

Abstract
Cerebrotendinous xanthomatosis (CTX) is a rare autosomal recessive disorder of bile acid biosynthesis. Clinically, CTX patients present with tendon xanthomas, juvenile cataracts, and progressive neurological dysfunction and can be diagnosed by the detection of elevated plasma cholestanol levels. CTX is caused by mutations affecting the sterol 27-hydroxylase gene (CYP27 ). CTX has been identified in a number of populations, but seems to have a higher prevalence in the Japanese, Sephardic Jewish, and Italian populations. We have assembled 12 previously unreported pedigrees from the United States. The CYP27 locus had been previously mapped to chromosome 2q33-qter. We performed linkage analyses and found no evidence of genetic heterogeneity. All CTX patients showed segregation with the CYP27 locus, and haplotype analysis and recombinant events allowed us to precisely map CYP27 to chromosome 2q35, between markers D2S1371 and D2S424. Twenty-three mutations were identified from 13 probands analyzed thus far; 11 were compound heterozygotes and 2 had homozygous mutations. Of these, five are novel mutations [Trp100Stop, Pro408Ser, Gln428Stop, a 10-base pair (bp) deletion in exon 1, and a 2-bp deletion in exon 6 of the CYP27 gene]. Three-dimensional structural modeling of sterol 27-hydroxylase showed that, while the majority of the missense mutations disrupt the heme-binding and adrenodoxin-binding domains critical for enzyme activity, two missense mutations (Arg94Trp/Gln and Lys226Arg) are clearly located outside these sites and may identify a potential substrate-binding or other protein contact site.
Supplementary key words : genetics, cholesterol, cholestanol, bile acids
Abbreviations: CTX, cerebrotendinous xanthomatosis; PCR, polymerase chain reaction; YAC, yeast artificial chromosome
The conversion of cholesterol into bile acids by the liver represents the major mechanism for the removal of cholesterol from the body (1). In the biosynthesis of bile acids, involving microsomal, mitochondrial, and peroxisomal compartments, enzymes modify both the ring structure and the side chain of cholesterol by oxidation, resulting in the formation of the primary bile acids, cholic acid and chenodeoxycholic acid (1).
Sterol 27-hydroxylase (EC 1.14.13.15) is a member of the mitochondrial cytochrome P450 family catalyzing the initial oxidation of the side chain of sterol intermediates in hepatic bile acid synthesis (2, 3). Together with two protein cofactors, adrenodoxin and adrenodoxin reductase, sterol 27-hydroxylase hydroxylates a variety of sterol substrates at the C-27 position, and in addition can hydroxylate vitamin D3 in the C-1 and C-25 positions (3).
Cerebrotendinous xanthomatosis (CTX) is a rare inherited sterol storage disorder caused by a deficiency of sterol 27-hydroxylase (4). CTX is characterized by abnormal deposition of cholesterol and cholestanol in body tissues (5, 6). Clinical progression of the disease is variable. Some patients develop severe symptoms early in infancy (7–10), whereas others first present clinical symptoms in middle age (4). In general, characteristic clinical features of CTX are the presence of juvenile cataracts and tendon xanthomas (4). Progressive neurological dysfunction may be present, if diagnosis has not been made early in life. The latter includes behavioral abnormalities; dementia; pyramidal paresis; cerebellar, brainstem, spinal, and peripheral nerve disorders; and epileptic seizures (4, 11). In addition, osteoporosis with frequent bone fractures and premature atherosclerosis have been documented (4).
The isolation and characterization of the sterol 27-hydroxylase gene (CYP27) allows for the early detection of CTX patients and the identification of healthy carriers at the molecular level (12–14). CYP27 has been mapped to the distal portion (q33-qter) of the long arms of chromosome 2, although its fine map has not been reported (13).
Sterol 27-hydroxylase, encoded by nine exons, consists of a 33-amino acid mitochondrial signal sequence (12), followed by a mature protein of 498 amino acids (14). It is a member of a large mitochondrial cytochrome P450 family (15) containing an adrenodoxin-binding site (residues 351–365) and the heme-binding site (residues 435–464) (12, 14, 16). About 37 different mutations of the CYP27 gene have been identified in CTX patients drawn from various populations (13, 14, 17–39). A large majority of these affect splice sites and are predicted to affect message stability or lead to abnormal splicing. Others lead to a nonsense stop codon and premature translational stop. Mutations that disrupt splicing often lead to rapid degradation of the aberrant mRNA, or lead to frame shift and translation of a polypeptide that would be predicted to be enzymatically inactive, because the heme or adrenodoxin domains are absent. All of these are therefore “null mutations.” Only 16 mutations are missense mutations that could lead to expression of the CYP27 protein. Of these, 10 are predicted to disrupt either the heme-binding or the adrenodoxin-binding domain, as they map within the identified binding sites (29). The other missense mutations are apparently located outside these domains and their functional effects have yet to be determined. It is possible that similarly occurring mutations might provide insights into the active center and/or the tertiary structure of sterol 27-hydroxylase.
In the present study, we have assembled 12 previously unreported pedigrees, and one other proband, from the United States that were clinically diagnosed with CTX. We initially fine mapped CYP27 to a yeast artificial chromosome (YAC) contig and used a set of flanking microsatellite markers to genotype all the CTX families. On the basis of the haplotype and linkage analysis, no evidence of genetic heterogeneity was found. Recombination analyses allowed us to refine the mapping data. Mutations of the sterol 27-hydroxylase gene from the 13 CTX probands were identified. All probands, except two, were heterozygous for mutations. Twenty-three mutations in total were identified, five of which are novel. Using structural modeling, we have mapped all the known missense mutations onto a putative three-dimensional model of sterol 27-hydroxylase. Most of the missense mutations appear to disrupt the heme- or adrenodoxin-binding sites, on the basis of their location in these domains. However, two missense mutations map well outside these areas and thus may be important in either substrate entry into the active site, or in an unidentified biological function that is critical for enzyme activity.
MATERIALS AND METHODS
Pedigrees
Twelve unrelated families with CTX were identified (Fig. 1) on the basis of clinical finding of tendon and tuberous xanthomas and elevated serum cholesterol and cholestanol levels. A total of 12 parents, 19 affected individuals, and 25 unaffected siblings were genotyped. The family tree of one proband, DB, was not available, but DNA from the proband was available as a fibroblast cell line. Informed consent was obtained from all participants in accordance with institutional review board requirements. Clinical characteristics of affected subjects are summarized in Table 1. Severity was scored depending on a clinical classification of the signs and symptoms, assigning a score of 0 for absent, 1 for mild, 2 for moderate, 3 for severe, and 4 for very severe (40). Only the approximate age of onset, on the basis of clinical history, was available.
Fig. 1.
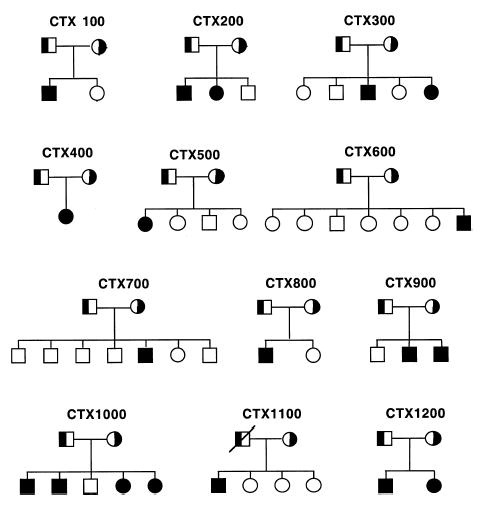
CTX pedigree trees assembled for this study. Pedigrees were assembled on the basis of identification of one or more previously diagnosed subjects with CTX. None of the marriages was consanguineous. The parents are indicated as obligate heterozygotes. All the families are white and of mixed European ancestry, except for pedigrees 400, 500, and 1200, which are of Italian ancestry.
TABLE 1.
Clinical characteristics and severity scores of CTX patients
| Patient | Current Age | Age at Onset | Tendon Xanthomas | Cataracts | Dementia | Cerebellar Ataxia | Spastic Paresis | Peripheral Neuropathy | Osteoporosis | Total Score | Plasma Cholestanol |
|---|---|---|---|---|---|---|---|---|---|---|---|
| years | mg/dL | ||||||||||
| 100-3 | 18 | Childhood | 2 | 2 | 2 | 0 | 0 | 0 | 0 | 6 | 2.6 |
| 200-3 | 46 | Childhood | 32 | 2 | 2 | 2 | 2 | 2 | 0 | 13 | 5.8 |
| 200-4 | 48 | Childhood | 3 | 4 | 2 | 4 | 4 | 3 | 2 | 22 | 6.2 |
| 300-5 | 36 | Childhood | 2 | 2 | 2 | 2 | 2 | 1 | 0 | 11 | 3.6 |
| 300-7 | 32 | Adolescence | 1 | 2 | 0 | 0 | 0 | 0 | 0 | 3 | 5.8 |
| 400-3 | 65a | Adulthood | 3 | 4 | 3 | 4 | 4 | 2 | 2 | 22 | 2.7 |
| 500-3 | 48 | Adulthood | 4 | 0 | 2 | 3 | 3 | 3 | 0 | 15 | 6.1 |
| 600-9 | 34 | Adulthood | 3 | 0 | 0 | 2 | 3 | 0 | 0 | 8 | 3.2 |
| 700-7 | 48 | Adulthood | 4 | 2 | 2 | 2 | 2 | 0 | 0 | 12 | 3.6 |
| 800-3 | 52 | Adulthood | 4 | 3 | 2 | 2 | 2 | 0 | 0 | 13 | 3.2 |
| 900-4 | 33 | Adolescence | 4 | 2 | 2 | 2 | 3 | 1 | 0 | 14 | 3.6 |
| 900-5 | 35 | Adolescence | 4 | 2 | 2 | 2 | 3 | 0 | 0 | 13 | 3.2 |
| 1000-3 | 48 | Adulthood | 2 | 0 | 2 | 2 | 2 | 2 | 0 | 10 | 1.5 |
| 1000-4 | 40 | Adolescence | 4 | 4 | 3 | 4 | 4 | 4 | 0 | 23 | 3.8 |
| 1000-6 | 39 | Adulthood | 2 | 4 | 2 | 2 | 2 | 2 | 0 | 14 | 1.5 |
| 1000-7 | 41 | Adulthood | 4 | 2 | 2 | 2 | 3 | 2 | 0 | 15 | 2.6 |
| 1100-3 | 45 | Adulthood | 2 | 0 | 2 | 1 | 1 | 0 | 0 | 6 | 6.4 |
| 1200-3 | 43 | Adolescence | 4 | 4 | 3 | 3 | 2 | 2 | 2 | 21 | 3.9 |
| DB | 47 | Childhood | 2 | 0 | 4 | 3 | 3 | 2 | 3 | 17 | 2.4 |
Deceased.
YAC mapping
The Centre d’Etude du Polymorphisme Humaine megaYAC library was screened by polymerase chain reaction (PCR) with primers ctx 5a/5b and ctx 6a/9b for the amplification of exons 5 and exons 6–9 of the CYP27 gene (14). One positive yeast artificial chromosome (YAC), 779F9, was identified. YAC 779F9 is 760 kb in size and it appears to be chimeric. There were three known DNA markers, and one expressed sequence tag, which have been unambiguously mapped to this YAC. Among these markers is a cooperative human linkage center tetranucleotide repeat polymorphic marker, CHLC.GATA3F05 (D2S433). There are several other available polymorphic markers in the databases that have been mapped around D2S433, but none of these are contained within YAC 779F9. A BLAST search, using the CYP27 cDNA sequence, identified a working draft sequence of a bacterial artificial chromosome (BAC), 459-I-19, GenBank ID AC009974, containing the CYP27 gene. This working draft sequence contains the CYP27 gene derived from a 31,056-bp contig. These sequences have nine exons and eight introns and confirm the gene structure of CYP27 previously reported (14). We identified a microsatellite repeat sequence identified within this BAC, CYPMFR, that was found to be polymorphic and is thus the closest informative marker to CYP27. The BAC was also screened by PCR for markers D2S2179, D2S2249, D2S433, and D2S424 and was found to be negative for all these markers, indicating that all of these markers are at least 33 kb away from CYP27. Because marker D2S433 is placed on a YAC positive for CYP27, we placed the marker CYPMFR adjacent to this marker (see Results).
Genotyping and linkage analysis
Genomic DNA was extracted from whole-blood cells as previously described (41). Genotyping was performed with 19 markers, 15 of which are located within 10 cM of D2S433 as previously described (41). The following microsatellite repeat markers (with sex-averaged distances in centimorgans as shown) were used for genotyping: D2S1399 - 34 - D2S1391 - 14 - D2S1384 - 2.5 - D2S1782 -3.2 - D2S1369 - 4.3 - D2S1649 - 0 - D2S1380 - 0 - D2S1345 - 0 -D2S1327 - 4.3 - D2S164 - 0.5 - D2S1371- 0.5 - D2S434 - 0 - D2S1338 -0 - D2S2210 - 0 - D2S2179 - 0 - D2S2249 - 0.5 - D2S433 - 2 - D2S424 -8.6 - D2S1363 (Research Genetics, Birmingham, AL). Reverse primers from each set were end labeled with [γ-32P]ATP, using standard techniques. After PCR amplification of the microsatellite repeats, the products were separated by 6% denaturing urea-acrylamide gels, dried, and analyzed by PhosphorImager (Molecular Dynamics, Sunnyvale, CA) (41). Multipoint linkage analysis was performed using GeneHunter (42). For multipoint analyses, markers that showed no separation by recombination were assigned an arbitrary value, based upon uniform distribution within such a region.
Exon amplification and mutational analysis
All nine exons, with immediate flanking intronic sequences, of the CYP27 gene were amplified from 13 CTX probands to identify mutations causing CTX. Oligonucleotide sequences and PCR conditions were as previously described (14). In addition, oligonucleotide Cf1 (5′-CTAGCTGGCCTTTGCTCGGC), located in the 5′ flanking region, and oligonucleotides 8F (5′-CCAGTTTGTGTTCTGCCAC) and 8R (5′-CAGGCTCAGAGAAGGCAGTG) were used for amplification of exons 1 and 8, respectively. DNA sequence analysis was performed with the ABI-PRISM™ 310 Genetic Analyzer or by manual sequencing with Amplicycle™ sequencing kit (Perkin Elmer, Norwalk, CT). Both strands were sequenced to confirm the identified mutations. In the case of deletions, PCR products were cloned and individual clones were sequenced to verify deletions, as previously described (43). The primers used for sequencing were the same as those used for PCR amplification. Sequence alignment was aided by the use of MacVector software running on an Apple iMac.
Structural modeling
A homology-based three-dimensional model was constructed from the CYP27 peptide sequence PIR2:A39740 (the full 531-amino acid sequence was truncated to 498 amino acids to reflect the mature protein) sterol 27-monooxygenase, using as a template the model structure P450SCC (44). This model is derived from a 2.6-Å crystal structure of Pseudomonas putida cytochrome P-450cam (45). Several procedures were attempted. The SwissModel resource was accessed via Web browser and SwissPDBViewer program. This process used P450SCC as the template and the alignment was performed using the BLAST and GCG GAP programs. This model was passed through one refinement cycle on the SwissModel server (http://www.expasy.ch/spdbv/mainpage.htm). A second model using P450SCC as template was constructed via the LOOK program (Molecular Applications Group, Palo Alto, CA; http://www.mag.com/products/look.html). This model was constructed and minimized internally by the program on the basis of automatic alignment of CYP27 sequence with the sequence of the template structures. The alignment was identical to the GCG Gap alignment and identical to the alignment used by SwissModel. The GeneFold module of SYBYL, which uses Composer (46 –50) and MatchMaker (51) approaches, built an additional model of CYP27 on the basis of P450SCC. A loop from positions 272 to 278 (QVSGY), which did not conform to structurally conserved regions, was added manually. The resulting structure was minimized to 0.05 kcal/A with the Kollman All_atom force field as implemented within SYBYL. As an analysis tool the PROTABLE feature of SYBYL and the MatchMaker energy plot of each of the above-described models were computed. Root mean square deviation (RMSD) calculations comparing the models with P450 SCC in the regions homologous to the Prosite heme motif (416 –426 P450SCC; 436–446 CYP27), the adrenodoxin (Arg347 P450SCC; Arg362 CYP27)-binding site, and a series of sites where mutations are known to occur (82, 92, 214, 353, 357, 393, 421, 426 P450 SCC; 94, 104, 226, 368, 372, 408, 441, 446 CYP27) were recorded.
RESULTS
Fine mapping and linkage analysis
Previous studies had localized CYP27 to chromosome 2q31-ter (13); a more localized position was not available. We initially screened a YAC library and identified a YAC that was positive for CYP27. A polymorphic marker, D2S433, was unambiguously assigned to it. On screening, this YAC was found to be negative for the remaining markers used in this study, suggesting that these markers were a minimum of 0.5 Mb away from CYP27, or that the YAC was chimeric. Subsequently, a BAC sequence was also identified by BLAST searches of the databases that contained CYP27, but by none of the other identified markers (see Materials and Methods). We identified three microsatellite repeat sequences within the sequenced BAC that could be used as potential polymorphic markers, only one of which was polymorphic (CYPMFR; see Materials and Methods). On the basis of the identification of a YAC that contained both D2S433 and CYP27 and a BAC that contained CYP27 and CYPMFR, we tentatively placed CYPMFR next to D2S433, mapping CYP27 to chromosome 2q35. Fifteen other markers were selected from the Whitehead and Marshfield databases, spanning an approximately 40-cM region, for genotype and linkage analyses.
Multipoint analysis is shown in Fig. 2. The x axis does not accurately depict the distances between the markers, an estimate of which is given in Materials and Methods. Two peak logarithm of the odds (LOD) scores of 2.8 and 3.0 between D2S1371 to D2S1338 and D2S2249 to D2S433, respectively, were obtained (Fig. 2). Note, however, that the marker order or the distance separating some of the markers, D2S434, D2S1338, D2S2210, D2S2179, and D2S2249, has not been established. Some of these markers have been placed at the same map position in the databases. Haplotype analyses and informative recombinant events of the affected subjects (solid vertical bars, Fig. 3A) further confirmed the two possible locations for CYP27 (hatched horizontal bars, Fig. 3A) between D2S1371 and D2S1338 or D2S2249 and D2S433. In addition, we noted that a number of unaffected siblings showed evidence of informative recombination in this area. For example, some of the siblings shared one of the mutant alleles and thus would be predicted to be carriers (Fig. 3B). Other siblings shared only a part of the mutant allele and exhibited a recombinant event in one of the two possible areas. Such individuals could either be carriers or wild-type normal individuals. Once the mutations in each of the pro-bands were identified (see below), we screened these siblings for the mutations. This allowed us to narrow the CPY27 locus to between D2S2249 and D2S433, placing CYPMFR in this interval (Fig. 3B, family CTX1100). For example, for family CTX1000, sibling 5 was unaffected, but shared the region bounded by markers D2S2210–D2S2249. Thus CYP27 cannot be located in this region and must be distal to D2S2249. For family CTX1100, both siblings 5 and 6 showed recombination for marker D2S2249 on the paternal mutant haplotype. Knowing the mutations for the proband, individual 3 (see Table 2), we screened both siblings and found that they were carriers for the intron 6 1205+1G→A mutation. This further localized CYP27 to lay distal to marker D2S2179. On the other hand, sibling 4 had a recombinant event on the paternal mutant haplotype (Fig. 3B) and was found not to carry the paternal mutation (exon 6, C1204T, Table 2), placing CYP27 distal to D2S2249 (cf. Fig. 3A with Fig. 3B). No recombinants for the marker CYPMFR were found in our cohorts, thus placing this marker between D2S2249 and D2S433, and localizing the CYP27 locus to between these markers. Therefore, these three markers (D2S2249, CYPMFR, and D2S433), being highly polymorphic and thus likely to be informative, may be useful for haplotype analyses and screening of carriers in CTX families.
Fig. 2.
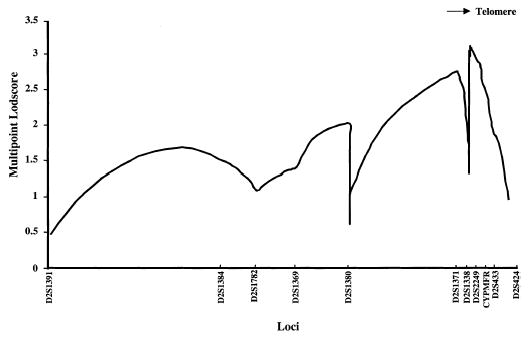
Multipoint analysis of informative markers to localize the CYP27 gene. Multipoint analysis between markers D2S1391 and D2S1363 was performed (spanning 40 cM) as described in Materials and Methods. A maximum LOD score of 3.0 was obtained for microsatellite marker D2S2249. There are two possible areas for the localization of CYP27 as indicated, between D2S164–D2S1388 and D2S2249–D2S433.
Fig. 3.
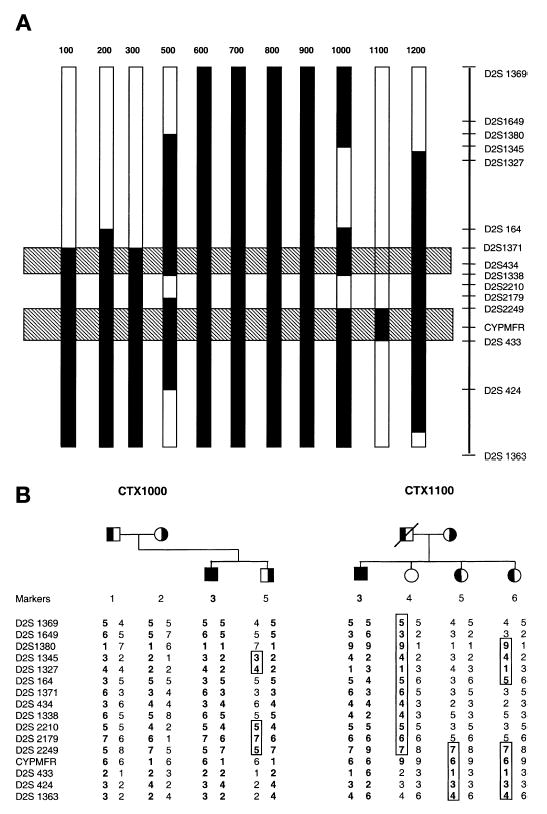
Haplotype analyses and fine-mapping of CYP27. A: Summary of the haplotype analyses from the 12 pedigrees, using all available individuals. The vertical bars represent haplotypes, with recombinant events depicted by the solid bars. The hatched horizontal bars indicate the two maximal areas of possible CYP27 localization, on the basis of recombination in affected and unaffected individuals. The distance between markers is not drawn to exact scale. When combined with the presence or absence of the mutation in “carriers” (B), CYP27 could be further localized to between D2S2249 and D2S433. In family CTX1000 (B), sibling 5 shows a recombinant event between D2S2210 and D2S2249 and shares this region with his affected brother. Because sibling 5 is a carrier for at least one defective allele, but is unaffected (on the basis of clinical and biochemical examination), the disease locus cannot lie between these two markers. Similarly, both siblings 5 and 6 in family CTX1100 (B) were found to be carriers for the paternal mutant allele, detected by direct PCR and sequencing, but share only the D2S2249–D2S1363 haplotype with their affected sibling. Haplotype analyses showed that both had a recombination event affecting D2S2249, suggesting that the mutant allele was at or below this marker. Sibling 4 shares this allele, but was not a carrier of the paternal mutation. This, therefore, placed CYP27 between D2S2249 and D2S433 (cf. A with B).
TABLE 2.
CYP27 mutations causing CTX
| Patient | Mutation | Effect of Mutation | Reference |
|---|---|---|---|
| 100-3 | Ex2 C → T | Arg94Trp | Present study; ref. 38 |
| Ex5 C → T | Splice site mutation | Present study; ref. 18 | |
| 200-3 | Ex6 C → T | Splice site mutation | Present study; ref. 13 |
| Ex8 C → T | Pro408Cys | Present study | |
| 300-5 | Ex8 C → T | Gln428stop | Present study |
| Ex8 C → T | Arg446Cys | Present study; ref. 13 | |
| 400-3 | In7 G+5 → T
Homozygous |
Frame shift → stop | Present study; ref. 30 |
| 500-3 | Ex2 C →T | Arg 94Trp | Present study; 38 |
| Ex3 G → C | Splice site mutation | Present study; ref. 30 | |
| 600-9 | Ex8 C → T | Arg446Cys | Present study; ref. 13 |
| Ex 1 Δ 10 bp | Frame shift → stop | Present study | |
| 700-7 | Ex4 A → G | Lys226Arg | Present study; ref. 39 |
| Ex6 C → T | Splice site mutation | Present study; ref. 13 | |
| 800-3 | Ex2 G → A | Arg94Gln | Present study; ref. 28 |
| In6 G+1 → A | Frame shift → stop | Present study; ref. 30 | |
| 900-4 | Ex 3 G → C
Homozygous |
Splice site mutation | Present study; ref. 30 |
| Ex4 C → T | Arg237stop | Present study; ref. 31 | |
| 1000-3 | In6 G+1 → A | Frame shift →stop | Present study; ref. 30 |
| 1100-3 | Ex6 C → T | Splice site mutation | Present study; ref. 23 |
| In6 G+1 →A | Frame shift → stop | Present study; ref. 30 | |
| 1200-3 | Ex 6 Δ 2bp
Second mutation not found |
Frame shift | Present study |
| DB | Ex2 G → A | Trp100Stop | Present study |
| Ex6 G → A | Splice site mutation | Present study; refs. 24 and 33 |
Mutational analysis of CYP27 gene
Mutational analysis was carried out in all 13 CTX probands, using PCR amplification and direct sequencing. All nine exons of the CYP27 gene were amplified and sequenced for each proband. From 13 CTX probands, 21 point mutations and 2 deletions were detected (Table 2). Two deletion mutations, suspected from the results of the sequence traces, were confirmed by cloning of PCR products and sequencing of selected cloned (Fig. 4). Both of the deletion mutations led to frame-shift and premature chain termination. The 10-bp deletion in exon 1 (proband 600-9; Table 2) led to a premature codon soon after the deletion site and no protein was expected to be synthesized, as it would code for a putative 5-amino acid polypeptide. Although the 2-bp deletion in exon 6 (proband 1200-3; Table 2) also led to premature chain termination, a theoretical 410-amino acid polypeptide (~46 kDa) could result, but would be devoid of the cofactor binding domains and thus would lead to a protein with no enzyme activity. In total, 21 point mutations were identified from 12 probands containing 7 missense mutations, 3 nonsense mutations, and 11 point mutations in exon/intron boundary sites (Table 2). Eleven probands carried heterozygous mutations and two probands carried homozygous mutations.
Fig. 4.
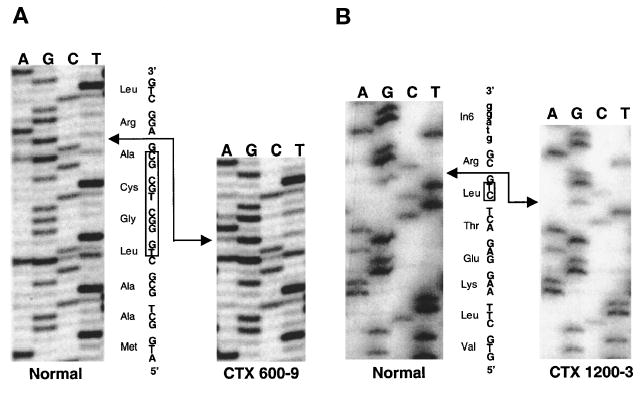
Deletion mutations affecting CYP27. Where deletions were suspected (see text), PCR products were cloned and sequenced. Representative sequence analyses for the 10-base pair (bp) (A) and the 2-bp (B) deletions are shown. Upper case indicates the coding region and lower case (B) indicates the intronic region. The boxes show the deleted bases.
A summary of the spectrum of mutations seen in our cohort, as well as those previously reported, is presented (see Table 2 and Fig. 5). Many of the mutations affected either mRNA splicing, or led to frame-shift and nonsense mutations that led to truncated translated products. A few of the mutations were missense mutations; these are indicated above the gene structure, Fig. 5. Note that although A183P (exon 3, 667G→C) and R372W/Q (exon 6, 1263+1G→A) were potential missense mutations, they have been shown to disrupt normal splicing (25, 30), and a full-length protein may not be synthesized. Five mutations affecting Pro351, Gly439, Arg441 (2 mutations), and Arg446, based solely on the primary polypeptide, affected the known adrenodoxin or heme-binding domains (shaded boxes, exons 6 and 8; Fig. 5). The remaining mutations lay outside these areas. To examine for a potential functional effect of all the missense mutations, a three-dimensional structure of CYP27 was constructed, on the basis of published high resolution crystal structures of homologous P450 enzymes.
Fig. 5.
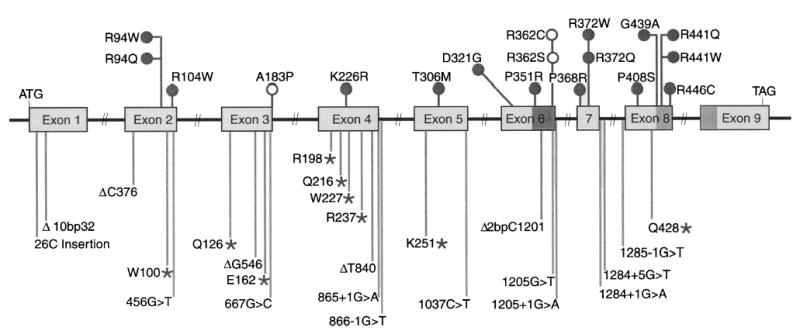
Mutations affecting CYP27 in CTX. Schematic summary of all published mutations affecting CYP27. Major deletions affecting the gene have been omitted for clarity. The adrenodoxin-binding (exon 6) and heme-binding (exons 8 and 9) domains are indicated by the shaded boxes. Missense mutations (open and closed circles) are indicated above the gene structure, and mutations that lead to frame shift, premature termination of translation (asterisks), or splicing abnormalities are indicated below. The mutations below the gene structure are likely to be null mutations. The mutations above the gene structure should lead to expressed protein (see text). Note that A183P and R362C or R362S (shown as open circles) are potentially missense mutations, but their effect is to disrupt splicing and thus the frame shift, resulting in truncated proteins [see text and ref. (25)].
Structural modeling
A multiple alignment between CYP27 and a number of closely related P450s was performed (Fig. 6). The 33-amino acid mitochondrial targeting sequence was removed and the CYP27 amino acid numbering was as previously utilized (52). Note that although Arg94 was not part of a previously recognized motif, it appeared to be highly conserved (Fig. 6 and see below).
Fig. 6.
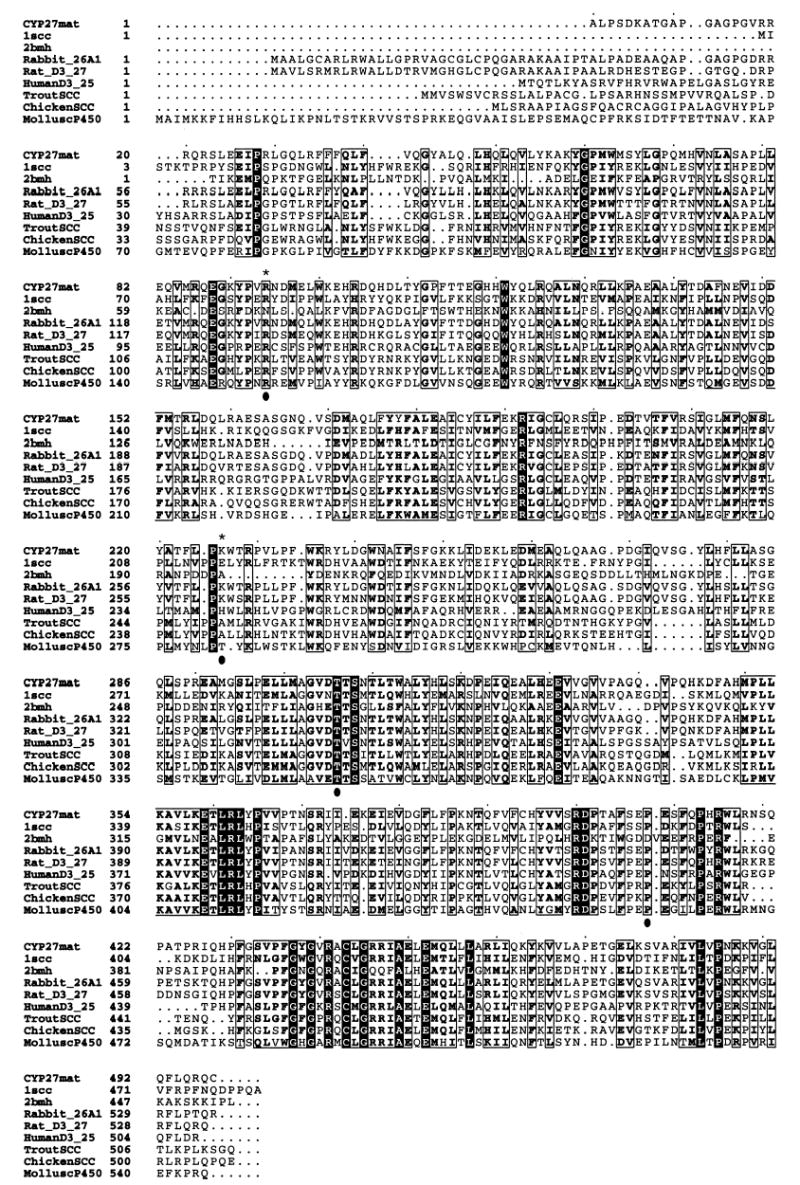
Multiple alignment of sequences homologous to CYP27. Multiple alignment using human CYP27 (CYP27mat), human cytochrome P450 side chain-cleavage enzyme (1scc), Pseudomonas putida cytochrome P450cam (2bmh), Oryctolagus cuniculus CYP26 (Rabbit_26A1), Rattus norvegicus CYP27 (Rat_D3_27), 25-hydroxyvitamin D3-α-hydroxylase (HumanD3_25), Oncorhynchus mykiss P450 monooxygenase (TroutSCC), chicken P450 monooxygenase (ChickenSCC), and Lymnaea stagnalis cytochrome P450 (MolluscP450) was performed using the GCG Pileup program. An asterisk indicates two residues, Arg94 and Lys226, using the human CYP27mat numbering. The adrenodoxin domain is contained between residues 351 and 365 and the heme-binding domain is contained between residues 435 and 464. Boxed residues indicate high areas of homology, with darkened areas indicating completely conserved residues. Note that Arg94, Thr306, and Pro408 are highly conserved, but Lys226 is not (indicated by filled circles). However, Lys226 is adjacent to a highly conserved proline at residue 225 (see also Fig. 7B).
Using P450SCC as the 3-D template, the various 3-D modeling algorithms agreed in the overall predicted fold pattern for CYP27 (Fig. 7A). The RMSD comparing backbone atoms for all three areas of interest was 1.2558 for the GeneFold model, 0.6252 for the SwissModel, and 1.1201 for the LOOK model. All the models preserved the heme-binding pocket. The RMSD comparing backbone atoms of the heme site were 1.0918 for GeneFold, 0.1235 for SwissModel, and 0.8339 for the LOOK structure. The MatchMaker energy score for SwissModel structure was −0.04 kT. For the GeneFold model the MatchMaker score was 0.0 kT. For the LOOK model the MatchMaker score was −0.02 kT. For comparison, the MatchMaker energy score for the P450SCC model (used as a template for CYP27 by all the programs) was −0.03 kT. The models therefore agreed with one another reasonably well. Figure 7A shows a superimposed image of the predicted sterol 27-hydroxylase on the crystal structure of P450SCC. The predicted structure, except for two areas (Fig. 7A), agreed well. Of the two discrepant areas, one of these was a putative membrane association loop for P450SCC (44); membrane association of CYP27 has been previously reported (53). The second area involved a short loop for which there were insufficient data as to whether this domain may or may not be biologically important.
Fig. 7.
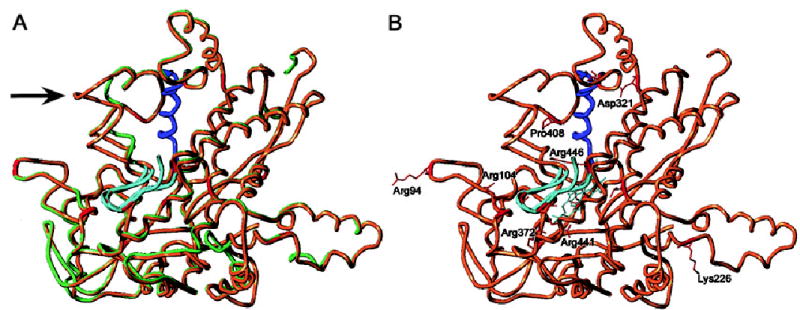
Structural modeling and mapping of missense mutations of CYP27. A three-dimensional structure was constructed, using P450SCC as template. A: The P450SCC structure (light green) superimposed on the predicted CYP27 structure (brown), to examine for goodness-of-fit. The arrow highlights an area where there is a discrepancy in the goodness-of-fit (A, see text for discussion). B: The sites of the missense mutations are shown in red and the two conserved domains, the adrenodoxin-binding site (residues 351 –365) and the heme-binding site (residues 435–464), are shown in blue and cyan, respectively. All but two of the missense mutations (indicated in red) are located in these two domains. Only a handful of the missense mutations are labeled for clarity. Two missense mutations, Arg94Trp and Lys226Arg, are located outside of these domains (see text for discussion). Note that Pro408 (highly conserved, see Fig. 6) and Asp321are now located in an area that could affect adrenodoxin binding, and Thr306 (highly conserved, not labeled) is likely to disrupt the heme domain.
Known point mutations were mapped onto the models and demonstrate that several missense mutations occurred in the two areas, the adrenodoxin- and heme-binding domains, predicted a priori to be critical to protein function (see Figs. 6 and 7B). Arg104, Arg368, and Arg372 (which on a linear polypeptide map outside the identified functional domains) lined the interior of the heme-binding region of the models. Arg94 and Pro408 appeared to be at the surface of the molecule on the same hemisphere as the heme-binding site. More significantly, Pro408 lay within hydrogen-bonding distance of Arg362, which has been identified as the adrenodoxin-binding site (35), and this mutation can be predicted to affect the active site. On the other hand, Lys226 was also a surface residue but was modeled as lying on the opposite face of the model from the heme cavity. This suggests either flaws in the models (GeneFold, SwissModel, and LOOK all place these residues similarly, however, when using P450SCC as a template; and see Fig. 7) or perhaps indicate additional functional properties of the protein (particularly for the Arg94 and Lys226 sites), which remain to be elucidated (see below).
Overall, the pattern of missense mutations, when mapped onto the model, demonstrated that various areas predicted a priori to be critical for enzyme activity did indeed map at functional sites. Figure 7B shows that most of the missense mutations occurred in the heme-binding site (Arg104Trp, Thr306Met, Pro368Arg, Arg372Trp, Arg441Glu, Arg441Trp, and Arg446Cys) or in the adrenodoxin-binding site (Asp321Gly, Pro351Leu, and Pro408Ser). All of these residues were displayed (indicated in red; Fig. 7B), although only a few labeled for clarity.
However, there were two missense mutations, Arg94Trp and Lys226Arg, that were located outside these areas, and the three-dimensional structure of CYP27 shows that these missense mutations did not seem to be involved in the heme-binding or adrenodoxin-binding domain (Fig. 7B). Interestingly, Arg94 appeared to be highly conserved among the various P450s (see Fig. 6), although this residue has not been previously implicated in the active site of the enzyme. Although Lys226 was not conserved (Fig. 6), it was preceded by a stretch of hydrophobic residues (see below).
DISCUSSION
There are only two ways to remove cholesterol from the body: either by direct excretion of cholesterol into bile (a minor pathway), or by conversion of cholesterol and its metabolites to bile acids and excretion into bile. Disorders of bile acid synthesis, although rare, are important in the study of cholesterol loss from the body. One such disorder is CTX. This rare, autosomal, recessively inherited condition is caused by mutations affecting a mitochondrial cytochrome P450 enzyme, sterol 27-hydroxylase, encoded by the CYP27 gene. Suspicion of this diagnosis is raised when a patient presents with tendon xanthomas, but does not exhibit elevated plasma cholesterol levels diagnostic of familial hypercholesterolemia; or when the patient manifests neurological dysfunction; or through screening of family members, when one affected sibling has been identified. Diagnosis is based both on clinical presentation and detection of elevated blood cholestanol levels and the absence of elevated plant sterols. The latter is important as another genetic condition, phytosterolemia, can also present in a similar manner, although neurological symptoms are not a feature of phytosterolemia (4). Diagnosis of CTX is based on elevations of plasma cholestanol levels, a cholesterol metabolite that is normally present in low concentrations, and more specifically by the demonstration of 25-hydroxylated C-27 bile alcohols, such as 5β-cholestane-3α,7α,12α,23R,25-pentol and 5β-cholestane-3α,7α,12α,24R,25-pentol in blood, urine, and feces (4). Untreated, patients with CTX accumulate cholesterol, as well as cholestanol in tissues, including brain and macrophages. Progressive neurological dysfunction, as well as atherosclerotic disease, may result. The pathophysiology, in part, involves the elevation of toxic bile alcohols in the plasma, damaging the blood–brain barrier. Inhibition of the bile alcohol production by the liver through exogenous bile acid supplementation results in restoration of the blood–brain barrier and an improvement of the neurological symptoms in many cases (4).
We have identified 12 new families with CTX; the patients/probands were diagnosed as having CTX on the basis of clinical and biochemical criteria, and were found to have mutations in CYP27. Genetic heterogeneity is an issue, as the bile acid biosynthesis involves a number of enzymes, many of which have not been fully characterized. Theoretically, it is possible that any one of these other enzymes could be genetically deficient, leading to a similar phenotype. On the basis of haplotype analyses of 15 microsatellite markers and mutation analysis in these pedigrees, there was no evidence of genetic heterogeneity for this rare disease. Using recombination and linkage analyses, we further fine-mapped the CYP27 gene locus to between markers D2S2249-D2S433. These markers are highly polymorphic and allow for rapid haplotype analyses. This information may be useful in segregation of future clinically diagnosed patients to examine for any evidence of genetic heterogeneity, as well as for carrier identification in the families of affected individuals. Genetic heterogeneity remains a possibility, because the clinical presentation and severity for CTX are variable and, to date, there appears to be little correlation between genotype and phenotype (39, 54). Thus patients may be diagnosed at an early age (7–10), or well into adulthood, and the symptoms/signs may range from being asymptomatic, to the presence of tendon xanthomas only, or to early neurological dysfunction (39, 54). In the present study, 23 mutations were identified from 13 probands with CTX; 10 probands were compound heterozygotes, and 2 probands were homozygous for mutations. Five of these mutations were novel and not previously reported.
A better understanding of the structure-function relationships may also allow for more insight into this disease. One approach is to identify missense mutations together with structural modeling to predict functional domains. CYP27 is a member of the larger cytochrome P450 family and two functional domains, the adrenodoxin-binding site (residues 384–395) and the heme-binding site (residues 468–497), have been identified (14–16, 55). In vitro transfection studies of mutant cDNAs that carry the Arg446Cys mutation (heme-binding domain) in COS cells showed low or undetectable levels of CYP27 enzyme activity (13, 19). Structural mapping, using computer modeling, of the mutated missense amino acid residues in three-dimensional structure of CYP27 allowed for prediction of the effects of these mutations on function. Most missense mutations that initially mapped to outside the conserved heme- and adrenodoxin-binding sites were shown to affect these domains. However, we found two missense mutations that were located outside of the adrenodoxin- and heme-binding domains and may throw some light on the structural complexities and enzyme-substrate specificities of CYP27.
Arg94 is a highly conserved residue that does not appear to map into the putative active site. Vijaykumar and Salerno (44) have reported that all three enzymatic reactions for P450SCC (hydroxylation of C-22, hydroxylation at C-20, and oxidative cleavage between C-20 and C-22 to yield pregnenolone) occur at the heme active site. By extrapolation, the multiple enzymatic reactions known for CYP27 are also predicted to involve the heme site. Thus the identification of Arg94 and its predicted map positioning at a site well away from the active site suggests that either the substrate pocket for CYP27 involves this surface residue, or this area has an unidentified biological function.
The mutation Lys226Arg is interesting. In the homologous region in P450SCC, this region is known to mediate membrane association. Membrane association has been reported for sterol 27-hydroxylase (53), but whether this association is necessary for enzyme action is not known. By inference, CYP27 should be associated with the membrane and this association is critical for enzyme function, if the biological effect of the mutation were to disrupt membrane association.
In vitro expression studies, using site-directed mutagenesis, should be conducted to understand whether these mutations affect any additional functional domains or interaction with the enzyme substrate specificities at the tertiary structure level.
The analyses reported in this study shed light on some of the missense mutations, as they map into the adrenodoxin- and heme-binding domains, crucial for enzymatic activity. In addition, two sites, Arg94 and Lys226, would suggest that these residues play a important role in enzyme activity. Further experiments will be needed to explain why these inactivate CYP27 and may reveal new aspects of this interesting enzyme.
ELECTRONIC DATABASES
BLAST server: http://www.ncbi.nlm.nih.gov/BLAST/
Protein data: http://www.ncbi.nlm.nih.gov/
Rabbit sterol 26-monooxygenase, accession number A33813; rat cholesterol 26-hydroxylase, accession number S09198; human 25-hydroxyvitamin D3-α-hydroxylase, accession number JC5713; trout cholesterol monooxygenase (side chain-cleaving), accession number S32197; chicken cytochrome P450scc, accession number JC6200; mollusk cytochrome P450 (CYPIO), accession number JX0225.
SwissModel Server: http://www.expasy.ch/spdbv/mainpage.htm
LOOK program (Molecular Applications Group): http://www.mag.com/products/look.html
Acknowledgments
The authors thank Sijing Niu and Teresa Kearns for help with the DNA sequencing, and the reviewers for many helpful suggestions. This work was supported by National Institutes of Health grants HL53917 ( J.C.), HL60613 (S.B.P.), and MO1 RR01070-25 (MUSC GCRC), and by the American Heart Association Scientist Development Award 9730087N (S.B.P.).
References
- 1.Bjorkhem I. Mechanism of degradation of the steroid side chain in the formation of bile acids. J Lipid Res. 1992;33:455–471. [PubMed] [Google Scholar]
- 2.Wikvall K. Hydroxylations in biosynthesis of bile acids. Isolation of a cytochrome P-450 from rabbit liver mitochondria catalyzing 26-hydroxylation of C27-steroids. J Biol Chem. 1984;259:3800–3804. [PubMed] [Google Scholar]
- 3.Okuda KI. Liver mitochondrial P450 involved in cholesterol catabolism and vitamin D activation [review] J Lipid Res. 1994;35:361–372. [PubMed] [Google Scholar]
- 4.Bjorkhem, I., and K. Muri-Boberg. 1995. Inborn errors in bile acid biosynthesis and storage of sterols other than cholesterol. In Metabolic Basis of Inherited Diseases. C. R. Scriver, A. L. Beaudet, W. S. Sly, and D. Valle, eds. McGraw-Hill, New York. 2073–2099.
- 5.Philippart M, Van Bogaert L. Cholestanolosis (cerebrotendinous xanthomatosis). A follow-up study on the original family. Arch Neurol. 1969;21:603–610. doi: 10.1001/archneur.1969.00480180059004. [DOI] [PubMed] [Google Scholar]
- 6.Salen G. Cholestanol deposition in cerebrotendinous xanthomatosis. A possible mechanism. Ann Intern Med. 1971;75:843–851. doi: 10.7326/0003-4819-75-6-843. [DOI] [PubMed] [Google Scholar]
- 7.Cruysberg JR, Wevers RA, Tolboom JJ. Juvenile cataract associated with chronic diarrhea in pediatric cerebrotendinous xanthomatosis [letter] Am J Ophthalmol. 1991;112:606–607. doi: 10.1016/s0002-9394(14)76874-6. [DOI] [PubMed] [Google Scholar]
- 8.Wevers RA, Cruysberg JR, van Heijst AF, Janssen-Zijlstra FS, Renier WO, van Engelen BG, Tolboom JJ. Paediatric cerebrotendinous xanthomatosis. J Inherit Metab Dis. 1992;15:374–376. doi: 10.1007/BF02435980. [DOI] [PubMed] [Google Scholar]
- 9.van Heijst AF, Wevers RA, Tangerman A, Cruysberg JR, Renier WO, Tolboom JJ. Chronic diarrhoea as a dominating symptom in two children with cerebrotendinous xanthomatosis. Acta Paediatr. 1996;85:932–936. doi: 10.1111/j.1651-2227.1996.tb14189.x. [DOI] [PubMed] [Google Scholar]
- 10.van Heijst AF, Verrips A, Wevers RA, Cruysberg JR, Renier WO, Tolboom JJ. Treatment and follow-up of children with cerebrotendinous xanthomatosis. Eur J Pediatr. 1998;157:313–316. doi: 10.1007/s004310050818. [DOI] [PubMed] [Google Scholar]
- 11.Verrips A, van Engelen BG, ter Laak H, Gabreels-Festen A, Janssen A, Zwarts M, Wevers RA, Gabreels FJ. Cerebrotendinous xanthomatosis. Controversies about nerve and muscle: observations in ten patients. Neuromuscul Disord. 2000;10:407–414. doi: 10.1016/s0960-8966(00)00112-7. [DOI] [PubMed] [Google Scholar]
- 12.Andersson S, Davis DL, Dahlback H, Jornvall H, Russell DW. Cloning, structure, and expression of the mitochondrial cytochrome P-450 sterol 26-hydroxylase, a bile acid biosynthetic enzyme. J Biol Chem. 1989;264:8222–8229. [PubMed] [Google Scholar]
- 13.Cali JJ, Hsieh CL, Francke U, Russell DW. Mutations in the bile acid biosynthetic enzyme sterol 27-hydroxylase underlie cerebrotendinous xanthomatosis. J Biol Chem. 1991;266:7779–7783. [PMC free article] [PubMed] [Google Scholar]
- 14.Leitersdorf E, Reshef A, Meiner V, Levitzki R, Schwartz SP, Dann EJ, Berkman N, Cali JJ, Klapholz L, Berginer VM. Frameshift and splice-junction mutations in the sterol 27-hydroxylase gene cause cerebrotendinous xanthomatosis in Jews of Moroccan origin. J Clin Invest. 1993;91:2488–2496. doi: 10.1172/JCI116484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gonzalez FJ. The molecular biology of cytochrome P450s [review] [published erratum appears in Pharmacol. Rev. 1989; 41:91–92] Pharmacol Rev. 1988;40:243–288. [PubMed] [Google Scholar]
- 16.Tuls J, Geren L, Millett F. Fluorescein isothiocyanate specifically modifies lysine 338 of cytochrome P-450scc and inhibits adrenodoxin binding. J Biol Chem. 1989;264:16421–16425. [PubMed] [Google Scholar]
- 17.Nakashima N, Sakai Y, Sakai H, Yanase T, Haji M, Umeda F, Koga S, Hoshita T, Nawata H. A point mutation in the bile acid biosynthetic enzyme sterol 27-hydroxylase in a family with cerebrotendinous xanthomatosis. J Lipid Res. 1994;35:663–668. [PubMed] [Google Scholar]
- 18.Reshef A, Meiner V, Berginer VM, Leitersdorf E. Molecular genetics of cerebrotendinous xanthomatosis in Jews of north African origin. J Lipid Res. 1994;35:478–483. [PubMed] [Google Scholar]
- 19.Kim KS, Kubota S, Kuriyama M, Fujiyama J, Bjorkhem I, Eggertsen G, Seyama Y. Identification of new mutations in sterol 27-hydroxylase gene in Japanese patients with cerebrotendinous xanthomatosis (CTX) J Lipid Res. 1994;35:1031–1039. [PubMed] [Google Scholar]
- 20.Meiner V, Meiner Z, Reshef A, Bjorkhem I, Leitersdorf E. Cerebrotendinous xanthomatosis: molecular diagnosis enables presymptomatic detection of a treatable disease. Neurology. 1994;44:288–290. doi: 10.1212/wnl.44.2.288. [DOI] [PubMed] [Google Scholar]
- 21.Meiner V, Marais DA, Reshef A, Bjorkhem I, Leitersdorf E. Premature termination codon at the sterol 27-hydroxylase gene causes cerebrotendinous xanthomatosis in an Afrikaner family. Hum Mol Genet. 1994;3:193–194. doi: 10.1093/hmg/3.1.193. [DOI] [PubMed] [Google Scholar]
- 22.Segev H, Reshef A, Clavey V, Delbart C, Routier G, Leitersdorf E. Premature termination codon at the sterol 27-hydroxylase gene causes cerebrotendinous xanthomatosis in a French family. Hum Genet. 1995;95:238–240. doi: 10.1007/BF00209413. [DOI] [PubMed] [Google Scholar]
- 23.Verrips A, Steenbergen-Spanjers GC, Luyten JA, van den Heuvel LP, Keyser A, Gabreels FJ, Wevers RA. Two new mutations in the sterol 27-hydroxylase gene in two families lead to cerebrotendinous xanthomatosis. Hum Genet. 1996;98:735–737. doi: 10.1007/s004390050294. [DOI] [PubMed] [Google Scholar]
- 24.Chen W, Kubota S, Nishimura Y, Nozaki S, Yamashita S, Nakagawa T, Kameda-Takemura K, Menju M, Matsuzawa Y, Bjorkhem I, Eggertsen G, Seyama Y. Genetic analysis of a Japanese cerebrotendinous xanthomatosis family: identification of a novel mutation in the adrenodoxin binding region of the CYP 27 gene. Biochim Biophys Acta. 1996;1317:119–126. doi: 10.1016/s0925-4439(96)00043-9. [DOI] [PubMed] [Google Scholar]
- 25.Garuti R, Lelli N, Barozzini M, Tiozzo R, Dotti MT, Federico A, Ottomano AM, Croce A, Bertolini S, Calandra S. Cerebrotendinous xanthomatosis caused by two new mutations of the sterol-27-hydroxylase gene that disrupt mRNA splicing. J Lipid Res. 1996;37:1459–1467. [PubMed] [Google Scholar]
- 26.Okuyama E, Tomita S, Takeuchi H, Ichikawa Y. A novel mutation in the cytochrome P450(27) (CYP27) gene caused cerebrotendinous xanthomatosis in a Japanese family. J Lipid Res. 1996;37:631–639. [PubMed] [Google Scholar]
- 27.Garuti R, Lelli N, Barozzini M, Dotti MT, Federico A, Bertolini S, Calandra S. Partial deletion of the gene encoding sterol 27-hydroxylase in a subject with cerebrotendinous xanthomatosis. J Lipid Res. 1996;37:662–672. [PubMed] [Google Scholar]
- 28.Watts GF, Mitchell WD, Bending JJ, Reshef A, Leitersdorf E. Cerebrotendinous xanthomatosis: a family study of sterol 27-hydroxylase mutations and pharmacotherapy. Q J Med. 1996;89:55–63. doi: 10.1093/oxfordjournals.qjmed.a030138. [DOI] [PubMed] [Google Scholar]
- 29.Chen W, Kubota S, Kim KS, Cheng J, Kuriyama M, Eggertsen G, Bjorkhem I, Seyama Y. Novel homozygous and compound heterozygous mutations of sterol 27-hydroxylase gene (CYP27) cause cerebrotendinous xanthomatosis in three Japanese patients from two unrelated families. J Lipid Res. 1997;38:870–879. [PubMed] [Google Scholar]
- 30.Garuti R, Croce MA, Tiozzo R, Dotti MT, Federico A, Bertolini S, Calandra S. Four novel mutations of sterol 27-hydroxylase gene in Italian patients with cerebrotendinous xanthomatosis. J Lipid Res. 1997;38:2322–2334. [PubMed] [Google Scholar]
- 31.Ahmed MS, Afsar S, Hentati A, Ahmad A, Pasha J, Juneja T, Hung WY, Ahmad A, Choudhri A, Saya S, Siddique T. A novel mutation in the sterol 27-hydroxylase gene of a Pakistani family with autosomal recessive cerebrotendinous xanthomatosis. Neurology. 1997;48:258–260. doi: 10.1212/wnl.48.1.258. [DOI] [PubMed] [Google Scholar]
- 32.Verrips A, Steenbergen-Spanjers GC, Luyten JA, Wevers RA, Wokke JH, Gabreels FJ, Wolthers BG, van den Heuvel LP. Exon skipping in the sterol 27-hydroxylase gene leads to cerebrotendinous xanthomatosis. Hum Genet. 1997;100:284–286. doi: 10.1007/s004390050506. [DOI] [PubMed] [Google Scholar]
- 33.Chen W, Kubota S, Teramoto T, Nishimura Y, Yonemoto K, Seyama Y. Silent nucleotide substitution in the sterol 27-hydroxylase gene (CYP 27) leads to alternative pre-mRNA splicing by activating a cryptic 5′ splice site at the mutant codon in cerebrotendinous xanthomatosis patients. Biochemistry. 1998;37:4420–4428. doi: 10.1021/bi972940a. [DOI] [PubMed] [Google Scholar]
- 34.Chen W, Kubota S, Seyama Y. Alternative pre-mRNA splicing of the sterol 27-hydroxylase gene (CYP 27) caused by a G to A mutation at the last nucleotide of exon 6 in a patient with cerebrotendinous xanthomatosis (CTX) J Lipid Res. 1998;39:509–517. [PubMed] [Google Scholar]
- 35.Chen W, Kubota S, Ujike H, Ishihara T, Seyama Y. A novel Arg362Ser mutation in the sterol 27-hydroxylase gene (CYP27): its effects on pre-mRNA splicing and enzyme activity. Biochemistry. 1998;37:15050–15056. doi: 10.1021/bi9807660. [DOI] [PubMed] [Google Scholar]
- 36.Shiga K, Fukuyama R, Kimura S, Nakajima K, Fushiki S. Mutation of the sterol 27-hydroxylase gene (CYP27) results in truncation of mRNA expressed in leucocytes in a Japanese family with cerebrotendinous xanthomatosis. J Neurol Neurosurg Psychiatry. 1999;67:675–677. doi: 10.1136/jnnp.67.5.675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wakamatsu N, Hayashi M, Kawai H, Kondo H, Gotoda Y, Nishida Y, Kondo R, Tsuji S, Matsumoto T. Mutations producing premature termination of translation and an amino acid substitution in the sterol 27-hydroxylase gene cause cerebrotendinous xanthomatosis associated with parkinsonism. J Neurol Neurosurg Psychiatry. 1999;67:195–198. doi: 10.1136/jnnp.67.2.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Verrips A, Nijeholt GJ, Barkhof F, Van Engelen BG, Wesseling P, Luyten JA, Wevers RA, Stam J, Wokke JH, van den Heuvel LP, Keyser A, Gabreels FJ. Spinal xanthomatosis: a variant of cerebrotendinous xanthomatosis. Brain. 1999;122:1589–1595. doi: 10.1093/brain/122.8.1589. [DOI] [PubMed] [Google Scholar]
- 39.Verrips A, Hoefsloot LH, Steenbergen GC, Theelen JP, Wevers RA, Gabreels FJ, van Engelen BG, van den Heuvel LP. Clinical and molecular genetic characteristics of patients with cerebrotendinous xanthomatosis. Brain. 2000;123:908–919. doi: 10.1093/brain/123.5.908. [DOI] [PubMed] [Google Scholar]
- 40.Berginer VM, Salen G, Shefer S. Long-term treatment of cerebrotendinous xanthomatosis with chenodeoxycholic acid. N Engl J Med. 1984;311:1649–1652. doi: 10.1056/NEJM198412273112601. [DOI] [PubMed] [Google Scholar]
- 41.Patel SB, Honda A, Salen G. Sitosterolemia: exclusion of genes involved in reduced cholesterol biosynthesis. J Lipid Res. 1998;39:1055–1061. [PubMed] [Google Scholar]
- 42.Kruglyak L, Daly MJ, Reeve-Daly MP, Lander ES. Parametric and nonparametric linkage analysis: a unified multi-point approach. Am J Hum Genet. 1996;58:1347–1363. [PMC free article] [PubMed] [Google Scholar]
- 43.Nemeth-Slany A, Talmud P, Grundy SM, Patel SB. Activation of a cryptic splice-site in intron 24 leads to the formation of apolipoprotein B-27.6. Atherosclerosis. 1997;133:163–170. doi: 10.1016/s0021-9150(97)00105-6. [DOI] [PubMed] [Google Scholar]
- 44.Vijayakumar S, Salerno JC. Molecular modeling of the 3-D structure of cytochrome P-450scc. Biochim Biophys Acta. 1992;1160:281–286. doi: 10.1016/0167-4838(92)90089-v. [DOI] [PubMed] [Google Scholar]
- 45.Poulos TL, Finzel BC, Gunsalus IC, Wagner GC, Kraut J. The 2.6-Å crystal structure of Pseudomonas putida cytochrome P-450. J Biol Chem. 1985;260:16122–16130. [PubMed] [Google Scholar]
- 46.Blundell TL, Elliott G, Gardner SP, Hubbard T, Islam S, Johnson M, Mantafounis D, Murray-Rust P, Overington J, Pitts JE, Sali A, Sibanda BL, Singh J, Sternberg MJE, Sutcliffe MJ, Thornton JM, Travers P. Protein engineering and design. Philos Trans R Soc Lond B Biol Sci. 1989;324:447–460. doi: 10.1098/rstb.1989.0059. [DOI] [PubMed] [Google Scholar]
- 47.Blundell TL. Problems and solutions in protein engineering-towards rational design [editorial] Trends Biotechnol. 1994;12:145–148. doi: 10.1016/0167-7799(94)90073-6. [DOI] [PubMed] [Google Scholar]
- 48.Johnson MS, Srinivasan N, Sowdhamini R, Blundell TL. Knowledge-based protein modeling. Crit Rev Biochem Mol Biol. 1994;29:1–68. doi: 10.3109/10409239409086797. [DOI] [PubMed] [Google Scholar]
- 49.Zhang L, Godzik A, Skolnick J, Fetrow JS. Functional analysis of the Escherichia coli genome for members of the alpha/beta hydrolase family. Folding Design. 1998;3:535–548. doi: 10.1016/s1359-0278(98)00069-8. [DOI] [PubMed] [Google Scholar]
- 50.Zhang B, Rychlewski L, Pawlowski K, Fetrow JS, Skolnick J, Godzik A. From fold predictions to function predictions: automation of functional site conservation analysis for functional genome predictions. Protein Sci. 1999;8:1104–1115. doi: 10.1110/ps.8.5.1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Godzik A, Skolnick J. Sequence-structure matching in globular proteins: application to supersecondary and tertiary structure determination. Proc Natl Acad Sci USA. 1992;89:12098–12102. doi: 10.1073/pnas.89.24.12098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cali JJ, Russell DW. Characterization of human sterol 27-hydroxylase. A mitochondrial cytochrome P-450 that catalyzes multiple oxidation reaction in bile acid biosynthesis. J Biol Chem. 1991;266:7774–7778. [PubMed] [Google Scholar]
- 53.Taniguchi S, Hoshita N, Okuda K. Enzymatic characteristics of CO-sensitive 26-hydroxylase system for 5beta-cholestane-3alpha,7alpha,12alpha-triol in rat-liver mitochondria and its intramitochondrial localization. Eur J Biochem. 1973;40:607–617. doi: 10.1111/j.1432-1033.1973.tb03233.x. [DOI] [PubMed] [Google Scholar]
- 54.Leitersdorf E, Safadi R, Meiner V, Reshef A, Bjorkhem I, Friedlander Y, Morkos S, Berginer VM. Cerebrotendinous xanthomatosis in the Israeli Druze: molecular genetics and phenotypic characteristics. Am J Hum Genet. 1994;55:907–915. [PMC free article] [PubMed] [Google Scholar]
- 55.Gonzalez FJ. Molecular genetics of the P-450 superfamily [review] Pharmacol Ther. 1990;45:1–38. doi: 10.1016/0163-7258(90)90006-n. [DOI] [PubMed] [Google Scholar]