

Abstract
Objective
To evaluate the parameters that mediate fibrogenesis in chronic pancreatitis (CP).
Background
Connective tissue growth factor (CTGF), which is regulated by transforming growth factor β (TGF-β), has recently been implicated in skin fibrosis and atherosclerosis. In the present study, the authors analyzed the concomitant presence of TGF-β1 and its signaling receptors—TGF-β receptor I, subtype ALK5 (TβR-IALK5), and TGF-β receptor II (TβR-II)—as well as CTGF and collagen type I in the pancreatic tissue of patients undergoing surgery for chronic pancreatitis.
Patients and Methods
CP tissue samples were obtained from 40 patients (8 women, 32 men) undergoing pancreatic resection. Tissue samples of 25 previously healthy organ donors (12 women, 13 men) served as controls. The expression of TGF-β1, TβR-IALK5, TβR-II, CTGF, and collagen type I was studied by Northern blot analysis. By in situ hybridization and immunohistochemistry, the respective mRNA moieties and proteins were localized in the tissue samples.
Results
Northern blot analysis showed that CP tissue samples exhibited concomitant enhanced mRNA expression of TGF-β1 (38-fold), TβR-II (5-fold), CTGF (25-fold), and collagen type I (24-fold) compared with normal controls. In addition, TβR-IALK5 mRNA was increased in 50% of CP tissue samples (1.8-fold). By in situ hybridization, TGF-β1, TβR-IALK5, and TβR-II mRNA were often seen to be colocalized, especially in the ductal cells and in metaplastic areas where atrophic acinar cells appeared to dedifferentiate into ductal structures. In contrast, CTGF was located in degenerating acinar cells and principally in fibroblasts surrounding these areas. Moreover, CTGF mRNA expression levels correlated positively with the degree of fibrosis in CP tissues.
Conclusion
The concomitant overexpression of CTGF, collagen type I, TGF-β1, and its signaling receptors in CP suggests that these proteins contribute to enhanced extracellular matrix synthesis and accumulation, resulting finally in the fibrogenesis observed in CP.
Connective tissue growth factor (CTGF) is a novel cysteine-rich peptide originally identified from human endothelial cells. 1 It belongs to a family of immediate-early genes that are especially needed for the coordination of complex biologic processes such as differentiation and tissue repair. 1 It exhibits platelet-derived growth factor (PDGF)-like chemotactic and mitogenic activities for mesenchymal cells and appears to be related to PDGF, although it has little peptide sequence homology to either PDGF A or B chain peptides. 2,3 In cultured human foreskin fibroblasts, CTGF mRNA and protein are upregulated after stimulation with transforming growth factor β1 (TGF-β1). In contrast, other growth factors—including PDGF, epidermal growth factor, and basic fibroblast growth factor—do not upregulate CTGF, suggesting that it is under regulatory control of TGF-β. 4–6 In addition, Igarashi et al 6 demonstrated, using an in vitro model for wound healing, that there is a coordinate expression of TGF-β1 before CTGF in the regenerating tissue, indicating that CTGF may be one of the downstream effectors of TGF-β activation. 6 The pathogenetic function of CTGF in fibrotic disorders has also been demonstrated in atherosclerosis, where its expression is markedly enhanced. Moreover, this increase in CTGF is regulated by TGF-β1. 7 These findings are important in light of the fact that TGF-β1, which strongly stimulates the synthesis of extracellular matrix components, including collagen, fibronectin, and proteoglycan, has recently been proposed as a major candidate in the fibrogenesis that occurs in different chronic inflammatory diseases, including chronic pancreatitis (CP). 8–10
CP represents an irreversible alteration of the exocrine parenchyma and is histologically characterized by destruction of acinar cells, accumulation of fibroblasts and extracellular matrix, and calcification in pancreatic ducts. 11 In addition, typical foci of chronic inflammatory cells are found in regions where changes appear to be ongoing in damaged parenchyma and around nerve bundles. 12,13 Nevertheless, the biochemical and molecular mechanisms underlying these histologic changes are poorly understood.
There is increasing knowledge concerning mediators participating in CP. Lee et al 8 have observed, in transgenic mice overexpressing TGF-β1, that an intense fibroblast proliferation and abnormal deposition of extracellular matrix occur in the pancreas. In addition, enhanced pancreatic TGF-β1 expression in patients with CP suggests a role of this growth factor in the pathogenesis and evolution of CP, especially in the accumulation of extracellular matrix. 9
It has also been demonstrated that the growth stimulatory effects of TGF-β on connective tissue cells appear to be mediated by an indirect mechanism of autocrine-induced growth factors, such as PDGF-like peptides. 14 Building on these findings, we hypothesized in the present study that CTGF, which is a downstream target of TGF-β1 activation, might play a pivotal role in the development and progression of fibrosis in CP.
To assess this hypothesis, we used Northern blot, in situ hybridization, and immunohistochemistry to evaluate concomitant gene expression and localization in the tissue samples of CTGF, TGF-β1, and its two signaling receptors—TGF-β receptor I, subtype ALK5 (TβR-IALK5), and TGF-β receptor II (TβR-II)—in both CP and the normal pancreas. Because TGF-β1 and CTGF are both inducers of collagen synthesis, we also analyzed collagen type I mRNA expression in the same tissue samples. We found that both TGF-β and CTGF pathways are upregulated in CP and that a significant relation exists between these molecular changes and the degree of pancreatic fibrosis.
MATERIALS AND METHODS
Patients and Tissue Sampling
Human pancreatic tissue samples were obtained from 40 patients with CP (8 women, 32 men; median age 45 years [range 27 to 72]) who underwent pancreatic resection because of CP-related complications at the University Hospital of Bern (Bern, Switzerland). The etiology of CP was alcohol overconsumption in all patients, and surgical procedures consisted of either a partial duodenopancreatectomy (Whipple operation, 8 patients) or a duodenum-preserving pancreatic head resection (32 patients). Histologically, CP was graded as moderate to severe in all the patients.
Normal human pancreatic tissue samples were obtained through an organ donor program from 25 previously healthy individuals (12 female patients, 13 male patients; median age 35 years [range 14 to 55]) from whom other organs were taken for transplantation and no recipients for the pancreas were present. Histologically, these pancreases were normal.
In all cases, freshly removed tissue samples were fixed in paraformaldehyde solution in the operating room, maintained for 12 to 24 hours, and then paraffin-embedded for histologic analysis. In addition, the part of the tissue samples destined for RNA extraction was snap-frozen in liquid nitrogen immediately on surgical removal and maintained at −80°C until use.
The studies were approved by the Human Subjects Committee of the University of Bern.
Probe Synthesis
The CTGF cDNA probe consisted of a 600-base pair EcoRI/PstI fragment of human CTGF cDNA (kindly provided by Dr. Barry S. Oemar, Cardiovascular Research Laboratory, Institute of Physiology, University of Zürich, Switzerland). 7 The TGF-β1 cDNA probe consisted of a 280-base pair EcoRI/XbaI fragment of human TGF-β1 cDNA. 15 The TβR-I cRNA probe, specifically detecting the ALK-5 subtype, consisted of a 377-base pair Bam-HI fragment of human TβR-I cDNA corresponding to nucleotides 442 to 818. 16 The TβR-II cRNA probe consisted of a 476-base pair Bam-HI fragment of human TβR-II cDNA corresponding to nucleotides 42 to 519. 16 The collagen cDNA probe consisted of a 1.8-kilobase EcoRI fragment of human fibroblast type I collagen cDNA (ATCC, Rockville, MD). 17 The amylase cDNA probe consisted of a 545-base pair EcoRI/Sau3AI fragment of human amylase (ATCC). 18
A 0.19-kilobase BamHI fragment of the mouse 7S cytoplasmic cDNA that cross-hybridizes with human 7S RNA 15,16 was used to verify equivalent RNA loading in the Northern blot experiments.
The CTGF, TGF-β1, TβR-IALK5, and TβR-II cDNA fragments were subcloned into the pGEM3Zf vector (Promega Biotechnology, Madison, WI) carrying promoters for the DNA-dependent SP6 and T7 RNA polymerases.
For the Northern blot analyses, cDNA probes (TGF-β1, CTGF, collagen, amylase, 7S) were labeled with [alpha-32P]dCTP (Du Pont International, Regensdorf, Switzerland), prepared by using a random primer labeling system (Boehringer-Mannheim, Mannheim, Germany). The cRNA probes used (TβR-IALK5, TβR-II) were radiolabeled with [alpha-32P]CTP (Du Pont International).
For in situ hybridization analysis, cRNA probes of TGF-β1, TβR-IALK5, TβR-II, and CTGF were labeled with digoxigenin. After linearization, the probes were transcribed using the Ribomax System (Promega Biotechnology). The transcription resulted in digoxigenin-labeled antisense riboprobes specific for TGF-β1, TβR-IALK5, TβR-II, and CTGF mRNA. The corresponding sense probes were prepared in an analogous manner. The probes were shortened to a length of about 150 bases according to the procedure of Cox et al 19 and stored in diethylpyrocarbonate-treated water at −80°C until further use.
Northern Blot Analysis
Total RNA was extracted by the guanidine isothiocyanate method, size-fractionated on 1.2% agarose/1.8 M formaldehyde gels, and stained with ethidium bromide for verification of RNA integrity and loading equivalency. 20,21 The RNA was electrotransferred onto nylon membranes (Gene Screen, Du Pont, Boston, MA) and cross-linked by ultraviolet irradiation. The filters were then prehybridized, hybridized, and washed under conditions appropriate for specific antisense cRNA riboprobes (TβR-IALK5, TβR-II) or cDNA probes (TGF-β1, CTGF, collagen, amylase, 7S), as previously reported. 21,22
In the case of antisense riboprobes, the blots were prehybridized overnight at 65°C in 50% formamide, 0.5% sodium dodecyl sulfate (SDS), 5× SSC (sodium chloride/sodium citrate buffer), 5× Denhardt’s solution (1× Denhardt’s = 0.02% ficoll, 0.02% polyvinylpyrrolidone, and 0.02% bovine serum albumin), 250 mg/L salmon sperm DNA, and 50 mM sodium phosphate buffer, pH 6.5. The blots were then hybridized for 18 hours at 65°C in the presence of 1 × 106 cpm/ml of the [alpha-32P]CTP-labeled antisense riboprobe, washed twice at 65°C in a solution containing 1× SSPE (150 mM NaCl, 10 mM NaH2PO4, and 1 mM EDTA) and 0.5% SDS, and washed twice at 65°C in a solution containing 0.1× SSPE and 0.5% SDS. 21,22
In the case of cDNA probes, blots were prehybridized overnight at 42°C in a prehybridization buffer that contained 50% formamide, 1% SDS, 0.75 M NaCl, 5 mM EDTA, 5× Denhardt’s solution, 100 mg/L salmon sperm DNA, 10% dextran sulfate, and 50 mM Na2PO4, pH 7.4. The hybridization was carried out at 42°C for 18 hours with the [alpha-32P]dCTP-labeled cDNA probe (1 × 106 cpm/ml for TGF-β1, CTGF, 1 × 105 cpm/ml for 7S) and then washed twice at 50°C in 2× SSC and three times at 55°C in 0.2× SSC and 2% SDS. 21,22
Membranes were then exposed at −80°C to Fuji x-ray films with intensifying screens for 1 to 12 days, and the intensity of the radiographic bands was quantified by video densitometry (Media Cybernetics, Silver Spring, MD). All membranes were rehybridized with the 7S cDNA probe to assess equivalent RNA loading, as previously reported. 20 Levels of TGF-β1, TβR-IALK5, TβR-II, CTGF, collagen, and amylase mRNA were expressed as the signal intensity of the mRNA signals normalized to the 7S signals. 21,22
In Situ Hybridization
In situ hybridization was performed as previously reported. 23,24 Briefly, normal pancreas and CP tissue samples were fixed in paraformaldehyde and paraffin-embedded. For each sample, several consecutive sections were analyzed. The tissue sections (4 μm) were deparaffinized, rehydrated with 1× phosphate-buffered saline (PBS), and incubated in 0.2 mol/L HCl for 20 minutes at room temperature. After the slides were rinsed in 2× SSC, the sections were treated with proteinase K (Boehringer Mannheim) at a concentration of 30 μg/ml (TGF-β1 and CTGF), 50 μg/ml (TβR-IALK5), or 35 μg/ml (TβR-II) for 15 minutes at 37°C. After acetylation, postfixation with 4% paraformaldehyde in PBS (5 minutes), and washing in 2× SSC, the samples were prehybridized at 63°C (CTGF), 67°C (TGF-β1), 64°C (TβR-IALK5), or 50°C (TβR-II) for ≥1 hour in 50% formamide (v/v), 4× SSC, 2× Denhardt’s reagent, and 250 μg RNA/ml. Hybridization was performed overnight at 63°C (CTGF), 67°C (TGF-β1), 64°C (TβR-IALK5), or 50°C (TβR-II) in 50% (v/v) formamide, 4× SSC, 2× Denhardt’s reagent, 500 μg RNA/ml, and 10% dextran sulfate (w/v). The final concentrations of the labeled probes were approximately 0.5 ng/μl. After hybridization, excess probe was removed by washing in 2× SSC and by RNase treatment: 40 U/ml RNase T1 and 0.2 μg/ml RNase (DNase-free) (both Boehringer Mannheim) at 37°C for 30 minutes. After washing for 20 minutes in 2× SSC at 65°C (CTGF), 68°C (TGF-β1), 66°C (TβR-IALK5), or 60°C (TβR-II), and then for 20 minutes in 0.2× SSC under the same stringent conditions, the tissue sections were incubated with an antidigoxigenin antibody conjugated with alkaline phosphatase (Boehringer Mannheim). For color reaction, 5-bromo-4-chloro-3-indolyl phosphate and nitro blue tetrazolium (Sigma, Buchs, Switzerland) were used.
Immunohistochemistry
Paraffin-embedded tissue sections (2- to 4-μm thick) were subjected to immunostaining using the streptavidin–peroxidase technique (Kirkegaard & Perry Laboratories, Gaithersburg, MD). Several consecutive tissue sections were submerged for 15 minutes in Tris buffered saline (TBS; 10 mM Tris-HCl, 0.85% NaCl, pH 7.4) containing 0.1% (v/v) Triton X-100, and then washed for 5 minutes in TBS solution, as previously reported. 15,22 Endogenous peroxidase activity was quenched by incubating the slides in methanol and in methanol/0.6% hydrogen peroxide, followed by washings in methanol and TBS containing 0.1% bovine serum albumin. 15,22 After treatment with hyaluronidase (1 mg/ml in 100 mM sodium acetate, 0.85% NaCl), the sections were incubated for 30 minutes at 23°C with 10% normal goat serum and then incubated overnight at 4°C with polyclonal antibodies detecting TGF-β1 (2.5 μg/ml), TβR-IALK5 (2.5 μg/ml), and TβR-II (2.5 μg/ml) (Santa Cruz Biotechnology, Santa Cruz, CA) diluted in 5% normal goat serum. Bound antibody was detected with a biotinylated goat antirabbit IgG secondary antibody and a streptavidin–peroxidase complex (Kirkegaard & Perry Laboratories), followed by incubation with diaminobenzidine tetrahydrochloride (0.05%) as the substrate and counterstaining with Mayer’s hematoxylin.
To ensure antibody specificity, control slides were incubated either in the absence of primary antibody or with an nonspecific IgG antibody. In both cases, no immunostaining was detected.
The slides were analyzed by two independent observers blinded to patient status; any differences were resolved by joint review and consultation with a third observer.
Assessment of the Degree of Fibrosis
To assess the degree of fibrosis in the CP samples, tissue sections stained with hematoxylin and eosin were analyzed. The percentage of fibrosis or parenchyma was measured on five different tissue sections in each CP tissue sample, and the mean value was calculated, as previously reported in detail. 25,26
Statistical Analysis
Results are expressed as median and range or as mean ± SEM. For statistical analysis, the Mann–Whitney test and the Spearman rank correlation test were used. Significance was defined as p < 0.05.
RESULTS
Northern Blot Analysis
In the normal pancreatic tissue samples, low levels of TGF-β1, TβR-IALK5, TβR-II, CTGF, and collagen type I mRNA were present (Fig. 1). All four mRNA moieties, but especially TGF-β1, CTGF, and collagen type I, were only faintly visible on the original autoradiographs in all normal pancreas samples.
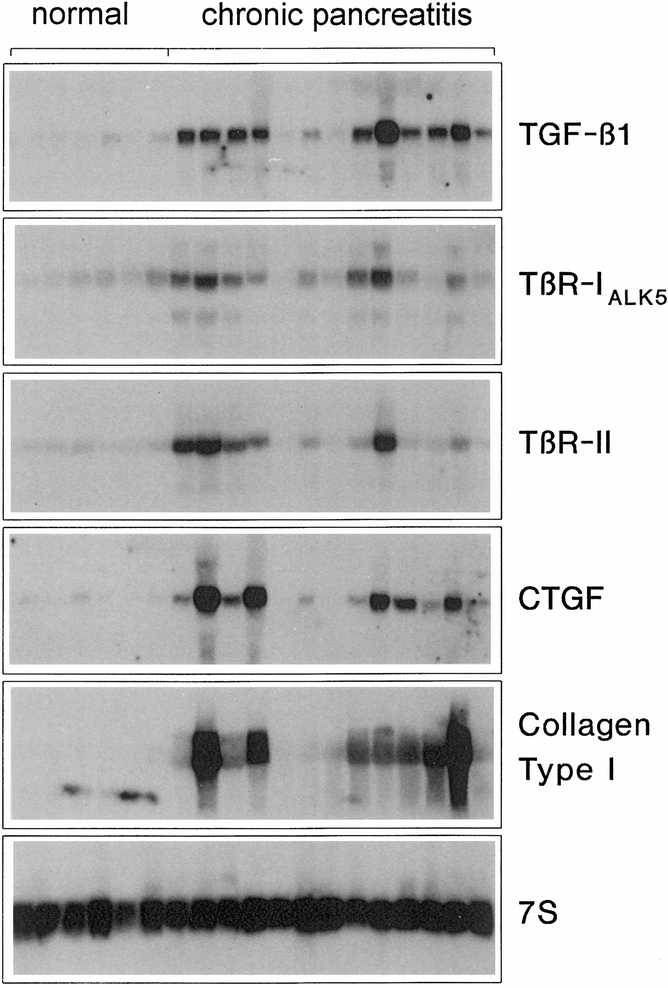
Figure 1. Northern blot analysis. TGF-β1, TβR-IALK5, TβR-II, CTGF, and collagen type I mRNA expression in the normal pancreas (lanes 1 to 6) and in chronic pancreatitis (CP) samples (lanes 7 to 18). In normal controls, low levels of these mRNA moieties were detectable. In contrast, in CP samples, enhanced levels of TGF-β1, TβR-IALK5, TβR-II, CTGF, and collagen type I mRNA were concomitantly present in most of the tissue samples. Filters were rehybridized with 7S cDNA to verify equivalent RNA loading. TGF-β1 mRNA migrates as a single 2.4-kilobase band, TβR-IALK5 as a single 6-kilobase band, TβR-II as a single 5.5-kilobase band, CTGF mRNA as a single 2.4-kilobase band, collagen type I mRNA as 3.0- and 2.2-kilobase bands, and 7S RNA as a 0.4-kilobase band.
In contrast to the findings in the normal pancreas, CP samples showed marked increases in TGF-β1, TβR-II, CTGF, and collagen type I mRNA levels (see Fig. 1). Densitometric analysis of the Northern blots indicated that in CP, TGF-β1 was increased by a factor of 38 (p < 0.001), TβR-II by a factor of 5 (p < 0.05), CTGF by a factor of 25 (p < 0.002), and collagen type I by a factor of 24 (p < 0.001) when compared with normal controls (Fig. 2). In addition, 50% of CP tissue samples showed a 1.8-fold (p < 0.05) increase in the level of TβR-IALK5 mRNA. However, when all CP samples were statistically analyzed together, the expression of TβR-IALK5 mRNA was not different compared with that of the normal controls. CTGF mRNA levels were not increased in 28% of the CP tissue samples. However, 36% of the patients with no increase in CTGF mRNA exhibited marked increases in TGF-β1 and TβR-II mRNA expression. In addition, TGF-β1, CTGF, and collagen type I mRNA were concomitantly increased in 65% of the CP tissue samples.
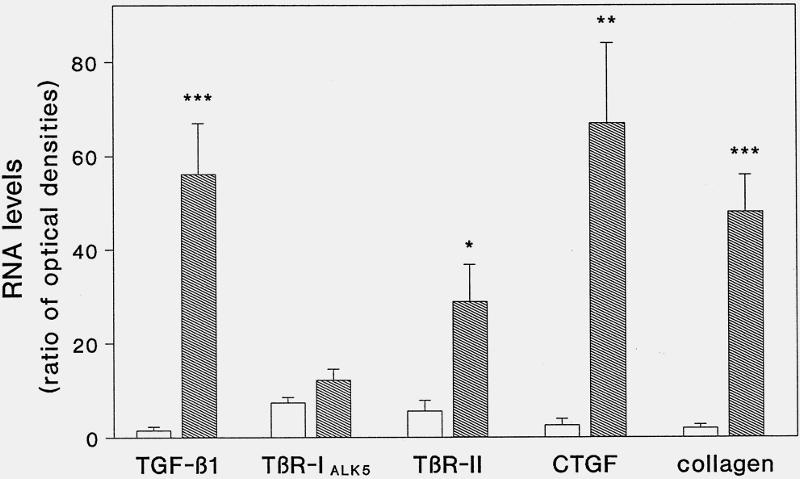
Figure 2. Densitometric analysis. TGF-β1, TβR-IALK5, TβR-II, CTGF, collagen type I, and 7S mRNA levels were analyzed by laser densitometry. The ratio of the optical density between TGF-β1, TβR-IALK5, TβR-II, CTGF, collagen type I, and corresponding 7S signal was calculated (bars = mean ± SD for the calculated ratios; * p < 0.05; ** p < 0.002; *** p < 0.0001; white bars, normal pancreas; shaded bars, chronic pancreatitis).
In contrast to TGF-β1, TβR-II, CTGF, and collagen type I, amylase mRNA expression in CP samples was markedly decreased (data not shown).
In Situ Hybridization
In situ hybridization was performed to localize the exact sites of TGF-β1, TβR-IALK5, TβR-II, and CTGF mRNA production in the normal and CP tissue samples. Consecutive tissue sections of each sample were used for TGF-β1, TβR-IALK5, TβR-II, and CTGF analysis.
In the normal pancreas, faint mRNA signals for TGF-β1 (Fig. 3A), TβR-IALK5 (Fig. 3C), TβR-II (Fig. 3E), and CTGF (Fig. 3G) were found. Islets of Langerhans and small ductal cells showed weak to moderate signals for TGF-β1 (Fig. 3A), TβR-IALK5 (Fig. 3C), and TβR-II (Fig. 3E) mRNA, whereas these structures were devoid of any signal for CTGF. Weak to moderate TGF-β1 mRNA signals were also localized in some acinar cells in a focal pattern. In addition, faint CTGF mRNA signals were present in some smooth muscle and endothelial cells of small and medium-sized arteries.
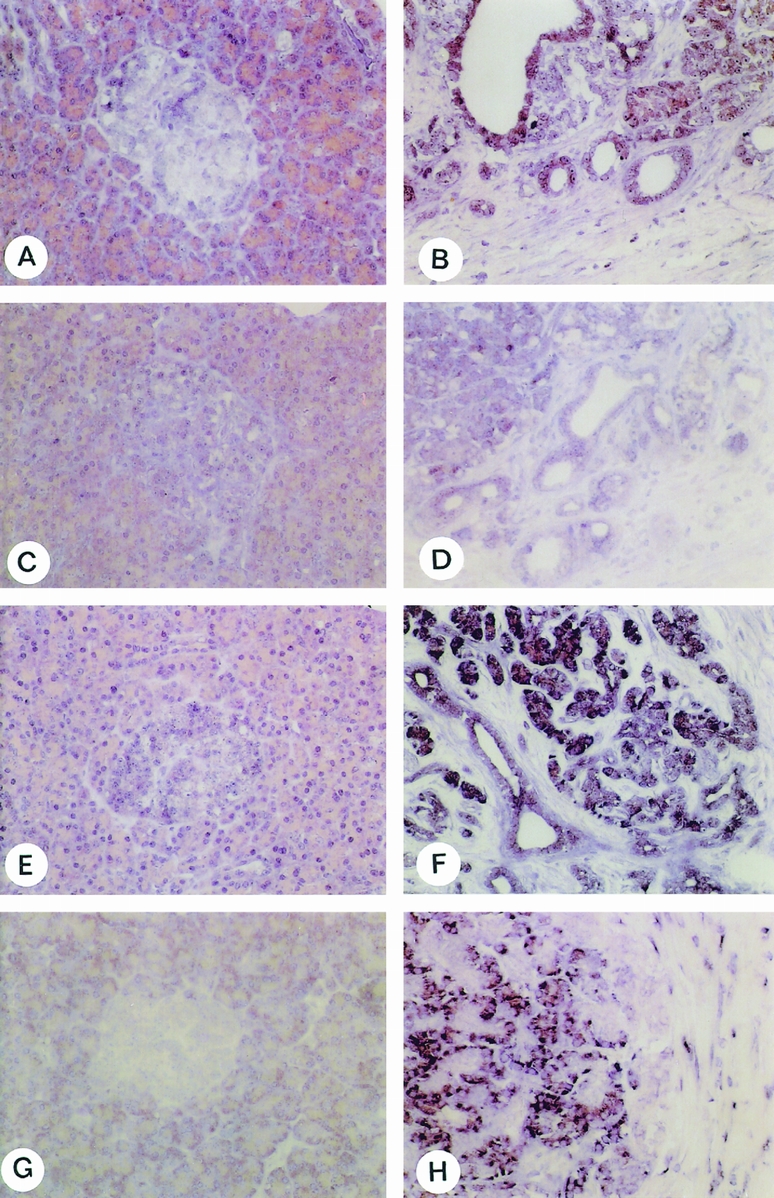
Figure 3. In situ hybridization. TGF-β1 (A, B), TβR-IALK5 (C, D), TβR-II (E, F) and CTGF (G, H) in situ hybridization analysis in the normal pancreas (A, C, E, G) and in chronic pancreatitis (CP) samples (B, D, F, H). In the normal pancreas, TGF-β1, TβR-IALK5, and TβR-II mRNA moieties were faintly visible in the islets of Langerhans and in a few acinar cells. The CTGF mRNA signal was detected mainly in a few acinar cells, whereas the islets were devoid of any mRNA signal. In contrast, in CP tissue samples, moderate to strong mRNA staining for TGF-β1, TβR-IALK5, and TβR-II was found in areas with ductal metaplasia and where the atrophic acinar cells appeared to dedifferentiate in ductal structures. CTGF mRNA signals were present in remaining acinar cells (left part of panel H) and in fibroblasts (right part of panel H) surrounding these areas (original magnification ×200).
In contrast to the normal pancreas, CP samples showed higher CTGF mRNA in situ hybridization signals (Fig. 3H). Weak to moderate CTGF mRNA signals were found in the remaining acinar cells (Fig. 3H), whereas metaplastic ductal cells were devoid of any CTGF mRNA signal. Primarily fibroblasts localized in areas with a high degree of pancreatic damage exhibited CTGF mRNA expression. This was especially evident where fibroblasts surrounded degenerating acinar cells and metaplastic ductal cells. TGF-β1 (Fig. 3B), TβR-IALK5 (Fig. 3D), and TβR-II (Fig. 3F) mRNA signals were often colocalized, especially in ductal cells and in areas with metaplastic lesions where the atrophic acinar cells appeared to dedifferentiate into ductlike structures. In contrast to the intense signal for CTGF in fibroblasts, only weak to moderate mRNA expression for TGF-β1, TβR-IALK5, and TβR-II was observed in these cells.
In situ hybridization experiments using the digoxigenin-labeled sense probes corresponding to the antisense probes failed to produce a signal.
Immunohistochemistry
To localize TGF-β1, TβR-IALK5, and TβR-II, immunohistochemical analysis was performed in normal pancreas and CP tissue samples (Fig. 4). For our purposes, specific antibodies were available for TGF-β1, TβR-IALK5, and TβR-II but not for CTGF.
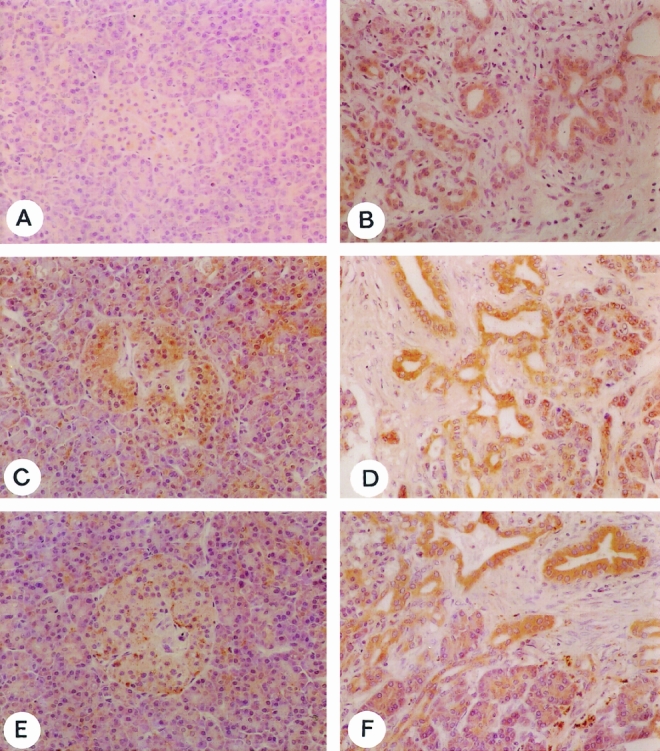
Figure 4. Immunohistochemical analysis. TGF-β1 (A, B), TβR-IALK5 (C, D), and TβR-II (E, F) immunoreactivity in the normal pancreas (A, C, E) and in chronic pancreatitis (CP) tissue samples (B, D, F). In the normal pancreas, islets of Langerhans were immunopositive for all factors. Only some isolated acinar cells showed mild immunoreactivity for TGF-β1, TβR-IALK5, and TβR-II. In contrast, CP tissue sections exhibited intense immunoreactivity for all factors, with moderate to strong immunoreactivity, especially in ductal cells and in acinar cells that appeared to dedifferentiate into tubular structures. The slides are counterstained with Meyer’s hematoxylin (original magnification ×200).
Normal pancreas samples exhibited weak TGF-β1 (Fig. 4A), TβR-IALK5 (Fig. 4C), and TβR-II (Fig. 4E) immunoreactivity, often colocalized in a few islet cells. The exocrine parenchyma showed weak to moderate immunoreactivity for TβR-IALK5 and TβR-II, especially in the small ductal cells and in a few acinar cells. TGF-β1 was present in a focal pattern in some acinar cells. In contrast to the normal pancreas, CP tissue sections exhibited intense immunoreactivity for TGF-β1 (Fig. 4B), TβR-IALK5 (Fig. 4D), and TβR-II (Fig. 4F). Intense staining signals were primarily colocalized in metaplastic ductal cells, often surrounded by immunoreactive inflammatory cells and fibroblasts. Also, the remaining acinar cells showed intense immunoreactivity, especially for TβR-IALK5 and TβR-II, and predominantly in areas close to pancreatic fibrosis.
Correlation Between Northern Blot Results and Histomorphologic Analysis
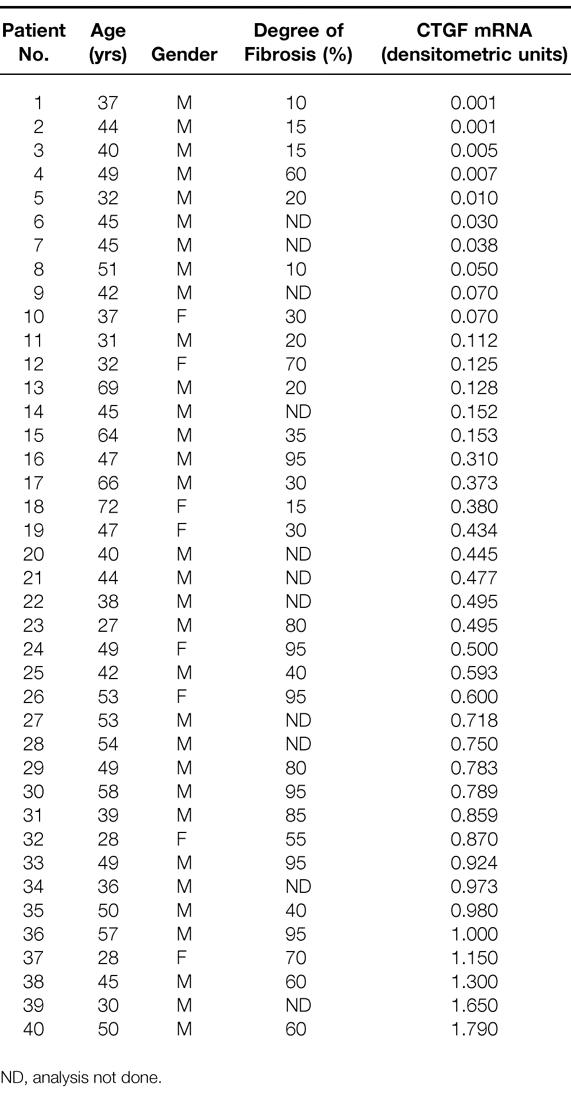
All CP tissue specimens were evaluated to determine whether the degree of fibrosis correlated with the level of expression of CTGF mRNA. Histomorphologic analysis of CP tissue sections showed a significant positive correlation (p < 0.001) between the degree of fibrosis and CTGF mRNA levels (r = 0.65). Thus, samples with higher levels of CTGF mRNA expression also exhibited higher degrees of pancreatic fibrosis (Table 1).
Table 1. Relation between Degree of Fibrosis and CTGF mRNA Expression
DISCUSSION
A disease primarily affecting the exocrine pancreas, CP is characterized by continuous destruction of the pancreatic acinar and ductal cells and replacement by fibrous tissue. 27–29 The increase of extracellular matrix deposition can lead to a pancreatic mass and subsequent compression or stenosis of structures adjacent to the pancreatic gland, generating complications that often require surgery. Overproduction of extracellular matrix often compromises the remaining vital parenchyma, leading to exocrine and sometimes endocrine insufficiency. 30 Several hypotheses have been postulated to explain the development of CP, but so far the pathogenesis of this disease remains unclear.
In the present study, we analyzed TGF-β1 and for the first time its two signaling receptors (TβR-IALK5 and TβR-II) as well as CTGF and collagen type I gene expression in CP and compared them with normal pancreatic tissue samples. Most CP tissue samples exhibited concomitant enhanced expression of TGF-β1, TβR-IALK5, TβR-II, CTGF, and collagen type I mRNA. Interestingly, in situ hybridization showed that the TGF-β1 and its signaling receptors TβR-IALK5 and TβR-II colocalized in metaplastic ductal cells and in remaining acinar cells. In contrast, CTGF mRNA was found mainly in fibroblasts surrounding these areas of tissue remodeling. These findings are important because they underscore the potential role for TGF-β in CP by demonstrating concomitant enhanced expression of TGF-β and its signaling receptors, both of which are required for effective signal transduction. In addition, TGF-β signaling might trigger CTGF expression in the surrounding fibroblasts, which is an important factor in fibroblast activation and extracellular matrix synthesis. In fact, histomorphologic analysis showed a positive correlation between the degree of fibrosis in CP tissue samples and respective levels of CTGF mRNA expression obtained by Northern blot analysis. In addition, tissue samples with enhanced mRNA levels of TGF-β1, CTGF, and collagen type I exhibited a marked decrease in amylase mRNA levels, demonstrating that overexpression of these factors is not due to a generalized increase in pancreatic mRNA expression.
Moreover, as previously reported in CP, 25,31 urokinase-dependent plasminogen activation occurs in regions characterized by dedifferentiation of acinar cells in ductular structures, thereby probably contributing to remodeling of pancreatic parenchyma. In addition, in hyperplastic ductal cells, we also reported enhanced expression and synthesis of acidic (aFGF) and basic (bFGF) fibroblast growth factors. 32 These findings are of interest if we consider that bFGF can induce the activation of the plasminogen activator system in endothelial cells, which results in a marked increase in urokinase plasminogen activator-induced plasmin. 33 Further, urokinase plasminogen activator activates latent TGF-β1 in its mature form, which in turn induces plasminogen activator inhibitor-1 and thus downregulates this proteolytic system; this favors fibrosis by inhibiting the extracellular matrix turnover. 34
In summary, there seems to be activation both of profibrotic factors and the plasminogen activation system, primarily in regions with ductal hyperplasia and in areas where acinar cells dedifferentiate into ductal structures. In addition, the upregulation of CTGF, which occurs predominantly in fibroblasts surrounding these regions, may contribute to this complex remodeling cascade, which is probably a major factor leading to fibrosis generation.
Therefore, we might suppose that the intense release of TGF-β1 from metaplastic ductal cells and degenerating acinar cells might induce CTGF expression in fibroblasts in a paracrine fashion, which subsequently may stimulate extracellular matrix synthesis and accumulation, to replace atrophic acinar cells. Alternatively, activation of fibroblasts surrounding metaplastic ductal cells and atrophic and degenerating acinar cells may lead to increased deposition of connective tissue. The presence of this increased collagen mass may cause mechanical compression of the exocrine pancreatic parenchyma, with obliteration of small and large pancreatic ducts, promoting disease progression and evolution; this has been postulated by Klöppel and Maillet. 35
The concomitant expression of TGF-β1, CTGF, and collagen type I in CP indicates the activation of a profibrotic cascade in which initiators induce the subsequent synthesis of secondary factors. Consequently, this activation might regulate and sustain specific cellular processes in tissue repair and remodeling, such as fibrosis induction and replacement of atrophic cells. CTGF exhibits chemotactic and mitogenic activities for mesenchymal cells and also stimulates collagen synthesis. 2–5 This hypothesis is supported by various studies that have demonstrated prolonged CTGF activation after brief exposure of the cells to TGF-β1 but not after stimulation with PDGF, FGF, and epidermal growth factor. 4 In our study, a subgroup of patients exhibited no increase in CTGF mRNA expression. These findings indicate that in CP additional pathways that initiate and promote fibrogenesis might be activated. 25,32,36,37 However, TGF-βs and their signaling receptors that upregulate CTGF have a preponderant function in extracellular matrix synthesis in a variety of benign and malignant disorders. 15,18,38
The present data indicate that TGF-β signaling is activated in CP by enhanced expression of the ligand and its signaling receptors. This upregulation seems to enhance the expression of CTGF, a downstream target of TGF-β, which subsequently induces fibroblast proliferation and extracellular matrix synthesis. These findings support the hypothesis that the generation of pancreatic fibrosis in CP is mediated by alterations of growth factors and their receptors. 26,36,37 In addition, the intense correlation between CTGF and the degree of fibrosis in CP tissue samples indicates that CTGF might be a critical marker for fibrotic tissue generation in CP.
In summary, the simultaneous presence of CTGF and TGF-β, both strong inducers of collagen synthesis, suggests that these proteins may act synergistically in fibrogenesis. At present, there are no animal models for CP that, in terms of etiology and pathophysiology, simulate the situation in humans. Therefore, no direct proof can be given that activation of TGF-β leads synergistically to CTGF induction in CP. However, experimental studies in other fibrotic disorders strongly support our hypothesis that CTGF is a downstream target of TGF-βs in CP. 3,4,6
Footnotes
Correspondence: Helmut Friess, MD, Dept. of Visceral and Transplantation Surgery, University of Bern, Inselspital, CH-3010 Bern, Switzerland. Email: helmut.friess@insel.ch
Supported by Swiss National Foundation grants (SNF Grant 32-39529 and SNF Grant 32-49494).
Accepted for publication February 9, 1999.
References
- 1.Ryseck RP, Macdonald BH, Mattei MG, Bravo R. Structure, mapping, and expression of fisp-12, a growth factor-inducible gene encoding a secreted cysteine-rich protein. Cell Growth Differ 1991; 2: 225–233. [PubMed] [Google Scholar]
- 2.Bradham DM, Igarashi A, Potter RL, Grotendorst GR. Connective tissue growth factor: a cysteine-rich mitogen secreted by human vascular endothelial cells is related to SRC-induced immediate early gene product CEF-10. J Cell Biol 1991; 114: 1285–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Franzier K, Williams S, Kothapalli D, Klapper H, Grotendorst GR. Stimulation of fibroblast cell growth, matrix production, and granulation tissue formation by connective tissue growth factor. J Invest Dermatol 1996; 107: 404–411. [DOI] [PubMed] [Google Scholar]
- 4.Grotendorst GR, Okochi H, Hayashi N. A novel transforming growth factor β response element controls the expression of the connective tissue growth factor gene. Cell Growth Differ 1996; 7: 469–480. [PubMed] [Google Scholar]
- 5.Igarashi A, Nashiro K, Kikuchi K, Sato S, Ihn H, Grotendorst GR. Significant correlation between connective tissue growth factor gene expression and skin sclerosis from patients with systemic sclerosis. J Invest Dermatol 1995; 105: 280–284. [DOI] [PubMed] [Google Scholar]
- 6.Igarashi A, Okochi H, Bradham DM, Grotendorst GR. Regulation of connective tissue growth factor gene expression in human skin fibroblasts during wound repair. Mol Biol Cell 1993; 4: 637–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oemar BS, Werner A, Garnier J-M, et al. Human connective tissue growth factor is expressed in advanced atherosclerotic lesions. Circulation 1997; 95: 831–839. [DOI] [PubMed] [Google Scholar]
- 8.Lee MS, Gu D, Feng L, et al. Accumulation of extracellular matrix and developmental dysregulation in the pancreas by transgenic production of transforming growth factor-β1. Am J Pathol 1995; 147: 42–52. [PMC free article] [PubMed] [Google Scholar]
- 9.Van Laethem JL, Deviere J, Resibois A, et al. Localization of transforming growth factor β-1 and its latent binding protein in human chronic pancreatitis. Gastroenterology 1995; 108: 1873–1881. [DOI] [PubMed] [Google Scholar]
- 10.Slater SD, Williamson RCN, Foster CS. Expression of transforming growth factor β-1 in chronic pancreatitis. Digestion 1995; 56: 237–241. [DOI] [PubMed] [Google Scholar]
- 11.Bockman DE, Boydston WR, Anderson MC. Origin of tubular complexes in human chronic pancreatitis. Am J Surg 1982; 144: 243–249. [DOI] [PubMed] [Google Scholar]
- 12.Bockman DE, Kennedy RH, Multigner L, Decaro A, Sarles H. Fine structure of the organic matrix of human pancreatic stones. Pancreas 1986; 1: 204–210. [DOI] [PubMed] [Google Scholar]
- 13.Büchler M, Weihe E, Friess H, et al. Changes in peptidergic innervation in chronic pancreatitis. Pancreas 1992; 7: 183–192. [DOI] [PubMed] [Google Scholar]
- 14.Battegay EJ, Raines EW, Seifert RA, Bowen-Pope DF, Ross R. TGF-β induces bimodal proliferation of connective tissue cells via complex control of an autocrine PDGF loop. Cell 1990; 63: 515–524. [DOI] [PubMed] [Google Scholar]
- 15.Friess H, Yamanaka Y, Büchler M, et al. Enhanced expression of transforming growth factor-β isoforms in pancreatic cancer correlates with decreased survival. Gastroenterology 1993; 105: 1846–1856. [DOI] [PubMed] [Google Scholar]
- 16.Friess H, Lu Z, Riesle E, et al. Enhanced expression of TGF-βs and their receptors in human acute pancreatitis. Ann Surg 1998; 227: 95–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chu ML, Myers JC, Bernard MP, Ding JF, Ramirez F. Cloning and characterization of five overlapping cDNAs specific for the human pro alpha 1(I) collagen chain. Nucleic Acids Res 1982; 10: 5925–5934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Riesle E, Friess H, Wagner M, et al. Enhanced expression of TGF-βs following acute edematous pancreatitis in rats suggests a role in pancreatic repair. Gut 1997; 40: 73–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cox KH, Deleon, DV, Augerer LM, Augerer RC. Detection of mRNAs in sea urchin embryos by in situ hybridization using asymmetric RNA probes. Devel Bio 1984; 101: 485–502. [DOI] [PubMed] [Google Scholar]
- 20.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem 1987; 162: 156–159. [DOI] [PubMed] [Google Scholar]
- 21.Friess H, Yamanaka Y, Büchler MW, et al. A subgroup of patients with chronic pancreatitis overexpress the c-erbB-2 protooncogene. Ann Surg 1994; 220: 183–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yamanaka Y, Friess H, Büchler M, et al. Synthesis and expression of transforming growth factor β1, β2, and β3 in the endocrine and exocrine pancreas. Diabetes 1993; 42: 746–756. [DOI] [PubMed] [Google Scholar]
- 23.Graber HU, Müller CF, Vandevelde M, Zurbriggen A. Restricted infection with canine distemper virus leads to down-regulation of myelin gene transcription in cultured oligodendrocytes. Acta Neuropathol 1995; 90: 312–318. [DOI] [PubMed] [Google Scholar]
- 24.Guo XZ, Friess H, Graber HU, et al. KAI1 expression is up-regulated in early pancreatic cancer and decreased in the presence of metastasis. Cancer Res 1996; 56: 4876–4880. [PubMed] [Google Scholar]
- 25.Friess H, Cantero D, Graber H, et al. Enhanced urokinase plasminogen activation in chronic pancreatitis suggests a role in its pathogenesis. Gastroenterology 1997; 113: 902–912. [DOI] [PubMed] [Google Scholar]
- 26.Friess H, Malfertheiner P, Isenmann R, Kühne H, Beger HG, Büchler MW. The risk of the pancreatico-intestinal anastomosis can be predicted preoperatively. Pancreas 1996; 13: 202–208. [PubMed] [Google Scholar]
- 27.Kashiwagi M, Friess H, Uhl W, et al. Phospholipase A2 isoforms are altered in chronic pancreatitis. Ann Surg 1998; 227: 220–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bockman DE. Surgical anatomy of the pancreas and adjacent structures. In: Beger HG, Büchler M, Malfertheiner P, eds. Standards in pancreatic surgery. Heidelberg: Springer-Verlag; 1993: 1–9.
- 29.Oertel JE, Heffess CS, Oertel YC. Pancreas. In: Sternberg SS, ed. Diagnostic surgical pathology. New York: Raven; 1989: 1057–1093.
- 30.Büchler MW, Friess H, Baer HU, Neoptolemos JP. Surgical treatment of chronic pancreatitis: new standards. In: Digestive surgery, vol. 13. Basel: Karger; 1996.
- 31.Gress TM, Müller-Pillasch F, Lerch MM, et al. Balance of expression of genes coding for extracellular matrix proteins and extracellular matrix degrading proteases in chronic pancreatitis. Z Gastroenterol 1994, 32: 221–225. [PubMed] [Google Scholar]
- 32.Friess H, Yamanaka Y, Büchler M, et al. Increased expression of acidic and basic fibroblast growth factors in chronic pancreatitis. Am J Pathol 1994; 144: 117–128. [PMC free article] [PubMed] [Google Scholar]
- 33.Flaumenhaft R, Abe M, Mignatti P, Rifkin DB. Basic fibroblast growth factor beta in endothelial cells: regulation of plasminogen activator activity. J Cell Biol 1992; 118: 901–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yee JA, Yan L, Dominguez JC, Allan EH, Martin TJ. Plasminogen-dependent activation of latent transforming growth factor beta by growing cultures of osteoblast-like cells. J Cell Physiol 1993; 157: 528–534. [DOI] [PubMed] [Google Scholar]
- 35.Klöppel G, Maillet B. Pseudocysts in chronic pancreatitis: a morphological analysis of 57 resection specimens and 9 autopsy pancreata. Pancreas 1991; 6: 266–274. [PubMed] [Google Scholar]
- 36.Bockman DE, Merlino G. Cytological changes in the pancreas of transgenic mice overexpressing transforming growth factor alpha. Gastroenterology 1992; 103: 1883–1892. [DOI] [PubMed] [Google Scholar]
- 37.Ebert M, Kasper HU, Hernenberg S, et al. Overexpression of platelet-derived growth factor (PDGF) chain and type β PDGF receptor in human chronic pancreatitis. Dig Dis Sci 1998; 43: 567–574. [DOI] [PubMed] [Google Scholar]
- 38.Horvath LZ, Friess H, Schilling M, et al. Altered expression of transforming growth factor-βs in chronic renal rejection. Kidney Intl 1996; 50: 489–498. [DOI] [PubMed] [Google Scholar]