-1 colocalizes with PKCζ in the nuclear compartment of fibroblasts. PKCζ association with MKP-1 in the nucleus may constitute the critical replication repressor system in fibroblasts.
Manipulation of PKCζ levels, either by inhibition or overexpression, leads to subsequent alteration in MKP-1 levels, suggesting that PKCζ tightly regulates MKP-1 expression in fibroblasts. MKP-1 expression is also regulated by PKC, albeit by a different isozyme, PKCε, in bone marrow macrophages (Valledor et al., 2000). In H41E rat hepatoma cells, insulin-induced MKP-1 expression is blocked by the myristoylated PKCζ pseudosubstrate peptide inhibitor (Lornejad-Schafer et al., 2003). However, in that report PKCζ-mediated MKP-1 expression is not correlated with any cellular response. We believe that our data are the first demonstration of nuclear colocalization of MKP-1 and PKCζ, which together function as terminators of proliferative signals in hypoxic fibroblasts. Our future studies will focus on evaluating the mechanism of PKCζ-mediated regulation of MKP-1 expression in vascular fibroblasts.
Excessive proliferation of fibroblasts is associated with a number of vascular diseases (Stenmark et al., 1987; Stenmark and Mecham, 1997; Stenmark et al., 2000; Rey and Pagano, 2002), asthma, chronic obstructive pulmonary diseases, pulmonary fibrosis (Zhong et al., 2005), and also cancer (Bhowmick et al., 2004). Multiple factors including growth factors, cytokines, mechanical stress, hypoxia, neurotransmitters, and hormones are believed to contribute to the processes leading to fibroblast proliferation (Sartore et al., 2001), wherein PKCζ might serve as a common second messenger mediating the termination signals for proliferative responses. One of the primary steps in the orchestrated “emergency stop” cascade may be the up-regulation of MKP-1 expression that dephosphorylates ERK1/2 and consequently attenuates proliferation. PKCζ exerts its suppressing effect on ERK activation and proliferation in vascular fibroblasts primarily through interactions that involve MKP-1. Thus, we postulate that the balance between PKCζ-mediated MKP-1 expression and ERK1/2 activities stimulated by hypoxia is critical for maintaining cellular homeostasis. Further understanding of the mechanisms by which hypoxia stimulates the PKCζ-mediated induction of MAP kinase phosphatase might lead to strategies for the prevention and treatment of fibroproliferative diseases resulting from chronic hypoxic exposure.
Acknowledgments
We thank Steve Hofmeister and Sandi Walchak for harvesting bovine pulmonary artery tissue and Dr. Michael Zawada, Dr. Mary Reyland, and Dr. Raphael Nemenoff for critical review of the manuscript. This study was supported by National Heart, Lung, and Blood Institute Grant HL 64917 (M. Das), HL57144-09, and HL14985-33 (K.R. Stenmark).
This article was published online ahead of print in MBC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E05-09-0869) on February 8, 2006.
References
- Atamas, S. P. (2002). Complex cytokine regulation of tissue fibrosis. Life Sci. 72, 631-643. [DOI] [PubMed] [Google Scholar]
- Berra, E., DiazMeco, M. T., Dominguez, I., Municio, M. M., Sanz, L., Lozano, J., Chapkin, R. S., and Moscat, J. (1993). Protein kinase C zeta isoform is critical for mitogenic signal transduction. Cell 74, 555-563. [DOI] [PubMed] [Google Scholar]
- Bhowmick, N. A., Neilson, E. G., and Moses, H. L. (2004). Stromal fibroblasts in cancer initiation and progression. Nature 432, 332-337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourbon, N. A., Sandirasegarane, L., and Kester, M. (2002). Ceramide-induced inhibition of Akt is mediated through protein kinase C zeta: implications for growth arrest. J. Biol. Chem. 277, 3286-3292. [DOI] [PubMed] [Google Scholar]
- Braun, M. U., and Mochly-Rosen, D. (2003). Opposing effects of δ- and ζ-protein kinase C isozymes on cardiac fibroblast proliferation: use of isozyme-selective inhibitors. J. Mol. Cell Cardiol. 35, 895-903. [DOI] [PubMed] [Google Scholar]
- Brunet, A., Brondello, J. M., L'Allemain, G., Lenormand, P., McKenzie, F., Pages, G., and Pouyssegur, J. (1995). MAP kinase module: role in the control of cell proliferation. CR Seances Soc. Biol. Fil. 189, 43-57. [PubMed] [Google Scholar]
- Buchner, K., Otto, H., Hilbert, R., Lindschau, C., Haller, H., and Hucho, F. (1992). Properties of protein kinase C associated with nuclear membranes. Biochem. J. 286, 369-375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capitani, S., Girard, P. R., Mazzei, G. J., Kuo, J. F., Berezney, R., and Manzoli, F. A. (1987). Immunochemical characterization of protein kinase C in rat liver nuclei and subnuclear fractions. Biochem. Biophys. Res. Commun. 142, 367-375. [DOI] [PubMed] [Google Scholar]
- Carlin, S., Poronik, P., Cook, D. L., Carpenter, L., Biden, T. J., Johnson, P.R.A., and Black, J. L. (2000). An antisense of protein kinase C-zeta inhibits proliferation of human airway smooth muscle cells. Am. J. Respir. Cell Mol. Biol. 23, 555-559. [DOI] [PubMed] [Google Scholar]
- Chang, F., Steelman, L. S., Shelton, J. G., Lee, J. T., Navolanic, P. M., Blalock, W. L., Franklin, R., and McCubrey, J. A. (2003). Regulation of cell cycle progression and apoptosis by the Ras/Raf/MEK/ERK pathway. Int. J. Oncol. 22, 469-480. [PubMed] [Google Scholar]
- Charles, C. H., Sun, H., Lau, L. F., and Tonks, N. K. (1993). The growth factor-inducible immediate-early gene 3CH134 encodes a protein-tyrosine-phosphatase. Proc. Natl. Acad. Sci. USA 90, 5292-5296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiarugi, V., Magnelli, L., Pasquali, F., Vannucchi, S., Bruni, P., Quattrone, A., Basi, G., Capaccioli, S., and Ruggiero, M. (1990). Transformation by ras oncogene induces nuclear shift of protein kinase C. Biochem. Biophys. Res. Commun. 173, 528-533. [DOI] [PubMed] [Google Scholar]
- Cho, Y., and Ziboh, V. A. (1995). Nutritional modulation of guinea pig skin hyperproliferation by essential fatty acid deficiency is associated with selective down regulation of protein kinase C-beta. J. Nutr. 125, 2741-2750. [DOI] [PubMed] [Google Scholar]
- Cogolludo, A., Moreno, L., Lodi, F., Tamargo, J., and Perez-Vizcaino, F. (2005). Postnatal maturational shift from protein kinase C zeta and voltage-gated K+ channels to RhoA/Rho kinase in pulmonary vasoconstriction. Cardiovasc. Res. 66, 84-93. [DOI] [PubMed] [Google Scholar]
- Corbit, K. C., Soh, J. W., Yoshida, K., Eves, E. M., Weinstein, I. B., and Rosner, M. R. (2000). Activation of Raf-1 signaling by protein kinase C through a mechanism involving Raf kinase inhibitory protein. Mol. Cell. Biol. 20, 5392-5403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dada, L., Chandel, N. S., Ridge, K. M., Pedemonte, C., Bertorello, A. M., and Sznajder, J. I. (2003). Hypoxia-induced endocytosis of Na, K-ATPase in alveolar epithelial cells is mediated by mitochondrial reactive oxygen species and protein kinase C-zeta. J. Clin. Invest. 111, 1057-1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das, M., Dempsey, E. C., Bouchey, D., Reyland, M. R., and Stenmark, K. R. (2000). Chronic hypoxia induces exaggerated growth responses in pulmonary artery adventitial fibroblasts: potential contribution of specific protein kinase C isozymes. Am. J. Respir. Cell Mol. Biol. 22, 15-25. [DOI] [PubMed] [Google Scholar]
- Das, M., Bouchey, D. M., Moore, M. J., Hopkins, D. C., Nemenoff, R. A., and Stenmark, K. R. (2001). Hypoxia-induced proliferative response of vascular adventitial fibroblasts is dependent on G-protein-mediated activation of mitogen-activated protein kinases. J. Biol. Chem. 276, 15631-15640. [DOI] [PubMed] [Google Scholar]
- Das, M., Dempsey, E. C., Reeves, J. T., and Stenmark, K. R. (2002). Selective expansion of fibroblast subpopulations from pulmonary artery adventitia in response to hypoxia. Am. J. Physiol. Lung Cell Mol. Physiol. 282, L976-L986. [DOI] [PubMed] [Google Scholar]
- Datta, K., Li, J., Bhattacharya, R., Gasparian, N., Wang, E., and Mukhopadhyay, D. (2004). Protein kinase C zeta transactivates hypoxia-inducible factor alpha by promoting its association with p300 in renal cancer. Cancer Res. 64, 456-462. [DOI] [PubMed] [Google Scholar]
- Fields, A. P., Pincus, S. M., Kraft, A. S., and May, W. S. (1989). Interleukin-3 and bryostatin 1 mediate rapid nuclear envelope protein phosphorylation in growth factor-dependent FDC-P1 hematopoietic cells. A possible role for nuclear protein kinase C. J. Biol. Chem. 264, 21896-21901. [PubMed] [Google Scholar]
- Hirai, T., and Chida, K. (2003). Protein kinase C zeta (PKCzeta): activation mechanisms and cellular functions. J. Biochem. (Tokyo) 133, 1-7. [DOI] [PubMed] [Google Scholar]
- Hug, H., and Sarre, T. F. (1993). Protein kinase C isoenzymes: divergence in signal transduction? Biochem. J. 291, 329-343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain, S., Assender, J. A., Bond, M., Wong, L. F., Murphy, D., and Newby, A. C. (2002). Activation of protein kinase C zeta is essential for cytokine-induced metalloproteinase-1, -3, and -9 secretion from rabbit smooth muscle cells and inhibits proliferation. J. Biol. Chem. 277, 27345-27352. [DOI] [PubMed] [Google Scholar]
- Kim, S. J., Chang, Y. Y., Kang, S. S., and Chun, J. S. (1997). Phorbol ester effects in atypical protein kinase C zeta overexpressing NIH3T3 cells: possible evidence for crosstalk between protein kinase C isoforms. Biochem. Biophys. Res. Commun. 237, 336-339. [DOI] [PubMed] [Google Scholar]
- Kent, K. C., Mii, S., Harrington, E. O., Chang, J. D., Mallette, S., and Ware, J. A. (1995). Requirement for protein kinase C activation in basic fibroblast growth factor-induced human endothelial cell proliferation. Circ. Res. 77, 231-238. [DOI] [PubMed] [Google Scholar]
- Keyse, S. M., and Emslie, E. A. (1992). Oxidative stress and heat shock induce a human gene encoding a protein-tyrosine phosphatase. Nature 359, 644-647. [DOI] [PubMed] [Google Scholar]
- Keyse, S. M. (1995). An emerging family of dual specificity MAP kinase phosphatases. Biochim. Biophys. Acta 1265, 152-160. [DOI] [PubMed] [Google Scholar]
- Lacasa, D., Agli, B., and Giudicelli, Y. (1995). Zeta protein kinase C in rat preadipocytes: modulation by insulin and serum mitogenic factors and possible role in adipogenesis. Biochem. Biophys. Res. Comm. 217, 123-130. [DOI] [PubMed] [Google Scholar]
- Laderoute, K. R., Mendonca, H. L., Calaoagan, J. M., Knapp, A. M., Giaccia, A. J., and Strok, P. J. (1999). Mitogen-activated protein kinase phosphatase-1 (MKP-1) expression is induced by low oxygen conditions found in solid tumor microenvironments. A candidate MKP for the inactivation of hypoxia-inducible stress-activated protein kinase/c-Jun N-terminal protein kinase activity. 1999. J. Biol. Chem. 274, 12890-12897. [DOI] [PubMed] [Google Scholar]
- Le Good, J. A., Ziegler, W. H., Parekh, D. B., Alessi, D. R., Cohen, P., and Parker, P. J. (1998). Protein kinase C isotypes controlled by phosphoinositide 3-kinase through the protein kinase PDK1. Science 281, 2042-2045. [DOI] [PubMed] [Google Scholar]
- Liu. C., Shi, Y., Ha, Z., Pan, Y., Liu, N., Han, S., Chen, Y., Lan, M., Qiao, T., and Fan, D. (2003). Suppression of the dual-specificity phosphatase MKP-1 enhances HIF-1 trans-activation and increases expression of EPO. Biochem. Biophys. Res. Commun. 312, 780-786. [DOI] [PubMed] [Google Scholar]
- Lornejad-Schafer, M. R., Schafer, C., Graf, D., Haussinger, D., and Schliess, F. (2003). Osmotic regulation of insulin-induced mitogen-activated protein kinase phosphatase (MKP-1) expression in H4IIE rat hepatoma cells. Biochem. J. 371, 609-619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mas, V. M., Hernandez, H., Plo, I., Bezombes, C., Maestre, N., Quillet-Mary, A., Filomenko, R., Demur, C., Jaffrezou, J. P., and Laurent, G. (2003). Protein kinase C zeta mediated Raf-1/extracellular-regulated kinase activation by daunorubicin. Blood 101, 1543-1550. [DOI] [PubMed] [Google Scholar]
- Neri, L. M., Billi, A. M., Manzoli, L., Rubbini, S., Gilmour, R. S., Cocco, L., and Martelli, A. M. (1994). Selective nuclear translocation of protein kinase C alpha in Swiss 3T3 cells treated with IGF-I, PDGF and EGF. FEBS Lett. 347, 63-68. [DOI] [PubMed] [Google Scholar]
- Nishida, E., and Gotoh, Y. (1993). The MAP kinase cascade is essential for diverse signal transduction pathways. Trends Biochem. Sci. 18, 128-131. [DOI] [PubMed] [Google Scholar]
- Nishizuka, Y. (1992). Intracellular signaling by hydrolysis of phospholipids and activation of protein kinase C. Science 258, 607-614. [DOI] [PubMed] [Google Scholar]
- Noguchi, T., Metz, R., Chen, L., Mattei, M. G., Carrasco, D., and Bravo, R. (1993). Structure, mapping, and expression of erp, a growth factor-inducible gene encoding a nontransmembrane protein tyrosine phosphatase, and effect of ERP on cell growth. Mol. Cell. Biol. 13, 5195-5205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal, S., Datta, K., Khosravi-Far, R., and Mukhopadhyay, D. (2001). Role of protein kinase C zeta in Ras-mediated transcriptional activation of vascular permeability factor/vascular endothelial growth factor expression. J. Biol. Chem. 276, 2395-2403. [DOI] [PubMed] [Google Scholar]
- Plows, D., Briassouli, P., Owen, C., Zoumpourlis, V., Garrett, M. D., and Pintzas, A. (2002). Ecdysone-inducible expression of oncogenic Ha-Ras in NIH 3T3 cells leads to transient nuclear localization of activated extracellular signal-regulated kinase regulated by mitogen-activated protein kinase phosphatase-1. Biochem. J. 362, 305-315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pouyssegur, J., Volmat, V., and Lenormand, P. (2002). Fidelity and spatiotemporal control in MAP kinase (ERKs) signalling. Biochem. Pharmacol. 64, 755-763. [DOI] [PubMed] [Google Scholar]
- Powell, D. J., Turban, S., Gray, A., Hajduch, E., and Hundal, H. S. (2004). Intracellular ceramide synthesis and protein kinase C zeta activation play an essential role in palmitate-induced insulin resistance in rat L6 skeletal muscle cells. Biochem. J. 382, 619-629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reffas, S., and Schlegel, W. (2000). Compartment-specific regulation of extracellular signal-regulated kinase (ERK) and c-Jun N-terminal kinase (JNK) mitogen-activated protein kinases (MAPKs) by ERK-dependent and non-ERK-dependent inductions of MAPK phosphatase (MKP)-3 and MKP-1 in differentiating P19 cells. Biochem. J. 352, 701-708. [PMC free article] [PubMed] [Google Scholar]
- Rey, F. E., and Pagano, P. J. (2002). The reactive adventitia: fibroblast oxidase in vascular function. Arterioscler. Thromb. Vasc. Biol. 22, 1962-1971. [DOI] [PubMed] [Google Scholar]
- Sartore, S., Chiavegato, A., Faggin, E., Franch, R., Puato, M., Ausoni, S., and Pauletto, P. (2001). Contribution of adventitial fibroblasts to neointima formation and vascular remodeling: from innocent bystander to active participant. Circ. Res. 89, 1111-1121. [DOI] [PubMed] [Google Scholar]
- Seta, K. A., Kim, R., Kim, H. W., Millhorn, D. E., and Beitner-Johnson, D. (2001). Hypoxia-induced regulation of MAPK phosphatase-1 as identified by subtractive suppression hybridization and cDNA microarray analysis. J. Biol. Chem. 276, 44405-44412. [DOI] [PubMed] [Google Scholar]
- Slack, D. N., Seternes, O. M., Gabrielsen, M., and Keyse, S. M. (2001). Distinct binding determinants for ERK2/p38alpha and JNK map kinases mediate catalytic activation and substrate selectivity of map kinase phosphatase-1. J. Biol. Chem. 276, 16491-16500. [DOI] [PubMed] [Google Scholar]
- Short, M., Nemenoff, R. A., Zawada, W. M., Stenmark, K. R., and Das, M. (2004). Hypoxia induces differentiation of pulmonary artery adventitial fibroblasts into myofibroblasts. Am. J. Physiol. Cell Physiol. 286, C416-C425. [DOI] [PubMed] [Google Scholar]
- Sodhi, C. P., Batlle, D., and Sahai, A. (2000). Osteopontin mediates hypoxia-induced proliferation of cultured mesangial cells: Role of protein kinase C and MAPK. Kidney Int. 58, 691-700. [DOI] [PubMed] [Google Scholar]
- Stawowy, P., Goetze, S., Margeta, C., Fleck, E., and Graf, K. (2003). LPS regulate ERK1/2-dependent signaling in cardiac fibroblasts via protein kinase C-mediated MKP-1 induction. Biochem. Biophys. Res. Commun. 303, 74-80. [DOI] [PubMed] [Google Scholar]
- Stenmark, K. R., Fasules, J., Voelkel, N. F., Henson, J., Tucker, A., Wilson, H., and Reeves, J. T. (1987). Severe pulmonary hypertension and arterial adventitial changes in newborn calves at 4300 m. J. Appl. Physiol. 62, 821-830. [DOI] [PubMed] [Google Scholar]
- Stenmark, K. R., and Mecham, R. P. (1997). Cellular and molecular mechanisms of pulmonary vascular remodeling. Annu. Rev. Physiol. 59, 89-144. [DOI] [PubMed] [Google Scholar]
- Stenmark, K. R., Bouchey, D., Nemenoff, R., Dempsey, E. C., and Das, M. (2000). Hypoxia-induced pulmonary vascular remodeling: contribution of the adventitial fibroblasts. Physiol. Res. 49, 503-517. [PubMed] [Google Scholar]
- Theodore, L., Derossi, D., Chassaing, G., Llirbat, B., Kubes, M., Jordan, P., Chneiweiss, H., Godement, P., and Prochiantz, A. (1995). Intraneuronal delivery of protein kinase C pseudosubstrate leads to growth cone collapse. J. Neurosci. 15, 7158-7167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toker, A. (1998). Signaling through protein kinase C. Front, Biosci. 3, D1134-D1147. [DOI] [PubMed] [Google Scholar]
- Tuteja, R., and Tuteja, N. (1998). Nucleolin: a multifunctional major nucleolar phosphoprotein. Crit. Rev. Biochem. Mol. Biol. 33, 407-436. [DOI] [PubMed] [Google Scholar]
- Valledor, A. F., Xaus, J., Comalada, M., Soler, C., and Celada, A. (2000). Protein kinase C epsilon is required for the induction of mitogen-activated protein kinase phosphatase-1 in lipopolysaccharide-stimulated macrophages. J. Immunol. 164, 29-37. [DOI] [PubMed] [Google Scholar]
- Xaus, J., Comalada, M., Valledor, A. F., Cardo, M., Herrero, C., Soler, C., Lloberas, J., and Celada, A. (2001). Molecular mechanisms involved in macrophage survival, proliferation, activation or apoptosis. Immunobiology 204, 543-550. [DOI] [PubMed] [Google Scholar]
- Yano, K., Bauchat, J. R., Liimatta, M. B., Clemmons, D. R., and Duan, C. (1999). Down-regulation of protein kinase C inhibits insulin-like growth factor I-induced vascular smooth muscle cell proliferation, migration, and gene expression. Endocrinology 140, 4622-4632. [DOI] [PubMed] [Google Scholar]
- Zhang, B., Hosaka, M., Sawada, Y., Torii, S., Mizutani, S., Ogata, M., Izumi, T., and Takeuchi, T. (2003). Parathyroid hormone-related protein induces insulin expression through activation of MAP kinase-specific phosphatase-1 that dephosphorylates c-Jun NH2-terminal kinase in pancreatic beta-cells. Diabetes 5, 2720-2730. [DOI] [PubMed] [Google Scholar]
- Zhong, H., Belardinelli, L., Maa, T., and Zeng, D. (2005). Synergy between A2B Adenosine receptors and hypoxia in activating human lung fibroblasts. Am. J. Respir. Cell Mol. Biol. 32, 2-8. [DOI] [PubMed] [Google Scholar]