

Abstract
Objective
To determine if cross-tolerance to septic shock could be induced by a previous insult with sublethal hemorrhage (SLH) and to characterize the mechanisms involved in this induced protective response.
Background Data
It is possible to condition animals by prior SLH such that they tolerate an otherwise lethal hemorrhage. It is also possible to condition animals with low doses of lipopolysaccharide (LPS) so that they survive a “lethal” septic insult. However, a paucity of information exists on cross-tolerance between hemorrhage and sepsis.
Methods
Rats were made tolerant by conditioning SLH or sham operation. Twenty-four hours later, tolerant and sham rats were exposed to a lethal dose of LPS. To explore the mechanism of tolerance induction, rats were given the macrophage (Mφ) inhibitor CNI-1493 or saline carrier before SLH. Survival and pulmonary vascular injury were determined after LPS. Serum tumor necrosis factor (TNF) levels and splenic Mφ TNF gene expression were measured at several time points.
Results
Prior SLH indeed made rats tolerant and imparted a significant survival benefit and reduction in pulmonary vascular injury after LPS. The tolerance induced by SLH was reversed by Mφ inhibition. Tolerant animals had low serum TNF levels immediately after SLH and reduced circulating TNF levels after LPS. SLH, however, did not inhibit the augmentation of TNF gene expression after LPS.
Conclusions
Sublethal hemorrhage bestows protection against a lethal LPS challenge. Inhibition of the Mφ attenuated the benefit of the tolerance induced by SLH. Circulating TNF but not TNF gene after LPS is lessened by SLH. This implicates changes in Mφ intracellular signaling in induction of the tolerant state.
Alterations in immune function are thought to play a pivotal role in the development and perpetuation of the systemic inflammatory response syndrome and multiple organ dysfunction syndrome. Alterations in the immune response that occur after an initial insult are central to the concept of “priming” or cellular reprogramming. 1–3 The changes in immune function that occur during priming can be either detrimental or beneficial to the host organism when it is exposed to a subsequent systemic insult. It is possible to condition animals by prior sublethal hemorrhage (SLH) such that they tolerate what should be a subsequent lethal hemorrhage. 4 It is also possible to condition animals in vivo with low doses of lipopolysaccharide (LPS) so that they survive an otherwise lethal septic insult. 5 Also, pretreatment of monocytes or macrophages in vitro induces a state of tolerance characterized by alterations in cytokine production in response to subsequent septic stimuli (LPS). 6–9 However, a paucity of information exists on cross-tolerance between hemorrhage and sepsis. 10,11 We therefore investigated whether cross-tolerance to a septic challenge could be induced by SLH and attempted to define the immune mechanisms involved in this response.
MATERIALS AND METHODS
All experiments were conducted in accordance with the guidelines of the University of South Florida Laboratory Animal Medical Ethics Committee. All animals were exposed to 12-hour light/dark cycles, were given food and water ad libitum, and were allowed to acclimate for ≥5 days before experimental manipulation.
Experimental Model
Male Sprague Dawley rats (325 to 375 g) underwent anesthesia with intraperitoneal pentobarbital (40 mg/kg). After induction of anesthesia, the groins were shaved bilaterally and the left femoral artery was dissected and distally ligated. The femoral artery was then cannulated with a 22-gauge heparinized angiocath (Terumo, Elkton, MD). The cannula was connected by intravenous tubing to a Wiggers column, the height of which had been precalibrated to decrease the mean arterial pressure of the experimental rats to 30 mmHg. This column was connected by a stopcock to an arterial pressure transducer and monitor for pressure measurement. All rats received 50 units of heparin through the arterial cannula. Conditioned rats then were allowed to bleed freely into the Wiggers column. The rats underwent 15 minutes of sustained hypotension once a mean arterial pressure of 30 mmHg was achieved. After this conditioning hemorrhage (SLH), the rats received shed blood only without additional “resuscitative” fluids. All cannulas were removed, the femoral artery was clipped, and the groin incision was sutured closed. Sham conditioned animals underwent cannulation of the femoral artery, heparin dose, and 15 minutes of monitoring, followed by femoral artery clipping. Rats were recovered from anesthesia, returned to their cages, and given food and water ad libitum.
Twenty-four hours after the conditioning SLH or sham operation, rats were given a dose of LPS previously shown to be uniformly lethal (40 mg/kg intraperitoneal). 11 Survival was subsequently determined.
Conditioning in the Presence of Macrophage Inhibition
To explore the role of the macrophage (Mφ) in the induction of tolerance, 20 rats were prospectively randomized to receive CNI-1493 (1 mg/kg intraperitoneal) or saline vehicle 2 hours before undergoing their conditioning SLH on day 1. CNI-1493 (N,N′-bis [3,5-diacetylphenyl]decanediamide tetrakis[amidinohydrazone] tetrahydro-chloride) is a synthesized tetravalent guanylhydrazone compound that inhibits cytokine-inducible arginine transport and nitric oxide production in the Mφ as well inhibiting the production of proinflammatory cytokines, including tumor necrosis factor (TNF), interleukin (IL)-1, and IL-6. 12–14
Measurement of Pulmonary Vascular Injury
To determine how induction of tolerance affects the development of lung injury, Evans blue dye extravasation was used to quantitate the degree of pulmonary vascular injury (PVI). 15,16 Rats underwent conditioning SLH or sham operation with or without Mφ inhibition. Twenty-four hours later, they received intraperitoneal LPS. Four and one-half hours after LPS, the rats were anesthetized, the right groin was opened, and 30 mg/kg of Evans blue dye ((6,6′-[3,3′-dimethyl[1,1′-biphenyl]-4,4′-diyl)bis(azo)]bis[[4-amino-5-hydroxy-1,3-naphthalenedi-sulfonic acid])(Sigma Chemical, St. Louis, MO) was injected into the femoral vein. After 30 minutes, the rats were killed, the pulmonary artery was cannulated, and a left atriotomy was performed for pulmonary vascular lavage with 60 ml Krebs solution (9.5 mg/ml) (Sigma). After resection, the right lower lobe was homogenized for 30 seconds in 5 ml formamide (Sigma). The tissue suspension was then incubated at 37°C for 16 hours, followed by centrifugation of the homogenate at 3900g for 10 minutes. The supernatant was subsequently analyzed for optical density by spectrophotometry (Bausch and Lomb Spectronic 1001, Rochester, NY) at a wavelength of 620 nm.
Measurement of Serum TNF Levels
Serum was collected from experimental rats 15 minutes after the conditioning hemorrhage on day 1, 24 hours after the conditioning hemorrhage before LPS on day 2, and 4 hours after administration of LPS. These samples were evaluated for TNF levels using an enzyme-linked immunoabsorbent assay for rat TNF-α (Biosource International, Camarillo, CA).
Measurement of TNF Gene Expression
RNA Isolation
Splenic tissue was harvested from rats after sham conditioning, conditioning SLH, and conditioning in the presence of CNI-1493. Spleens were harvested 2 hours after the conditioning hemorrhage on day 1 and 4 hours after the administration of LPS on day 2. These tissues were snap-frozen in liquid nitrogen. The splenic tissue was thawed, and 2 ml Trizol (Gibco, Grand Island, NY) was added. The tissue was then homogenized and 0.9 ml cold chloroform was added for each milliliter of homogenized tissue. Samples were centrifuged at 12,000g for 15 minutes at 4°C. Five hundred microliters of the aqueous phase was then added to an equal volume of cold isopropyl alcohol, vortexed, and stored overnight at 4°C. The following day, the samples were centrifuged for 15 minutes at 12,000g. The supernatant was discarded and 500 μl of cold 75% ethanol was added to the RNA precipitate, vortexed, and centrifuged at 7500g for 8 minutes at 4°C. The supernatant was discarded and 250 μl of fresh cold 75% ethanol was added, followed by centrifugation at 7500g for 8 minutes at 4°C. After discarding the supernatant, the pellet was resuspended in 50 to 100 μl of 1 mM EDTA in DEPC-treated water. A 5-μl aliquot was taken for spectrophotometric verification of RNA presence at 260 and 280 angstrom wavelength.
Reverse Transcription
Oligodeoxythymidine primer (1 μl) was added to 4 μg RNA and DEPC water to make a total volume of 10 μl, and this was placed in the thermocycler (Biometra TRIO Thermoblock, Tampa, FL) at 70°C for 10 minutes, with a pause at 4°C. After mixing, 4 μl 5X synthesis buffer, 2 μl 0.1 M DTT, 1 μl Rnasin (Promega, Madison, WI), 1 μl 10 mM deoxynucleotide triphosphate, and 1 μl Superscript II (Gibco, Gaithersburg, MD) were added. These Eppendorf tubes were then incubated in the thermocycler for 15 minutes at 25°C, 50 minutes at 42°C, and 15 minutes at 70°C, with a pause at 4°C. One microliter Rnase H (Gibco) was then added, and the tubes were incubated in the thermocycler for 20 minutes at 37°C.
Polymerase Chain Reaction
The reverse-transcribed complementary DNA strands (0.4 μg) were added to a mix of 2 μl dNTP, 10 μl 5X synthesis buffer, 31 μl DEPC water, and 2.5 μl TNF primer. The TNF primer sequence was sense 5′-GTAGCCCACGTCGTAGCAAA-3′ and antisense 5′-CCCTTCTCCAGCTGGAAGAC-3′. The prepared complementary DNA strands were coamplified with their specific primers for 25 cycles, with each cycle consisting of 1 minute at 95°C, 1 minute at 57°C, and 1 minute at 75°C, with Taq DNA polymerase (0.5 μl) added after the first cycle and rat β-2 microglobulin (BMG) primer mix (2 μl) added after 10 cycles. After amplification, 15 μl cDNA sample and 3 μl DNA loading buffer were combined. The reaction products were subsequently visualized by electrophoresis in 1.5% Metaphor agarose (Fischer Biotech, Pittsburgh, PA) containing ethidium bromide. Ultraviolet illumination was used to visualize the DNA bands, and the gels were digitally photographed and stored on computer disk. Band intensity was determined and analyzed using Sigma Scan software (Jandel Scientific, San Rafael, CA). Coamplification of BMG cDNA was used as an internal control, and the ratio of TNF to BMG intensity was used to control for possible differences in loading or amplification. 17
Statistical Methods
Results are expressed as mean ± SEM. Statistical analysis was performed using the EPISTAT statistical program (Epistat Services, Richardson, TX), applying the Student t test, with significance being assigned to p < 0.05.
RESULTS
Survival
This model of conditioning by SLH resulted in a 100% survival rate immediately after the conditioning insult. All animals were alive before LPS administration on day 2. Conditioning by SLH, however, results in significant physiologic stress, because these conditioned rats are acidotic (arterial pH 7.1) at the end of the 15-minute SLH. Sham-operated rats showed no evidence of acidosis during the sham procedure.
Rats subjected to conditioning SLH were indeed tolerant to a subsequent (24 hours later) septic insult. This tolerance was manifested by a significant increase in the survival of tolerant rats in response to the LPS challenge when compared to sham-operated rats (p < 0.05) (Fig. 1). This survival benefit was partially reversed in rats that received the Mφ inhibitor CNI-1493 before conditioning SLH (p < 0.05) (Fig. 2).
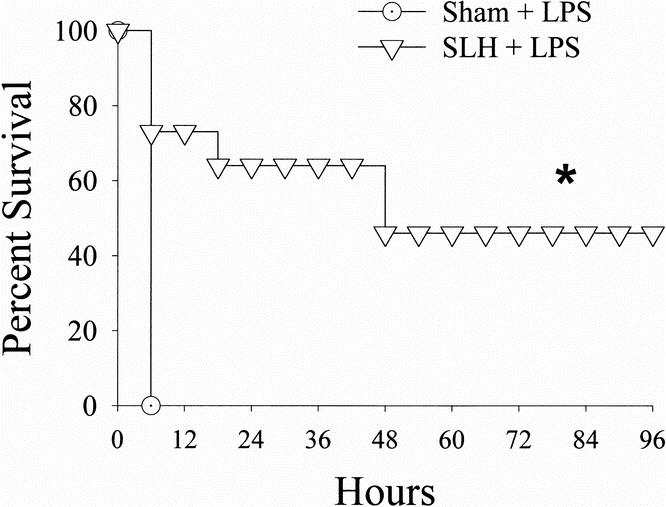
Figure 1. Survival curves comparing rats undergoing lethal intraperitoneal LPS (40 mg/kg) after either sham operation or conditioning SLH. *, p < 0.05 SLH vs. sham.
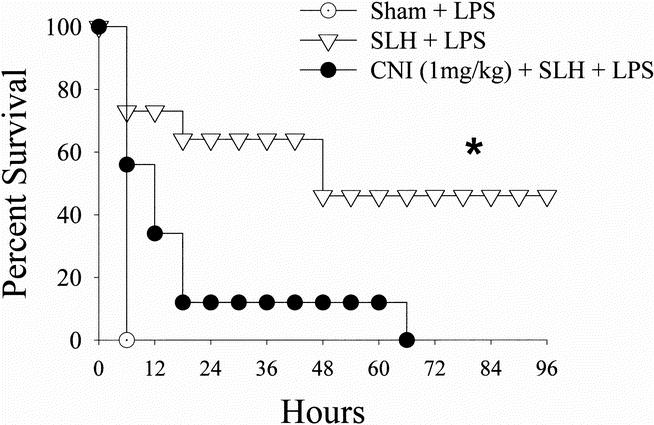
Figure 2. Survival curves comparing rats undergoing lethal intraperitoneal LPS (40 mg/kg) after sham operation, conditioning SLH, or conditioning SLH in the presence of the macrophage inhibitor CNI-1493. *, p < 0.05 SLH vs.sham and SLH + CNI-1493.
Pulmonary Vascular Injury
The PVI measured by extravasation of Evans blue dye into the lung was increased by LPS exposure in both sham-operated and SLH conditioned rats when compared with normal (noninstrumented/no LPS) rats. However, the degree of PVI was significantly less in tolerant rats when compared with both sham-operated rats and rats conditioned in the presence of Mφ inhibition by CNI-1493 (p < 0.05) (Fig. 3).
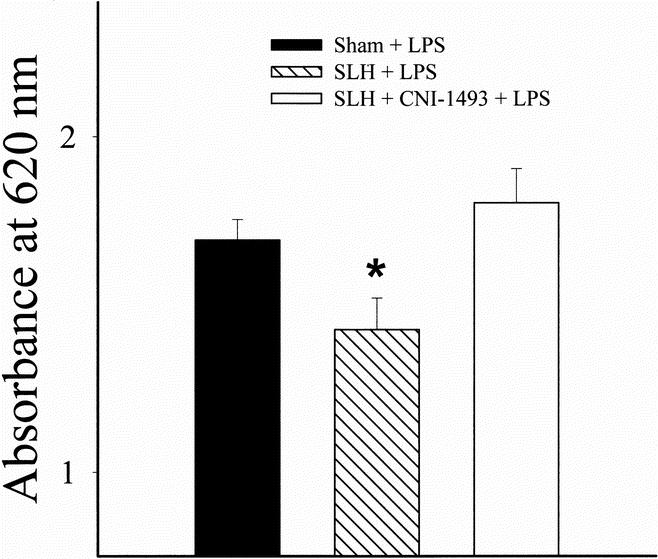
Figure 3. PVI quantitated by Evans blue dye extravasation after intraperitoneal LPS (40 mg/kg) after sham operation, conditioning SLH, or conditioning SLH in the presence of the macrophage inhibitor CNI-1493. *, p < 0.05 SLH vs. sham and SLH + CNI-1493.
Serum TNF Levels
Two hours after the conditioning insult of SLH, there was a significant elevation of TNF in the serum of tolerant rats when compared with the TNF levels in sham-operated rats. The levels of serum TNF in the tolerant rats remained elevated 24 hours later, before giving the LPS on day 2. In contrast, 4 hours after the administration of LPS on day 2, the sham rats exhibited a large rise in serum TNF; this increase in TNF was significantly abrogated in tolerant rats. Macrophage inhibition before SLH inhibited the rise in serum TNF seen 2 hours after SLH. When animals subjected to SLH in the presence of Mφ inhibition by CNI-1493 were given LPS as a second challenge, the rise in serum TNF was higher than that seen in tolerant animals but not as high as that seen in sham animals (p < 0.05) (Fig. 4).
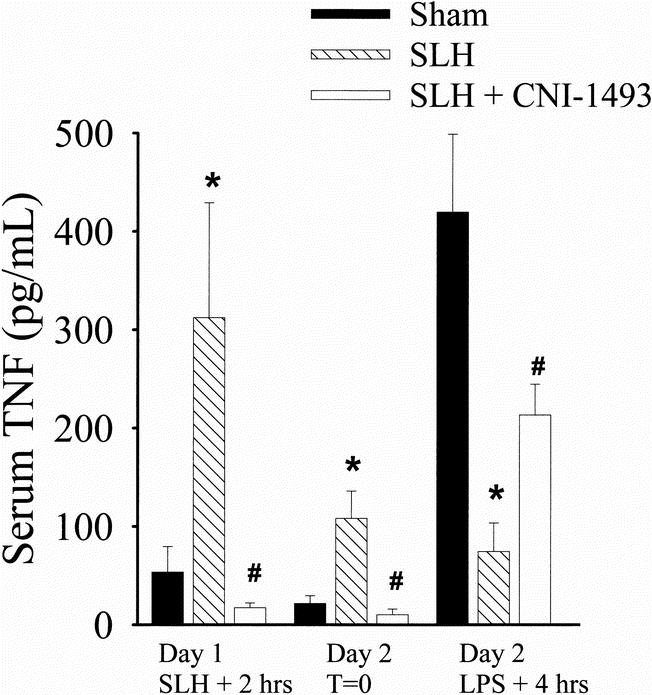
Figure 4. Serum TNF collected on day 1, 2 hours after sham or conditioning SLH, day 2 before LPS (T = 0), and day 2, 4 hours after intraperitoneal LPS (40 mg/kg). *, p < 0.05 SLH vs. sham. #, p < 0.05 SLH vs. SLH + CNI-1493.
TNF Gene Expression
TNF gene expression in response to the conditioning SLH itself was consistent with the early TNF rise seen in the serum of tolerant rats. Two hours after SLH, there was a statistically significant increase in the expression of TNF mRNA in the spleens of conditioned rats when compared with sham-operated rats. Pretreatment with CNI-1493 inhibited the increase in splenic TNF mRNA after SLH (p < 0.05) (Fig. 5). Although serum TNF levels on day 2 after the administration of LPS was less in tolerant animals than in shams, TNF mRNA was increased to a similar degree in all experimental groups 4 hours after LPS exposure (Fig. 6) .
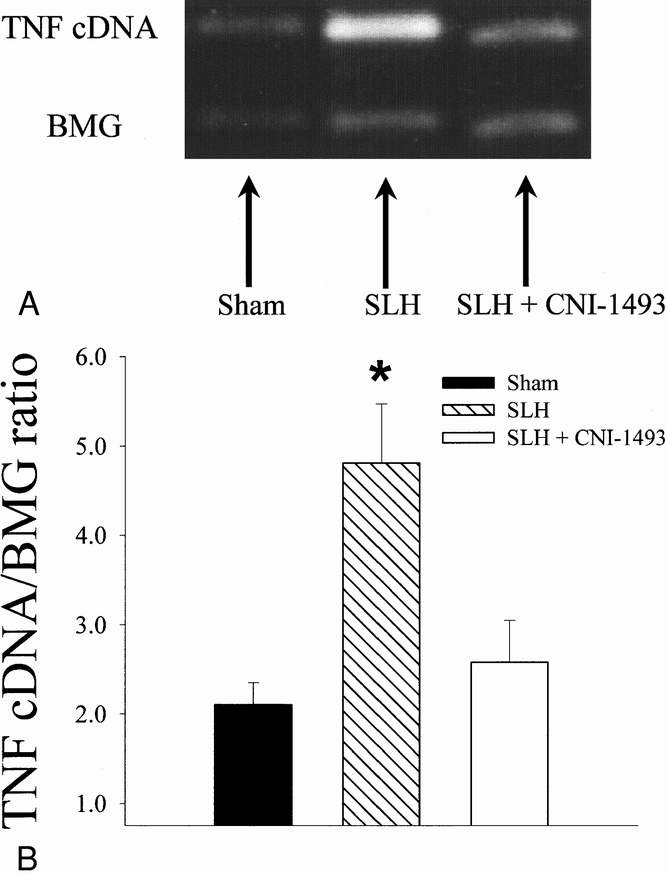
Figure 5. A. Reverse transcription–polymerase chain reaction of mRNA for TNF in splenic tissue collected 2 hours after sham conditioning, conditioning SLH, and SLH preceded by macrophage inhibition with CNI-1493. BMG indicates the housekeeper gene β-2 microglobulin. B. Graphic representation of above reverse transcription–polymerase chain reaction. Ratio of TNF and BMG cDNA is used to control for loading and variations in amplification. *, p < 0.05 SLH vs. sham and SLH + CNI-1493.
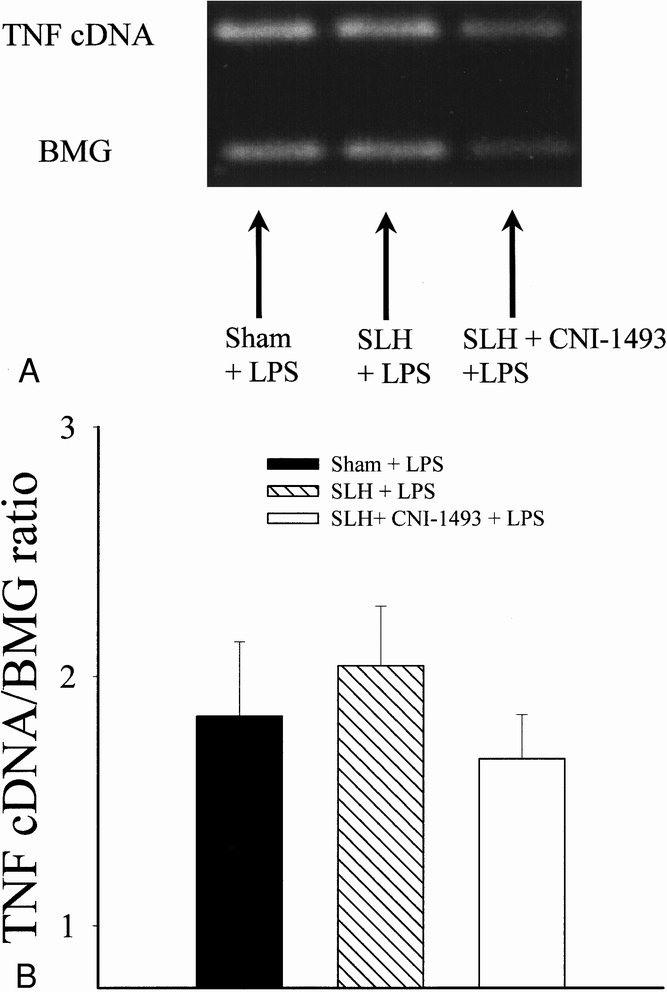
Figure 6. A. Reverse transcription–polymerase chain reaction of mRNA for TNF in splenic tissue collected 4 hours after LPS administration on day 2 in rats that had previously undergone sham conditioning, conditioning SLH, and SLH preceded by macrophage inhibition with CNI-1493. BMG indicates the housekeeper gene β-2 microglobulin. B. Graphic representation of above reverse transcription–polymerase chain reaction. Ratio of TNF and BMG cDNA is used to control for loading and variations in amplification.
DISCUSSION
The sequence of hemorrhagic shock followed by exposure to septic stimuli is often seen in the same critically injured trauma patients who are at risk for the development of adult respiratory distress syndrome, systemic inflammatory response syndrome, and multiple organ dysfunction syndrome. Thus, the elucidation of the alterations that occur in the inflammatory response in the setting of sequential systemic insults or “hits” has vast therapeutic potential. The goal of investigations in this setting should be to gain the knowledge necessary to modulate the immune system so that the beneficial responses, such as those required to resist infections, are optimized and the deleterious responses involved in the “autoimmune” destruction of tissues seen in adult respiratory distress syndrome and multiple organ dysfunction syndrome are inhibited. Although it has been postulated that the changes in cellular immune response (priming) that occur after an initial insult or “hit” are necessarily harmful to the organism as a whole, 1–3 recent studies have explored how alterations in the immune response after stressors can be beneficial. 4,18 The concepts of priming and tolerance are thus two sides of the same coin—cellular reprogramming.
Previous studies in our laboratory have shown that a sublethal but physiologically significant hemorrhage can induce tolerance to a subsequent severe hemorrhage. 4 With the current series of experiments, we have been able to demonstrate that cross-tolerance to a septic insult can be induced by previous SLH. Improved survival and decreases in pulmonary capillary leak (PVI) after LPS exposure are manifestations of this tolerance. These same advantages were also seen when tolerant animals were exposed to hemorrhage as a second insult.
The improved outcomes seen in the tolerant animals were associated with alterations in the cytokine response. The process of SLH, which was used to induce tolerance, led to a significant augmentation in the production of both gene (mRNA) and protein for the proinflammatory cytokine TNF. After the second insult with LPS, the tolerant rats had a blunting in their TNF response, whereas the nontolerant (sham) rats produced significantly more TNF protein. Interestingly, after LPS, there was no significant difference in the level of TNF mRNA seen between tolerant and nontolerant animals. The production of TNF occurs at multiple levels (pre- and posttranscriptional), 19–21 and this suggests that the induction of the tolerant state in our model may lead to posttranscriptional changes in the regulation of this particular cytokine (TNF) in response to endotoxin.
To try to dissect the immune mechanisms responsible for induction of the tolerant state, we investigated the effects of giving the Mφ inhibitor CNI-1493 before the induction of tolerance by SLH. Although the exact mechanisms by which CNI-1493 inhibits Mφ activation is still being defined, it is believed that its effects are mediated at least in part by inhibition of p38 MAP kinases. Suppression of the Mφ reversed the ability of SLH to induce tolerance, as evidenced by attenuation in both the benefit on survival and PVI after LPS challenge. However, if a second dose of CNI-1493 is given before LPS administration, no further differences are seen in survival and lung injury. Also, CNI-1493 given only before LPS administration (after conditioning) does not duplicate the attenuation in survival and lung injury seen when CNI-1493 is given before the tolerizing stimulus (SLH) (data not shown). This implicates Mφ-derived mediator(s) elaborated during the tolerizing stimulus as having a central role in the induction of the tolerant state. The increase in TNF seen after SLH also supports the pivotal role of Mφ in tolerance. Although TNF itself may be involved, it is not likely that TNF is the sole mediator in the induction of tolerance. Studies in our laboratory have not been able to reproduce the tolerance induced by SLH with exogenous administration of TNF. In vitro models of Mφ LPS tolerance have also failed to reproduce the tolerance caused by LPS pretreatment by giving exogenous TNF. 6 Other in vitro models of LPS tolerance have found associated increases in IL-10 and prostaglandin E2, both of which have antiinflammatory properties. 6,22 Whether the induction of the tolerant state in our model is modulated by inhibition of proinflammatory mediators or the production of counterinflammatory mediators such as IL-10 or prostaglandin E2 has yet to be determined. These issues are being pursued in our laboratory.
An issue worth discussing is the differential results in TNF gene and the effects of CNI in our model of tolerance when the “second hit” is severe hemorrhage versus LPS. After conditioning with SLH, survival and lung injury are improved irrespective of whether the second insult is severe hemorrhage or LPS. In addition, inhibition in serum TNF protein is seen in both circumstances. Surprisingly, although the response in serum TNF is the same, TNF mRNA was significantly less in tolerant animals after severe hemorrhage than when LPS was the second insult. Also, whereas Mφ inhibition with CNI-1493 during the induction of tolerance by SLH was associated with intermediate levels (between tolerant and nontolerant) of serum TNF in response to LPS as the second insult, serum TNF and TNF mRNA levels were both very low (less than in tolerant rats) in CNI-1493–treated rats after severe hemorrhage as a second insult. These differential results suggest both an overlap and divergence in the inflammatory pathways for tolerance to hemorrhage and sepsis. It is likely that these alterations occur at several levels in the regulation of the inflammatory cascade (including pre- and posttranscriptional). Additional studies to characterize the alterations in cytokine signal transduction pathways associated with tolerance are under way.
In summary, we have shown that tolerance to a septic insult can be induced by prior SLH. This tolerance is associated not only with changes in survival and end organ injury (PVI) but also with alterations in the inflammatory cytokine response. The Mφ is implicated as having a central role in the induction of the tolerant state, because its inhibition by CNI-1493 leads to a reversal of the benefits of tolerance. The alterations in the inflammatory response after tolerance are likely to be the result of the “reprogramming” that occurs at various levels in the signal transduction pathway. Further characterizations of these changes would allow for “therapeutic reprogramming” of the immune response in a way that would allow clinicians to optimize patient outcomes.
Discussion
Dr. James M. Seeger (Gainesville, Florida): This paper confirms some similar studies done by members of our group in vascular surgery at the University of Florida. Dr. Martin Back, our senior fellow last year, who now actually is a faculty member at the University of Florida, and Dr. Thomas Huber from our division at Florida, found a similar improved survival in rats who underwent visceral ischemia reperfusion 24 hours prior to LPS administration. Like you, they also found decreased TNF levels after LPS administration in the conditioned animals, which correlated directly with improved survival.
In their model, they also found that this tolerance to LPS appeared to be IL-10–dependent and that visceral ischemia reperfusion induced high circulating levels of IL-10 that persisted to 24 hours at the time of LPS administration. And that administration of anti–IL-10 eliminated this tolerance to LPS and, in fact, increased the mortality in the animals that were given LPS. This stimulates my question.
It is my understanding that IL-10 can decrease monocyte NF-κB levels by stabilizing I-κB. And I wonder if you measured IL-10 levels in your study and if you feel that the induction of IL-10 by sublethal hemorrhage may play a role in your results.
Secondly, since IL-10 can be given exogenously, might this have a role in inducing this kind of tolerance that we all seek in the traumatically injured patient?
Dr. Basil A. Pruitt, Jr. (San Antonio, Texas): I have several questions, the answers to which may help in evaluating your conclusions:
You note that the conditioned rats were acidotic at the end of the 15-minute sublethal hemorrhage. How long were these animals acidotic? Had the acidosis been corrected by the time of the LPS challenge? And is the development of acidosis essential to the induction of tolerance?
Is the extravasation of Evans blue dye pressure sensitive? That is, can it be influenced by pulmonary artery pressure? If it can be, then we need to be assured that the differences in pulmonary vascular injury that you observed were not simply due to differences in pulmonary vascular pressure.
Is the tolerance mechanism you propose specific for endotoxin-producing organisms, or applicable to all bacterial challenges? Have you in fact subjected the tolerant animals to an infectious challenge and observed the protective effect in the tolerant animals?
One monocyte product which may produce lung injury by promoting intravascular extravasation of neutrophils is IL-8. Have you monitored that chemokine and correlated changes in pulmonary injury to changes in IL-8 levels?
The differential effect of LPS and severe hemorrhage as the second insult on TNF messenger RNA is interesting. Do you think that that difference merely reflects the effect of tissue and cell hypoxia associated with severe hemorrhage?
Since the activation of nuclear factor-κB can be impaired by glucocorticoids, have you correlated tolerance with a glucocorticoid response in the treated and untreated animals subjected to the sublethal hemorrhage? Would glucocorticoid antagonists prevent the development of tolerance or obliterate its protective effect at the time of secondary challenge?
Lastly, before we have a divisional or battalion pre-battle bleed, are these findings species-specific? That is, in man even sublethal hemorrhage, which we see in virtually every trauma patient, seems to predispose the patient to infection rather than protect him or her from infection.
Dr. Cynthia Mendez (Closing Discussion): In answer to Dr. Seeger’s question as far as IL-10 in our model, we have actually checked the serum levels of IL-10 in our model, and there is no elevation of IL-10 in the conditioned animals. As far as whether or not we have checked exogenous IL-10, we have not done that yet in our model.
In response to Dr. Pruitt’s questions, as far as the first question in response to acidosis, the animals, by the second day, no longer have systemic acidosis. However, recent literature has implied that the acidosis in and of itself may alter the response of macrophage. So it may be that the conditioning stimulus that the acidosis reprograms the macrophage somehow, at the time of the conditioning stimulus, but by the time they get the LPS—originally they are not acidotic. They do have a systemic acidosis before they die, but it’s the same for both the conditioned and unconditioned animals.
In regard to the sensitivity of Evans blue dye, we did not actually check pulmonary vascular pressures in these animals. We have looked at the histology of the lungs, and the conditioned animals have less neutrophils, which would imply that it is related to the vascular permeability. And we do try to be very specific in how we infuse the Evans blue dye.
Additionally, these animals are not—even though they do have a severe acidosis—they are not hypoxic by the time that they die.
As far as whether or not this response is specific for endotoxin, our previous studies have shown that the tolerance can also be induced to hemorrhage. We have not used either whole organisms or any other infectious challenges.
In regard to IL-8, we have not actually measured IL-8 in our model. We have measured other cytokines. Like I said, we have measured IL-10, and IL-10 is not elevated in the conditioned animals. IL-8 we have yet to measure. IL-1 is not consistent. And IL-6 is also elevated, the same as 10.
In regard to whether the message differences in LPS and hemorrhage is a second insult, whether it’s an issue of organ hypoxia, neither of those animals before they die are actually hypoxic. But they are hypotensive, and that is their terminal event. This may be an issue not so much of the specific insult but the degree of insult. The endotoxin is actually a more severe insult than the severe hemorrhage—those animals look sicker. So it may be a matter of LPS being more of a shotgun than endotoxin.
In regard to whether or not this is species-specific, I think the issue of tolerance right now is highly discussed. It may be species-specific, it may be insult-specific, it may be timing. I think that there is an issue of degree in timing in regards to tolerance. Some people say that the neutrophils, for example, get primed and overreact to the second insult. The lymphocyte folks say that they get immunosuppressed, and thus, it’s bad. As far as the macrophage, I think that the macrophage can have differential effects. I think this is more of a mechanistic issue, and then maybe we could target our therapies at altering the inflammation so that we get just enough inflammation to protect the patient from an infectious insult and not so much that they get autoimmune destruction of their lungs and liver and kidneys.
Footnotes
Correspondence: Cynthia Mendez, MD, James A. Haley VA Hospital, Department of Surgery (112), Tampa, FL 33612.
Presented at the 110th Annual Meeting of the Southern Surgical Association, December 6–9, 1998, The Breakers, West Palm Beach, Florida.
Supported by AT&T Surgical Scholars Program.
Accepted for publication December 1998.
References
- 1.Meldrum DR, Cleveland JC Jr, Rowland RT, et al. Early and delayed preconditioning: differential mechanisms and additive protection. Am J Physiol 1997; 273: H725–733. [DOI] [PubMed] [Google Scholar]
- 2.Meldrum DR, Cleveland JC Jr, Moore EE, et al. Adaptive and maladaptive mechanisms of cellular priming. Ann Surg 1997; 226 (5): 587–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moore F, Moore E. Evolving concepts in the pathogenesis of postinjury multiple organ failure. Surg Clin North Am 1995; 75 (2): 257–277. [DOI] [PubMed] [Google Scholar]
- 4.Kramer A, Salhab K, Carey L, et al. Protection by sublethal conditioning hemorrhage is reversed by macrophage inhibition in a rat model of hemorrhagic shock. J Trauma (in press). [Google Scholar]
- 5.Johnston C, Greisman S. Mechanisms of endotoxin tolerance. In Proctor R, Hinshaw L. Pathophysiology of endotoxin. Amsterdam: Elsevier; 1985: 359–401.
- 6.Li M, Seatter S, Manthei R, et al. Macrophage endotoxin tolerance: effect of TNF or endotoxin pretreatment. J Surg Res 1994; 57: 85–92. [DOI] [PubMed] [Google Scholar]
- 7.Seatter S, Bennet T, Li M, et al. Macrophage endotoxin tolerance, tumor necrosis factor and interleukin-1 regulation by lipopolysaccharide pretreatment. Arch Surg 1994; 129: 1263–1270. [DOI] [PubMed] [Google Scholar]
- 8.Seatter S, Li M, Bubrick M, et al. Endotoxin pretreatment of human monocytes alters subsequent endotoxin-triggered release of inflammatory mediators. Shock 1995; 3 (4): 252–258. [DOI] [PubMed] [Google Scholar]
- 9.West M, Seatter S, Bellingham J, et al. Mechanisms of reprogrammed macrophage endotoxin signal transduction after lipopolysaccharide pretreatment. Surgery 1995; 118 (2): 220–228. [DOI] [PubMed] [Google Scholar]
- 10.Drucker W, Schlatter J, Drucker R. Metabolic factors associated with endotoxin-induced tolerance for hemorrhagic shock. Surgery 1968; 64 (1): 75–84. [PubMed] [Google Scholar]
- 11.Zervos E, Norman J, Denham D, et al. Cytokine activation through sublethal hemorrhage is protective against early lethal endotoxic challenge. Ann Surg 1997; 132: 1216–1221. [DOI] [PubMed] [Google Scholar]
- 12.Bianchi M, Ulrich P, Bloom O, et al. An inhibitor of macrophage arginine transport and nitric oxide production (CNI-1493) prevents acute inflammation and endotoxin lethality. Mol Med 1995; 1 (3): 254–266 [PMC free article] [PubMed] [Google Scholar]
- 13.Bianchi M, Bloom O, Raabe T, et al. Suppression of proinflammatory cytokines in monocytes by a tetravalent guanylhydrazone. J Exp Med 1996; 183: 927–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cohen P, Dennis HN, Caragine T, et al. CNI-1493 inhibits monocyte/macrophage tumor necrosis factor by suppression of translation efficiency. Proc Natl Acad Sci 1996; 93: 3967–3971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Patterson C, Rhoades R, Garcia J. Evans blue dye as a marker of albumin clearance in cultured endothelial monolayer and isolated lung. J Appl Physiol 1992; 72 (3): 865–873. [DOI] [PubMed] [Google Scholar]
- 16.Dallal M, Chang S. Evans blue dye in the assessment of permeability-surface area product in perfused rat lungs. J Appl Physiol 1994; 77 (2): 1030–1035. [DOI] [PubMed] [Google Scholar]
- 17.Santagati S, Garnier M, Carlo P, et al. Quantitation of low abundance mRNAs in glial cells using different polymerase chain reaction (PCR)-based methods. Brain Res Protocols 1997; 1: 217–223. [DOI] [PubMed] [Google Scholar]
- 18.Zervos E, Bloomston M, Carey L, et al. Conditioning to hemorrhagic shock improves survival and attenuates inflammatory cytokine gene induction in the lung, liver and spleen of rats. Surg Forum 1997; 48: 127–129. [Google Scholar]
- 19.Beutler B, Krochin N, Milsark I, et al. Control of cachectin (tumor necrosis factor) synthesis: mechanisms of endotoxin resistance. Science 1986; 232 (4753): 977–980. [DOI] [PubMed] [Google Scholar]
- 20.Han J, Brown T, Beutler B. Endotoxin-responsive sequences control cachectin/tumor necrosis factor biosynthesis at the translational level. J Exp Med 1990; 171 (2): 465–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Han J, Beutler B, Huez G. Complex regulation of tumor necrosis factor mRNA turnover in lipopolysaccharide-activated macrophages. Biochem Biophys Acta 1991; 1090 (1): 22–28. [DOI] [PubMed] [Google Scholar]
- 22.Frakenberger M, Pechumer H, Ziegler-Heitbrock H. Interleukin-10 is upregulated in LPS tolerance. J Inflam 1995; 45: 56–63. [PubMed] [Google Scholar]