

Abstract
NBL2 is a tandem 1.4-kb DNA repeat, whose hypomethylation in hepatocellular carcinomas was previously shown to be an independent predictor of disease progression. Here, we examined methylation of all cytosine residues in a 0.2-kb subregion of NBL2 in ovarian carcinomas, Wilms tumors, and diverse control tissues by hairpin-bisulfite PCR. This new genomic sequencing method detects 5-methylcytosine on covalently linked complementary strands of a DNA fragment. All DNA clones from normal somatic tissues displayed symmetrical methylation at seven CpG positions and no methylation or only hemimethylation at two others. Unexpectedly, 56% of cancer DNA clones had decreased methylation at some normally methylated CpG sites as well as increased methylation at one or both of the normally unmethylated sites. All 146 DNA clones from ten cancers could be distinguished from all 91somatic control clones by assessing methylation changes at three of these CpG sites. The special involvement of DNMT3B in NBL2 methylation was indicated by analysis of cells from ICF syndrome patients (immunodeficiency, centromeric region instability, facial anomalies), who have mutations in the gene encoding DNA methyltransferase 3B. Blot hybridization of 33 cancer DNAs digested with CpG methylation-sensitive enzymes confirmed that NBL2 arrays are unusually susceptible to cancer-linked hypermethylation and hypomethylation, consistent with our novel genomic sequencing findings. The combined Southern blot and genomic sequencing data indicate that some of the cancer-linked alterations in CpG methylation are occurring with considerable sequence specificity. NBL2 is an attractive candidate for an epigenetic cancer marker and for elucidating the nature of epigenetic changes in cancer.
Keywords: DNA hypermethylation, DNA hypomethylation, DNMT3B, ICF syndrome, genomic sequencing
Introduction
Both hypermethylation (1,2) and hypomethylation (3) of DNA have been observed in most tested cancers, but in different sequences (4–7). Many specific gene regions become hypermethylated, and some other gene regions and many non-coding DNA repeats become hypomethylated during carcinogenesis (8–10). Nonetheless, hypomethylation and hypermethylation in different parts of the genome in various cancers have been found not to be significantly associated with each other (4, 6, 11). Therefore, cancer-linked DNA hypomethylation is not simply a response to cancer-linked hypermethylation nor vice versa.
Here we show for the first time that relative to normal somatic tissues, cancers can display both hypomethylation and hypermethylation within the same small region on the same DNA molecule. We analyzed NBL2, a tandem 1.4-kb repeat with a complex sequence. We recently found by Southern blot (SB) analysis that NBL2 exhibits either predominant hypermethylation or hypomethylation at HhaI sites in 89% of 18 studied ovarian carcinomas and 84% of 51 Wilms tumors (12). We also observed hypomethylation at NotI sites in some ovarian carcinomas. Itano et al. (13) demonstrated that hypomethylation at NotI sites was an independent prognostic indicator in hepatocellular carcinoma patients. NBL2 is often hypomethylated at NotI sites in neuroblastomas (hence, the name NBL2) and hepatocellular carcinomas (14, 15). This primate-specific sequence (15) is CpG-rich (61% C+G; 5.7% CpG). It is present in about 200–400 copies per haploid human genome, mostly in the vicinity of the centromeres of four of the five acrocentric chromosomes (12), repeat-rich regions for which only little sequence information is available.
The powerful method that we used to analyze NBL2 methylation in ovarian carcinomas, Wilms tumors, and various control cell populations is the hairpin-bisulfite PCR variant of genomic sequencing (hairpin GS) developed by Laird and colleagues (16). In bisulfite-based genomic sequencing, bisulfite causes deamination of unmethylated C residues while methylated C residues (usually only in CpG sequences) are resistant (17). Hairpin GS allows analysis of methylation of every CpG dinucleotide-pair (dyad) in a given region on covalently linked DNA strands of a restriction fragment. Analysis of both CpG’s of a dyad is of special interest because CpG dyads are generally assumed to be symmetrically methylated or symmetrically unmethylated, except during DNA replication. Hairpin GS also unambiguously differentiates naturally occurring sequence variation from bisulfite- and PCR-mediated C-to-T conversions at unmethylated cytosines. Our results show that NBL2, which does not appear to be a gene (12), is especially sensitive to multiple diverse DNA methylation changes during oncogenesis, even within the same 0.2-kb region on the same DNA molecule.
Results
Hairpin-bisulfite genomic sequencing strategy and validation
We analyzed the 1.4-kb NBL2 repeat by hairpin GS (16) as shown in Fig. 1. In DNA clones resulting from bisulfite treatment and PCR, a genomic m5CpG, the predominant site of vertebrate DNA methylation, will appear as CpG because it escaped bisulfite deamination, and an unmethylated CpG will become TpG due to cytosine deamination followed by amplification. In hairpin GS, strand ligation results in the sequence information from both genomic strands of a DNA fragment being present in each strand of the resulting DNA clone (Fig. 1A). Corresponding CpG positions in the two halves of one strand of a DNA clone are compared to determine the methylation status of CpG dyads in the template DNA molecule. For simplicity, we will use the following terms for the DNA clones, which describe the CpG dyad methylation status of the molecule that gave rise to the clone (Fig. 1B): M/M (symmetrically methylated), U/U (symmetrically unmethylated), M/U (hemimethylated with the lower strand unmethylated), and U/M (hemimethylated with the upper strand unmethylated). Not only does hairpin GS resolve a symmetrical methylation pattern at a CpG dyad from hemimethylation, but also it allows an unmethylated CpG to be unambiguously distinguished from germline C-to-T changes (Fig. 1B). This is especially useful for DNA repeats because of their appreciable sequence variation (16).
Figure 1.
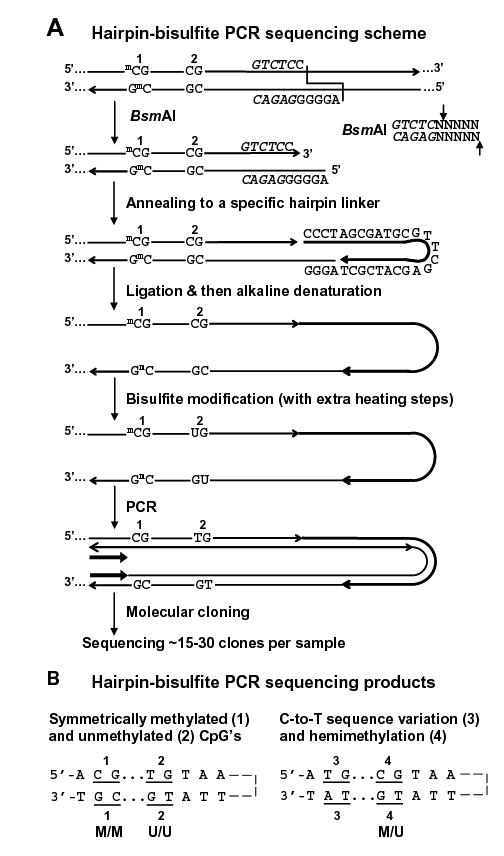
Hairpin genomic sequencing methodology. (A) Hairpin GS of the NBL2 repeat with the covalently linked upper and lower strands (not to scale) diagrammed as a hairpin to illustrate their complementarity before bisulfite deamination of all unmethylated C residues. mC, 5-methylcytosine. The recognition site for BsmAI is in italics and its cleavage specificity is shown on the right. (B) Examples of discrimination between different methylated configurations of CpG dyads and C-to-T changes in a genomic sequence. Part of one strand of each molecular clone is depicted in the hairpin configuration to align CpG positions that were part of a genomic dyad.
From hairpin GS of each normal tissue or cancer DNA, we obtained a single or predominant PCR band of the expected size (508 bp) from which 12 to 32 clones were generated and sequenced (Fig. 2). Given the originally self-complementary nature of the ligated DNA for bisulfite treatment and the specificity of bisulfite for denatured DNA, various controls were done to make sure that hairpin GS did not yield artifacts. First, we should see essentially only CpG methylation (16) because we were examining postnatal tissues (18). As expected, we found only 0 – 0.3% of non-CpG C residues per tissue sample (0.1% overall; data not shown). Also, only 0.6% of C residues persisted as C in hairpin GS clones from an NBL2-containing E. coli plasmid (Fig. 2B). In addition, we checked the completeness of bisulfite modification by digesting all hairpin PCR products with Tsp509I (recognizing AATT). Gel electrophoresis of the digests indicated complete digestion due to bisulfite-mediated C deamination at genomic AACC or AACT in NBL2 (data not shown). Hairpin GS of an NBL2 plasmid methylated in vitro at most CpG sites by M.SssI showed that most of the CpG C residues were retained in the clones.
Figure 2.
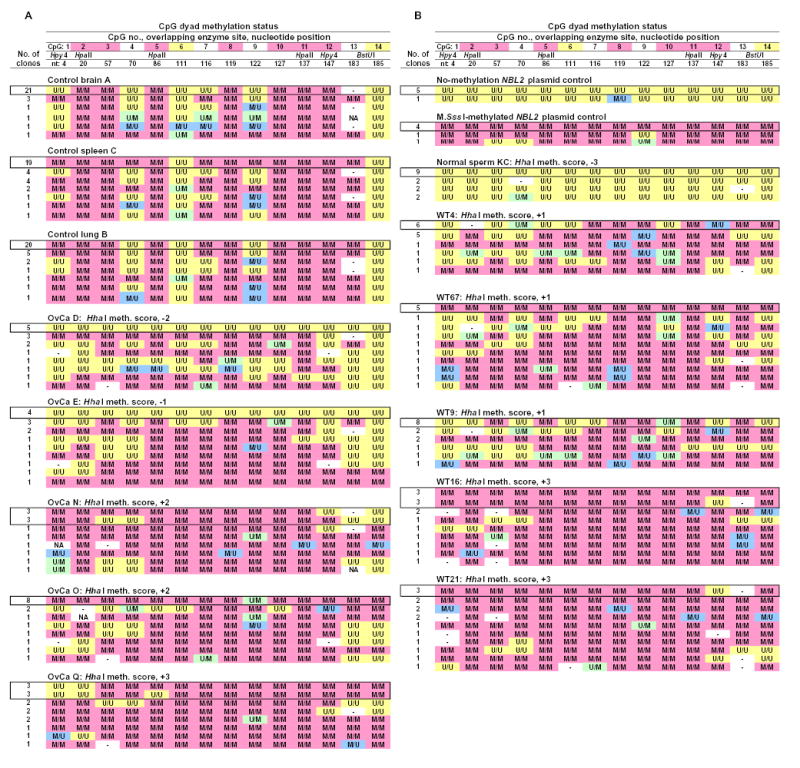
Hairpin genomic sequencing results from NBL2 for normal tissues, cancers, and in vitro-methylated (at CpG’s) or unmethylated NBL2 plasmid. Hairpin-bisulfite PCR-derived genomic sequences are shown for each clone. Each observed epigenetic pattern for a given sample illustrated only once. The most abundant pattern for each sample is boxed. M/M, a symmetrically methylated CpG dyad; U/U, a symmetrically unmethylated CpG dyad; M/U and U/M, the two orientations of hemimethylated CpG dyads; -, no CpG was present at that site due to sequence variation; NA, methylation could not be analyzed due to sequencing mistakes. Upon alignment of the two halves of the strands of the clones, sequencing mistakes were rare. At the top of each column, the following are given: the CpG site number (pink highlighting for sites always M/M in somatic controls and yellow for sites never M/M in somatic controls), nucleotide position within the post-primer sequenced region, and restriction sites. Hpy4, HpyCH4IV.
Also, we also verified that we were not amplifying only a few template molecules by using a 1:20 dilution of the bisulfite-treated DNA instead of the undiluted sample for PCR. A strong PCR product band was obtained with or without dilution from each sample. We conclude that the sequenced molecular clones represent the heterogeneity in the sample DNA, which is consistent with their epigenetic and genetic sequence diversity (Fig. 2). Lastly, we did a mixing experiment with hairpin PCR products amplified as 1:0, 1:3, 1:1, 3:1, and 0:1 mixtures of M.SssI-methylated and unmethylated NBL2 plasmid. We obtained approximately the expected ratios of BstUI-sensitive (CGCG) to BstUI-resistant sites in the PCR product mixtures. Therefore, there was no appreciable selection during the PCR for templates that were methylated or unmethylated, as is sometimes found for bisulfite-treated DNA even when methylation-specific primers are avoided (19).
Hypomethylation and hypermethylation within the same molecular clones from cancers
The normal cell subtype of origin for ovarian epithelial carcinomas is an uncertain minor ovarian epithelial component (20) and for Wilms tumors, an embryonic kidney remnant. Because they are not available, we compared these diverse cancers to various normal postnatal somatic tissues. First, we had to identify CpG sites with invariant methylation status in somatic control tissues (brain, spleen, and lung from different normal individuals). There was a surprisingly high degree of conservation of a complex methylation pattern at NBL2 in the normal somatic tissues (Figs. 2 and 3A) in contrast to the usual findings of either very heterogeneous methylation patterns from molecule to molecule or almost complete methylation or lack of methylation in a given DNA region (21–23). Among the 91 NBL2 DNA clones from somatic controls subject to hairpin GS, 7 of the 14 CpG sites were always symmetrically methylated (CpG2, 3, 5, 8, 10, 11, and 12). Two nonadjacent CpG’s were never symmetrically methylated (CpG6 and 14). One of these, CpG14, was always U/U, and the other, CpG6, was usually U/U but occasionally U/M or M/U. CpG13, which is exactly adjacent to always-unmethylated CpG14 was often replaced by GpG, and hence could not be methylated. However, whenever it was not replaced, it was always M/M despite its immediate U/U neighbor (Fig. 2 and 3A). Normal sperm had no M/M sites in this subregion (Fig.2B), consistent with previous results from various tandem DNA repeats (9).
Figure 3.
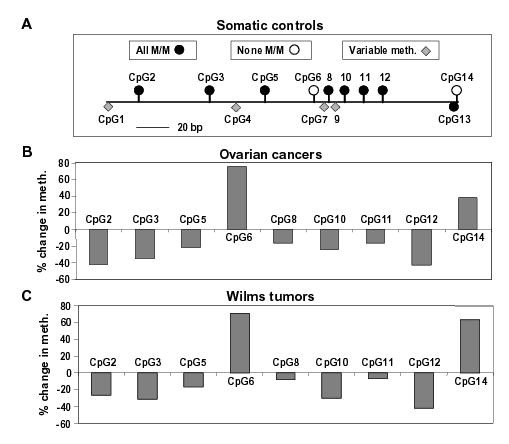
Comparison of methylation in somatic control tissues (brain, spleen, and lung), ovarian carcinomas, and Wilms tumors. (A) Cartoon illustrating to scale the positions of the CpG sites in the hairpin GS region and their methylation status in the somatic controls. The 7 CpG’s that were always M/M and the 2 CpG’s that were either always U/U (CpG14) or usually U/U and occasionally hemimethylated (CpG6) are shown above the horizontal line. The variably methylated CpG’s are shown as diamonds below the line. The filled-in circle below the line represents CpG13, which was always methylated when present, but often not present due to germline sequence variation. (B) and (C) show the overall change in methylation in five ovarian carcinomas and five Wilms tumors at CpG’s that were either always M/M or never M/M in somatic controls. The % change in methylation at CpG2, 3, 5, 8, 10, 11, and 12 is shown as the negative of the percentage of cancer clones with hypomethylation (loss of M/M status) at that position; for CpG6 and 14, it is the percentage of cancer clones with hypermethylation (gain of M/M status).
None of the 146 DNA clones from the ten cancers had the conserved methylation pattern of normal somatic controls (Fig. 2). Moreover, 56% of the cancer clones had a mixture of both hypomethylated and hypermethylated CpG sites. These methylation changes were defined by the loss of the normally conserved M/M status at CpG2, 3, 5, 8, 10, 11, or 12 and the gain of M/M status at CpG6 or 14, sites never normally symmetrically methylated (Figs. 2 and 3). The overall methylation status at each of these nine CpG sites in the cancers was significantly different from that in the somatic controls (p<0.005; p-value adjusted for multiple comparisons).
The contribution of spreading of methylation or demethylation to DNA methylation changes in cancer
Spreading of de novo methylation along a DNA region has been observed (24–27). We did not see evidence of predominant spreading of de novo methylation or demethylation because a pairwise comparison of neighboring CpG sites in NBL2 in the cancers indicated that there was no statistically significant bias towards adjacent sites having the same methylation status. Furthermore, at CpG6 and CpG8, which are separated by only 6 bp, there were seven clones from four cancers (OvCaN and WT4, 9, 21, and 67) that exhibited opposite methylation changes, namely hypermethylation at CpG6 (M/M) and hypomethylation at CpG8 (M/U; Fig. 2). Overall, the methylation changes in many of the clones suggest multiple discontinuous hits of demethylation and de novo methylation within a 0.2-kb region during carcinogenesis.
Nonetheless, there were some DNA clones whose methylation patterns suggested spreading of methylation or demethylation. Some had all 14 CpG dyads unmethylated or all methylated (Fig. 2). Others had the first five or six CpG sites unmethylated on at least one strand. The frequency of clones with no methylation in the first five CpG sites was significantly higher than expected if the methylation at each site was independent, as was the combined frequency of fully methylated or fully unmethylated clones (both p-values<0.0001). In summary, there seems to be spreading of altered DNA methylation patterns in some, but not most, of the copies of NBL2 in the examined cancers.
Hemimethylation in the cancers
In the examined NBL2 subregion in the somatic controls, 1.6% of the CpG sites and 15% of the somatic control clones displayed hemimethylation. The hemimethylation frequencies rose in the cancers to 3.4% of the CpG sites in 47% of the ovarian cancer clones and 6.6% of the CpG sites in 71% of the Wilms tumor clones. Because incomplete bisulfite modification was observed at only ~0.1% of the non-CpG C residues, we are truly describing hemimethylation at CpG’s. There was also a change in the distribution of hemimethylation in the cancer DNAs. Many more of the 14 CpG positions were subject to hemimethylation in the cancers than in the somatic controls. In addition, some of the hemimethylated CpG’s at a given position in the cancer clones displayed a strong bias for demethylation of the top or the bottom strand (Table 1). Hemimethylated CpG dyads in cancer and control clones usually did not occur as runs, but rather had the closest CpG on either side as an M/M or U/U dyad. Furthermore, of the 27 cancer clones containing more than one hemimethylated CpG site, 15 had hemimethylated dyads of opposite polarity with respect to which strand was unmethylated.
Table 1.
Examples of asymmetry in hemimethylation of NBL2 sites in cancers
| No. of clones with indicated type of demethylation at:
|
|||
|---|---|---|---|
| Dyad methylation status | CpG8 | CpG10 | CpG12 |
| Symmetrically demethylated (demeth. in both strands) | 9 | 22 | 49 |
| Hemimethylated (demeth. in only one strand)* | 9 | 18 | 11 |
| Hemimethylated as U/M (demeth. only in top strand) | 1 | 18 | 0 |
| Hemimethylated as M/U (demeth. only in bottom strand) | 8 | 0 | 11 |
U/M status at CpG10 was seen in five cancers. M/U at CpG8 was seen in five cancers and a CpG12 in four cancers.
CpG sites with preferred methylation changes in cancers
Some normally M/M CpG sites appeared to be more likely to become demethylated in both the ovarian carcinomas and Wilms tumors than others (Fig. 3). To test the significance of this finding, we did a pairwise comparison of methylation changes in cancer clones at the seven normally M/M sites and also at the two normally unmethylated CpG dyads. In both the Wilms tumor group and the ovarian carcinoma group, the following significant differences were observed: demethylation at CpG12 was more frequent than at CpG8 or 11, demethylation at CpG2 was more frequent than at CpG5; and demethylation at CpG3 was more frequent than at CpG11 (p <0.05 after adjustment for multiple comparisons). With respect to the two positions that were never normally symmetrically methylated, CpG6 was significantly more prone to cancer-associated hypermethylation (conversion to M/M) than CpG14 (p<0.00001) in ovarian carcinomas, although not in the Wilms tumors.
There was also evidence of preferential epigenetic patterning involving multiple CpG positions in the sequenced NBL2 region from the cancers. Eleven cancer clones derived from four cancers had the following methylation status: CpG4, U/M; CpG12, M/U; CpG1, 3, 5, 6, and 10, U/U; CpG7, 8, 9, 11, 13, and 14, M/M (Fig. 2: first row for WT4, second row for WT9 and OvCaO, and third row for WT67). This methylation pattern constitutes changes from the normally conserved methylation status of the five underlined CpG sites.
To try to explain site preferences for cancer-linked methylation changes or for the conserved methylation patterns in somatic controls, we looked for possible effects of the sequence 1–3 bp on either side of each CpG. No rules for predicting the methylation status in the somatic controls or cancers based upon adjacent sequences could be deduced just from the region subjected to genomic sequencing. However, SB analysis of NBL2 arrays gave us further insights, as described below.
Distinguishing all cancer and somatic control clones by methylation status at several CpG’s
We searched for a few CpG sites whose methylation status could be used to distinguish all the cancer-derived molecular clones from all the somatic control clones. We found such sites with 100% predictive power by generating a classification tree from the data. All but two of the cancer-derived clones displayed symmetrical methylation at CpG6 (M/M) or demethylation at CpG10 (U/U or U/M); none of the somatic control-derived clones had these epigenetic attributes. The two exceptional tumor clones could not display hypomethylation because CpC or CpT replaced CpG6 (Fig. 2B, WT67 and 21, hyphen at the CpG6 position). Those two clones exhibited hypermethylation at CpG14, which distinguishes them from all somatic control-derived clones. Our ability to distinguish all NBL2 cancer clones from all NBL2 somatic control clones also demonstrates the purity of the cancer DNA samples used for this analysis.
Analysis of CpG methylation by Southern blotting
All cancers in this study and an additional 13 ovarian carcinomas and 46 Wilms tumors had been examined by SB analysis for methylation at HhaI and NotI sites with a 1.4-kb NBL2 probe (12). HhaI digests of DNAs from various postnatal somatic control tissues from 15 individuals gave very similar distributions of intermediate-molecular weight hybridizing fragments (e.g., see Fig. 4A), while NotI digests all gave very high-molecular-weight hybridizing fragments (12). A comparison of cancers and somatic controls revealed predominant hypermethylation at HhaI sites in 81% of the 69 cancers and hypomethylation in 4% of them (e.g., Fig. 4A). Advantages of SB analysis are that it can show long-range methylation patterns not identifiable by genomic sequencing, especially in tandem repeats, and it provides results from the population average of all the copies of the examined sequence.
Figure 4.
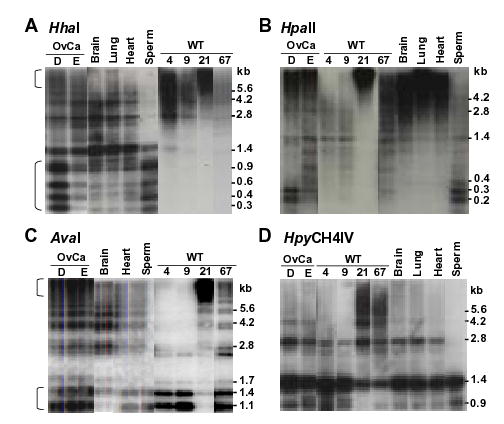
Representative SB analysis of NBL2 hyper- and hypomethylation in cancer DNAs. Ovarian carcinoma, Wilms tumor, and control DNAs were digested with the indicated CpG methylation-sensitive enzymes and probed with the 1.4-kb NBL2 sequence. The brackets in panels A and C indicate the separate hypermethylated and hypomethylated fractions of NBL2 repeats in OvCaD and in OvCaE although the hypomethylated repeats were more prominent, especially for HhaI digests. Different exposures from the same blot were used for these panels.
In this study, we compared the above SB results describing methylation at all HhaI sites throughout the NBL2 arrays and the hairpin GS results for methylation at every CpG in a 0.2-kb NBL2 subregion (Table 2). HhaI-site methylation scores were approximated from phosphorimager quantitation of SB results (+1 to +3, increasing hypermethylation relative to somatic controls; −1 to −3, increasing hypomethylation). The genomic sequencing data for each cancer clone were quantified as the weighted average of hypermethylation at the two normally unmethylated CpG’s and hypomethylation at the seven normally methylated CpG’s. There was a significant association between NBL2 methylation changes in the cancers determined by these two assays (p<0.001). Therefore, both are monitoring similar methylation changes. Also, a comparison of the overall m5C content of the DNA (by HPLC analysis ) and the total proportion of methylated sites in the NBL2 0.2-kb subregion indicated a significant association between these two epigenetic parameters (p=0.001) as well as between global DNA methylation and NBL2 HhaI-site methylation. Therefore, NBL2 methylation changes in cancer are linked to global DNA methylation changes.
Table 2.
Methylation changes in NBL2 repeats in Wilms tumors and ovarian carcinomas relative to controls somatic controls as determined by hairpin sequencing or Southern blot analysis
| Summary of genomic sequencing resultsa |
NBL2 methylation scores from SB with the indicated DNA digestsb |
Global DNA methylationc |
|||||
|---|---|---|---|---|---|---|---|
| Sample | Hypermeth. (%) | Hypometh. (%) | Hha I | Ava I | Hpa II | Bst UI | %C methylated |
| Ovarian carc. D | 22 | 50 | −2 | ↓ | ↓ | ↓ | 3.31 |
| Ovarian carc. E | 33 | 47 | −1 | ↓ | ↓ | ↓ | 2.94 |
| Wilms tumor 9 | 33 | 26 | +1 | ↓ | ↓ | ↓ | 3.09 |
| Wilms tumor 4 | 50 | 30 | +1 | ↓ | ↓ | NC | 2.88 |
| Ovarian carc. N | 63 | 11 | +2 | NC | ↓ | ↑ | 3.76 |
| Wilms tumor 67 | 78 | 9.3 | +1 | ↓ | ↓ | ↓ | 3.45 |
| Ovarian carc. O | 81 | 9.3 | +2 | ↓ | ↓ | ↑ | 3.73 |
| Wilms tumor 21 | 86 | 4.9 | +3 | ↑ | ↑ | ↑ | 3.90 |
| Ovarian carc. Q | 87 | 14 | +3 | NC | ↓ | ↑ | 3.57 |
| Wilms tumor 16 | 89 | 5.3 | +3 | ↑ | ↑ | ↑ | 3.67 |
| ICF B LCL | 19 | 53 | −3 | ↓ | ↓ | ND | ND |
| Pat C LCL | 63 | 12 | +2 | ↓ | NC | ND | ND |
Hypermethylation in NBL2 at the the normally unmethylated CpG6 and CpG14 was calculated as the overall percentage of these two sites with symmetrical methylation and hypomethylation, as the overall loss of symmetrical methylation at the normally methylated CpG2, 3, 5, 8, 10, 11, and 12.
Hha I site methylation scores were from previous SB analyses with phosphorimager quantitation (12). Negative values, overall hypomethylation at Hha I sites; positive values, overall hypermethylation at these sites. For the other CpG methylation-sensitive enzymes, downward and upward arrows denote hypomethylation and hypermethylation, respectively. NC, no change in methylation relative to the somatic controls; ND, not determined.
Global genomic methylation levels determined by HPLC analysis of DNA digested to mononucleosides (11). Depending on the tissue, somatic controls have 3.43–4.04% of genomic C residues methylated.
To enable us to analyze methylation changes at HpaII, AvaI, HpyCH4IV, or BstUI sites in NBL2 arrays from cancer samples, we first characterized methylation of normal somatic DNAs digested with these CpG methylation-sensitive enzymes and hybridized to an NBL2 probe. All somatic controls from various postnatal somatic tissues derived from different individuals gave very similar results with a given enzyme (Fig. 4 and data not shown). However, there was much more resistance of NBL2 arrays in all these controls to cleavage by some of these enzymes than by others. These results could not be explained by the frequency of the restriction sites in NBL2. There are an average of about 9–10 HpaII sites vs. 5–6 HhaI, 2–4 AvaI, 3–5 HpyCH4IV, and 6 BstUI sites per NBL2 monomer (GenBank Y10752 and AC0128692). Nonetheless, NBL2 arrays in somatic controls were much more resistant to digestion by HpaII than by the other enzymes, and HpyCH4IV gave more cleavage than the other enzymes (Fig.4). We disproved the possibility that the low extent of digestion of NBL2 arrays by HpaII in somatic control DNAs was due to sequence variation by showing complete digestion of all tested samples to <0.4-kb fragments by MspI, an isoschizomer of HpaII. MspI is resistant to CpG methylation except at GGCCGG sites (28). Also, internal controls for all digests showed that no inhibitors were present. The preferential methylation of HpaII sites in NBL2 from somatic controls observed in these SB assays was consistent with genomic sequencing data (Fig. 2, CpG2, 5, and 11).
All 18 ovarian cancer DNAs and 13 of the 15 Wilms tumor DNAs examined with at least 3 of the above enzymes exhibited altered SB patterns of NBL2 methylation relative to somatic controls (Table 2 and data not shown). We compared SB data from cancer DNAs digested with different enzymes (Fig. 2) with the caveats that HpaII digests give an underestimate of hypermethylation and HpyCH4IV digests give an underestimate of hypomethylation. Importantly, HhaI sites appeared to undergo de novo methylation during carcinogenesis more frequently than AvaI, HpyCH4IV, and BstUI sites despite all of these enzymes giving mostly intermediate-molecular-weight NBL2-hybridizing bands in somatic controls (Fig. 4, Table 2, and data not shown). This suggests some sequence specificity to cancer-linked hypermethylation. In addition, the distribution of NBL2-containing restriction fragments in HhaI digests and AvaI digests of ovarian carcinomas D and E indicated that NBL2 arrays can be bifurcated into two epigenetic components differing in the extent of methylation at a given restriction site in several of the cancers (brackets in Fig. 4A and C). Long tandem regions of hypermethylation at these two kinds of restriction sites were observed as increases in NBL2 signal in >10-kb fragments even though those tumors also displayed increases in low-molecular weight signal relative to the somatic controls. Individual fractions of NBL2 repeats with respect to long-range methylation patterns might correspond to NBL2 arrays on different acrocentric chromosomes.
Involvement of DNMT3B in methylation of NBL2
ICF syndrome patients usually have missense DNMT3B mutations in both alleles (29), which greatly reduce enzymatic activity (30). To examine the involvement of DNMT3B in methylation of NBL2, we did SB analysis of DNA digests from six ICF B-cell lines, known to have DNMT3B mutations, and ten control B-cell lines. Relative to normal somatic tissues, hypomethylation at NBL2 HhaI sites was seen in four of the six ICF lymphoblastoid cell lines (LCLs) but none of the ten control LCLs. Instead, the control LCLs were hypermethylated in NBL2 arrays compared to normal somatic tissues, including leukocytes (Fig. 5A and data not shown). This indicates that NBL2 underwent de novo methylation at HhaI sites during generation or passage of LCLs only if the LCLs had normal DNMT3B activity.
Figure 5.
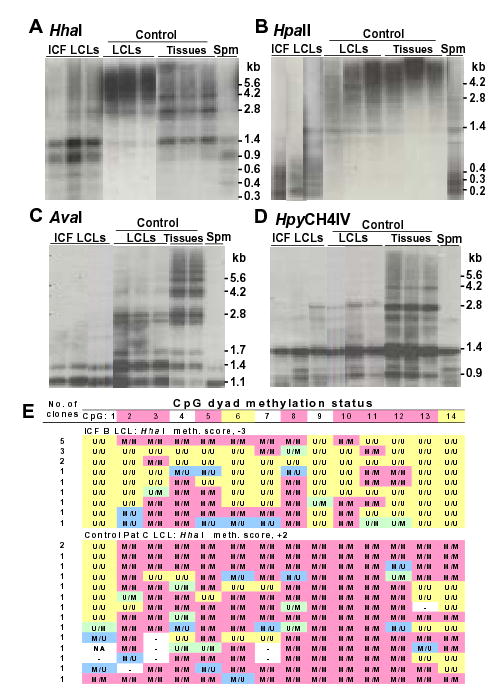
Analysis of methylation in ICF and control LCLs. (A–D), Southern blot analysis as described for Fig. 5. (E) genomic sequencing as for Fig. 2. The ICF LCLs were ICF B, C, and S and the control LCLs were maternal B, maternal C, and paternal C, respectively, from phenotypically normal parents of ICF patients (45,46). The somatic control tissues for panels A and B were brain, lung, and heart; in panel C, lung and spleen; and in panel D, brain, lung, and spleen. Spm, normal sperm.
All but one of the ICF LCLs displayed hypomethylation at NotI sites in NBL2 arrays while none of the control LCLs did (data not shown). In addition, the ICF LCLs showed hypomethylation at HpaII sites compared with control LCLs and control somatic tissues (Fig. 5B). However, both control and ICF LCLs exhibited hypomethylation at AvaI and HpyCH4IV sites compared to control somatic tissues, although the extent of AvaI site hypomethylation was greater for the ICF LCLs (Fig. 5C and D). By hairpin GS of NBL2, there was hypomethylation at some CpG’s and hypermethylation at others in LCLs from ICF patients B and C and in a control LCL (Fig. 5E and data not shown). Nonetheless, there was more hypomethylation in the ICF cells and more hypermethylation in the control cells relative to normal somatic tissues (Table 2).
NBL2 transcription
We previously demonstrated that eight diverse somatic tissues and 16 out of 20 cancers (ovarian carcinomas and Wilms tumors) were negative by RT-PCR for NBL2 transcripts (12). The four positive cancers and one tested LCL (ICF B) evidenced transcripts by both real-time and semi-quantitative RT-PCR, but only at low levels. There was no relationship to hypomethylation at HhaI sites in NBL2, and NBL2 RNA was shown to probably result from run-through transcription. Here we tested five more ICF LCLs and ten control LCLs for NBL2 transcripts by real-time RT-PCR with GAPDH transcripts as the internal standard. Low levels of NBL2 RNA were seen in four of the ICF LCLs that displayed hypomethylation at HhaI sites (data not shown). Neither of the other two ICF LCLs and none of the control LCLs gave a signal appreciably above background, and also none of these displayed hypomethylation at HhaI sites. Duplicate cDNAs prepared from each LCL with random primers or oligo(dT) gave similar results in real-time RT-PCR. Also, semi-quantitative RT-PCR confirmed that the correct size product was obtained from an ICF LCL (ICF C) using either oligo(dT) or random priming, and no product was obtained from a control LCL (Pat C). Product formation from ICF LCLs was shown to be dependent on reverse transcription. An unspecified promoter adjacent to one of the NBL2 arrays might be hypomethylated in the NBL2 RNA-positive ICF LCLs and cancers and thereby activated for run-through transcription.
Discussion
The tandem 1.4-kb NBL2 repeat provided new insights into several aspects of epigenetics in normal tissues and cancers. In the 0.2-kb subregion of NBL2 from diverse control somatic tissues that was examined by hairpin GS, there was a completely conserved pattern of undermethylation at two non-adjacent CpG’s and full methylation at seven other CpG’s (Fig. 3A). This methylation pattern was lost in all 146 DNA clones from ten cancers (ovarian carcinomas and Wilms tumors). Moreover, all but two of the cancer clones were hypomethylated at one specific CpG dyad or hypermethylated at another dyad only 14 bp away (CpG6 and 10). None of the normal DNA clones had this epigenetic signature. The two exceptional cancer clones lacked one of these CpG’s because of sequence variation, but the methylation status of a third CpG (CpG14) allowed those two clones to be distinguished from all normal clones. Hypermethylation at CpG6 and/or CpG14 as well as hypomethylation at CpG2, 3, 5, 8, 10, 11, and/or 12 were seen in the majority of cancer DNA clones. Although hypermethylation of CpG6 or hypomethylation of CpG10 were the most diagnostic epigenetic changes for cancer, there was only one cancer DNA clone (in WT67, Fig. 2) that displayed both hypermethylation and hypomethylation at precisely these positions. This observation and the non-random nature of many of the other DNA methylation changes observed by hairpin GS and SB analyses indicate that losses and gains of methylation in NBL2 during carcinogenesis are often targeted to specific CpG positions and in specific patterns within the repeat.
The targeting of NBL2 for non-random hypermethylation and hypomethylation cannot be explained by transcription-related binding of sequence-specific DNA binding proteins, as is the case for certain promoters (31). NBL2 underwent extensive cancer-linked alterations in methylation despite its lack of transcription in normal tissues and in most analyzed cancers and the absence of an in silico-predicted gene structure (12). Therefore, silencing of transcription is not necessary for all cancer-associated DNA hypermethylation although it has been implicated in promoter hypermethylation (32). Moreover, an in silico search for consensus sites for sequence-specific DNA-binding proteins in NBL2 (www.cbil.upenn.edu/tess) did not yield putative sites that could explain the observed methylation patterns. Whether gene regions and other DNA repeats incur the same type of interspersed hyper- and hypomethylation, which might occur especially in the early stages of tumorigenesis, remains to be determined.
Cancer-linked demethylation of NBL2 was often observed in more than one of the seven normally methylated CpG positions with intervening CpG’s that retained methylation. With respect to hemimethylation, cancer clones had a higher frequency of hemimethylated CpG sites than somatic control clones, and 15 clones had two hemimethylated sites with opposite strands unmethylated. These results indicate that demethylation by inhibition of maintenance methylation after DNA replication is not the major source of cancer-linked hypomethylation. Instead they suggest some kind of active demethylation. However, there was a small, but statistically significant, percentage of cancer DNA clones containing runs of unmethylated CpG’s on one or both strands. An overall decrease in fidelity of maintenance methylation could contribute to this cancer-associated demethylation, but the finding that most hypomethylation was intermittent along the examined cancer DNA molecules and the considerable sequence-specificity observed in the cancer-linked CpG hypomethylation suggest more than just the previously reported inaccurate methylation maintenance (33). The mechanism for demethylation in cancer is uncertain. However, it is clear that mammals have the capacity for active demethylation, as seen in the male pronucleus of the mouse zygote (34). Hemimethylated sites in vertebrate DNAs were described previously (16, 35). There is evidence for demethylation specifically of one strand in a transcription regulatory region followed by demethylation of the other during normal vertebrate development (36, 37). Our results suggest that hemimethylated sites are a rather stable intermediate in demethylation of DNA during carcinogenesis and that this demethylation of one strand of a CpG dyad occurs with preferences for certain CpG’s and with strand preferences at some of these CpG’s.
With regard to de novo methylation of NBL2 in cancer, DNMT3B is likely to be the main enzyme involved, as determined by our analysis of B-cell LCLs from controls and from ICF patients who have inactivating mutations in DNMT3B that eliminate most DNMT3B activity (30). The much lower levels of NBL2 methylation in ICF LCLs than in control LCLs indicate that DNMT3B is necessary for establishing the normal NBL2 methylation pattern during development. The hypermethylation of NBL2 at HhaI sites in control LCLs relative to somatic control tissues could be explained by overexpression of DNMT3B (as well as DNMT3A and DNMT1) during transformation with Epstein-Barr virus (38) and/or by the oncogenic transformation-associated loss of fidelity of DNA methyltransferases (33). In vitro transformation of lymphocytes by Epstein-Barr virus may provide a model for understanding NBL2 methylation changes during malignant transformation in vivo because both hypomethylation and hypermethylation relative to control somatic tissues was observed in NBL2 in normal LCLs. In the two ICF LCLs subject to genomic sequencing, despite the overall hypomethylation of NBL2, some hypermethylation was observed at CpG6, although not at CpG14, the other site at which we could analyze cancer-linked hypermethylation. Also, the control LCL displayed more methylation at CpG6 than CpG14. Similarly, CpG6 was hypermethylated significantly more frequently than CpG14 in ovarian carcinomas. Moreover, CpG6 was occasionally hemimethylated in somatic controls while CpG14 was always symmetrically unmethylated. These findings might be related to the dynamic system of normal maintenance methylation and de novo methylation proposed by Pfeifer et al. (39). At NBL2, there may be infrequent de novo methylation of CpG6 in one strand in normal cells, which is not followed by maintenance methylation. In contrast, there may be frequent hemimethylation at this site with subsequent maintenance methylation upon oncogenic transformation.
An in vitro study of methylation by Dnmt3b indicated strong sequence preferences for de novo methylation (40). Our findings, especially from SB analysis, support this idea although the specificities that we found do not match the in vitro ones. This enzyme may have its sequence preferences strongly altered in vivo. Both our genomic sequencing and SB analysis indicate that HpaII sites (CCGG) have an especially high level of methylation in NBL2 in normal somatic tissues. Also, our SB analysis suggests that HhaI sites (GCGC; which were missing from the bisulfite-sequenced region) were more frequently de novo methylated in the cancers than HpyCH4IV (ACGT), AvaI (CYCGRG), and BstUI (CGCG) sites.
Evidence had been provided for cross-talk between demethylation and de novo methylation pathways in tumorigenesis (41) and in Arabidopsis containing an antisense DNA methyltransferase transgene (42). However, hypermethylation of 5′ regions of tumor suppressor genes and hypomethylation of LINE1 interspersed repeats, satellite DNA, and promoter regions of cancer-testes antigen genes (4, 6, 11, 43) have been shown to be statistically independent of each other, even though all such changes are linked to cancer. Although hypermethylation at certain DNA sequences and hypomethylation at others in cancer are not associated with one another, this study shows that both cancer-linked hypo- and hypermethylation are somehow targeted to NBL2 repeats. We hypothesize that a chromatin structure change in NBL2 arrays occurs during oncogenesis that predisposes to both demethylation and de novo methylation in cis. Alternatively, NBL2 arrays, which have a high overall m5CpG content, might first be demethylated during tumorigenesis and the resulting chromatin structure change might favor further demethylation as well as de novo methylation.
Previous bisulfite-based genomic sequencing studies of cancer DNA usually involved unmethylated CpG-rich promoter regions that become hypermethylated mostly homogeneously (22, 23, 44). There may be several reasons for NBL2 displaying surprisingly complex, non-random patterns of altered methylation during carcinogenesis. It is apparently not a gene, and its methylation status probably confers no selective advantage to a developing tumor. This is unlike the situation with promoters of tumor suppressor genes whose almost complete methylation can benefit the growing tumor by repressing transcription or stabilizing this repression. Also, unlike most DNA regions from cancers analyzed by genomic sequencing, NBL2 normally has very low levels of methylation at some CpG’s and complete methylation at many others so that both cancer-linked increases and decreases of DNA methylation can be observed. Furthermore, it seems to be an unusually frequent target for multiple methylation changes during carcinogenesis. As such, it is a good candidate for a cancer marker as well as a source of insight into cancer-linked epigenetic alterations without the skewing of DNA methylation patterns by oncogenic selection pressures.
Materials and Methods
Patients and DNA samples
With IRB approval, primary tumor samples were obtained from surgery patients prior to chemotherapy or radiation therapy. Informed consent was given by all patients or unlinked samples were used. The LCLs were previously described (45–47) or available from the Coriell Institute (GM17900, AG14836, AG14953, AG15022). Control somatic tissues were autopsy specimens of trauma victims (individuals A, B, and C, all males of 56, 19, and 68 y, respectively). DNA was purified as previously described (11).
Hairpin-bisulfite PCR and cloning
Hairpin-bisulfite PCR was done basically as described by Laird et al. (16) using an NBL2 sequence (Y10752, GenBank) to design primers and the hairpin linker. Human DNA (0.5 μg) or NBL2-containing pDMHD-1 (50 ng) (14) plus 450 ng of λ DNA carrier were digested with 10 U of BsmAI and ligated to 5′-CCCTAGCGATGCGTTCGAGCATCGCT-3′. The DNA was denatured with 0.6 M NaOH at 37°C for 15 min followed by incubation in boiling water for 1 min. At hourly intervals during the 5-h bisulfite treatment, the sample was incubated 4 times in boiling water for 1 min. In an ultrafiltration device (Microcon-100; Millipore), bisulfite-modified DNA was washed 3 times with water, desulfonated with 0.3 M of NaOH at 37°C for 15 min, and eluted in 50 μl of 10 mM Tris-HCl, 1 mM EDTA, pH 7.5. The primers for subsequent PCR had a 3′ T or A corresponding to deamination products from a non-CpG C residue or its complement. The primers were F2-1, 5′-TTTTTGTGGGTTTGTGTTAGT-3′, and R2-2, 5′-CAAAAACATCTTTATTCCTCTA-3′. F2-1 was replaced by F2-2, 5′-AYGTGGTTTGGGTTAGGTAT-3′, in the second round of PCR. Only the F2-2 primer had a CpG in the analogous unmodified genomic sequence (at positions 2 and 3). After denaturation at 94°C for 15 min, PCR was performed (Hotstar, Qiagen) for 30 cycles on 2 μl of the bisulfite-treated DNA (94°C, 15 sec; 52°C, 15 sec, 72°C 1 min, and a final extension at 72°C for 5 min). Then, 1 μl of the product was amplified analogously for an additional 35 cycles. Purified fragments obtained by electrophoresis in a 1.5% agarose gel were used for cloning (TA Cloning Kit, Invitrogen), transformation (E. coli, Top10F), and sequencing (Translational Genomics Research Institute). Due to much sequence variation, there were three CpG positions that were much less frequently present in the clones than the 14 CpG positions referred to above. They are omitted for simplicity, but their methylation status did not change any of the conclusions of this study.
Southern blot analysis
For Southern blot analysis, 1.5 μg of human DNA was digested with 15–30 U of restriction endonuclease overnight according to the manufacturer’s procedures (New England Biolabs), all with parallel internal controls as previously (12). At least three diverse somatic control tissues and sperm DNA were included as references in each blot.
RT-PCR
Using random primers in one set and oligo(dT) in a duplicate set, cDNA was synthesized from 3 μg of total RNA that had been treated with 3 U of DNase I (Amplification Grade, Invitrogen) for 45 min at room temperature. Real-time PCR (SYBR green PCR Master Mix, Applied Biosystems) was done with previously described primers and conditions (12). Semi-quantitative RT-PCR with evaluation of the product by gel electrophoresis was also done as previously described (12).
Statistical methods
Genomic sequencing methylation data were analyzed using R version 2.0.1 (http://www.r-project.org). Chi-square test statistics were used to assess differences of proportions, and strengths of association for continuous and ordinal variables were evaluated using the standard Pearson’s correlation and Kendall’s tau statistics, respectively. Where appropriate, p-values were adjusted for multiple tests using the Holm procedure. Classification trees were generated using the RPART library (48).
Acknowledgments
We are very grateful to Drs. Louis Dubeau, Murali Chintagumpala, Jeffrey Dome, Lisa Diller, Martin Champagne, Hisaki Nagai for supplying the cancer samples and the NBL2 plasmid and Kathryn E. Donahoe for valuable editorial assistance.
Footnotes
Grant support: This research was supported in part by NIH grant no. CA 81506 and a Louisiana Cancer Research Consortium Grant.
References
- 1.Baylin SB, Hoppener JW, de Bustros A, Steenbergh PH, Lips CJ, Nelkin BD. DNA methylation patterns of the calcitonin gene in human lung cancers and lymphomas. Cancer Res. 1986;46:2917–2922. [PubMed] [Google Scholar]
- 2.Issa JP. CpG island methylator phenotype in cancer. Nat Rev Cancer. 2004;4:988–993. doi: 10.1038/nrc1507. [DOI] [PubMed] [Google Scholar]
- 3.Feinberg AP, Vogelstein B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature. 1983;301:89–92. doi: 10.1038/301089a0. [DOI] [PubMed] [Google Scholar]
- 4.Ehrlich, M., Woods, C., Yu, M., Dubeau, L., Yang, F., Campan, M., Weisenberger, D., Long, T. I., Youn, B., Fiala, E., and Laird, P. Quantitative analysis of association between DNA hypermethylation, hypomethylation, and DNMT RNA levels in ovarian tumors. Oncogene, in press, 2005. [DOI] [PMC free article] [PubMed]
- 5.Narayan A, Ji W, Zhang XY, Marrogi A, Graff JR, Baylin SB, Ehrlich M. Hypomethylation of pericentromeric DNA in breast adenocarcinomas. Int J Cancer. 1998;77:833–838. doi: 10.1002/(sici)1097-0215(19980911)77:6<833::aid-ijc6>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 6.Santourlidis S, Florl A, Ackermann R, Wirtz HC, Schulz WA. High frequency of alterations in DNA methylation in adenocarcinoma of the prostate. Prostate. 1999;39:166–174. doi: 10.1002/(sici)1097-0045(19990515)39:3<166::aid-pros4>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 7.Bariol C, Suter C, Cheong K, Ku SL, Meagher A, Hawkins N, Ward R. The relationship between hypomethylation and CpG island methylation in colorectal neoplasia. Am J Pathol. 2003;162:1361–1371. doi: 10.1016/S0002-9440(10)63932-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.De Smet C, De Backer O, Faraoni I, Lurquin C, Brasseur F, Boon T. The activation of human gene MAGE-1 in tumor cells is correlated with genome-wide demethylation. Proc Natl Acad Sci USA. 1996;93:7149–7153. doi: 10.1073/pnas.93.14.7149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ehrlich M. DNA methylation in cancer: too much, but also too little. Oncogene. 2002;21:5400–5413. doi: 10.1038/sj.onc.1205651. [DOI] [PubMed] [Google Scholar]
- 10.Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet. 2002;3:415–428. doi: 10.1038/nrg816. [DOI] [PubMed] [Google Scholar]
- 11.Ehrlich M, Jiang G, Fiala ES, Dome JS, Yu MS, Long TI, Youn B, Sohn OS, Widschwendter M, Tomlinson GE, Chintagumpala M, Champagne M, Parham DM, Liang G, Malik K, Laird PW. Hypomethylation and hypermethylation of DNA in Wilms tumors. Oncogene. 2002;21:6694–6702. doi: 10.1038/sj.onc.1205890. [DOI] [PubMed] [Google Scholar]
- 12.Nishiyama R, Qi L, Tsumagari K, Dubeau L, Weissbecker K, Champagne M, Sikka S, Nagai H, Ehrlich M. A DNA repeat, NBL2, is hypermethylated in some cancers but hypomethylated in others. Cancer Biol Ther. 2005;4:440–448. doi: 10.4161/cbt.4.4.1622. [DOI] [PubMed] [Google Scholar]
- 13.Itano O, Ueda M, Kikuchi K, Hashimoto O, Hayatsu S, Kawaguchi M, Seki H, Aiura K, Kitajima M. Correlation of postoperative recurrence in hepatocellular carcinoma with demethylation of repetitive sequences. Oncogene. 2002;21:789–797. doi: 10.1038/sj.onc.1205124. [DOI] [PubMed] [Google Scholar]
- 14.Nagai H, Kim YS, Yasuda T, Ohmachi Y, Yokouchi H, Monden M, Emi M, Konishi N, Nogami M, Okumura K, Matsubara K. A novel sperm-specific hypomethylation sequence is a demethylation hotspot in human hepatocellular carcinomas. Gene. 1999;237:15–20. doi: 10.1016/s0378-1119(99)00322-4. [DOI] [PubMed] [Google Scholar]
- 15.Thoraval D, Asakawa J, Wimmer K, Kuick R, Lamb B, Richardson B, Ambros P, Glover T, Hanash S. Demethylation of repetitive DNA sequences in neuroblastoma. Genes Chromosomes Cancer. 1996;17:234–244. doi: 10.1002/(SICI)1098-2264(199612)17:4<234::AID-GCC5>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 16.Laird CD, Pleasant ND, Clark AD, Sneeden JL, Hassan KM, Manley NC, Vary JC, Jr, Morgan T, Hansen RS, Stoger R, Hairpin-bisulfite PCR: assessing epigenetic methylation patterns on complementary strands of individual DNA molecules. Proc Natl Acad Sci USA. 2004;101:204–209. doi: 10.1073/pnas.2536758100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Frommer M, McDonald LE, Millar DS, Collis CM, Watt F, Grigg GW, Molloy PL, Paul CL. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc Natl Acad Sci USA. 1992;89:1827–1831. doi: 10.1073/pnas.89.5.1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dodge JE, Ramsahoye BH, Wo ZG, Okano M, Li E. De novo methylation of MMLV provirus in embryonic stem cells: CpG versus non-CpG methylation. Gene. 2002;289:41–48. doi: 10.1016/s0378-1119(02)00469-9. [DOI] [PubMed] [Google Scholar]
- 19.Warnecke PM, Stirzaker C, Melki JR, Millar DS, Paul CL, Clark SJ. Detection and measurement of PCR bias in quantitative methylation analysis of bisulphite-treated DNA. Nucleic Acids Res. 1997;25:4422–4426. doi: 10.1093/nar/25.21.4422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dubeau L. The cell of origin of ovarian epithelial tumors and the ovarian surface epithelium dogma: does the emperor have no clothes? Gynecol Oncol. 1999;72:437–442. doi: 10.1006/gyno.1998.5275. [DOI] [PubMed] [Google Scholar]
- 21.Millar DS, Paul CL, Molloy PL, Clark SJ. A distinct sequence (ATAAA)n separates methylated and unmethylated domains at the 5′-end of the GSTP1 CpG island. J Biol Chem. 2000;275:24893–24899. doi: 10.1074/jbc.M906538199. [DOI] [PubMed] [Google Scholar]
- 22.Melki JR, Vincent PC, Clark SJ. Concurrent DNA hypermethylation of multiple genes in acute myeloid leukemia. Cancer Res. 1999;59:3730–3740. [PubMed] [Google Scholar]
- 23.Amoreira C, Hindermann W, Grunau C. An improved version of the DNA Methylation database (MethDB) Nucleic Acids Res. 2003;31:75–77. doi: 10.1093/nar/gkg093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Toth M, Lichtenberg U, Doerfler W. Genomic sequencing reveals a 5-methylcytosine-free domain in active promoters and the spreading of preimposed methylation patterns. Proc Natl Acad Sci U S A. 1989;86:3728–3732. doi: 10.1073/pnas.86.10.3728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Turker MS. Gene silencing in mammalian cells and the spread of DNA methylation. Oncogene. 2002;21:5388–5393. doi: 10.1038/sj.onc.1205599. [DOI] [PubMed] [Google Scholar]
- 26.Nguyen C, Liang G, Nguyen TT, Tsao-Wei D, Groshen S, Lubbert M, Zhou JH, Benedict WF, Jones PA. Susceptibility of nonpromoter CpG islands to de novo methylation in normal and neoplastic cells. J Natl Cancer Inst. 2001;93:1465–1472. doi: 10.1093/jnci/93.19.1465. [DOI] [PubMed] [Google Scholar]
- 27.Yan PS, Shi H, Rahmatpanah F, Hsiau TH, Hsiau AH, Leu YW, Liu JC, Huang TH. Differential distribution of DNA methylation within the RASSF1A CpG island in breast cancer. Cancer Res. 2003;63:6178–6186. [PubMed] [Google Scholar]
- 28.Busslinger M, deBoer E, Wright S, Grosveld FG, Flavell RA. The sequence GGCmCGG is resistant to MspI cleavage. Nucleic Acids Res. 1983;11:3559–3569. doi: 10.1093/nar/11.11.3559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hansen RS, Wijmenga C, Luo P, Stanek AM, Canfield TK, Weemaes CM, Gartler SM. The DNMT3B DNA methyltransferase gene is mutated in the ICF immunodeficiency syndrome. Proc Natl Acad Sci USA. 1999;96:14412–14417. doi: 10.1073/pnas.96.25.14412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gowher H, Jeltsch A. Molecular enzymology of the catalytic domains of the Dnmt3a and Dnmt3b DNA methyltransferases. J Biol Chem. 2002;277:20409–20414. doi: 10.1074/jbc.M202148200. [DOI] [PubMed] [Google Scholar]
- 31.Hornstra IK, Yang TP. High-resolution methylation analysis of the human hypoxanthine phosphoribosyltransferase gene 5′ region on the active and inactive X chromosomes: correlation with binding sites for transcription factors. Mol Cell Biol. 1994;14:1419–1430. doi: 10.1128/mcb.14.2.1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Clark SJ, Melki J. DNA methylation and gene silencing in cancer: which is the guilty party? Oncogene. 2002;21:5380–5387. doi: 10.1038/sj.onc.1205598. [DOI] [PubMed] [Google Scholar]
- 33.Ushijima T, Watanabe N, Shimizu K, Miyamoto K, Sugimura T, Kaneda A. Decreased fidelity in replicating CpG methylation patterns in cancer cells. Cancer Res. 2005;65:11–17. [PubMed] [Google Scholar]
- 34.Santos F, Hendrich B, Reik W, Dean W. Dynamic reprogramming of DNA methylation in the early mouse embryo. Dev Biol. 2002;241:172–182. doi: 10.1006/dbio.2001.0501. [DOI] [PubMed] [Google Scholar]
- 35.Liang G, Chan MF, Tomigahara Y, Tsai YC, Gonzales FA, Li E, Laird PW, Jones PA. Cooperativity between DNA methyltransferases in the maintenance methylation of repetitive elements. Mol Cell Biol. 2002;22:480–491. doi: 10.1128/MCB.22.2.480-491.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Saluz HP, Jiricny J, Jost JP. Genomic sequencing reveals a positive correlation between the kinetics of strand-specific DNA demethylation of the overlapping estradiol/glucocorticoid-receptor binding sites and the rate of avian vitellogenin mRNA synthesis. Proc Natl Acad Sci U S A. 1986;83:7167–7171. doi: 10.1073/pnas.83.19.7167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Singal R, vanWert JM. De novo methylation of an embryonic globin gene during normal development is strand specific and spreads from the proximal transcribed region. Blood. 2001;98:3441–3446. doi: 10.1182/blood.v98.12.3441. [DOI] [PubMed] [Google Scholar]
- 38.Tsai CN, Tsai CL, Tse KP, Chang HY, Chang YS. The Epstein-Barr virus oncogene product, latent membrane protein 1, induces the downregulation of E-cadherin gene expression via activation of DNA methyltransferases. Proc Natl Acad Sci USA. 2002;99:10084–10089. doi: 10.1073/pnas.152059399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pfeifer GP, Steigerwald SD, Hansen RS, Gartler SM, Riggs AD. Polymerase chain reaction-aided genomic sequencing of an X chromosome-linked CpG island: methylation patterns suggest clonal inheritance, CpG site autonomy, and an explanation of activity state stability. Proc Natl Acad Sci USA. 1990;87:8252–8256. doi: 10.1073/pnas.87.21.8252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Handa V, Jeltsch A. Profound flanking sequence preference of Dnmt3a and Dnmt3b mammalian DNA methyltransferases shape the human epigenome. J Mol Biol. 2005;348:1103–1112. doi: 10.1016/j.jmb.2005.02.044. [DOI] [PubMed] [Google Scholar]
- 41.Pogribny IP, Miller BJ, James SJ. Alterations in hepatic p53 gene methylation patterns during tumor progression with folate/methyl deficiency in the rat. Cancer Lett. 1997;115:31–38. doi: 10.1016/s0304-3835(97)04708-3. [DOI] [PubMed] [Google Scholar]
- 42.Jacobsen SE, Meyerowitz EM. Hypermethylated SUPERMAN epigenetic alleles in Arabidopsis. Science. 1997;277:1100–1103. doi: 10.1126/science.277.5329.1100. [DOI] [PubMed] [Google Scholar]
- 43.Kaneda A, Tsukamoto T, Takamura-Enya T, Watanabe N, Kaminishi M, Sugimura T, Tatematsu M, Ushijima T. Frequent hypomethylation in multiple promoter CpG islands is associated with global hypomethylation, but not with frequent promoter hypermethylation. Cancer Sci. 2004;95:58–64. doi: 10.1111/j.1349-7006.2004.tb03171.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rush LJ, Raval A, Funchain P, Johnson AJ, Smith L, Lucas DM, Bembea M, Liu TH, Heerema NA, Rassenti L, Liyanarachchi S, Davuluri R, Byrd JC, Plass C. Epigenetic profiling in chronic lymphocytic leukemia reveals novel methylation targets. Cancer Res. 2004;64:2424–2433. doi: 10.1158/0008-5472.can-03-2870. [DOI] [PubMed] [Google Scholar]
- 45.Ehrlich M, Buchanan K, Tsien F, Jiang G, Sun B, Uicker W, Weemaes C, Smeets D, Sperling K, Belohradsky B, Tommerup N, Misek D, Rouillard JM, Kuick R, Hanash S. DNA methyltransferase 3B mutations linked to the ICF syndrome cause dysregulation of lymphocyte migration, activation, and survival genes. Hum Mol Genet. 2001;10:2917–2931. doi: 10.1093/hmg/10.25.2917. [DOI] [PubMed] [Google Scholar]
- 46.Tuck-Muller CM, Narayan A, Tsien F, Smeets D, Sawyer J, Fiala ES, Sohn O, Ehrlich M. DNA hypomethylation and unusual chromosome instability in cell lines from ICF syndrome patients. Cytogenet Cell Genet. 2000;89:121–128. doi: 10.1159/000015590. [DOI] [PubMed] [Google Scholar]
- 47.Gisselsson D, Shao C, Tuck-Muller C, Sogorovic S, Palsson E, Smeets D, Ehrlich M. Interphase chromosomal abnormalities and mitotic missegregation of hypomethylated sequences in ICF syndrome cells. Chromosoma. 2005;114:118–126. doi: 10.1007/s00412-005-0343-7. [DOI] [PubMed] [Google Scholar]
- 48.Breiman, L., Friedman, J. H., Olshen, R. A., and Stone, C. J. Classication and Regression Trees. Belmont, Ca.: Wadsworth, 1984.