

Abstract
The activation peptide of vertebrate trypsinogens contains a highly conserved tetra-aspartate sequence (Asp19–22 in humans) preceding the Lys-Ile scissile bond. A large body of research has defined the primary role of this acidic motif as a specific recognition site for enteropeptidase, the physiological activator of trypsinogen. In addition, the acidic stretch was shown to contribute to the suppression of autoactivation. In the present study we determined the relative importance of these two activation peptide functions in human cationic trypsinogen. Individual Ala-replacements of Asp19–22 had minimal or no effect on trypsinogen activation catalyzed by human enteropeptidase. Strikingly, a tetra-Ala19–22 trypsinogen mutant devoid of acidic residues in the activation peptide was still a highly specific substrate for human, but not for bovine, enteropeptidase. In contrast, an intact Asp19–22 motif was critical for autoactivation control. Thus, single-Ala mutations of Asp19, Asp20 and Asp21 resulted in 2–3-fold increased autoactivation, whereas the Asp22→Ala mutant autoactivated at a 66-fold increased rate. These effects were multiplicative in the tri-Ala19–21 and tetra-Ala19–22 mutants. Structural modeling revealed that the conserved hydrophobic S2 subsite of trypsin and the unique Asp218, which forms part of the S3-S4 subsite, participate in distinct inhibitory interactions with the activation peptide. Finally, mutagenesis studies confirmed the significance of the negative charge of Asp218 in autoactivation control. The results demonstrate that in human cationic trypsinogen the Asp19–22 motif per se is not required for enteropeptidase recognition, whereas it is essential for maximal suppression of autoactivation. The evolutionary selection of Asp218, which is absent in the large majority of vertebrate trypsins, provides an additional mechanism of autoactivation control in the human pancreas.
Digestive trypsins are synthesized and secreted by the pancreas as inactive precursors. Physiological activation of trypsinogen takes place in the duodenum, where enteropeptidase (enterokinase) specifically cleaves the Lys23-Ile24 peptide bond [see refs 1, 2, and references therein], which corresponds to Lys15-Ile16 in the chymotrypsin-based numbering system (chymo#). The activating cleavage removes a typically 8 amino-acid long activation peptide. In vertebrate trypsinogens, the activation peptide contains a highly conserved tetra-aspartate sequence next to the scissile peptide bond (Fig 1). Experiments with synthetic peptides and protein substrates indicated that the acidic residues are required for enteropeptidase recognition and cleavage [1–7]. Due to its highly specific extended subsite interactions, in analogy to restriction endonucleases, enteropeptidase became known as a restriction protease, and the Asp-Asp-Asp-Asp-Lys recognition site has been widely utilized as a protein engineering tool for the specific cleavage of fusion proteins. Definitive evidence that the Asp19–22 motif of the activation peptide participates in essential subsite interactions with enteropeptidase came from the crystal structure of the bovine enteropeptidase catalytic subunit complexed with an inhibitor analog of the activation peptide, Val-Asp-Asp-Asp-Asp-Lys-chloromethane [8]. The structure demonstrated that the P2 and P4 Asp residues (corresponding to Asp22 and Asp20 in Fig 1) formed salt-bridges with Lys99 (chymo#), a unique basic exosite on the catalytic subunit of enteropeptidase. In addition, the P3 Asp (Asp21 in Fig 1) was hydrogen bonded to the hydroxyl group of Tyr174 (chymo#). The P5 Asp (Asp19 in Fig 1) was disordered in the structure. In accordance with structural predictions, mutation of Lys99 in the catalytic subunit of enteropeptidase abolished trypsinogen activation. Although the crystal structure suggested an important role for at least 3 of the 4 Asp residues in enteropeptidase recognition, a number of studies using various protein substrates or synthetic peptides indicated that a minimal recognition sequence for enteropeptidase consists of a Lys/Arg at P1 and an Asp/Glu at P2, while the P3-P5 acidic residues might enhance activity [1–7]. Consistent with the critical role of the P2 Asp was the recent observation that the D22G mutant of human cationic trypsinogen was resistant to activation by bovine enteropeptidase [9].
Figure 1.
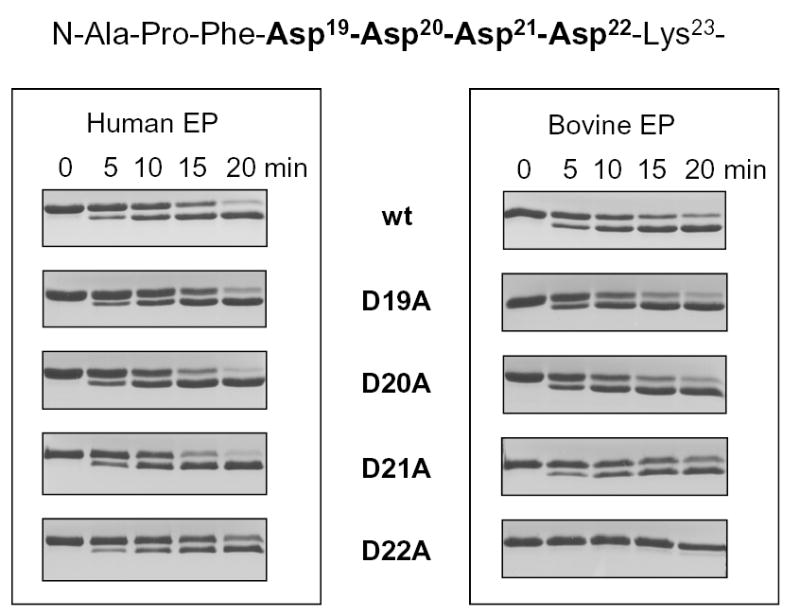
Activation of single-alanine mutants in the Asp19–22 motif by enteropeptidase (EP). The activation peptide sequence of wild-type human cationic trypsinogen is indicated, with the mutated positions highlighted. Activation of wild-type (wt) and D19A, D20A, D21A and D22A trypsinogens (2 μM concentration) was carried out with 50 ng/mL (0.45 nM) bovine or 10 ng/mL (0.13 nM) human enteropeptidase at 37 °C, in 0.1 M Tris-HCl (pH 8.0), 1 mM CaCl2 and 120 nM soybean trypsin inhibitor (final concentrations). Samples were precipitated with trichloroacetic acid, and analyzed by 13 % reducing SDS-PAGE gels and Coomassie-blue staining. The relevant portions of representative gels (n=3–5), demonstrating the trypsinogen→trypsin mobility shift are shown. The calculated activation rates by 0.13 nM human enteropeptidase were 85 nM/min for wild-type, D19A, D20A and D21A trypsinogens; and 70 nM/min for the D22A mutant. Using 0.45 nM bovine enteropeptidase, these values were 96 nM/min and 4 nM/min, respectively. Note that the wild-type and mutant trypsinogens all contained the inactivating S200A mutation, therefore autoactivation did not interfere with the assay.
The inhibitory function of the trypsinogen activation peptide on trypsin-mediated trypsinogen activation (autoactivation) was first demonstrated by chemical modification of the Asp residues in the activation peptide, which greatly enhanced autoactivation [10]. Subsequently, tryptic digestion of synthetic model peptides indicated that Asp residues are not favored in the P2-P5 positions [11, 12]. More recently, biochemical characterization of pancreatitis-associated activation peptide mutations in human cationic trypsinogen confirmed the importance of Asp residues in the activation peptide in autoactivation control. A model peptide with the D22G mutation was cleaved by bovine trypsin at a higher rate compared to the wild type activation peptide [13], and recombinant human trypsinogens carrying the D19A or D22G mutations exhibited markedly increased autoactivation [9]. Taken together with previous data, these findings indicated that the tetra-Asp sequence in the mammalian trypsinogen activation peptides has evolved for both efficient inhibition of trypsinogen autoactivation within the pancreas and optimal enteropeptidase recognition in the duodenum. However, to provide further experimental support to this notion, quantitative comparison of enteropeptidase- and trypsin-mediated trypsinogen activation, combined with systematic mutagenesis of the trypsinogen activation peptide, was necessary.
In the present study, the role of the Asp19–22 motif in the dual functionality of the activation peptide was investigated by site-directed mutagenesis in human cationic trypsinogen. This human trypsinogen isoform exhibits an unusually high propensity for autoactivation, to the extent that inborn mutations which moderately increase autoactivation cause hereditary pancreatitis [14–18]. Our findings demonstrate that the primary function of the tetra-Asp sequence in the activation peptide of human cationic trypsinogen is suppression of autoactivation. Two inhibitory interactions have been identified; one between Asp22 of trypsinogen and the conserved hydrophobic S2 subsite of trypsin and another between the unique Asp218 exosite and Asp19–21 of the activation peptide. Together, these interactions can suppress the rate of autoactivation by more than 2 orders of magnitude. In contrast, the Asp19–22 sequence is not required for enteropeptidase recognition, and confers only a modest catalytic improvement to enteropeptidase mediated trypsinogen activation in humans.
EXPERIMENTAL PROCEDURES
Materials
N-CBZ-Gly-Pro-Arg-p-nitroanilide was from Sigma (St. Louis, Missouri, USA), and ultrapure bovine enterokinase from Biozyme Laboratories (San Diego, California, USA). Recombinant human pro-enteropeptidase was from R & D Systems (Minneapolis, Minnesota, USA). Human pro-enteropeptidase (0.07 mg/mL stock solution; 641 nM concentration) was activated with 50 nM human cationic trypsin in 0.1 M Tris-HCl (pH 8.0), 10 mM CaCl2 and 2 mg/ml bovine serum albumin (final concentrations) for 30 min at room temperature, and diluted 10-fold to obtain a working stock solution of 64 nM enteropeptidase concentration in 0.1 M Tris-HCl (pH 8.0), 1 mM CaCl2 and 2 mg/ml bovine serum albumin. The inclusion of bovine serum albumin was essential for the long-term stability of enteropeptidase. Soybean trypsin inhibitor was purchased from Fluka and purified to homogeneity on a bovine trypsin affinity column followed by MonoQ ion-exchange chromatography.
Nomenclature
The common names “cationic trypsinogen” and “anionic trypsinogen” are used to denote the two major human trypsinogen isoforms [19]. Note that these names reflect only the relative isoelectric points of the proenzymes, and, in fact, all human trypsinogens are anionic. Autoactivation of trypsinogen is a bimolecular self-amplifying process in which trypsin activates trypsinogen to trypsin (Trypsin + Trypsinogen → 2Trypsin). The term “autoactivation” is used throughout this study to describe trypsin-mediated trypsinogen activation. Amino acid residues in the human trypsinogen sequences are denoted according to their actual position in the native, wild-type preproenzyme, starting with Met1. Where indicated by the “chymo#” prefix, the chymotrypsin-based conventional numbering is also used.
Expression plasmids and mutagenesis
Construction of expression plasmids harboring the human cationic (protease, serine, 1 gene; PRSS1) and anionic (PRSS2) trypsinogen genes was described previously [16, 17, 20]. Single or combined mutations D20A, D21A, D22A, D19E, D20E, D21E, D22E, A3D (D19A/D20A/D21A), A4 (D19A/D20A/D21A/D22A), D218Y, D218H, D218S, D218E and S200A in the PRSS1 gene and mutation Y218D in the PRSS2 gene were introduced by oligonucleotide-directed site-specific PCR-mutagenesis. Mutant D19A was constructed previously [9]. The PCR products were digested with restriction endonucleases Xho I and Sac I (D218Y, D218H, D218S, D218E, Y218D and S200A) or with Nco I and EcoR I (D20A, D21A, D22A, D19E, D20E, D21E and D22E) and ligated into the similarly treated expression plasmids.
Expression and purification of recombinant trypsinogens
Trypsinogen mutants were expressed in the E. coli Rosetta(DE3) strain as cytoplasmic inclusion bodies. Trypsinogen was solubilized in guanidine-HCl, renatured in vitro, and purified to homogeneity using ecotin-affinity chromatography, as reported previously [16]. Trypsinogen preparations were stored on ice, in 50 mM HCl. Concentrations of cationic and anionic trypsinogen solutions were calculated from their ultraviolet absorbance at 280 nm, using theoretical extinction coefficients of 36,160 M−1 cm−1; 37,440 M−1 cm−1; 37,320 M−1 cm−1; and 36,040 M−1 cm−1 for wild-type cationic trypsinogen, D218Y cationic trypsinogen mutant, wild-type anionic trypsinogen, and Y218D anionic trypsinogen mutant, respectively. The molecular masses of recombinant cationic and anionic trypsinogens were 25,149.3 Da and 25,077 Da, respectively.
Autoactivation assays
Trypsinogen in 50 mM HCl was diluted to 2 μM final concentration in the appropriate buffer and the HCl was neutralized with NaOH, resulting in 15 mM NaCl concentration. Buffers were used at 0.1 M final concentration, Na-acetate at pH 4.0 and pH 5.0; Na-MES (2-morpholinoethanesulfonic acid) at pH 6.0, Na-HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) at pH 7.0, and Tris-HCl at pH 8.0. As inert protein, 2 mg/ml bovine serum albumin was included in the autoactivation mixtures. The albumin was extensively dialyzed against distilled water before use. Because commercial albumin preparations slightly inhibit human anionic trypsin, but not cationic trypsin [20], in the autoactivation experiments with anionic trypsinogen bovine serum albumin was omitted. Autoactivation reactions were initiated by addition of 10 nM trypsin (final concentration), and reaction-mixtures were incubated at 37 °C in the presence of 1 mM CaCl2 (wild-type and mutant cationic trypsinogens) or 10 mM CaCl2 (wild-type and mutant anionic trypsinogens). These Ca2+ concentrations were previously found optimal for autoactivation of the two trypsinogen isoforms [20]. At given times, 2 μL aliquots were removed for trypsin activity assay. Trypsin activity was determined using the synthetic chromogenic substrate, N-CBZ-Gly-Pro-Arg-p-nitroanilide (0.14 mM final concentration) in 200 μL volume. One-minute time-courses of p-nitroaniline release were followed at 405 nm in 0.1 M Tris-HCl (pH 8.0), 1 mM CaCl2, at room temperature using a Spectramax Plus 384 microplate reader (Molecular Devices).
Calculation of initial rates
Progress curve analysis with KINSIM and FITSIM computer programs [21, 22] was used to estimate the second-order rate constant of the “Trypsin + Trypsinogen → 2Trypsin” reaction [see discussion in ref 23]. Initial rates were then obtained by multiplying the rate constant with the initial concentrations of the reactants, i.e. 10 nM trypsin and 2000 nM trypsinogen, and rates were expressed as nM trypsin generated per min (nM/min). Under the conditions used in this study, degradation of cationic trypsin(ogen) during autoactivation was minimal, and this side-reaction was ignored in the calculations. Autoactivation of the less stable anionic trypsinogen was always measured in the presence 10 mM Ca2+, which provides sufficient protection against autolysis [20].
Activation of S200A trypsinogen mutants
Trypsinogens (2 μM concentration) carrying the S200A mutation were activated with 10 nM human cationic trypsin, 50 ng/mL (0.45 nM) bovine enteropeptidase or 10 ng/mL (0.13 nM) human enteropeptidase at 37 °C in 0.1 M Tris-HCl (pH 8.0) and 1 mM CaCl2 (final concentrations). To eliminate the effect of potential trypsin contamination, the enteropeptidase activation reactions were carried out in the presence of 0.12 μM soybean trypsin inhibitor (final concentration), unless indicated otherwise. Aliquots were withdrawn from the activation mixtures and trypsinogen was precipitated with trichloroacetic acid (10 % final concentration). The precipitate was recovered by centrifugation, dissolved in Laemmli sample buffer containing 100 mM dithiothreitol (final concentration) and samples were heat-denatured at 95 °C for 5 min. Electrophoretic separation was performed on 13 % SDS-PAGE mini gels in standard Tris-glycine buffer. For the A3D/S200A and A4/S200A mutants the conventional 13 % reducing SDS-PAGE gel did not resolve trypsinogen from trypsin, therefore 21% gels and non-reducing conditions were used for separation. Gels were stained with Brilliant Blue R for 30 min, destained with 30 % methanol, 10 % acetic acid and dried. Densitometric quantitation of bands was carried out as described in [20]. Activated trypsin concentrations were determined from the density of the trypsin band, plotted as a function of time, and rates were estimated from linear fits to the initial portions of the reactions.
RESULTS
Zymogen activation with human and bovine enteropeptidase
To study the significance of the Asp19–22 residues (Fig 1) in zymogen activation, individual Ala and Glu replacements were carried out in recombinant human cationic trypsinogen, yielding single-Ala mutants D19A, D20A, D21A, D22A and single-Glu mutants D19E, D20E, D21E, and D22E. In addition, tri-Ala replacement of the Asp19–21 segment (mutant A3D) and tetra-Ala replacement of the entire Asp19–22 motif (mutant A4) were also performed. Because of the strong autoactivation of cationic trypsinogen, specific rates of enteropeptidase activation could not be determined with catalytically competent proenzymes. Therefore, the catalytic Ser200 (chymo# Ser195) was replaced with Ala in wild-type (mutant S200A) and mutant trypsinogens.
Activation of S200A-trypsinogen mutants by the enteropeptidase holoenzyme was peformed at 37 °C, in 0.1 M Tris-HCl (pH 8.0), 1 mM CaCl2, followed by SDS-PAGE analysis and densitometric quantitation. To inhibit any possible trypsin carry-over or contamination from the enteropeptidase preparations, the activation experiments were carried out in the presence of 0.12 μM soybean trypsin inhibitor (Kunitz), which abolishes trypsin activity but does not affect enteropeptidase. Kinetic analysis of trypsinogen activation by human enteropeptidase yielded a KM of 1.4 μM and a kcat of 35.1 s−1 (Table 1). The KM value is in good agreement with previous reports, while the kcat is almost 10-fold higher, probably due to the ultrapure recombinant enteropeptidase preparation or to the different reaction conditions used (see Table 1). Subsequently, the activation assays were carried out with 2 μM trypsinogen concentration, which corresponds to the approximate KM value of the activation reaction. Unexpectedly, rates of activation of wild-type trypsinogen or mutants D19A, D20A and D21A were identical when activated with human enteropeptidase (Fig 1). Activation with bovine enteropeptidase proceeded approximately 4-fold slower (note the different enzyme concentrations used; also see Table 1), but no measurable difference was detected between wild-type or D19A, D20A and D21A trypsinogens. The data indicate that the Asp19-Asp20-Asp21 segment does not play any significant role in enteropeptidase recognition. However, a markedly different picture emerged when mutant D22A was activated with human or bovine enteropeptidase. Bovine enteropeptidase exhibited 24-fold decreased activity for the D22A mutant relative to wild-type trypsinogen, whereas activation by human enteropeptidase was only 1.2-fold slower. Clearly, Asp22 plays a critical role for recognition by bovine enteropeptidase, however, it contributes only minimally to activation by the cognate human enzyme. The remarkable species-specificity observed here underscores the importance of studying physiologically relevant enteropeptidase-trypsinogen pairs to understand the evolutionary processes that shaped zymogen activation.
Table 1.
Kinetic parameters of trypsinogen activation by human and bovine enteropeptidase. Rates of enteropeptidase-mediated S200A-trypsinogen activation were determined at 6 different substrate concentrations ranging from 0.1 μM to 14 μM. Results are listed as the mean ± S.E. For comparison, values reported in the literature are also shown. Reaction conditions were as follows.
| KM (μM) | kcat (s−1) | kcat/KM (M−1 s−1) | reference | |
|---|---|---|---|---|
|
1Human enteropeptidase
Human cationic trypsinogen |
1.4 ± 0.3 | 35.1 ± 4 | 2.5 x 107 | present study |
|
1Human enteropeptidase
A4 mutant trypsinogen |
2.1 ± 0.3 | 11.2 ± 0.2 | 5.3 x 106 | present study |
|
2Bovine enteropeptidase
Human cationic trypsinogen |
1.5 ± 0.2 | 8.8 ± 0.2 | 5.9 x 106 | present study |
|
3Human enteropeptidase
Human cationic trypsinogen |
7.2 | 2.3 | 3.3 x 105 | [31] |
|
4Bovine enteropeptidase
Bovine trypsinogen |
5.6 ± 0.9 | 4.0 ± 1.2 | 7.1 x 105 | [32] |
|
5Bovine enteropeptidase
Bovine trypsinogen |
1.2 ± 0.3 | 6.9 ± 0.4 | 5.7 x 106 | [32] |
37 °C, 100 mM Tris-HCl (pH 8.0), 1 mM CaCl2, 0.12 μM soybean trypsin inhibitor, 0.13 nM human enteropeptidase
37 °C, 100 mM Tris-HCl (pH 8.0), 1 mM CaCl2, 0.12 μM soybean trypsin inhibitor, 0.45 nM bovine enteropeptidase
25 °C, 28 mM Na-succinate (pH 5.6), 10 mM CaCl2
21 °C, 50 mM Na-citrate (pH 5.6); 1 nM bovine enteropeptidase
37 °C, 25 mM Tris-HCl (pH 8.4), 10 mM CaCl2, 40 μM ovomucoid, 0.3 nM bovine enteropeptidase
The results in Fig 1 strongly argue that, when examined individually, none of the Asp residues within the Asp19–22 motif is required for activation by human enteropeptidase and do not explain the strong evolutionary conservation of this sequence. The data also raise the possibility that, in fact, acidic residues within the activation peptide are entirely dispensable for enteropeptidase recognition and cleavage. To address this question, mutant A3D, in which only Asp22 is present as a single acidic residue, and mutant A4, which is completely devoid of Asp residues, were activated with enteropeptidase. A number of technical difficulties hindered these experiments. Firstly, removal of the Asp residues from the activation peptide abolished the mobility shift routinely observed on reducing SDS-PAGE gels and trypsinogen and trypsin migrated at identical positions. To visualize the activation reaction, activated trypsinogens were resolved on 21 % gels, under non-reducing conditions. As shown in Fig 2, in this electrophoresis system the mobility shift becomes apparent again. Using MALDI-TOF mass spectrometry, we have confirmed that the gel shift was caused by the removal of the activation peptide (not shown). Secondly, mutant A4 was extremely trypsin sensitive, and even minute amounts of trypsin(ogen) contamination originating from the affinity column or labware could result in significant trypsin-mediated activation. This problem was overcome by inclusion of soybean trypsin inhibitor in the activation reactions, and experiments were carried out with 0.12 μM, 0.5 μM, 1 μM and 2 μM inhibitor concentrations, with identical results. Fig 2A demonstrates that both human and bovine enteropeptidase activated A3D trypsinogen. When compared to activation of wild-type trypsinogen (cf. Fig 1), the rate of A3D trypsinogen activation by human enteropeptidase was identical; while activation by bovine enteropeptidase was 2-fold decreased. This observation was consistent with the findings in Fig 1, indicating that the Asp19–21 triplet is not required for recognition by enteropeptidase from either species. Strikingly, A4 trypsinogen was also activated by human enteropeptidase with a rate that was circa 2-fold decreased relative to wild-type trypsinogen. In contrast, bovine enteropeptidase did not cleave A4 trypsinogen to any detectable degree (Fig 2B). Kinetic analysis of A4 trypsinogen activation by human enteropeptidase revealed a slightly increased KM (2.1 μM) and an approximately 3-fold decreased kcat (11.2 s−1, Table 1). Therefore, A4 trypsinogen is still a specific enteropeptidase substrate, which is cleaved with high catalytic efficiency (kcat/KM = 5.3 x 106 M−1 s−1). Taken together, the results shown in Figs 1 and 2 clearly demonstrate that the Asp19–22 motif per se in human cationic trypsinogen is not essential for recognition and cleavage by human enteropeptidase.
Figure 2.
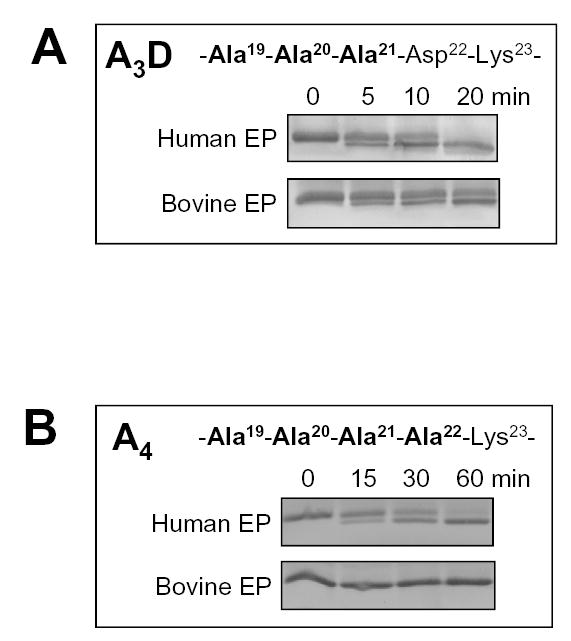
Activation of the tri-alanine (A3D) and tetra-alanine (A4) mutants in the Asp19–22 motif by enteropeptidase. The mutated activation peptide sequences are indicated. The mutants also carried the S200A mutation to prevent autoactivation. Reaction conditions are given in Fig 1. Samples were resolved under non-reducing conditions on 21 % SDS-PAGE gels and stained with Coomassie-blue. Representative experiments (n=5) are shown; note the longer time-course in panel B. The activation rates by 0.13 nM human enteropeptidase were 84 nM/min for A3D and 36 nM/min for A4 trypsinogen. Activation of the A3D mutant by 0.45 nM bovine enteropeptidase exhibited a rate of 51 nM/min.
Zymogen activation by trypsin (autoactivation)
Individual Ala mutations in all positions of the Asp19–22 motif stimulated autoactivation. At pH 8.0, 1 mM CaCl2, 37 °C, mutation D19A and D21A increased the autoactivation rate about 2-fold, while mutation D20A enhanced the rate more than 3-fold (Fig 3). When the combined effect of these 3 mutations was tested with A3D trypsinogen, the autoactivation rate was 13-fold increased, indicating that the inhibitory effects of the Asp residues within the Asp19-Asp20-Asp21 triplet are multiplicative. Remarkably, the D22A mutation stimulated autoactivation to such an extent that it was impossible to follow by activity assays. Therefore, trypsin-mediated activation of D22A-trypsinogen was analyzed with SDS-PAGE and densitometry using the catalytically deficient D22A/S200A double mutant (Fig 4). Relative to wild-type trypsinogen, mutant D22A exhibited a drastic increase in trypsin-mediated activation, which was 66-fold higher at pH 8.0. Although data are not shown, activation of the tetra-Ala A4/S200A mutant by trypsin was approximately 500-fold accelerated relative to wild-type (S200A) trypsinogen.
Figure 3.
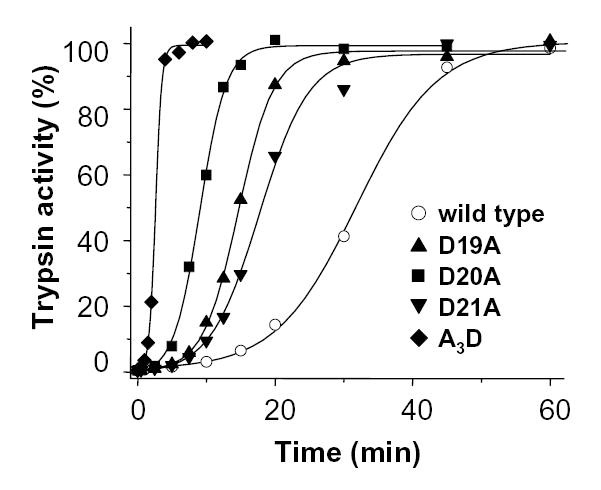
Autoactivation of single-alanine mutants D19A, D20A, D21A and the tri-alanine mutant A3D. Trypsin mediated trypsinogen activation was measured at 37 °C in 0.1 M Tris-HCl (pH 8.0), 1 mM CaCl2 and 2 mg/mL bovine serum albumin with 2 μM trypsinogen and 10 nM trypsin initial concentrations. Trypsin activity was expressed as percent of potential maximal activity, which was determined by activation with human enteropeptidase. The rates of autoactivation calculated from progress curve analysis were as follows. Wild type (open circles), 1.7 nM/min; D19A (triangles), 3.4 nM/min; D20A (squares), 5.4 nM/min; D21A (inverted triangles), 2.9 nM/min; A3D (diamonds), 22 nM/min.
Figure 4.
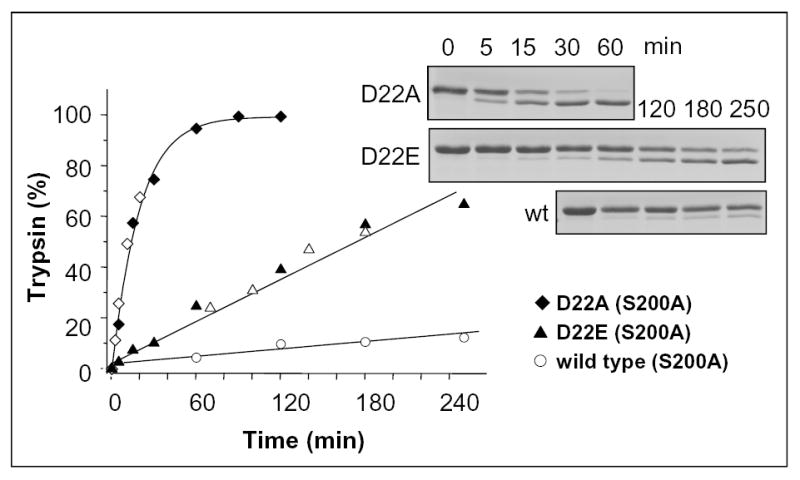
Activation of the D22A and D22E trypsinogen mutants by trypsin. Activation of wild-type/S200A (open circles), D22A/S200A (diamonds) and D22E/S200A (triangles) cationic trypsinogens (2 μM) with 10 nM trypsin was carried out in 0.1 M Tris-HCl (pH 8.0), 1 mM CaCl2 at 37 °C. The open and filled symbols represent independent experiments. Samples were analyzed by 13 % reducing SDS-PAGE, Coomassie blue staining and densitometry. The intensity of the trypsin band was expressed as percentage of the sum of the trypsin and trypsinogen bands. The initial rates were 1.4 nM/min for wild type; 6.4 nM/min for D22E and 93.2 nM/min for D22A.
The significance of the negative charges within the Asp19–22 motif was also assessed by conservative Glu substitutions. Individual replacements of Asp19, Asp20 and Asp21 by Glu had minimal, mostly inhibitory effects on autoactivation. Using 2 μM trypsinogen and 10 nM trypsin concentrations, at pH 8.0, autoactivation rates of 1.7 nM/min; 1.8 nM/min; 0.9 nM/min, and 0.9 nM/min were determined for wild-type, D19E, D20E and D21E trypsinogens, respectively (not shown). However, conservative Glu replacement of Asp22 in mutant D22E resulted in a 5-fold increased autoactivation rate (9.8 nM/min). Similarly, trypsin-mediated activation of the D22E/S200A mutant showed a 5-fold increased rate relative to S200A trypsinogen (Fig 4). The results not only identify Asp22 as the critical determinant of autoactivation control, but also establish that the entire intact Asp19–22 motif is mandatory for maximal suppression of autoactivation in human cationic trypsinogen.
Modeling the interactions between cationic trypsin and the Asp19–22 motif of the trypsinogen activation peptide
To visualize the subsite interactions between human cationic trypsin and the trypsinogen activation peptide, we superimposed the structures of human cationic trypsin [24] and bovine enteropeptidase light chain [8], which was crystallized in complex with an inhibitor analog of the activation peptide, Val-Asp-Asp-Asp-Asp-Lys-chloromethane (Fig 5). The high degree of structural similarity between the two chymotrypsin-like serine proteases allowed the virtual positioning of the Asp20-Asp21-Asp22-Lys23 sequence in complex with cationic trypsin. With respect to potential interactions of the Asp residues with cationic trypsin, two observations were made. First, the most important determinant of autoactivation, the P2 Asp22 faces a hydrophobic groove on trypsin; lined by His63 (chymo# His57), Leu104 (chymo# Leu99) and Trp216 (chymo# Trp215). This hydrophobic subsite is well conserved in vertebrate trypsins [25] and it is responsible for the documented hydrophobic P2 preference of trypsin [26]. Clearly, a negatively charged Asp residue is unfavored in this environment, while the small hydrophobic Ala in the D22A mutant would be readily accommodated. The 5-fold increased autoactivation of the D22E mutant is probably explained by the larger rotamer repertoire of the longer Glu side-chain, which might mitigate the conflict between the negative charge and the hydrophobic S2 subsite.
Figure 5.
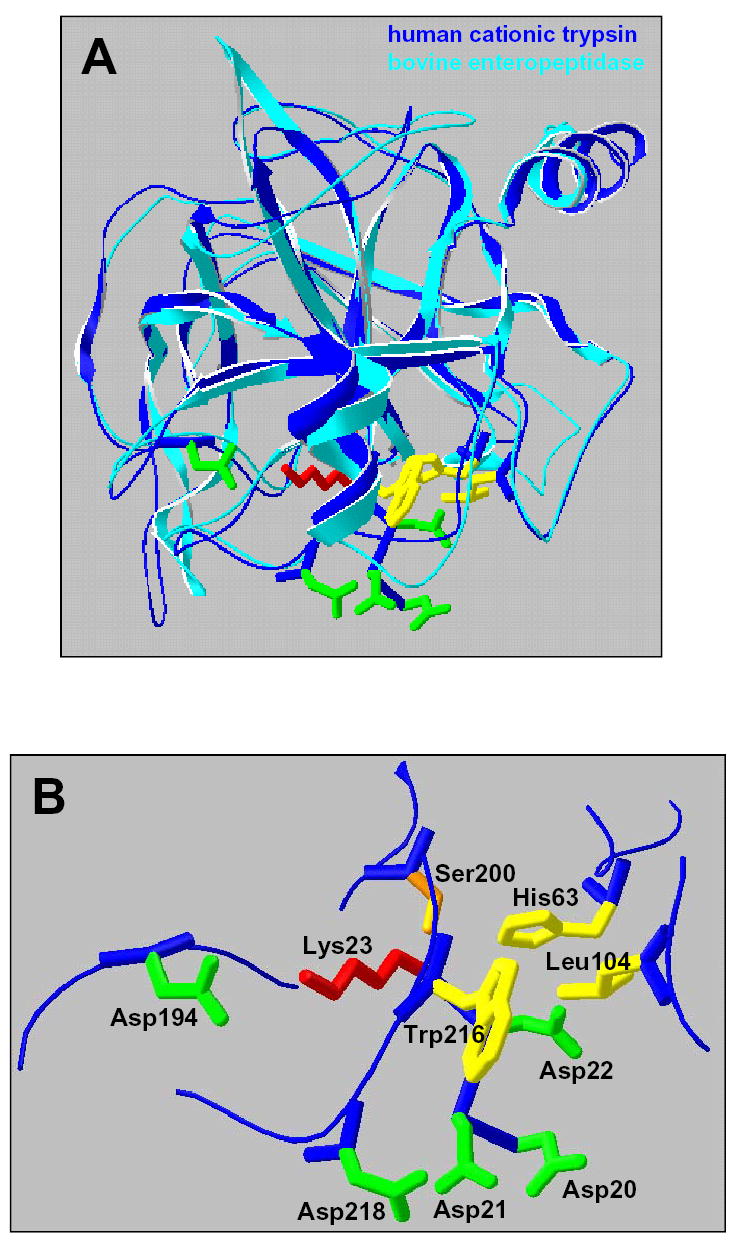
Interactions between the Asp19–22 motif in the trypsinogen activation peptide and human cationic trypsin. A. Ribbon diagram of the superimposed human cationic trypsin (Protein Data Bank file 1TRN, dark blue) and bovine enteropeptidase catalytic subunit complexed with an inhibitor analog of the activation peptide (Protein Data Bank file 1EKB, light blue). The structure of trypsin (chain B of the crystallographic dimer shown here) fits enteropeptidase with a root-mean-square deviation of 0.95 Å for 208 Cα positions. B. A model of the activation peptide bound to trypsin. The indicated portion of the activation peptide corresponds to the Asp20-Asp21-Asp22-Lys23 sequence in human cationic trypsinogen (see Fig 1). The P1 Lys23 (red) of the activation peptide interacts electrostatically with the S1 specificity determinant Asp194 (green; chymo# Asp189). The P2 Asp22 is facing a hydrophobic groove (yellow) formed by the catalytic His63 (chymo# His57), Leu104 (chymo# Leu99) and Trp216 (chymo# Trp215). The P3 Asp21 (green) appears to interact with the Asp218 (chymo# Asp217) surface residue. The catalytic Ser200 (chymo# Ser195) is shown in orange. The image was rendered using DeepView/Swiss-PdbViewer v. 3.7 (www.expasy.org/spdbv/) [33].
Second, in our model Asp218 (chymo# Asp217) on the surface of cationic trypsin came in close proximity to Asp21 of the activation peptide. In contrast to the conserved hydrophobic S2 subsite, the Asp218 negative exosite is absent in the vast majority of mammalian trypsins which typically contain Tyr at this position (see Discussion). The structural model in Fig 5 strongly suggests that the inhibitory action of the activation peptide in human cationic trypsinogen is partly due to an electrostatic clash between the negatively charged Asp218 and one or more of the Asp residues in the activation peptide, most likely Asp21. This hypothesis was particularly attractive, because it further emphasized the highly specialized aspects of the Asp19–22 motif in human cationic trypsinogen. To provide functional evidence for this inhibitory interaction, position 218 was subjected to mutagenesis studies.
A negative charge at position 218 is required for inhibition of autoactivation
First, Asp218 was replaced with Tyr (mutant D218Y), because human anionic trypsinogen and most other mammalian trypsinogens carry a tyrosine residue at this position. Because protonation of Asp residues might affect the electrostatic repulsion between Asp218 and Asp21, rates of autoactivation were measured over the pH range from 4.0 to 8.0. Autoactivation of wild-type and D218Y trypsinogen was essentially identical at pH 4.0 and 5.0, however, in the pH 6.0–8.0 range autoactivation of D218Y-trypsinogen was markedly stimulated, while wild-type cationic trypsinogen exhibited only a marginal increase (Fig 6AB). At the pH 7.0 optimum, the difference in the rates of autoactivation between wild-type and mutant D218Y trypsinogens amounted to 11-fold (32.3 vs. 2.9 nM/min). For comparison, the pH dependence of autoactivation was also determined for the single-Ala mutants D19A, D20A, and D21A as well as the tri-Ala mutant A3D (Fig 6C). Interestingly, the single-Ala mutations caused less pronounced changes (2–3-fold increase at the pH 7.0 optimum) than the D218Y mutation, suggesting that not only Asp21, but the full Asp19–21 sequence is required for the optimal inhibitory interaction with Asp218. Consistent with this interpretation, removal of the entire Asp19–21 triplet in the A3D mutant resulted in an autoactivation profile that was essentially identical to that of the D218Y mutant. Taken together, these results support the predicted electrostatic inhibitory interaction between Asp218 and the trypsinogen activation peptide.
Figure 6.
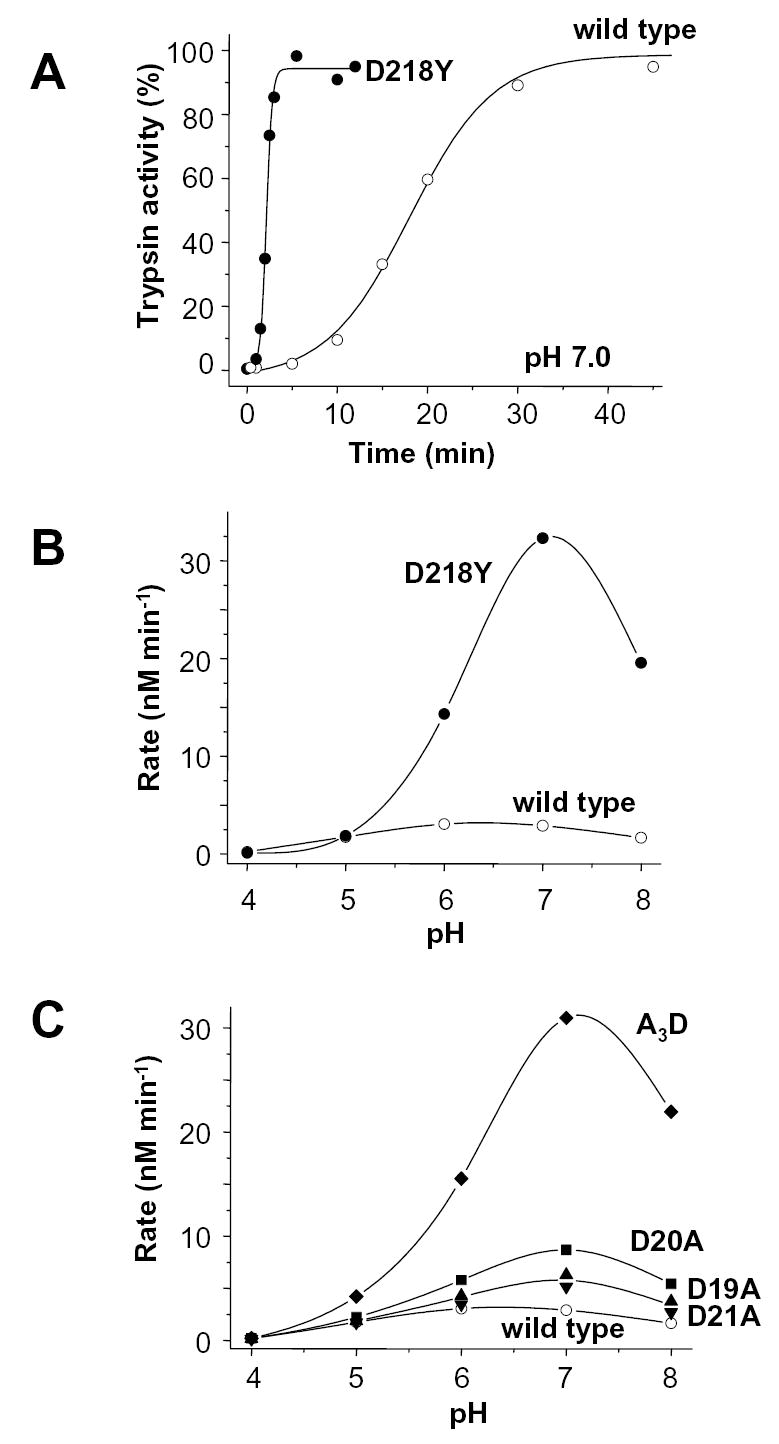
Autoactivation of wild-type (open symbols) and D218Y (solid symbols) human cationic trypsinogen. A. Time-courses of autoactivation were followed at 37 °C in 0.1 M Na-HEPES (pH 7.0), 1 mM CaCl2 and 2 mg/mL bovine serum albumin. Initial trypsinogen and trypsin concentrations were 2 μM and 10 nM, respectively. B. Effect of pH on autoactivation of wild-type and D218Y trypsinogen. C. For comparison, the pH dependence of autoactivation of the activation peptide mutants D19A, D20A, D21A and A3D are also shown (for time-courses at pH 8.0 see Fig 3). Initial rates were calculated from time-courses of autoactivation using progress curve analysis, as described in Experimental Procedures. The buffers used were Na-acetate, (pH 4.0 and 5.0); Na-MES (pH 6.0); Na-HEPES (pH 7.0) and Tris-HCl (pH 8.0).
To characterize further the side-chain requirement at position 218 for suppression of autoactivation, Asp218 was mutated to serine, histidine, or glutamate. These side-chains also occur naturally at position 218, e.g. in bovine cationic trypsin (Ser), in human mesotrypsin (His), or in a snake trypsin (Glu). As shown in Fig 7, a marked increase in autoactivation was observed with both the D218S and D218H mutations, indicating that the Ser and His side-chains, together with Tyr, are unable to suppress autoactivation of cationic trypsinogen. On the other hand, autoactivation of wild-type and D218E mutant cationic trypsinogens were indistinguishable, demonstrating that a negative charge at position 218 is necessary for inhibition of autoactivation.
Figure 7.
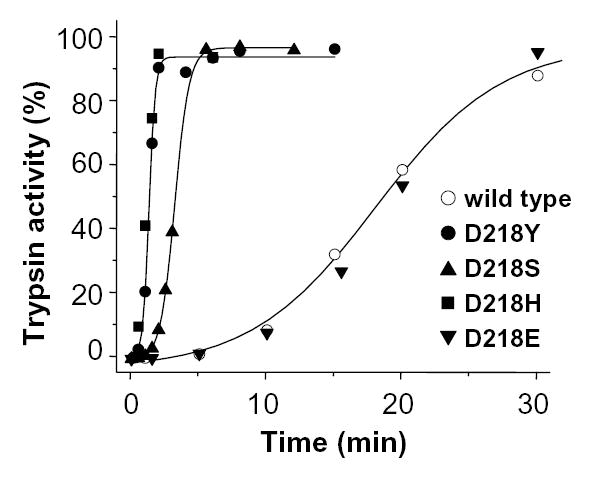
Effect of different amino-acid side-chains at position 218 on trypsinogen autoactivation. See Fig 6A for experimental conditions. The rates of autoactivation calculated from progress curve analysis were as follows. Wild type (open circles), 2.9 nM/min; D218Y (solid circles), 32.3 nM/min; D218S (triangles), 16.1 nM/min; D218H (squares), 31.2 nM/min; D218E (inverted triangles), 2.7 nM/min.
Introduction of Asp218 into anionic trypsinogen suppresses autoactivation
Human anionic trypsinogen carries a Tyr residue at position 218 and exhibits a pH profile of autoactivation that is similar to the D218Y cationic trypsinogen mutant (Fig 8, cf. Fig 6). Because the activation peptide sequence of anionic trypsinogen is identical to that of cationic trypsinogen, it seemed reasonable to assume that replacement of Tyr218 with Asp should reconstitute the same interaction that operates in cationic trypsinogen. Indeed, the Y218D mutation reduced autoactivation of anionic trypsinogen almost 6-fold at pH 7.0 (5.4 vs. 0.9 nM/min; Fig 8), indicating that the inhibitory repulsion between Asp218 and the tetra-aspartate tract of the activation peptide has been successfully engineered.
Figure 8.
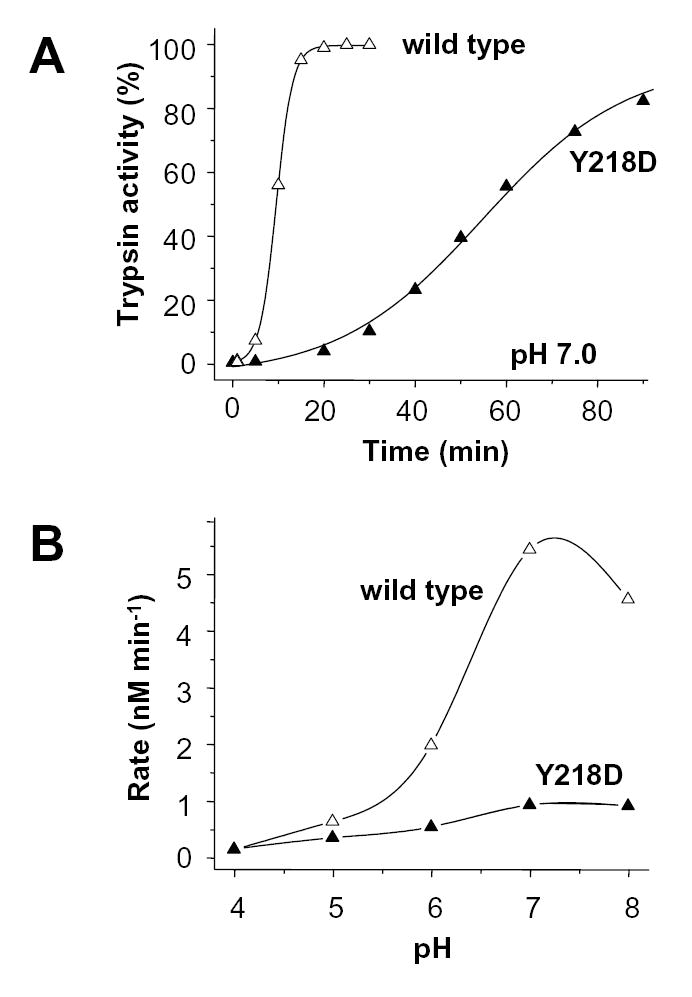
Effect of replacement of Tyr218 with Asp (Y218D, solid symbols) on the autoactivation of human anionic trypsinogen (wild type, open symbols). A. Autoactivation of 2 μM trypsinogen was initiated with 10 nM trypsin (final concentrations) and time-courses were followed at 37 °C in 0.1 M Na-HEPES (pH 7.0) and 10 mM CaCl2. B. pH-dependence of autoactivation was determined as described in Experimental Procedures and in Fig 6B. Note that autoactivation experiments with anionic trypsinogen were always performed in 10 mM Ca2+ and in the absence of bovine serum albumin for maximal stability and activity [20].
DISCUSSION
The most surprising finding of this study is that the highly conserved tetra-aspartate sequence in the activation peptide of human cationic trypsinogen is not required for enteropeptidase-mediated activation. Thus, not only were single-Ala mutants D19A, D20A, D21A and D22A all activated normally by human enteropeptidase, but the tetra-Ala replacement mutant A4, which is completely devoid of any acidic residues in the activation peptide, was also activated with somewhat reduced, but still remarkable efficiency. Therefore, the tenet that human enteropeptidase recognizes its physiological substrate through the Asp19–22 motif appears to be wrong. Instead, enteropeptidase recognition seems to be determined by so far uncharacterized distant subsite interactions, in which the heavy chain of enteropeptidase might play a significant role. Interestingly, however, the presence of the P2 Asp22 in the activation peptide was an almost absolute requirement for activation by bovine enteropeptidase (see Fig 1). This observation suggests that the importance of the tetra-Asp motif in enteropeptidase recognition might be species and isoform specific. Because only bovine and human cationic trypsinogens have been characterized in detail, it is difficult to generalize the results to other vertebrate trypsinogens. We favor the hypothesis that the diminished significance of Asp19–22 in enteropeptidase-mediated activation of human cationic trypsinogen might indicate a recent evolutionary change, and the acidic residues in the activation peptide are necessary for enteropeptidase recognition in the majority of vertebrate trypsinogens.
The critical role of the P2 Asp22 in activation by bovine enteropeptidase is also in agreement with the available crystallographic data and mutational analysis indicating an essential interaction between Lys99 (chymo#) of the bovine enteropeptidase catalytic subunit and the P2 Asp (Asp22 in Fig 1) of the activation peptide [8]. However, the crystal structure also shows that the P4 Asp (Asp20) participates in a salt-bridge with Lys99 (chymo#), and the P3 Asp (Asp21) is hydrogen bonded to the hydroxyl group of Tyr174 (chymo#). It is not readily apparent why in our study no functional role could be demonstrated for the P3 and P4 Asp residues in trypsinogen activation by bovine or human enteropeptidase. One possible explanation is that the trypsinogen activation peptide interacts differently with the enteropeptidase catalytic subunit (used in the crystallization studies) and the enteropeptidase holoenzyme (used here). Alternatively, the interactions observed in the crystal structure might be redundant and might not translate to better catalytic efficiency.
Our results indicate that the conserved Asp19–22 motif has been maintained in human cationic trypsinogen for reasons that are unrelated to enteropeptidase recognition and suggest that autoactivation control is the primary function of this acidic sequence. Indeed, experiments with single and multiple Ala-mutants confirmed that each Asp residue plays a role in autoactivation control, and their effects are synergistic in a multiplicative manner. The contribution of Asp22 is the most significant, as indicated by the 66-fold increased autoactivation of the D22A mutant, whereas mutants D19A, D20A and D21A exhibited 2-3-fold increased rates of autoactivation. Combination of the D19A, D20A and D21A mutations in the tri-Ala mutant A3D resulted in 13-fold increased autoactivation, whereas the tetra-Ala mutant A4 was activated by trypsin approximately 500-fold more rapidly than wild-type trypsinogen. Structural modeling revealed two distinct inhibitory interactions between the Asp19–22 motif of the activation peptide and cationic trypsin. Asp22 is oriented towards the conserved hydrophobic S2 subsite of trypsin, formed by His63 (chymo# His57), Leu104 (chymo# Leu99) and Trp216 (chymo# Trp215), resulting in an unfavorable subsite interaction that explains the large effect of the D22A mutation. Furthermore, the unique Asp218 surface residue, which forms part of the S3-S4 subsite on trypsin, appears to participate in an inhibitory electrostatic interaction with the P3 Asp21. Mutagenesis of Asp218 clearly confirmed that this acidic exosite is essential for inhibition of autoactivation, and removal of the negative charge at position 218 can result in an 11-fold increase. Although modeling suggested that Asp218 interacts with the P3 Asp21, we remain tentative about this interaction, because single-Ala mutation of Asp21 (D21A) stimulated autoactivation only 2-fold, and triple-Ala mutation of the Asp19–21 sequence was necessary to achieve the same degree of autoactivation stimulation as with the D218Y mutant. To reconcile the structural and functional data, a plausible explanation is that an intact tetra-Asp sequence is required to position Asp21 for optimal repulsion with Asp218. In single-Ala mutants D19A, D20A or D21A, the re-arrangement of the remaining Asp residues can maintain a partial inhibitory interaction with Asp218, whereas the combined tri-Ala mutant A3D exhibits full relief from the Asp218-dependent inhibition. An uninterrupted tetra-Asp sequence also appears to be essential for Ca2+ binding to the activation peptide. Although data are not shown, autoactivation of wild-type cationic trypsinogen was stimulated by Ca2+, whereas no effect was observed with mutants D19A, D20A, D21A and D22A. It is likely that Ca2+-mediated stimulation is important for physiological zymogen activation in vertebrates, and represents an additional selective pressure for the evolutionary conservation of the intact tetra-Asp motif in the activation peptide.
The present study also provides a molecular explanation to the previously described unique ability of human cationic trypsinogen to autoactivate at acidic pH [27]. This is clearly due to the electrostatic repulsion between Asp218 and the acidic activation peptide, which becomes prominent above pH 5.0 and suppresses autoactivation. As a result, despite the typical pH-dependent stimulation of trypsin activity, autoactivation remains essentially unchanged between pH 5.0 and pH 8.0 (see Fig 6). In contrast, bovine trypsinogen [28] or human anionic trypsinogen [20, see also Fig 8] autoactivate much better at pH 8.0 than at pH 5.0.
In evolutionary terms, the selective advantage of Asp218 seems to lie in protection against premature trypsinogen autoactivation at neutral or alkaline pH, which prevails in the pancreatic ducts. However, this mechanism of autoactivation control appears to be rare among vertebrate trypsinogens. This position (chymo# 217) can carry a variety of residues (Tyr, Ala, Ser, His, Ile, Asp, Glu), but there is a strong preference for Tyr, particularly in mammalian trypsinogens [25]. Besides human cationic trypsinogen, Asp is found in rat trypsinogen V, but in this minor isoform the tetra-Asp sequence of the activation peptide is disrupted by an Asn residue, suggesting that the electrostatic repulsion might not be optimal. In the recently released genomic sequence of the beta T-cell receptor locus from the rhesus macaque monkey (Macaca mulatta), the try9 and try13 trypsinogen isoforms contain Asp218 (GenBank entry AC149201). Finally, Glu was identified at this position in a partial trypsinogen cDNA of the snake Bothrops jararaca (GenBank entry AF190273), and in a genomic trypsinogen sequence of the green pufferfish Tetraodon nigroviridis (GenBank entry CAG00064).
A possible explanation why Asp218 is conspicuously missing from most vertebrate trypsinogens is that the true selective advantage is not suppression of autoactivation per se, but regulation of the pH dependence of autoactivation in such a manner that ensures essentially identical autoactivation over the pH range from pH 5.0 to pH 8.0. This could offer the obvious physiological benefit of enhanced zymogen activation in the duodenum when acidic gastric output lowers the pH transiently. It is noteworthy, that trypsinogen activation by enteropeptidase has a similarly broad pH optimum, suggesting a case of convergent evolution to cope with the inhibitory effect of gastric acid. We speculate that in most species enteropeptidase-mediated trypsinogen activation is sufficient to achieve rapid and complete trypsinogen activation in the duodenum, while in humans, and possibly in a handful of other species, trypsinogen autoactivation is also required for full zymogen activation.
The same mechanism that ensures efficient zymogen activation in the duodenum might have significant pathological consequences as well. Autoactivation of cationic trypsinogen in the acidic secretory compartment can lead to premature intra-acinar trypsinogen activation, even in the absence of cathepsin B activity [27]. The situation is further aggravated in hereditary pancreatitis, in which inborn mutations increase the propensity of cationic trypsinogen to autoactivate [14–18]. This notion is also supported by the fact that genetic variants of human anionic trypsinogen, which cannot autoactivate under acidic conditions, have not been found in association with hereditary pancreatitis or other forms of human pancreatitis [29, 30].
Acknowledgments
This work was supported by NIH grant DK058088 to M. S.-T. We thank Vera Sahin-Tóth for technical assistance and Miklós Tóth, Zoltán Kukor, Edit Szepessy and Jian-Min Chen for critical reading of the manuscript.
References
- 1.Light A, Janska H. Trends Biochem Sci. 1989;14:110–112. doi: 10.1016/0968-0004(89)90133-3. [DOI] [PubMed] [Google Scholar]
- 2.Lu, D., and Sadler, J. E. (2000) in Handbook of Proteolytic Enzymes, Eds. Barrett, A. J., Rawlings, N. D., and Woessner, J. F., Academic Press, 2000, pp 50–54.
- 3.Maroux S, Baratti J, Desnuelle P. J Biol Chem. 1971;246:5031–5039. [PubMed] [Google Scholar]
- 4.Anderson LE, Walsh KA, Neurath H. Biochemistry. 1977;16:3354–3360. doi: 10.1021/bi00634a011. [DOI] [PubMed] [Google Scholar]
- 5.Light A, Savithri HS, Liepnieks JJ. Anal Biochem. 1980;106:199–206. doi: 10.1016/0003-2697(80)90138-4. [DOI] [PubMed] [Google Scholar]
- 6.Mikhailova AG, Rumsh LD. FEBS Lett. 1999;442:226–230. doi: 10.1016/s0014-5793(98)01656-1. [DOI] [PubMed] [Google Scholar]
- 7.Mikhailova AG, Likhareva VV, Vaskovsky BV, Garanin SK, Onoprienko LV, Prudchenko IA, Chikin LD, Rumsh LD. Biochemistry (Mosc) 2004;69:909–917. doi: 10.1023/b:biry.0000040224.47278.3b. [DOI] [PubMed] [Google Scholar]
- 8.Lu D, Futterer K, Korolev S, Zheng X, Tan K, Waksman G, Sadler JE. J Mol Biol. 1999;292:361–373. doi: 10.1006/jmbi.1999.3089. [DOI] [PubMed] [Google Scholar]
- 9.Chen JM, Kukor Z, Le Maréchal C, Tóth M, Tsakiris L, Raguénès O, Férec C, Sahin-Tóth M. Mol Biol Evol. 2003;20:1767–1777. doi: 10.1093/molbev/msg183. [DOI] [PubMed] [Google Scholar]
- 10.Radhakrishnan TM, Walsh KA, Neurath H. Biochemistry. 1969;8:4020–4027. doi: 10.1021/bi00838a019. [DOI] [PubMed] [Google Scholar]
- 11.Delaage M, Desnuelle P, Lazdunski M. Biochem Biophys Res Comm. 1967;29:235–240. doi: 10.1016/0006-291x(67)90593-1. [DOI] [PubMed] [Google Scholar]
- 12.Abita JP, Delaage M, Lazdunski M. Eur J Biochem. 1969;8:314–324. doi: 10.1111/j.1432-1033.1969.tb00530.x. [DOI] [PubMed] [Google Scholar]
- 13.Teich N, Ockenga J, Hoffmeister A, Manns M, Mössner J, Keim V. Gastroenterology. 2000;119:461–465. doi: 10.1053/gast.2000.9312. [DOI] [PubMed] [Google Scholar]
- 14.Whitcomb DC, Gorry MC, Preston RA, Furey W, Sossenheimer MJ, Ulrich CD, Martin SP, Gates LK, Jr, Amann ST, Toskes PP, Liddle R, McGrath K, Uomo G, Post JC, Ehrlich GD. Nat Genet. 1996;14:141–145. doi: 10.1038/ng1096-141. [DOI] [PubMed] [Google Scholar]
- 15.Howes N, Lerch MM, Greenhalf W, Stocken DD, Ellis I, Simon P, Truninger K, Ammann R, Cavallini G, Charnley RM, Uomo G, Delhaye M, Spicak J, Drumm B, Jansen J, Mountford R, Whitcomb DC, Neoptolemos JP European Registry of Hereditary Pancreatitis and Pancreatic Cancer (EUROPAC) Clin Gastroenterol Hepatol. 2004;2:252–261. doi: 10.1016/s1542-3565(04)00013-8. [DOI] [PubMed] [Google Scholar]
- 16.Sahin-Tóth M. J Biol Chem. 2000;275:22750–22755. doi: 10.1074/jbc.M002943200. [DOI] [PubMed] [Google Scholar]
- 17.Sahin-Tóth M, Tóth M. Biochem Biophys Res Commun. 2000;278:286–289. doi: 10.1006/bbrc.2000.3797. [DOI] [PubMed] [Google Scholar]
- 18.Szilágyi L, Kénesi E, Katona G, Kaslik G, Juhász G, Gráf L. J Biol Chem. 2001;276:24574–24580. doi: 10.1074/jbc.M011374200. [DOI] [PubMed] [Google Scholar]
- 19.Chen, J .M. and Férec, C. (2003) in Nature encyclopedia of the human genome, Ed. Cooper D. N., Macmillan London, Nature Publishing Group, pp. 645–650.
- 20.Kukor Z, Tóth M, Sahin-Tóth M. Eur J Biochem. 2003;270:2047–2058. doi: 10.1046/j.1432-1033.2003.03581.x. [DOI] [PubMed] [Google Scholar]
- 21.Barshop BA, Wrenn RF, Frieden C. Anal Biochem. 1983;130:134–145. doi: 10.1016/0003-2697(83)90660-7. [DOI] [PubMed] [Google Scholar]
- 22.Zimmerle CT, Frieden C. Biochem J. 1989;258:381–387. doi: 10.1042/bj2580381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu JW, Wu Y, Wang ZX. Eur J Biochem. 2001;268:1547–1553. [PubMed] [Google Scholar]
- 24.Gaboriaud C, Serre L, Guy-Crotte O, Forest E, Fontecilla-Camps JC. J Mol Biol. 1996;259:995–1010. doi: 10.1006/jmbi.1996.0376. [DOI] [PubMed] [Google Scholar]
- 25.Roach JC, Wang K, Gan L, Hood L. J Mol Evol. 1997;45:640–652. doi: 10.1007/pl00006268. [DOI] [PubMed] [Google Scholar]
- 26.Hedstrom L. Chem Rev. 2002;102:4501–4524. doi: 10.1021/cr000033x. [DOI] [PubMed] [Google Scholar]
- 27.Figarella C, Miszczuk-Jamnska B, Barrett A. Biol Chem Hoppe-Seyler. 1988;369(Suppl):293–298. [PubMed] [Google Scholar]
- 28.Kunitz M. J Gen Physiol. 1939;22:293–310. doi: 10.1085/jgp.22.3.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen JM, Audrezet MP, Mercier B, Quere I, Ferec C. Scand J Gastroenterol. 1999;34:831–832. doi: 10.1080/003655299750025796. [DOI] [PubMed] [Google Scholar]
- 30.Idris MM, Bhaskar S, Reddy DN, Mani KR, Rao GV, Singh L, Chandak GR. Gut. 2005;54:728–729. doi: 10.1136/gut.2004.055335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brodrick JW, Largman C, Hsiang MW, Johnson JH, Geokas MC. J Biol Chem. 1978;253:2737–2742. [PubMed] [Google Scholar]
- 32.Lu D, Yuan X, Zheng X, Sadler JE. J Biol Chem. 1997;272:31293–31300. doi: 10.1074/jbc.272.50.31293. [DOI] [PubMed] [Google Scholar]
- 33.Guex N, Peitsch MC. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]