

Abstract
Objective
To analyze the presentation, localization, surgical management, pathology, and long-term outcome of a large series of patients with pheochromocytomas.
Summary Background Data
There are several areas of controversy pertaining to pheochromocytomas. Although many studies report a higher rate of malignancy for extraadrenal pheochromocytomas than for adrenal pheochromocytomas, the number of patients with the former tumor are small and statistical analysis is lacking. There has also been recent debate as to whether microscopic features of the tumor may be predictive of future behavior.
Methods
From 1950 to 1998, the authors observed 108 pheochromocytomas in 104 patients. The outcome of these patients has been followed prospectively. The medical records of these patients were reviewed for data on the presentation, localization, surgical management, pathology, and outcome. Patient survival was analyzed using Kaplan–Meier survival distributions.
Results
This study included 66 female patients and 38 male patients. The average age at surgery was 42.3 years. Sporadic cases accounted for 84% of the patients; the other 16% had multiple endocrine neoplasia type 2, von Recklinghausen’s disease, von Hippel-Lindau disease, or Carney’s syndrome. Of 64 adrenal tumors, 55 were initially considered benign, 6 had microscopic malignant features, and 3 had malignant disease. Mean patient follow-up was 12.6 years. To date, in five additional patients (none with microscopic disease) malignant disease developed (13% overall rate of malignancy). Recurrence occurred as late as 15 years after resection. Of 26 extraadrenal pheochromocytomas, 14 were initially considered benign, 8 had microscopic malignant features, and 4 had malignant disease. Thus, 46% of patients had either malignant disease or tumors with malignant features. Mean patient follow-up was 11.5 years. In one patient with benign disease and in one patient with malignant features, malignant disease developed (23% overall rate of malignancy). The difference in the rate of malignancy was not statistically significant between adrenal and extraadrenal pheochromocytomas. Patients with adrenal and extraadrenal pheochromocytomas also had similar rates of survival (p = NS).
Conclusions
The data suggest that patients with extraadrenal pheochromocytomas have the same risk of malignancy and the same overall survival as patients with adrenal pheochromocytomas. Lifelong follow-up of these patients is mandatory.
The management of adrenal and extraadrenal pheochromocytomas has challenged surgeons since 1926, when both Charles Mayo and Cesar Roux reported the resection of such tumors. 1 There is current debate as to whether extraadrenal pheochromocytomas are more likely to exhibit malignant behavior than the more commonly encountered tumor in the adrenal site. 2–4 In addition, as with other neuroendocrine tumors, the future behavior of apparently benign pheochromocytomas has been difficult to predict. The criteria that apply to many other types of tumors, including cellular atypia, increased mitotic activity, and vascular or capsular invasion, have not been thought to be reliable predictors of malignancy. 5,6 Three recent publications, however, have rekindled the debate as to whether microscopic features may be predictive of future behavior. 7–9
The primary aim of the present study was to analyze the presentation, localization, surgical management, pathology, and long-term outcome of a large series of patients with pheochromocytomas.
METHODS
From 1950 to 1998, we observed 108 pheochromocytomas in 104 patients. These patients have been followed prospectively through documented return visits, phone contact by physicians, and contact by the tumor registry. Formal contact with this group of patients as a whole has been made approximately every 5 to 7 years since 1980. The initial symptoms and clinical presentations of these patients were obtained retrospectively through physician charts and the medical records. Two patients were lost to follow-up after discharge, and two patients were lost to follow-up after 3 years because they moved out of the United States.
Records were reviewed for demographic information, presenting signs and symptoms, past medical history, family history, preoperative laboratory studies confirming the diagnosis of pheochromocytoma, tumor localization studies, preoperative preparation, surgical procedure, tumor pathology, and perioperative and postoperative complications. If past medical history or family history was not specifically addressed and recorded in the hospital chart, no assumptions were made for this study. In addition, all anesthetic records were reviewed by one of the authors. The intraoperative course was judged subjectively as smooth, moderate, or complicated.
Beginning in 1968, virtually all patients with a suspected pheochromocytoma had biochemical confirmation of pheochromocytoma by either increased catecholamine and catecholamine metabolites concentration in a 24-hour urine collection or elevated catecholamine concentration in the plasma. Before this date, provocative testing, including histamine and glucagon stimulation, was routinely performed, as was the regitine infusion test.
Beginning in 1963, once the diagnosis of pheochromocytoma was made, preoperative radiographic localization was attempted. For the analysis of preoperative localization studies, 10 a true-positive imaging study had to be confirmed by surgical and pathologic findings. A false-negative imaging study was defined as no lesion detected by the imaging study, although one was identified by surgical and pathologic findings. A false-positive imaging study was defined as a lesion detected by an imaging study and attributed to a pheochromocytoma that was not confirmed as such by surgical and pathologic findings. Sensitivity equals the number of true-positive results divided by the sum of the true-positive and false-negative results. Specificity equals the total number of patients imaged minus the number of false-positive results, all divided by the total number of patients imaged.
The preoperative management used at Vanderbilt has been extensively discussed by Robertson et al. 11 Preoperative α-blockade consisting of phenoxybenzamine was first used at our institution in 1967. Since then, patients were blocked between 1 day and several weeks before surgery. Currently, all patients undergoing surgical management of pheochromocytoma are blocked before surgery with phenoxybenzamine. Preoperative use of β-blockade began in 1968 but has been used sparingly for specific indications (significant supraventricular tachycardia or ventricular tachycardia). To date, only 13 patients have been placed on such long-acting agents. Alpha-methyl-tyrosine (metyrosine), which inhibits tyrosine hydroxylase, the rate-limiting step in the synthesis of catecholamines, was first used at our institution in 1973. It has been used as a blocking agent in 43 patients and has always been used in combination with phenoxybenzamine. Patients have been considered ready for surgery when plasma metanephrine levels have reverted to near-normal levels in response to pharmacologic blockade.
Before 1978, virtually all pheochromocytomas were resected using a thoracoabdominal approach. Since 1979, only one pheochromocytoma has been resected using a thoracoabdominal approach. After 1979, the midline abdominal incision became the preferred approach. Beginning in 1998, laparoscopy has been selectively used in three patients, and we expect this will be used much more frequently in the future. 12,13
Tumor dimensions and pathologic features were obtained from the pathology reports and were correlated with the surgical report. Tumor volume was calculated according to the formula: (π/6) × Dx × Dy × Dz, where Dx, Dy, and Dz are the three measured dimensions of the tumor ex vivo.
The final diagnosis of malignancy was strictly based on the finding of either metastatic disease or gross tumor infiltration into surrounding structures such as spine, liver, or kidney. All tumors were also assessed for microscopic capsular or vascular invasion. For the purposes of this analysis, these latter findings were considered suggestive of malignancy, and these patients were considered at risk.
Follow-up was defined as the time from surgery to last contact with the patient or the patient’s local physician if the patient was still living. If the patient was deceased, follow-up was defined as the time from surgery to death. Any perioperative deaths (within 90 days) were excluded from the calculation of follow-up. Two patients lost to follow-up within the first year were also excluded from the calculation of follow-up.
Statistical Analysis
Simple means between groups were compared using t tests. Survival distributions were determined by life-table analysis using the method of Kaplan and Meier. 14 Comparison of survival distributions was performed by log-rank analysis. Analysis of tumor volume was done by linear regression. 15 All analysis was performed using SPSS-MAC (SPSS Inc., Chicago, IL). A probability value of <0.05 was the limit of statistical significance.
RESULTS
Demographics
This study included 66 female patients and 38 male patients (p = NS). The average age at surgery was 42.3 years (median 44.5 years, range 9 to 79 years). The average age was 43.5 years in the female patients and 40.1 in the male patients (p = NS). Eight patients were 18 years of age or younger. Eighty-seven patients were white, 12 patients were black, and 9 patients were either “other” or not recorded.
Sporadic cases of pheochromocytoma accounted for 87 (84%) of the patients. Nine patients (8.6%) were either members of a multiple endocrine neoplasia type 2 (MEN2) kindred or were index cases of MEN2 (one patient). Two patients (1.9%) had von Recklinghausen’s disease, four patients (3.8%) had von Hippel-Lindau disease, and two patients had Carney’s syndrome (1.9%).
Family History and Past Medical History
Accurate information about family history of hypertension was available in 46 of 87 patients without MEN2, von Hippel-Lindau disease, or von Recklinghausen’s disease. There was a family history of hypertension in only 16 of the latter patients (35%), whereas in 30 instances (65%) there was clearly no family history of hypertension.
A past medical history of hypertension was defined as any episode of documented hypertension occurring at least 1 year before the diagnosis of pheochromocytoma was made. Careful documentation of past hypertension history was actually present in only 42 patients; hypertension was present in only 18 (42%) instances, whereas no past history of hypertension was documented in 24 (57%) of the patients.
Symptoms of Hypertension
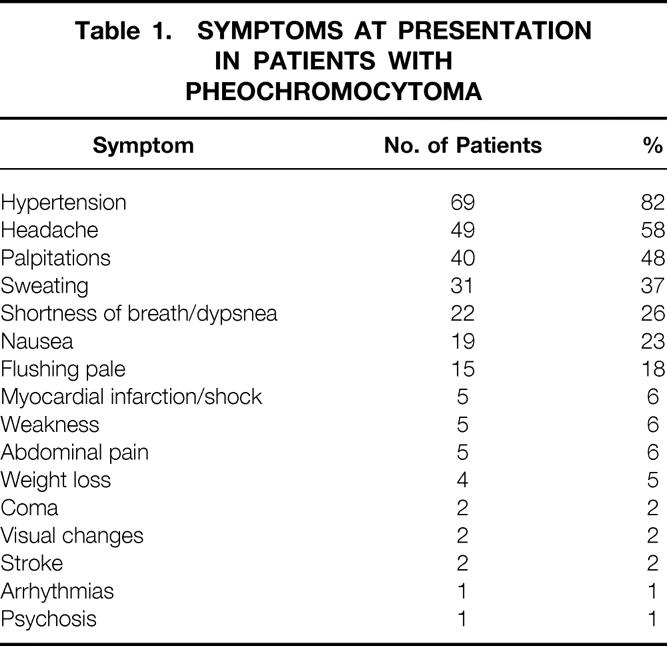
Symptoms were thought to be well documented in 84 cases (Table 1). The most common symptom was hypertension, which was present in 69 (82%) of the cases. Conversely, nine patients (11%) had clear documentation of no hypertension. This excluded those normotensive patients who were evaluated solely on the basis of a family history of MEN2.
Table 1. Symptoms at Presentation in Patients with Pheochromocytoma
Other Symptoms
Table 1 also lists the other most frequent signs and symptoms associated with pheochromocytoma besides hypertension. Headache, palpitations, and sweating were the three next most frequent symptoms, occurring in 49 (58%), 40 (48%), and 31 (37%) of the patients with pheochromocytoma.
Radiologic Imaging Studies
From 1968 to 1990, preoperative angiography was used extensively to localize these tumors. Twenty studies were performed (Table 2). The overall sensitivity of the test to detect these tumors was 84% and the specificity was 95%. There was one false-positive result, and in one case angiography detected one pheochromocytoma but missed a second tumor that was present (Fig. 1). Computed tomography (CT) has been the predominant imaging technique used at our institution since 1977 (Fig. 2). The majority of these studies were performed without intravenous contrast. The overall sensitivity was 94% and the specificity was 97%. Magnetic resonance imaging (MRI) has been used at our institution since 1985. The overall sensitivity was 83% and the specificity was 100%.
Table 2. Imaging Modalities in Patients with Pheochromocytoma
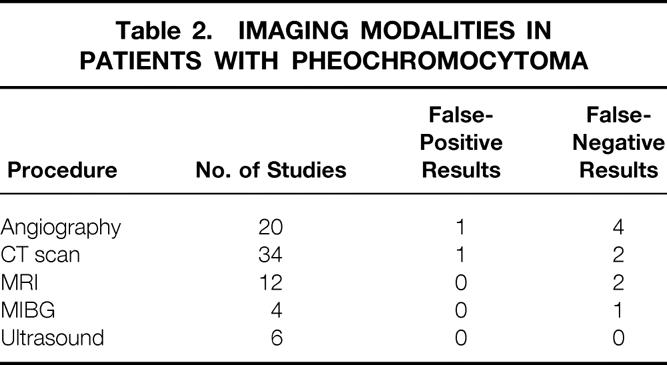
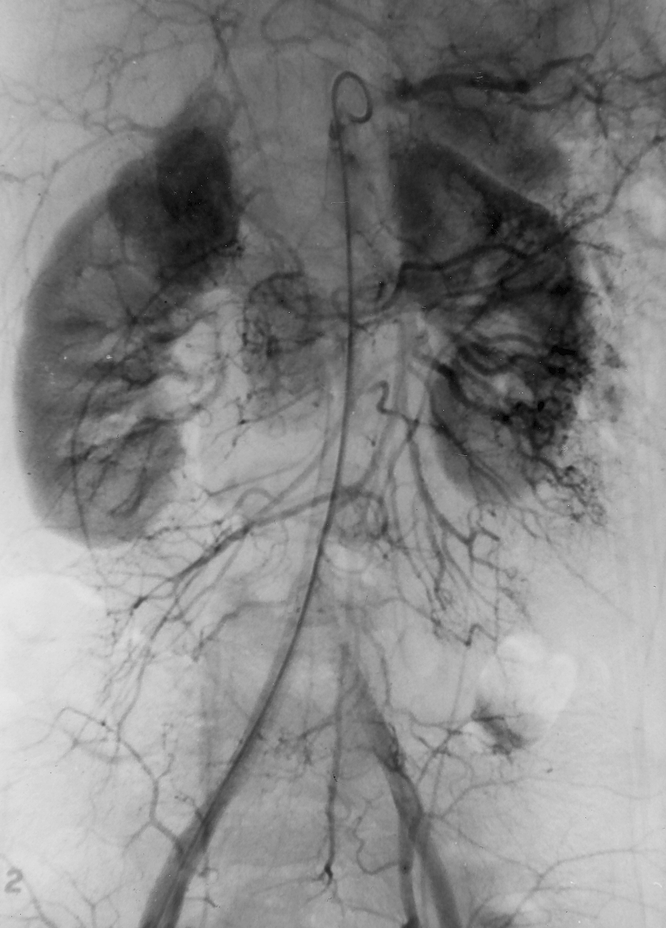
Figure 1. Angiogram demonstrating the typical radiographic blush of an extraadrenal pheochromocytoma to the patient’s right of the aorta. This is also the angiogram that did not detect a second extraadrenal pheochromocytoma located on the patient’s left side; it was successfully resected at a second operation.
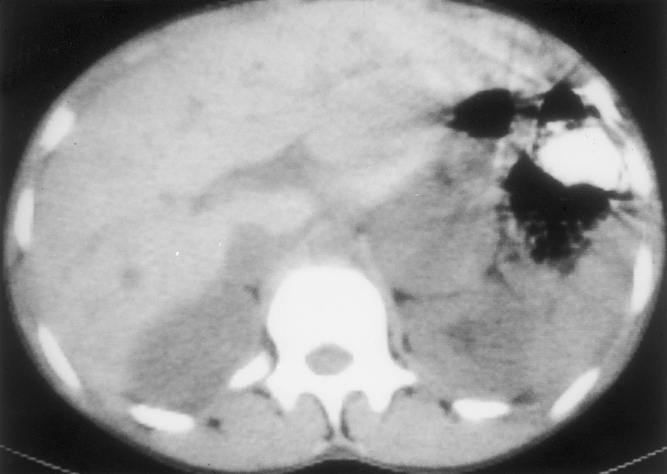
Figure 2. A representative noncontrast CT scan of the abdomen demonstrating a 5-cm left adrenal pheochromocytoma.
In only four patients has MIBG been used. The study yielded a true-positive result in three patients and a false-negative result in one. Ultrasound has been used six times and was correctly positive in all six cases. However, because of the low numbers, sensitivity and specificity were not calculated for these last two modalities.
No significant complications have been attributed to any of these studies. Several patients became significantly hypertensive during angiography, but these episodes were effectively managed pharmacologically.
Autopsy Diagnosis
In 11 patients, the diagnosis of pheochromocytoma was made at autopsy. Ten of these patients were among the first 17 patients included in this study. The last patient at our institution diagnosed by autopsy was in 1969; it was an incidental finding in a patient who died from prostate cancer. Of these 11 patients, death was most likely attributable to the tumor in six patients. The last known patient to die from an unrecognized pheochromocytoma at our institution was in 1956. Of these patients, one patient died during a histamine provocative test, three suffered strokes, one patient died immediately after a hysterectomy, and one patient died in the operating room from hemodynamic instability during a procedure for an ectopic pregnancy.
Surgical Procedures and Surgical Course
Between 1950 and 1998, 103 separate surgical procedures for pheochromocytoma have been performed at Vanderbilt and its VA-affiliated hospital. Some patients had two procedures. Sixty-four unilateral and nine bilateral adrenalectomies have been performed. Twenty-six resections of extraadrenal pheochromocytomas have been performed, as well as four biopsies of unresectable tumors.
Sufficient data were available to determine that at least 16 patients (all in the early years of this series) underwent surgical resection without preoperative blockade. Subjectively, the surgical course was classified as relatively smooth in 5 patients and complicated in 11 (69%). Nevertheless, there were no perioperative complications attributable to hemodynamic instability.
There was clear documentation of preoperative α-blockade in 67 cases. In 57 cases, the intraoperative course was very smooth. In three cases the surgical course was complicated: in one case, there was considerable hypertension during induction; in the other two cases, persistent hypotension after the resection of the pheochromocytoma required vigorous volume resuscitation. Again, there were no complications related to these events. In an additional seven cases, the reviewer thought that the intraoperative course was intermediate.
Metyrosine was used in 43 of the patients who were also blocked with phenoxybenzamine. In only two of these patients was the intraoperative course complicated, and that was related to hypotension after removal of the tumor, but the hypotension did respond to volume resuscitation. Preoperative β-blockade was initiated in only 13 patients, and no conclusions were made based on this small number of cases.
Pathology
Accurate tumor measurements could be obtained from the records of 68 patients. Tumor volumes were calculated for all pheochromocytomas except in patients with MEN2 who underwent bilateral adrenalectomy, because those tumors were often small and multiple. Tumor volumes averaged 121.2 cm3 in the first 34 patients and 49.2 cm3 in the last 34 patients. Tumor volumes across time were assessed by linear regression. The decrease in tumor size since 1950 was statistically significant (p = 0.021). The mean tumor diameter of malignant pheochromocytomas was 7.58 cm, and the mean tumor diameter of benign pheochromocytomas was 5.23 cm. This difference tended to be significant (0.05 < p < 0.10), suggesting that larger tumors have a greater propensity to exhibit malignant behavior.
Surgical Complications
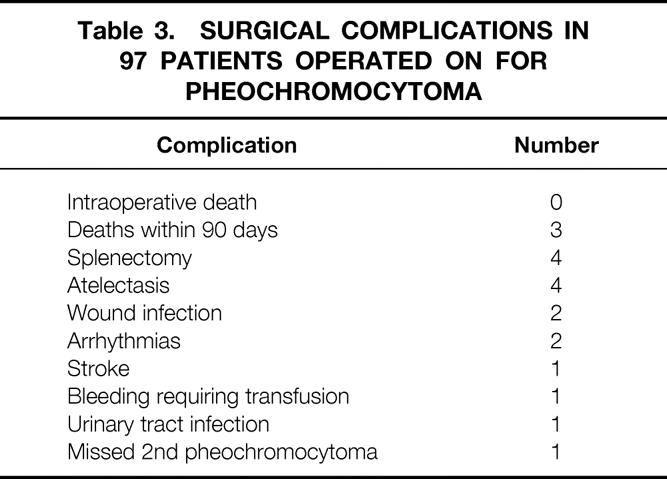
The surgical complications are listed in Table 3. One perioperative death occurred 6 weeks after successful resection of a unilateral pheochromocytoma in 1967. The death was attributed to postoperative renal failure secondary to methoxyflurane anesthesia, not intraoperative hemodynamic changes; this has been discussed in a previous publication. 6 There was another death at postoperative day 90 in 1965, most likely as a result of hypertension secondary to renal arterial disease. A third perioperative death occurred in 1970. The patient had an unresectable malignant pheochromocytoma and died from hypertension and seizures after an exploratory laparotomy and biopsy. There have been no perioperative deaths since 1970. Additional complications also listed in Table 3 include four incidental splenectomies, one postoperative stroke that did not result from significant hemodynamic changes, and one episode of postoperative bleeding requiring transfusion but not a second surgical procedure.
Table 3. Surgical Complications in 97 Patients Operated on for Pheochromocytoma
Outcome of Unilateral Adrenal Disease
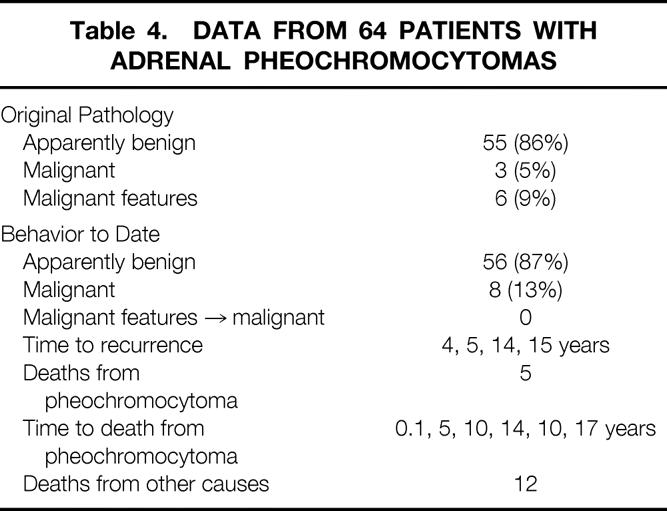
Table 4 summarizes the biologic behavior of the tumors and the outcomes of patients who underwent unilateral adrenalectomy. Of the 64 adrenal pheochromocytomas, 34 were in the left adrenal gland and 30 were in the right adrenal gland (p = NS). Based on strict histopathologic definitions, 55 of the lesions were initially consistent with a benign process, whereas three (5%) patients had malignant disease. An additional six tumors (9%) demonstrated microscopic vascular or capsular invasion, and these patients were considered at risk. Patient follow-up in this group has averaged 12.6 years (median 9 years). During this time, five patients who originally had no evidence of malignant disease (grossly or microscopically) returned with metastatic disease. Presentation has been as long as 15 years after the initial resection. None of the six at-risk patients with microscopic vascular or capsular invasion have demonstrated recurrence or metastases. Thus, to date, eight (13%) of these tumors have proven to be malignant. Five patients have died from pheochromocytoma. Excluding the one perioperative death, mean time to death was 11.5 years. An additional 12 patients with unilateral pheochromocytomas have died from other causes.
Table 4. Data from 64 Patients with Adrenal Pheochromocytomas
Extraadrenal Pheochromocytomas
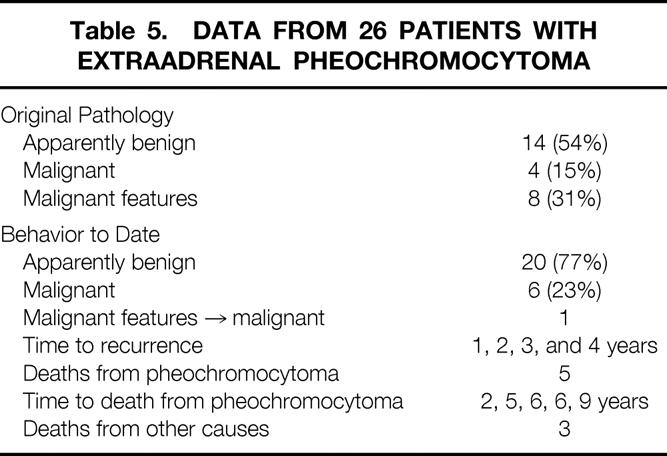
Table 5 summarizes the biologic behavior of the tumors and the outcomes of patients who underwent resection of extraadrenal pheochromocytomas. Twenty-six such pheochromocytomas were identified. Based on strict histopathologic definitions, 14 (54%) of the lesions were initially consistent with a benign process, whereas four (15%) patients had malignant disease with either metastatic disease or gross regional invasion. An additional eight tumors (31%) demonstrated microscopic vascular or capsular invasion, and patients were considered at risk. Thus, for the purposes of this study, 46% of the patients either had clearly malignant disease or tumors with classic malignant features. Patient follow-up in this group has averaged 11.5 years (range, 3 months to 26 years; median 9.0 years). During this time, one patient who originally had no evidence of malignant disease grossly or microscopically returned with metastatic disease 2 years after the initial resection. One of the eight at-risk patients with microscopic vascular or capsular invasion demonstrated metastases 1 year after surgery. Thus, to date, six (23%) of these tumors have proven to be malignant.
Table 5. Data from 26 Patients with Extraadrenal Pheochromocytoma
Malignancy rates between adrenal and extraadrenal pheochromocytomas were analyzed using the Mann–Whitney test. 15 The higher percentage of malignancy observed in the extraadrenal pheochromocytomas was not statistically significant (p = 0.31).
Five patients have died from pheochromocytoma; the mean time to death was 5.4 years. An additional three patients with extraadrenal pheochromocytomas have died from other causes.
Bilateral Pheochromocytomas
Nine patients underwent bilateral adrenalectomy, largely for pheochromocytoma in the setting of MEN2. None of these patients demonstrated malignant disease at the time of surgery, and in none of them has recurrent or metastatic disease developed. Mean follow-up has been 13.1 years (range 3 to 22 years). One patient died from metastatic medullary thyroid cancer 8 years after adrenalectomy.
Patterns of Recurrence
Of the six patients with extraadrenal pheochromocytomas proven to be malignant, bone metastases developed in four and two had grossly invasive recurrent disease at the site of the original tumor. Bone metastases also developed in both of these latter two patients. Of the eight patients with malignant adrenal pheochromocytomas, recurrent disease developed in four patients, metastases to bone occurred in two patients, gross invasive recurrent disease at the site of the original tumor occurred in one patient, and metastases to the liver occurred in one patient.
Survival Data
Overall location-specific survival is shown in Figure 3. This reflects patients with unilateral adrenal pheochromocytoma, extraadrenal pheochromocytoma, or bilateral disease who died of all causes. Using log-rank analysis, there was no statistical difference in survival between the groups (p = 0.82). Figure 4 depicts cause-specific survival. These are the survival curves reflecting patients who died specifically from their tumor. Again, analysis demonstrates no statistical difference in survival between groups (p = 0.31).
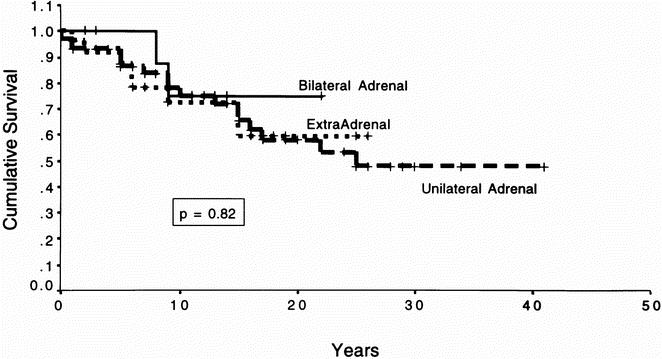
Figure 3. Overall actual survival. This figure depicts the Kaplan–Meier survival curve for all patients with pheochromocytoma dying from all causes. The patients were divided into categories based on whether they underwent bilateral adrenalectomies, unilateral adrenalectomies, or resection of extraadrenal pheochromocytomas. The survival times of the individual groups were compared using the log-rank test; p = 0.82, indicating that there was no statistical difference in the survival time between groups.
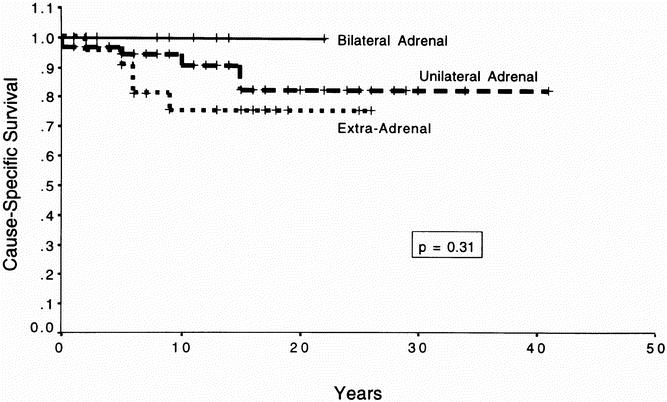
Figure 4. Cause-specific survival. This figure depicts the Kaplan–Meier survival curve for all patients with pheochromocytoma dying specifically from pheochromocytoma. The patients were divided into categories based on whether they underwent bilateral adrenalectomies, unilateral adrenalectomies, or resection of extraadrenal pheochromocytomas. The survival times of the individual groups were compared using the log-rank test; p = 0.31, indicating that there was no statistical difference in the survival time between groups.
DISCUSSION
This study represents 104 patients with pheochromocytomas treated since 1950. Overall, 13% of patients with a diagnosis of a unilateral adrenal pheochromocytoma and 23% of patients with an extraadrenal pheochromocytoma proved to have malignant disease. The primary findings of this study were as follows:
1. Although the rate of malignancy of extraadrenal pheochromocytoma was twice that of unilateral pheochromocytoma, this difference was not statistically significant.
2. Patients with extraadrenal and adrenal pheochromocytomas appear to have similar rates of survival.
3. Microscopic features appear to have little predictive value in determining the future biologic behavior of these tumors. However, patients with malignant pheochromocytomas tended to have larger tumors than patients with benign tumors.
Although generally obvious once the diagnosis of pheochromocytoma is made, the symptoms associated with this tumor can be deceptive. In the current series, hypertension, intermittent headaches, palpitations, and sweating were the four most common presenting symptoms. Similar presenting symptoms have been previously documented. 16 However, in a number of female patients in the range of 40 to 50 years of age, symptoms such as headache, sweating, palpitations, and flushing were falsely accounted for as perimenopausal symptoms, delaying the diagnosis in several patients for years. In addition, 11% of the patients had no episodes of hypertension. Careful consideration needs to be given to any patient complaining of “spells” that may or may not be accompanied by hypertension.
Although not discussed in the current study, beginning in 1968 the measurement of urinary catecholamines became integral to the evaluation and management of patients suspected of harboring a pheochromocytoma. Originally reported by Remine et al 17 and reconfirmed in the study by Orchard et al, 16 urinary metanephrines were the single best test, establishing the presence of the disease in 92% of patients tested. The combination of urinary metanephrines and vanillylmandelic acid (VMA) had a diagnostic sensitivity in detecting pheochromocytoma of 98%. Although Van Way et al from our institution reported favoring the measurement of urinary catecholamines in 1974, 18 it is now our general practice to obtain at least two sets of 24-hour urine collections for total and fractionated catecholamines, metanephrines, and VMA. We occasionally also use plasma catecholamine and metanephrine measurements; the latter test is used by one of the authors (JAO) to monitor the adequacy of preoperative blockade. An equivocal diagnosis sometimes requires a clonidine suppression test. 19
Since 1967, at our institution, preoperative catecholamine blockade has become standard in the management of patients with pheochromocytomas. 11 Phenoxybenzamine, an α-blocker, is initiated first in all patients with pheochromocytomas. In most patients, this has then been combined with metyrosine, which directly inhibits the synthesis of catecholamines. Thus, both the production and the actions of catecholamines are reduced. The use of these agents has been critical to the surgical success of the past 30 years. The Mayo Clinic has also reported very impressive results, largely using a combination of α- and β-blockade. 16 Although the approach may differ slightly, the point is that careful attention to the preoperative preparation of the patient plays an important role in the overall success of the surgical management of these patients. Most centers report that preoperative blockade is a standard component in the management of patients with pheochromocytoma, but some have recently suggested operating on these patients without preoperative α-blockade. 20 It is possible for a skilled anesthesiologist to conduct such a procedure with relative safety, but there is little need to take unnecessary risk when effective preoperative blockade can be accomplished so easily and the perioperative management can be simplified.
Once the diagnosis of pheochromocytoma is confirmed biochemically, localization of the tumor should be performed. In most of the initial patients making up the current series, localization of the tumor was performed as part of the surgical procedure. Beginning around 1968, preoperative angiography became a frequently used modality with a reasonable sensitivity and good specificity. However, angiography has been essentially replaced by CT and MRI. At our institution, both modalities are used, and the results from the current study suggest they are probably equivalent. Both demonstrated a specificity of ≥97%. Although the sensitivity of CT was greater than that of MRI, there was no statistical difference between these two imaging techniques.
The surgical approach to pheochromocytomas has changed markedly in 48 years. At our institution, a thoracoabdominal incision was used to approach most pheochromocytomas before 1979. The approach was based on the need for wide exposure so that a nonmanipulative dissection could be used in removal of the tumor, particularly in the years before preoperative blockade. 21 Through the use of preoperative localization as well as preoperative blockade, the midline abdominal incision has been the preferred approach for the last 20 years. As CT and MRI techniques have improved, the need to explore the abdomen thoroughly has diminished, 16 and the surgical approach continues to become more specifically targeted toward the tumor. Three of the last four pheochromocytomas at our institution have been resected using laparoscopy. There have been several recent series reporting the successful resection of a significant number of pheochromocytomas using this surgical technique. 12,13 It is likely that this approach will become the standard mode of resection, particularly for relatively small tumors.
A longstanding pathology question has been the definition of malignancy when classifying these tumors. Review of histopathologic findings by Scott and Halter 6 failed to show substantial differences between benign pheochromocytomas and those that proved to be malignant. Cellular hyperchromatism, bizarre mitotic figures, vascular invasion, and capsular invasion were features of both biologically benign and biologically malignant tumors. These authors defined true malignancy of pheochromocytoma as the occurrence of spread of tumor cells in or to anatomic areas where there is no known embryologic residue of paraganglionic tissue. This has remained the accepted definition of malignancy for pheochromocytomas.
Nevertheless, the presence of microscopic vascular or capsular invasion remains a traditional cause for concern. The issue of whether these microscopic features have predictive value has recently been raised by several reports. In the recent report by O’Riordain et al, 2 microscopic tumor invasion was reported to be a risk factor in patients with extraadrenal pheochromocytomas. In a recent letter from Gosset et al, 8 reporting on 91 patients with adrenal and extraadrenal pheochromocytomas, mitosis and vascular and capsular invasion were found in no benign tumors but in 32% of malignant tumors.
In the current study, of 64 adrenal pheochromocytomas, 3 met the strict definition of malignancy at the time of resection. An additional six had microscopic features of capsular or vascular invasion. In none of these six patients did metastatic disease occur. Of the patients with extraadrenal pheochromocytomas reported in the current study, four patients clearly had malignant disease at presentation. Eight additional patients had microscopic capsular or vascular invasion, but only one of those patients has returned with metastatic disease. Combining the observations from both extraadrenal and adrenal pheochromocytomas, only 1 patient out of 14 with malignant features has returned with true malignant disease. These observations are consistent with the conclusions of Scott and Halter 6 and suggest that microscopic vascular or capsular invasion is not predictive of the future biologic behavior of the tumor.
For patients with apparently benign disease, van Heerden et al 22 reported on 98 patients who underwent resection of apparently benign sporadic pheochromocytomas. In six patients (6.5%), recurrent pheochromocytoma consistent with malignancy developed. Although review of the histopathology demonstrated microscopic capsular and vascular invasion in 9.2% and 4.1% of these tumors, respectively, in none of these patients did recurrent disease develop.
In the current study, five patients with adrenal pheochromocytomas who had no histopathologically worrisome features, and apparently benign tumors, returned 14 and 15 years after adrenalectomy with liver metastases and gross recurrent invasion into spleen, and pancreas, respectively, defining these tumors as malignant. Thus, if at the time of initial resection there are no gross or histopathologic features of malignancy, the probability that the tumor will ultimately prove to be malignant could be considered to be 8% to 9%. This figure is very similar to that reported above by van Heerden et al. 22 Of 14 patients with extraadrenal pheochromocytomas and apparently benign disease, in only 1 of 14 (7%) has malignant disease developed. This rate is similar to that observed for adrenal pheochromocytomas.
Regarding the probability of malignancy in patients with adrenal pheochromocytoma, Remine et al 17 reported a malignancy rate of 13.1% in 138 cases of pheochromocytoma encountered at the Mayo Clinic from 1926 to 1970. In 90% of the 138 cases, the tumor was of adrenal origin. Scott and Halter 6 reported an 8.3% incidence of malignancy in 48 patients with adrenal pheochromocytoma. Proye et al 4 recently reported on the combined results from three European studies. In this combined series, 31 of 268 adrenal pheochromocytomas were malignant (11.5%). A series of 55 surgically treated patients has been reported by Favia et al. 23 All but five were adrenal in origin. Malignancy was found in five (9.1%) patients, and to date no patients with apparently benign disease have returned with metastases. Their mean follow-up, however, is only 7.3 years. The current study extends the series of Scott and Halter 6 with an overall rate of malignancy for adrenal pheochromocytomas of 13%, a rate very consistent with the reports cited above. However, the findings of the current study and the other studies cited above differ from those reported in the recent study of Pommier et al. 3 They reported on 73 pheochromocytomas treated at Memorial Sloan-Kettering Cancer Center, the National Cancer Institute, and Oregon Health Sciences University, in which 47% of adrenal tumors were malignant, as defined by the presence of metastatic disease. This rate is clearly greater than those reported from other centers but this is likely to be related to the type of referral two of the three centers in their study usually see.
Regarding the rate of malignancy observed in extraadrenal pheochromocytomas, Scott and Halter 6 reported that 5 of 12 extraadrenal pheochromocytomas were malignant (42%). O’Riordain et al 2 reported on 66 patients with extraadrenal pheochromocytomas with a median follow-up of 8.8 years. Metastatic disease was found in 21%. An additional 10 patients (15%) had locally invasive disease, of whom 4 died. In six of these patients, the locally invasive disease was not resectable. This generated an overall malignancy rate of 36%. In the report from Proye et al 4 combining the results from three European studies, 17 of 42 extraadrenal pheochromocytomas (40.5%) were malignant. In 1990, Scott et al 21 published an update on the results of the pheochromocytomas observed at our institution from 1950 to 1986. At that time, 19 extraadrenal pheochromocytomas had been observed, and 32% were classified as malignant. The current series adds seven additional extraadrenal pheochromocytomas, all of which are benign to date, yielding a malignancy rate of 23%. Follow-up in these additional patients is <10 years, so it is possible that the true malignancy rate will increase slightly over time. Median follow-up is, however, similar between the current study and that of O’Riordain et al. 2 In the current study, no statistically significant difference was noted between the rate of malignancy in adrenal and extraadrenal tumors, despite an almost twofold increase in the rate of malignancy. Even if all recent patients with follow-up of <4 years were eliminated from the current analysis, the difference in the rate of malignancy does not reach statistical significance. As was the case with adrenal lesions, the highest rate of malignancy for extraadrenal pheochromocytomas has been reported by Pommier et al. 3 Of 22 extraadrenal pheochromocytomas, 11 (50%) were malignant. There was no statistical increase in the probability that extraadrenal tumors were malignant, nor was there a statistical difference in survival between patients with adrenal tumors and those with extraadrenal tumors. Again, the high rate of malignancy is likely to be related to the type of referral two of the three centers in their study usually see.
A relation between tumor size and the probability of malignancy has been previously commented on by several investigators. Scott and Halter 6 have previously discussed this relation based on the first 69 patients in the current series. Each of the nine malignant tumors had a diameter of ≥6 cm. In the current study, the diameter of malignant tumors tended to be significantly greater than that of benign tumors (p = 0.056). In the study by O’Riordain et al 2 of extraadrenal tumors, tumor size >5 cm was a strong predictor of persistent or recurrent disease. No patient with a primary tumor of ≤5 cm died from disease. Since the publication by Scott and Halter, 6 there have only been 3 malignant pheochromocytomas in 39 cases. One was a large, unresectable extraadrenal pheochromocytoma in a patient with HIV, and in the other two instances the tumor diameters exceeded 5 cm. In the current study, analysis of the change in tumor volume of the resected specimens over time has demonstrated a statistically significant decrease of >50%. One could speculate that as the tumor grows, there is an increased likelihood of a late biologic event that promotes malignant behavior and metastatic spread. If tumor volume is truly decreasing, perhaps the diagnosis is being made earlier and smaller pheochromocytomas are being resected. It will be interesting to determine whether malignancy rates will decrease over the next 20 years.
In the absence of metastatic disease or gross local invasion, the future behavior of apparently benign pheochromocytomas remains difficult to predict. Flow cytometry may have some promise in predicting the future behavior of these tumors. 7 Of 22 patients in the study of Nativ et al who had disease progression, 21 had tumors with abnormal DNA ploidy. No patients with diploid tumors died from pheochromocytoma. Further studies incorporating a larger number of patients may be needed to evaluate whether this technique can identify patients with apparently benign tumors who are at high risk for recurrence and metastatic disease.
The current study included eight patients 18 years or younger, who underwent nine surgical procedures for pheochromocytomas. All patients but one had significant hypertension, and one patient had seizures and coma. Six of eight had headaches, and half had pallor with flushing. Two patients had bilateral tumors, two patients had unilateral adrenal tumors, and four patients had extraadrenal tumors. None of the pheochromocytomas were malignant (mean follow-up 11.0 years). One of the patients was normotensive but had “spells” of headaches and palpitations. This experience is similar to that reported by Caty et al. 24 In that series of 14 children, 6 of 18 tumors were extraadrenal, and hypertension was the dominant symptom. Malignancy was demonstrated in only one patient.
Although not the focus of the current report, consideration should also be given to screening all patients with pheochromocytoma for MEN2 25,26 and von Hippel-Lindau disease, 27 particularly young patients, those with a suspicious or positive family history, and those with a multiplicity of endocrine tumors.
The data from the current study suggest that extraadrenal pheochromocytomas may not carry greater malignant potential than adrenal pheochromocytomas, and that survival in patients with extraadrenal pheochromocytomas is similar to that in patients with adrenal pheochromocytomas. However, in the absence of highly reliable predictors, our data indicate that all patients with pheochromocytoma should continue to be followed closely, and that lifetime follow-up is mandatory. Biochemical screening should be performed if any of the symptoms associated with pheochromocytoma are noted. The need for lifetime follow-up is demonstrated by the current series, in which patients with apparently benign disease returned with metastases up to 15 years after resection.
Acknowledgment
The authors thank Kay Covington from the Vanderbilt Tumor Registry for her tireless work.
Discussion
Dr. Jon A. van Heerden (Rochester, New York): Data on 106 patients with pheochromocytomas is worthy of attention because of the large database with a tumor which is exceedingly rare.
What purpose, though, does a study which spans 48 years, almost half a century, serve? Would a study outlining the authors’ experience over the last 10 years have been more meaningful or more educational to us? The Vanderbilt group has, I believe, given us a timeline of the marked changes that have occurred in the management of these patients in the past half-century. And perhaps an emphasis and some questions about some of these changes are in order.
The authors’ surgical approach to the adrenal glands was unique, truly unique, in that 90 of their 106 patients had a thoracoabdominal incision. This approach in most other centers is extremely rare and is reserved for very large and obviously malignant tumors. Why was this radical approach so commonly employed? The authors have—and I was pleased to note—recently excised two tumors laparoscopically. This then is the value of a timeline study. Is this their current approach of choice?
Secondly, in their preoperative preparation, beta blockade with propranolol was used in only 20% of patients. This is in contrast to about 90% to 95% in our practice. What are the authors’ indications for preoperative beta blockade?
Thirdly, the authors correctly stress that despite preoperative blockade, a significant number of patients will experience intraoperative hemodynamic fluctuations which will require treatment, usually with nitroprusside. This occurred in only 13% of the patients in this study.
Utilizing the set points of a systolic blood pressure of greater than 180 mm of mercury, lasting for longer than 10 minutes and requiring treatment, we found that 31%, or 45 of 143, of a series of patients operated on by us between 1983 and 1996 fulfilled these criteria. Are the authors telling us that 87% of the patients in this study required no such treatment intraoperatively? This would be most unusual. What are their criteria for significant hypertension?
And, lastly, Dr. Goldstein reminded us that these tumors may be malignant. Twelve percent of their pheochromocytomas and 23% of their paragangliomas were thus classified. In the absence of verified distant metastases, the histological differentiation between benign and malignant pheochromocytomas and paragangliomas is a most inexact science. This fact mandates lifelong surveillance of all of these patients. In the absence of metastases, what were the authors’ criteria for the diagnosis of malignancy?
Today, I believe the surgical management of patients with these tumors is very safe if a multidisciplinary team approach is employed. Dr. Goldstein emphasized this team approach very well. The safety of current treatment is substantiated by our own experience with 143 consecutive patients in whom the operative mortality was zero, with only percent of patients being admitted to the ICU postoperatively and these most often for 23 hours of observation. A marked change, I’m sure you’ll agree.
We have indeed come a long way, and I thank our Vanderbilt colleagues for reminding us of the path that we and our patients have traveled.
Dr. Jeffrey F. Moley (St. Louis, Missouri): This report contributes to our understanding of pheochromocytomas on several levels. The series extends back to 1950, and diligent follow-up allowed the authors to identify two patients who developed recurrences of presumably benign disease 14 and 15 years after initial treatment. Their study confirms the observation that the benign or malignant potential of pheochromocytomas cannot be predicted from histologic evaluation.
These tumors are quite interesting on a molecular level. We have demonstrated that all pheochromocytomas from patients with multiple endocrine neoplasia type 2a and 2b have deletions of the short arm of chromosome 1. This finding is also present in one third of sporadic pheochromocytomas, but we have observed no correlation with malignant potential.
I have several questions for Dr. Goldstein. First of all, in Dr. O’Rearden’s and Dr. van Heerden’s study from Mayo Clinic of functional extraadrenal paragangliomas, they noted a 25% incidence of bilaterality or of multiple pheochromocytomas, even in patients with no family history. In your extended follow-up in these patients, did you observe this phenomenon?
Secondly, it’s been suggested by some authors, including Dr. Bruce Ponder from Cambridge, England, that all patients with pheochromocytomas should be screened genetically for von Hippel-Lindau disease, neurofibromatosis type 1, and multiple endocrine neoplasia type 1. What recommendations do you make regarding genetic counseling and testing of patients who present with apparently sporadic pheochromocytomas?
And, lastly, now that you are starting to incorporate laparoscopic adrenalectomy into your armamentarium, what are your size cutoff criteria for resecting a pheochromocytoma? We have been resecting pheochromocytomas up to 6 cm and 7 cm laparoscopically; other authors have removed them at even larger sizes.
Dr. George L. Irvin III (Miami, Florida): Dr. Griffen, Dr. Copeland. I rise for just a moment of history along Dr. van Heerden’s lines.
Ten years ago to the day, we presented a paper before the Association on the lateral approach to pheochromocytomas. Well, that was like heresy back in 1988, and the senior author on this paper, Dr. Scott, who we remembered very well this morning, got up to discuss my paper. And I’d like to read from the discussion exactly what he said: “I only rise to question the fundamental validity of his departure from the old, timeworn but time-honored operative approach to abdominal pheochromocytoma.” Then he goes on to say that “our attitude”—that is, the Nashville group’s attitude—“has been long to use an anterior transperitoneal approach to all large adrenal tumors with malignant potential and all abdominal pheochromocytomas without exception.”
You know, Dr. Scott had that booming voice and the chandeliers would shake, you know, and I got up as a relatively new member of the organization and my knees were shaking so bad you could hear them in the back of the room, and I thanked Dr. Scott for his criticism of our paper, which was based on the CT scan of the adrenal glands, which I thought at that time was better than me feeling around in the abdomen for the other adrenal gland to see if there was a tumor there. And I thanked Dr. Scott for his criticisms and suggested that maybe the Nashville group ought to buy one of those newfangled x-ray machines. And I’m very glad to see that they did and that they have now changed their approach to this tumor.
Dr. Armistead M. Williams (Richmond, Virginia): My comments are historical in nature primarily, as Buck Irvin said. In 1950, I was a third-year medical student in Charlottesville. I had the privilege of working up a patient whose several complaints included intermittent headaches, sweating, palpations, flushing, and nausea. With the help of Yader’s Symptom Diagnosis—which some of you may not even know because it probably has gone out of print—I was able to come across the term “pheochromocytoma.”
Not knowing how rare it was, not knowing much about it, I decided to put that as my only diagnosis on the work-up. Well, we had to present to Bill Parson and Ken Crispel, who as many of you know were very big in endocrinology. They brought in the benzodioxin test, and lo and behold, the test was positive, and the patient was operated upon by the chairman of urology.
And this is the only question I have to ask of Dr. Goldstein or whoever closes the paper: were the urologists excluded from this paper? I noticed they weren’t even listed in the end of the summary.
Dr. Richard E. Goldstein (Closing Discussion): First, addressing the comments by Dr. van Heerden, he asked whether it would be more worthwhile to look at patients over a shorter period of time. We actually believe that due to the fact that the issue of malignancy can take years to really sort out, as manifested by the fact that several patients with apparently benign disease did not manifest malignancies until 14 or 15 years later, that there is great value in this setting looking back over a series actually extending back years. I think that that generates accurate data.
The thoracoabdominal incision was used by Dr. Scott almost completely. I think that largely stemmed from the initial experience where these patients were operated on in an unblocked condition, and the fundamental approach was to essentially dissect the patient away from these tumors, and that operative approach allowed excellent exposure. That has been used only once at our institution since 1979, and I think for very large tumors it does have value.
I think that clearly, once preoperative imaging became better and blocking became better, that that led to the change to a midline operative approach. I do think that laparoscopy will become probably the major way that these tumors are technically taken out.
One of the interesting side issues is the fact that if these tumors that are truly being picked up earlier and therefore tumor volume is diminishing, I think it becomes easier to resect these tumors with the laparoscope, because we are probably operating on smaller lesions. Nevertheless, I think surgeons should do what is safe, and if it is not felt that it can be safely taken out using laparoscopic methods, it is absolutely appropriate and mandated that open approaches should be used.
Dr. van Heerden is correct when he says that we have used beta blockade very infrequently. Our major indications for beta blockade have been supraventricular or ventricular tachycardia. Beta blockade combined with alpha blockade has been used with very high frequency at the Mayo Clinic and their results have been absolutely excellent.
I think the point is that careful preoperative preparation, whether it’s done with alpha blockade, alpha blockade plus metyrosine, or alpha blockade plus beta blockade, when it’s done carefully by people who know what they’re doing should yield excellent operative results.
My personal worry—although I don’t think it will be borne out as I’ve looked back over the data—is that when you give alpha and beta blockade that you knock out several systems, and the ability of patients to compensate should there be a problem is diminished. However, our results as well as the results of the Mayo Clinic would indicate that alpha blockade and beta blockade are equally excellent.
As we look back over operative records, we tended to use a systolic pressure of greater than 200 or one of less than 80 for adults to indicate some degree of instability. Most of the patients operated on currently do receive some low-dose nipride during the operative procedure.
I think it is difficult, and I’m open to thoughts on how to quantitate operative stability, but it is just difficult to just look at maximum and minimal pressures and get a true sense of how hard the anesthesia team is having to work to keep that patient stable. And so I think purely using those factors can be a little bit misleading. We are currently looking at ways to get a better handle on just how stable those patients are.
Malignancy was defined as gross invasion into surrounding tissues, such as the spine, or clearly distant metastases in areas where there should be no paraganglionic tissue, such as bone.
I think laparoscopy is becoming our preferred approach, or I should say, at least the approach that we think of first. And I will address that a little bit more in answering the comments by Dr. Moley.
Getting on to the comments by Dr. Moley, he asked about whether we had seen multiple extraadrenal tumors in the same patient. In our experience with 26 extraadrenal pheochromocytomas, we have seen that twice, so we do not have a large experience to comment on.
As far as screening patients, we certainly believe that all patients with pheochromocytomas should be considered for screening for MEN2 as well as the von Hippel-Lindau, particularly those patients who are young, have multiple tumors, or have a family history that would make one think that they may have syndromes. I believe you mentioned MEN1 screening, but I don’t believe that is really on line yet. I think I previously have addressed the issues on laparoscopy.
For Dr. Irvin, I would like to thank you very much for your comments and for your thoughts on Dr. Scott. And, again, for Dr. Williams, I’d like to thank you very much for your comments and thoughts on this issue.
Footnotes
Correspondence: Richard E. Goldstein, MD, PhD, D-5217 MCN, Vanderbilt University, Nashville, TN 37232.
Presented at the 110th Annual Meeting of the Southern Surgical Association, December 6–9, 1998, The Breakers, West Palm Beach, Florida.
Accepted for publication December 1998.
References
- 1.Wilbourn RB. Early surgical history of phaeochromocytoma. Br J Surg 1987; 74: 594–596. [DOI] [PubMed] [Google Scholar]
- 2.O’Riordain DS, Young WF Jr, Grant CS, et al. Clinical spectrum and outcome of functional extraadrenal paraganglioma. World J Surg 1996; 20: 916–922. [DOI] [PubMed] [Google Scholar]
- 3.Pommier RF, Vetto JT, Billingsly K, et al. Comparison of adrenal and extraadrenal pheochromocytomas. Surgery 1993; 114: 1160–1166. [PubMed] [Google Scholar]
- 4.Proye CAG, Vix H, Jansson S, et al. “The” pheochromocytoma: a benign, intra-adrenal, hypertensive, sporadic unilateral tumor. Does it exist? World J Surg 1994; 18: 467–472. [DOI] [PubMed] [Google Scholar]
- 5.Scott HW Jr, Reynolds V, Green N, et al. Clinical experience with malignant pheochromocytomas. Surg Gynecol Obstet 1982; 154: 801–818. [PubMed] [Google Scholar]
- 6.Scott HW Jr, Halter SA. Oncologic aspects of pheochromocytoma: the importance of follow-up. Surgery 1984; 96: 1061–1066. [PubMed] [Google Scholar]
- 7.Nativ O, Grant CS, Sheps SG, et al. The clinical significance of nuclear DNA ploidy pattern in 184 patients with pheochromocytoma. Cancer 1992; 69: 2683–2687. [DOI] [PubMed] [Google Scholar]
- 8.Gosset P, Lecomte-Houcke M, Carnaille B, Proye C. Adrenal and extra-adrenal phaeochromocytomas. Contribution of histology, immunochemistry and DNA flow cytometry in the diagnosis of malignancy. Eur J Surg 1996; 162: 77. [PubMed] [Google Scholar]
- 9.Montresor E, Iacono C, Nifosai F, et al. Retroperitoneal paragangliomas: role of immunohistochemistry in the diagnosis of malignancy and in assessment of prognosis. Eur J Surg 1994; 160: 547–552. [PubMed] [Google Scholar]
- 10.Jalil ND, Pattou FN, Combemale F, et al. Effectiveness and limits of preoperative imaging studies for the localisation of pheochromocytomas and paragangliomas: a review of 282 cases. Eur J Surg 1998; 164: 23–28. [DOI] [PubMed] [Google Scholar]
- 11.Robertson D, Oates JA Jr, Berman ML. Preoperative and anesthetic management of pheochromocytoma. In: Scott HW Jr, ed. Surgery of the adrenal glands. Philadelphia: JB Lippincott; 1990: 225–240.
- 12.Gagner M, Pomp A, Heniford BT, et al. Laparoscopic adrenalectomy: lessons learned from 100 consecutive procedures. Ann Surg 1997; 226: 238–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Staren ED, Prinz RA. Adrenalectomy in the era of laparoscopy. Surgery 1996; 120: 706–711. [DOI] [PubMed] [Google Scholar]
- 14.Altman DG. Practical statistics for medical research. London: Chapman and Hall; 1995: 368–371.
- 15.Ferguson GA. Statistical analysis in psychology and education, 5th ed. New York: McGraw-Hill, Inc.; 1981.
- 16.Orchard T, Grant CS, van Heerden JA, Weaver A. Pheochromocytoma: continuing evolution of surgical therapy. Surgery 1993; 114: 1153–1159. [PubMed] [Google Scholar]
- 17.Remine WH, Chong GC, van Heerden JA, et al. Current management of pheochromocytoma. Ann Surg 1974; 179: 740–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Van Way CW III, Scott HW Jr, Page DL, Rhamy RK. Pheochromocytoma. Curr Prob Surg 1974; June: 1–59. [DOI] [PubMed] [Google Scholar]
- 19.Sjoberg RJ, Simcic KJ, Kidd GS. The clonidine suppression test for pheochromocytoma. Arch Intern Med 1992; 152: 1193–1197. [PubMed] [Google Scholar]
- 20.Boutros AR, Bravo E, Zanettin G, Straffon RA. Perioperative management of 63 patients with pheochromocytoma. Cleveland Clinic J Med 1990; 57: 613–617. [DOI] [PubMed] [Google Scholar]
- 21.Scott HW Jr, Van Way CW III, Gray GF Jr, Sussman CR. Pheochromocytoma. In: Scott HW Jr, ed. Surgery of the adrenal glands. Philadelphia: JB Lippincott; 1990: 187–223.
- 22.van Heerden JA, Roland CF, Carney J, et al. Long-term evaluation following resection of apparently benign pheochromocytoma(s)/paragangliomas(s). World J Surg 1990; 14: 325–329. [DOI] [PubMed] [Google Scholar]
- 23.Favia G, Lumachi F, Polistina F, D’Amico DF. Pheochromocytoma, a rare cause of hypertension: long-term follow-up of 55 surgically treated patients. World J Surg 1998; 22: 689–694. [DOI] [PubMed] [Google Scholar]
- 24.Caty MG, Coran AG, Geagen M, Thompson NW. Current diagnosis and treatment of pheochromocytoma in children. Arch Surg 1990; 125: 978–981. [DOI] [PubMed] [Google Scholar]
- 25.Wells SA Jr, Larimore TS. Genetic testing for multiple endocrine neoplasia. Br J Surg 1993; 80: 1092–1093. [DOI] [PubMed] [Google Scholar]
- 26.Decker RA, Peacock ML, Borst MJ, et al. Progress in genetic screening of multiple endocrine neoplasia type 2A: is calcitonin testing obsolete? Surgery 1995; 118: 257–264. [DOI] [PubMed] [Google Scholar]
- 27.Neumann HPH, Berger DP, Sigmund G, et al. Pheochromocytomas, multiple endocrine neoplasia type 2, and von Hippel-Lindau disease. N Engl J Med 1993; 329: 1531–1538. [DOI] [PubMed] [Google Scholar]