

Abstract
Objective
To assess outcome and prognostic factors for survival of adults with Ewing’s sarcoma/primitive neuroectodermal tumor (PNET).
Background
Ewing’s sarcoma/PNET is a disease of childhood rarely seen in adults. Accordingly, there is a relative paucity of published literature pertaining to outcome for adults with this disease.
Methods
Between 1979 and 1996, 37 patients with newly diagnosed Ewing’s sarcoma/PNET were evaluated and treated at the Adult Sarcoma Program at Dana-Farber Cancer Institute and Brigham & Women’s Hospital. Twenty-six patients had localized disease at presentation and 11 had metastatic disease. All but two patients received multiagent chemotherapy. Local treatment consisted of surgery (7 patients), surgery and radiation therapy (19), radiation therapy (6), or no local treatment (5). Median follow-up for living patients was 100 months (range 8 to 199).
Results
The 5-year survival rate for the group overall was 37% ± 9%. The 5-year local control rate was 85% ± 7%. Significant favorable predictors for survival on univariate analysis included localized disease at presentation, primary origin in bone, primary size <8 cm, and a favorable objective response to chemotherapy. Patients with localized disease had a 5-year survival rate of 49% ± 11% compared with 0% for those with metastatic disease at presentation. Multivariate analysis showed three significant independent predictors for death: metastatic disease at presentation, primary origin in extraosseous tissue versus bone, and age 26 years or older.
Conclusion
Adult patients with Ewing’s sarcoma/PNET at highest risk for death are those who are older than 26 years and have metastatic disease or an extraosseous primary tumor. The development of novel therapies should target these high-risk groups.
Ewing’s sarcoma and peripheral neuroectodermal tumor (PNET) are “small round blue cell tumors.” The majority of cases share the cytogenetic translocation t(11;22) (q24;q12), with occasional variations, and a characteristic immunohistochemical staining profile. 1–3 Both these tumors are believed to show neuroectodermal differentiation, albeit in different degree; Ewing’s sarcoma tends to be poorly differentiated, whereas PNET most often shows definite neuroectodermal differentiation. Although once viewed as distinct entities, Ewing’s sarcoma, Askin’s tumor, and PNET are now considered together as members of the Ewing family of tumors. 4,5 As such, they are increasingly grouped together for both treatment and prognostic factor analysis. 6–8
Ewing’s sarcoma/PNET has a marked propensity for systemic spread, and therefore intensive multiagent chemotherapy is the mainstay of treatment. Significant progress has been made in terms of defining active chemotherapy regimens, 9–12 and current multiinstitutional studies are underway to optimize these regimens. Local treatment usually consists of surgery, surgery plus radiation therapy, or radiation therapy alone. The relative efficacies of these local treatments remain controversial.
Most patients with Ewing’s sarcoma/PNET are 10 to 20 years old, 13 and therefore there is a relative paucity of literature relating to outcome for adults with this disease of childhood and adolescence. Some early reports had suggested that adults with Ewing’s sarcoma/PNET have a less favorable outcome than children, 14,15 but a recent study of adult patients from the Royal Marsden Hospital in London has suggested that adults may have a similar outcome to children. 16 The objective of the current study was to evaluate the outcomes and assess prognostic factors for a population of adult patients with Ewing’s sarcoma/PNET seen at our institution.
METHODS
Between 1979 and 1996, 44 patients with Ewing’s sarcoma/PNET were evaluated at the Adult (defined as 18 years or older) Sarcoma Program at Dana-Farber Cancer Institute and Brigham & Women’s Hospital. Seven patients were previously treated at other institutions and were excluded from this analysis because of lack of adequate treatment and follow-up data. The study population therefore comprises 37 adult patients with Ewing’s sarcoma/PNET. In all cases, the pathology was reviewed at Brigham & Women’s Hospital and diagnoses were confirmed by immunohistochemistry, electron microscopy, and cytogenetic analysis (when available). The diagnoses included are Ewing’s sarcoma of bone, extraosseous Ewing’s sarcoma, and PNET.
The standard patient evaluation included history and physical examination, complete blood count and serum chemistries, computed tomography (CT) and/or magnetic resonance (MR) scan of the primary site, chest CT scan, bone scan, and bone marrow biopsy. Based on these studies, patients were classified as having localized or metastatic disease. In addition, the size of the largest dimension of the primary tumor, as seen on CT or MR scan, was recorded. Before the treatment of any patient, informed consent was obtained.
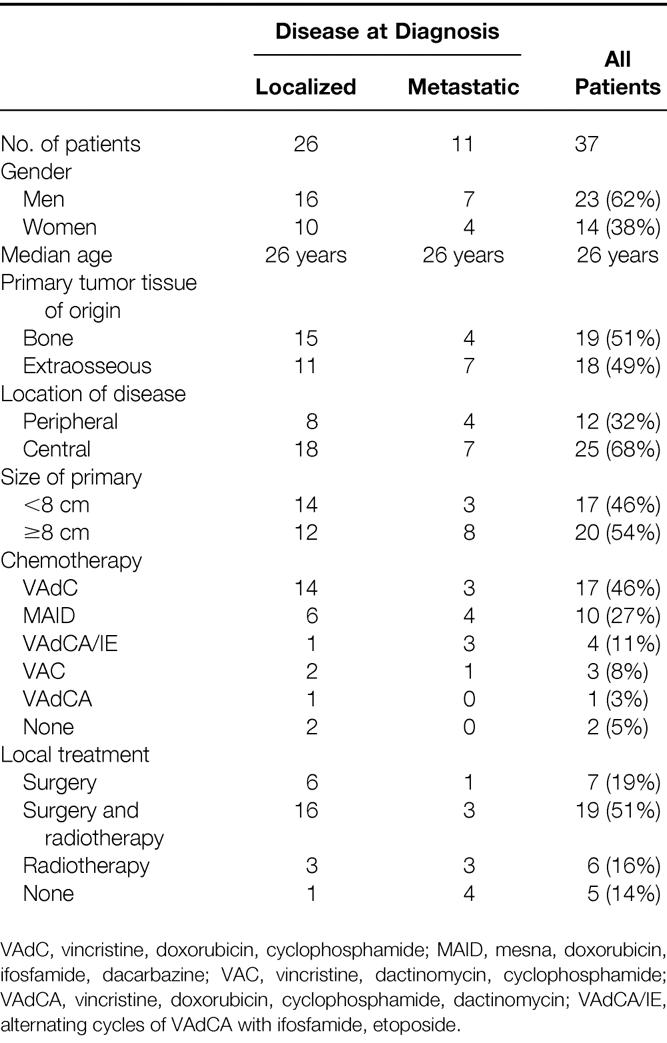
Patient characteristics are shown in Table 1. Among the 37 patients, 26 had localized disease and 11 had metastatic disease. The median age was 26 years (range 18 to 46 years). Nineteen patients had primary tumor origin in bone (51%) and 18 had an extraosseous primary tumor (49%). The sites of disease were grouped as peripheral or central. Peripheral disease was defined as all extremity lesions. There were 12 patients with the following peripheral sites: thigh (4 patients), lower leg (4), ankle (2), and foot (2). Central disease was defined as disease involving the head and neck, trunk, thorax, retroperitoneum, or pelvis. There were 25 patients with the following central sites of disease: chest wall (10 patients), retroperitoneum (5), flank/back (3), pelvis (3), shoulder (1), head and neck (1), groin (1), and mediastinum (1). The chest wall was the single most common site of involvement (ten patients, 27%). The size of the primary tumor ranged from 3 to 20 cm, with a median size of 8 cm. Among the 11 patients with metastatic disease, the sites of metastases were the lung in 9 patients and bone for 2 patients.
Table 1. Patient and Treatment Characteristics for Adults with Ewing’s Sarcoma/PNET (n = 37)
Systemic Treatment
Chemotherapy was recommended for all patients. Thirty-five of the 37 patients (95%) received chemotherapy, and 2 patients refused systemic treatment. (These two patients underwent surgery for localized disease of the ankle and foot, and subsequently died of distant disease at 18 and 33 months, respectively.) The chemotherapy regimens delivered included:
• VAdC (vincristine, doxorubicin, cyclophosphamide) for 17 patients
• MAID (mesna, doxorubicin, ifosfamide, dacarbazine) for 10 patients
• VAdCA/IE (VAdCA alternating with ifosfamide, etoposide) for 4 patients
• VAC (vincristine, dactinomycin, cyclophosphamide) for 3 patients
• VAdCA (vincristine, doxorubicin, cyclophosphamide, dactinomycin) for 1 patient.
Of the 35 patients who received chemotherapy, 25 could be evaluated for response. Ten patients with localized disease underwent resection of their primary tumor before initiation of chemotherapy and therefore could not be evaluated for response to chemotherapy.
Local Treatment
Surgery consisted of biopsy only for 11 patients, marginal excision for 1, wide excision for 22, radical compartmental excision for 2, and amputation (transmetatarsal) for 1. Radiation therapy was delivered to 25 patients (68%). Six patients received radiation therapy alone, and 19 received irradiation in conjunction with surgical resection (before surgery for 1, after surgery for 18). The median dose was 54 Gy (range 40 to 64.5). Five patients received no local treatment after initial chemotherapy. Among the 26 patients with localized disease, the local treatment consisted of surgery for 6 (5 wide excisions, 1 transmetatarsal amputation), surgery and irradiation for 16 (1 marginal excision, 13 wide excisions, 2 radical excisions), and irradiation for 3. One patient with localized disease of the retroperitoneum received neither surgery nor irradiation; she had a complete response to MAID chemotherapy and went on to receive a bone marrow transplant.
Patients were followed every 3 to 4 months for the first 3 years and every 6 months thereafter. Data were collected on local control, disease status, survival, and cause of death. The median follow-up time for all patients was 27 months (range 7 to 199), and the median follow-up for living patients was 100 months (range 8 to 199).
Of the 17 living patients, 3 were followed for <2 years and 8 were followed for <5 years.
Statistics
The following clinical and treatment-related factors were considered in the analysis of potential prognostic factors: extent of disease at presentation, age, gender, primary origin in bone versus extraosseous tissue, site of disease, size of the primary tumor, and response to chemotherapy. For the continuous variables, age and size of primary tumor, the median value was used as the breakpoint for statistical analyses. Actuarial local control and overall survival curves were calculated from date of diagnosis and estimated by the Kaplan–Meier product limit method. 17 The log rank test was used to compare curves for the univariate analysis. 18 The Cox proportional hazards model was used to assess independent prognostic factors for survival. 19
RESULTS
Response to Chemotherapy
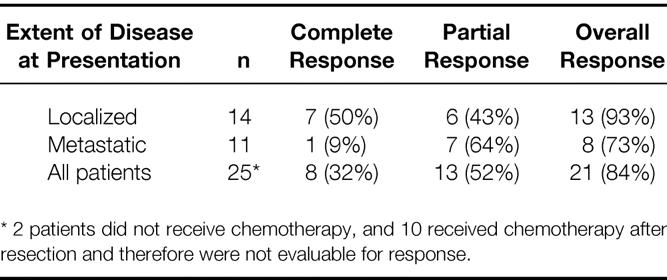
Twenty-five patients could be evaluated for response to chemotherapy. A complete response was noted in 8 patients (32%) and a partial response in 13 patients (52%), yielding an overall response rate of 84%. As shown in Table 2, the chemotherapy response rate was higher among patients with localized disease (93%), with a 50% complete response rate, than among those with metastatic disease (73%), with only a 9% complete response rate.
Table 2. Chemotherapy Response by Extent of Disease at Presentation (n = 25)
Patterns of Failure
Thirty-five patients could be evaluated for local and distant recurrence (two patients had initial progression of disease). Overall, there were four local recurrences (11%). All four represented the first site of failure, and one of the four patients had a synchronous distant relapse. All four of these patients had received chemotherapy. Two of these patients (with tumors of the chest wall and retroperitoneum) underwent a wide excision with close margins followed by irradiation (55 and 55.8 Gy, respectively). One patient with a 7-cm retroperitoneal tumor had a complete response to chemotherapy, refused surgery, and received 54 Gy. The final patient with a local recurrence received irradiation (60 Gy) for an 11-cm lower leg primary. The actuarial 5-year local control rate was 85% ± 7%, and the times to local recurrence for these four patients were 6, 7, 17, and 24 months. All four patients died of distant relapse. There were too few cases to perform statistical analyses assessing predictors for local recurrence.
Fourteen patients had distant metastases as the first site of failure (40%). The sites of distant relapse included the lung in 10 patients, bone in 2, bone and pelvis in 1, and unknown in 1. The median time to distant relapse was 13 months (range 5 to 38).
Survival
Overall, 17 (46%) patients remain alive and 20 (54%) have died. Twelve of the 26 patients with localized disease died; the cause of death was Ewing’s sarcoma/PNET for 11 and complications from bone marrow transplantation for 1 patient, who was without evidence of disease at the time of death. Among the 11 patients with metastatic disease, 8 died of Ewing’s sarcoma/PNET and 3 are alive with disease. The 5-year actuarial survival rate for all patients was 37% ± 9%.
The univariate analysis of prognostic factors for survival is summarized in Table 3. Statistically significant favorable predictors for survival included localized disease at presentation, primary tumor origin in bone, size of the primary <8 cm, and response to chemotherapy. The poorest survival rates were seen for patients with metastatic disease and for those whose disease did not respond to chemotherapy. As shown in Figure 1, patients with localized disease had a 5-year survival rate of 49% ± 11% compared with 0% for patients with metastatic disease (p = 0.002). For patients with metastatic disease at presentation, there have been no survivors longer than 4 years. Patients with a primary bone tumor had a 5-year survival rate of 52% ± 12% compared with 21% ± 12% for those with a primary extraosseous tumor (p = 0.01, Fig. 2). The 5-year survival rate for patients with a primary tumor <8 cm was 59% ± 13% compared with 20% ± 10% for those with tumors ≥8 cm (p = 0.03, Fig. 3). As depicted in Figure 4, the 5-year survival rate for patients who had a complete response to chemotherapy was 56% ± 20% compared with 35% ± 15% for those with a partial response; for those whose disease did not respond to chemotherapy, there were no survivors beyond 3 years (p = 0.01).
Table 3. Significant Predictors for Survival on Univariate Analysis (n = 37)
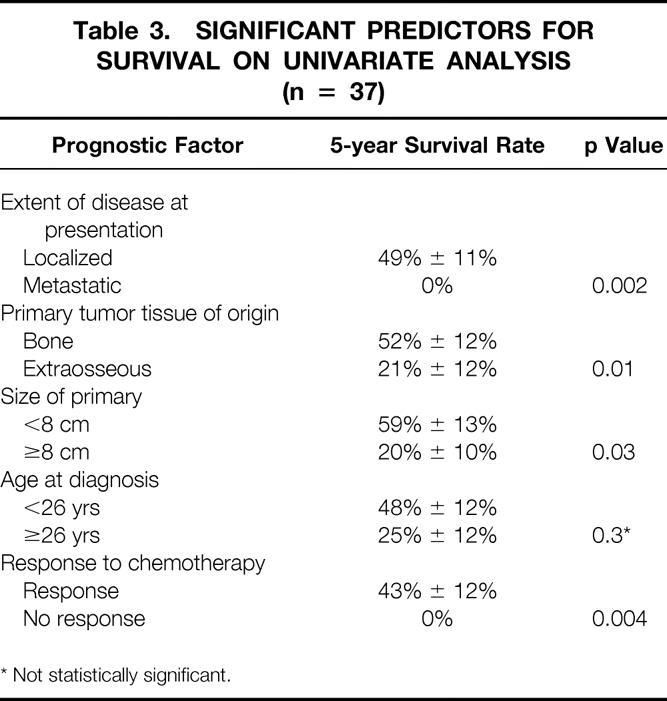
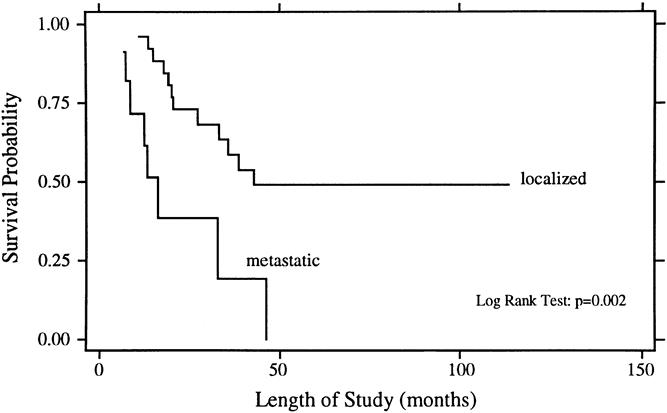
Figure 1. Overall survival rates for patients with localized (n = 26) and metastatic (n = 11) Ewing’s sarcoma/PNET at presentation.
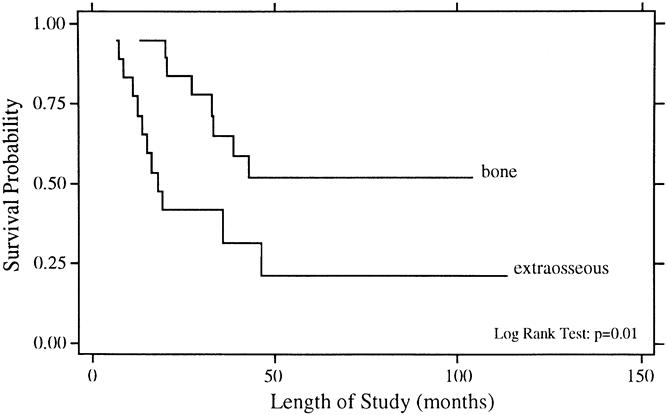
Figure 2. Overall survival rates for patients with primary tumor origin in bone (n = 19) and extraosseous tissue (n = 18).
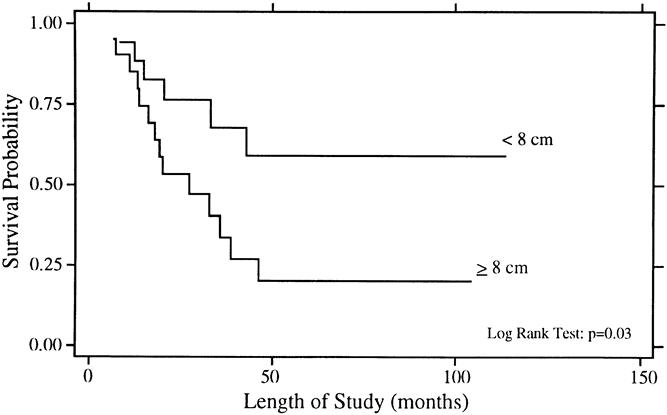
Figure 3. Overall survival rates for patients with size of primary <8 cm (n = 17) and ≥8 cm (n = 20).
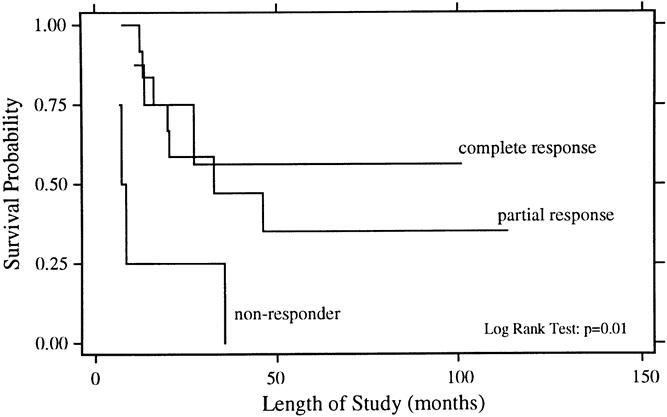
Figure 4. Overall survival rates for patients with a complete response to chemotherapy (n = 8), a partial response to chemotherapy (n = 13), and no response to chemotherapy (n = 4).
As shown in Figure 5, it appeared that patients younger than 26 years had a better 5-year survival rate than older adults, but this comparison was not statistically significant on univariate analysis (48% vs. 25%, p = 0.3). Further, gender and peripheral versus central site of disease were not significant predictors for survival.
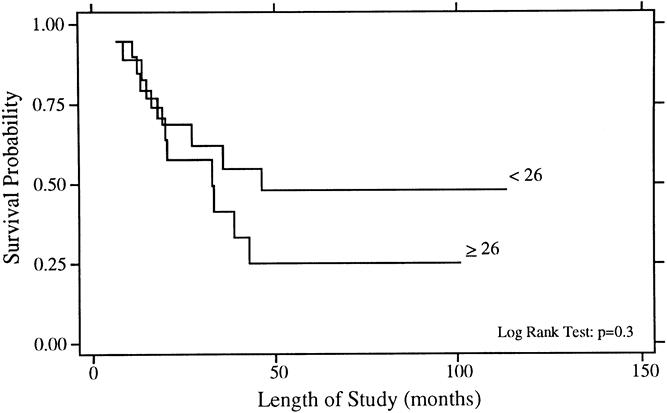
Figure 5. Overall survival rates for patients younger than 26 years (n = 19) and 26 years or older (n = 18).
The multivariate analysis is summarized in Table 4. Cox regression showed the following three factors to be significantly associated with death: metastatic disease at presentation (hazard ratio [HR] 3.4, p = 0.01), primary extraosseous tumor (HR 5.0, p = 0.005), and age 26 years or older (HR 3.7, p = 0.02). Size of the primary tumor was no longer significant in the multivariate model (HR 1.8, p = 0.3), and it appeared that tumor size and metastatic disease at presentation were correlated. Only 3 of 17 patients (18%) with tumors <8 cm had metastatic disease, whereas 8 of the 20 patients (40%) with tumors >8 cm had metastatic disease at initial presentation. Response to chemotherapy was not included in the multivariate analysis because the number of evaluable patients was too small (10 of the 26 patients with localized disease underwent initial surgery and could not be evaluated for response to chemotherapy).
Table 4. Significant Predictors for Death on Multivariate Analysis (n = 37)

Patients With Localized Disease at Presentation
The patient and treatment characteristics for the 26 patients with localized disease at presentation are depicted in Table 1. All but two patients received chemotherapy, and the local therapy consisted of surgery and irradiation for 16 patients, surgery for 6, irradiation for 3, and biopsy only for 1. Patients with localized disease had a 93% response rate to chemotherapy. Fifty percent had a complete response and 43% had a partial response (see Table 2). The patterns of first failure were an isolated local recurrence for three patients, a synchronous local and distant recurrence for one patient, and an isolated distant recurrence for seven patients. Twelve of the 26 patients (46%) with localized disease are dead; the cause of death was Ewing’s sarcoma/PNET in 11 and complications from bone marrow transplantation in 1 patient, who was without evidence of disease at the time of death. As shown in Figure 1, the 5-year actuarial survival rate for the patients with localized disease was 49% ± 11%. Univariate analysis showed borderline statistical significance favoring size of the primary tumor <8 cm (p = 0.05) and age younger than 26 years (p = 0.09), whereas there were no significant differences for primary tumor tissue of origin or location of disease.
Multivariate analysis showed a significant adverse effect for primary extraosseous tumor (HR 6.8, p = 0.01) and age 26 years or older (HR 5.0, p = 0.03). Primary tumor size ≥8 cm was associated with borderline statistical significance (HR 3.1, p = 0.07).
DISCUSSION
The 5-year overall survival rate for this series of adult patients with Ewing’s sarcoma/PNET was 37%, and the 5-year local control rate was 85%. Significant adverse predictors for survival included metastatic disease at presentation, primary extraosseous tumor, and age 26 years or older.
The survival outcome for this series of adult patients with Ewing’s sarcoma/PNET treated with combined-modality therapy is in the lower end of the range of survival rates reported for children. The 5-year survival rate for patients with localized disease in this series was 49%, and 5-year survival rates reported in the literature for pediatric patients with localized Ewing’s sarcoma/PNET range from 42% to 80%. 9,10,20–26 Most patients with Ewing’s sarcoma/PNET are children or teenagers; accordingly, the majority of the literature relates to outcome and prognostic factors for younger patients with this disease. There has been an impression that adults with childhood malignancies tend to fare worse than their pediatric counterparts. 14,15,27 However, a recent series from the Royal Marsden Hospital of 59 adults with Ewing’s sarcoma/PNET showed a 5-year survival rate for all patients of 38%, and for patients with localized disease of 52%. 16 (These rates are strikingly similar to those of the current series: the 5-year survival rate is 37% for all patients and 49% for those with localized disease.)
As stated above, the results from both of these series of adult patients fall in the lower end of the range of results reported for pediatric patients. The authors of the Royal Marsden report interpreted their findings as comparable to those seen for children. However, 23 of the 59 (39%) cases were in patients 19 years or younger, thus overlapping with the teenage category. In any case, because neither the current nor the Royal Marsden study directly compared the outcomes for adults and children, any such conclusions comparing the two age groups remain speculative.
The published data for adults with Ewing’s sarcoma/PNET are otherwise quite limited. A previous report of adult patients with Ewing’s sarcoma from this institution comprised only 16 patients. 14 Further, half of the group included patients with unfavorable characteristics: five had refractory or recurrent disease and an additional three had metastatic Ewing’s sarcoma/PNET. Seven of these eight patients died of disease, whereas only two of the eight patients with previously untreated localized Ewing’s sarcoma/PNET died of disease. These results are consistent with the known prognostic factors for Ewing’s sarcoma. However, as appropriately stated in the original report, the numbers were too small to draw any firm conclusions regarding outcome for adults versus children. A study of adult patients with Ewing’s sarcoma from M.D. Anderson included 34 patients with localized disease. 15 Of these, a crude rate of 38% of patients remained without disease progression. Note was made that all patients with pelvic primaries died of disease, but a more detailed analysis of potential prognostic factors was not performed. On the whole, the results appear to indicate a poorer-than-expected outcome for these adults, but the comparability of these data to other series is not clear.
Age has been studied as a potential prognostic factor in reports comprised largely of children, with conflicting results. In the present study, older age was not a statistically significant prognostic factor for survival on univariate analysis, but it became a significant adverse predictor on multivariate analysis. A statistically significant adverse outcome for older patients has also been reported for patients older than 25 years by Kinsella et al 25 and for patients older than 15 years by Picci et al 28 and Nesbit et al. 9 Further, Burgert et al 10 reported a nonsignificant trend for increased distant metastases with increasing age for patients enrolled in IESS II. Conversely, Wilkins et al 20 found no association between age and outcome, nor did Verrill et al 16 in their series of adult patients.
Measurable metastatic disease at presentation, not surprisingly, was associated with a fourfold increased risk for death. Several other authors have also found metastatic disease to confer poor survival. 6,16,25,29–32 This finding has prompted investigations of high-dose chemotherapy with stem cell support for patients with metastatic disease. 33–35 Moreover, in this series, among patients with metastatic disease, there were no survivors beyond 4 years. Some reports have shown equally poor survival rates for patients with metastatic disease at diagnosis, 16,30,31 whereas others have shown 3- and 5-year survival rates in the range of 30% to 50%. 6,20,29,36 The results of the current series suggest that adults with metastatic disease at diagnosis may fare worse than children with metastatic disease. This may be related to the fact that children have more bone marrow reserve than adults and therefore can better tolerate prolonged, intensive chemotherapy.
There is also a consistency in the literature with respect to the adverse effect of large tumor size. 16,21,23,28,37,38 Although tumor bulk has been defined in varying ways (>100 ml, >200 ml, >500 cc, >8 cm, >10 cm), it is clear that increasing tumor burden is associated with an increased risk of death. Hayes et al 23 used a cut-off value of 8 cm, which is similar to this analysis. In this series, size ≥8 cm was associated with an adverse outcome on univariate analysis, but it was no longer statistically significant on multivariate analysis. The hazard ratio on multivariate analysis still favored small primary tumor size, and the nonsignificant p value was probably a function of small patient numbers and a correlation between tumor size and the presence of metastatic disease at presentation.
The location of the primary tumor was not a significant predictor for survival in this series. Some reports have shown central or pelvic disease to be an adverse prognostic factor for survival 9,21,22,25 ; other reports have not. 23,39,40 The reason for these discordant results may be that size and location of disease are often covariables (e.g., central or pelvic tumors are often quite large at the time of diagnosis). In a large study from Europe that analyzed both site and size, site was not a significant predictor. 40
Response to chemotherapy was another factor in this analysis with a profound effect on survival. Among patients whose disease did not respond to chemotherapy, there were no survivors beyond 3 years. This is supported by several groups who have demonstrated that both radiologic and pathologic response to chemotherapy is associated with improved survival. 16,28,41,42
Finally, in this study, patients with primary bone tumors had a statistically better survival than those with extraosseous tumors on univariate and multivariate analyses. It is not intuitively clear why this would be true. The literature does not contain many reports assessing the prognostic importance of bone versus extraosseous tissue of origin, and this should be addressed in future studies. Interestingly, the series of adult patients reported by the Royal Marsden Hospital showed no statistically significant difference in outcome based on tissue of origin. 16
The 5-year local recurrence rate in this series was 15%; this is comparable to the local recurrence rates reported by other investigators, which range from 9% to 21%. 9,10,12,21–25 Local treatment for the patients in the current study consisted of surgery, irradiation, or a combination of both. There were only four local recurrences, occurring in two patients treated with wide excision and irradiation and two treated with irradiation alone. Given the small number of events, it was not possible to analyze potential prognostic factors for local recurrence. Several reports suggest that a higher local control rate is achieved with surgery or surgery plus irradiation compared with irradiation alone. 20,21,26,43 However, many of these reports are prone to bias in that the patients treated with irradiation were the ones with the larger, less favorable tumors. Others have demonstrated local control rates after irradiation to be similar to those achieved with surgery. 24,25 No prospective randomized trial has been performed comparing local treatment options.
Because there does not appear to be a large difference in outcome, many recommend that the choice of local therapy should be made with an emphasis on minimizing side effects. Generally, if a resection can be performed with minimal functional morbidity, this option is preferred to irradiation because of the concern about late radiation-induced side effects (most notably second tumors in the irradiated field). 22,44,45
In conclusion, we have found that older adults (26 years or older) with Ewing’s sarcoma/PNET have survival rates inferior to those of younger adults. Further, we have speculated that the effect of age on prognosis may be a continuum, and that children may have more favorable outcomes than adults. The reasons for this observation are not known. It may be that Ewing’s sarcoma/PNET is biologically a different disease in older adults. Alternatively, adults may not have enough bone marrow reserve to tolerate the long chemotherapy regimens that have produced good results for children. In addition to older age, metastatic disease at diagnosis and primary extraosseous tumor were adverse predictors for survival. These findings may contribute to the design of future studies; patients who meet these high-risk criteria may be appropriate candidates for novel therapeutic approaches using strategies such as stem cell transplantation, immunomodulation, and newer systemic agents.
Footnotes
Correspondence: Samuel Singer, MD, Division of Surgical Oncology, Brigham & Women’s Hospital, 75 Francis St., Boston, MA 02115.
Presented in part at the American Society of Clinical Oncology Meeting, Philadelphia, Pennsylvania, May 18–21, 1996.
Accepted for publication December 29, 1999.
References
- 1.Whang-Peng J, Triche TJ, Knutsen T, et al. Chromosome translocation in peripheral neuroepithelioma. N Engl J Med 1984; 311: 584–585. [DOI] [PubMed] [Google Scholar]
- 2.Turc-Carel C, Philip I, Berger MP, Lenoir GM. Chromosome translocation in Ewing’s sarcoma. N Engl J Med 1983; 309: 497–498. [Google Scholar]
- 3.Unni KK, Inwards CY. Osteoarticular tumors. In Fletcher CDM (ed). Diagnostic histopathology of tumors. London: Churchill Livingstone; 1995: 1104–1106.
- 4.Delattre O, Zucman J, Melot T, et al. The Ewing family of tumors: a sub-group of small-round-cell tumors defined by specific chimeric transcripts. N Engl J Med 1994; 331: 294–299. [DOI] [PubMed] [Google Scholar]
- 5.Grier HE. The Ewing family of tumors: Ewing’s sarcoma and primitive neuroectodermal tumors. Pediatr Clin North Am 1997; 44: 991–1004. [DOI] [PubMed] [Google Scholar]
- 6.Miser JS, Kinsella TJ, Triche TJ, et al. Treatment of peripheral neuroepithelioma in children and young adults. J Clin Oncol 1987; 5: 1752–1758. [DOI] [PubMed] [Google Scholar]
- 7.Bruckner JD, Conrad EU, Malo L, et al. Primitive neuroectodermal tumor and extraosseous Ewing’s sarcoma: a comparison of clinical features and management. Proc Am Soc Clin Oncol 1992; 11: 414. [Google Scholar]
- 8.Carter RL, Al-Sam SZ, Corbett RP, Clinton S. A comparative study of immunohistochemical staining for neuron-specific enolase, protein gene product 9.5, and S-100 protein in neuroblastoma, Ewing’s sarcoma, and other round cell tumors in children. Histopathology 1990; 16: 461–467. [DOI] [PubMed] [Google Scholar]
- 9.Nesbit ME, Gehan EA, Burgert EO, et al. Multimodal therapy for the management of primary, nonmetastatic Ewing’s sarcoma of bone: a long-term follow-up of the first intergroup study. J Clin Oncol 1990; 8: 1664–1674. [DOI] [PubMed] [Google Scholar]
- 10.Burgert EO, Nesbit ME, Garnsey LA, et al. Multimodal therapy for the management of nonpelvic, localized Ewing’s sarcoma of bone: intergroup study IESS II. J Clin Oncol 1990; 8: 1514–1524. [DOI] [PubMed] [Google Scholar]
- 11.Grier H, Krailo M, Link M, et al. Improved outcome in non-metastatic Ewing’s sarcoma and PNET of bone with the addition of ifosfamide and etoposide to vincristine, Adriamycin, cyclophosphamide, and actinomycin: a Children’s Cancer Group and Pediatric Oncology Group report. Proc Am Soc Clin Oncol 1994; 13: 421. [Google Scholar]
- 12.Dunst J, Sauer R, Burgers JMV, et al. Radiation therapy as local treatment in Ewing’s sarcoma: results of the Cooperative Ewing’s Sarcoma Studies CESS 81 and CESS 86. Cancer 1991; 67: 2818–2825. [DOI] [PubMed] [Google Scholar]
- 13.Young JL Jr, Miller RW. Incidence of malignant tumors in US children. J Pediatr 1975; 86: 254–258. [DOI] [PubMed] [Google Scholar]
- 14.Siegel RD, Ryan LM, Antman KH. Adults with Ewing’s sarcoma: an analysis of 16 patients at the Dana-Farber Cancer Institute. Am J Clin Oncol 1988; 11: 614–617. [PubMed] [Google Scholar]
- 15.Sinkovics JG, Plager C, Ayala AG, et al. Ewing sarcoma: its course and treatment in 50 adult patients. Oncology 1980; 37: 114–119. [DOI] [PubMed] [Google Scholar]
- 16.Verrill MW, Judson IR, Harmer CL, et al. Ewing’s sarcoma and primitive neuroectodermal tumor in adults: are they different from Ewing’s sarcoma and primitive neuroectodermal tumor in children? J Clin Oncol 1997; 15: 2611–2621. [DOI] [PubMed] [Google Scholar]
- 17.Kaplan El, Meier P. Nonparametric estimation from incomplete observations. J Am Stat Assoc 1958; 53: 457–481. [Google Scholar]
- 18.Peto R, Peto J. Asymptotically efficient rank invariant test procedures (with discussion). J Roy Stat Soc [A] 1972; 135: 185–206. [Google Scholar]
- 19.Cox DR. Regression models and life tables (with discussion). J Roy Stat Soc [B] 1972; 34: 187–220. [Google Scholar]
- 20.Wilkins RM, Pritchard DJ, Burgert EO, Unni KK. Ewing’s sarcoma of bone: experience with 140 patients. Cancer 1986; 58: 2551–2555. [DOI] [PubMed] [Google Scholar]
- 21.Sailer SL, Harmon DC, Mankin HJ, et al. Ewing’s sarcoma: surgical resection as a prognostic factor. Int J Radiat Oncol Biol Phys 1988; 15: 43–52. [DOI] [PubMed] [Google Scholar]
- 22.Bacci G, Toni A, Avella M, et al. Long-term results in 144 localized Ewing’s sarcoma patients treated with combined therapy. Cancer 1989; 63: 1477–1486. [DOI] [PubMed] [Google Scholar]
- 23.Hayes FA, Thompson EI, Meyer WH, et al. Therapy for localized Ewing’s sarcoma of bone. J Clin Oncol 1989; 7: 208–213. [DOI] [PubMed] [Google Scholar]
- 24.Marcus RB, Cantor A, Heare TC, et al. Local control and function after twice-a-day radiotherapy for Ewing’s sarcoma of bone. Int J Radiat Oncol Biol Phys 1991; 21: 1509–1515. [DOI] [PubMed] [Google Scholar]
- 25.Kinsella TJ, Miser JS, Waller B, et al. Long-term follow-up of Ewing’s sarcoma of bone treated with combined modality therapy. Int J Radiat Oncol Biol Phys 1991; 20: 389–395. [DOI] [PubMed] [Google Scholar]
- 26.Dunst J, Jurgens H, Sauer R, et al. Radiation therapy in Ewing’s sarcoma: an update of the CESS 86 trial. Int J Radiat Oncol Biol Phys 1995; 32: 919–930. [DOI] [PubMed] [Google Scholar]
- 27.Prestidge BR, Donaldson SS. Treatment results among adults with childhood tumors: a 20-year experience. Int J Radiat Oncol Biol Phys 1989; 17: 507–514. [DOI] [PubMed] [Google Scholar]
- 28.Picci P, Bohling T, Bacci G, et al. chemotherapy-induced tumor necrosis as a prognostic factor in localized Ewing’s sarcoma of the extremities. J Clin Oncol 1997; 15: 1553–1559. [DOI] [PubMed] [Google Scholar]
- 29.Hayes FA, Thompson EI, Parvey L, et al. Metastatic Ewing’s sarcoma: remission induction and survival. J Clin Oncol 1987; 5: 1199–1204. [DOI] [PubMed] [Google Scholar]
- 30.Jurgens H, Bier V, Harms D, et al. Malignant peripheral neuroectodermal tumors. A retrospective analysis of 42 patients. Cancer 1988; 61: 349–357. [DOI] [PubMed] [Google Scholar]
- 31.Marina NM, Etcubanas E, Parham DM, et al. Peripheral primitive neuroectodermal tumor (peripheral neuroepithelioma) in children. A review of the St. Jude experience and controversies in diagnosis and management. Cancer 1989; 64: 1952–1960. [DOI] [PubMed] [Google Scholar]
- 32.Kushner BH, Hajdu SI, Gulati SC, et al. Extracranial primitive neuroectodermal tumors, the Memorial Sloan-Kettering Cancer Center experience. Cancer 1991; 67: 1825–1829. [DOI] [PubMed] [Google Scholar]
- 33.Burdach S, Jurgens H, Peters C, et al. Myeloablative radiochemotherapy and hematopoietic stem-cell rescue in poor-prognosis Ewing’s sarcoma. J Clin Oncol 1993; 11: 1482–1488. [DOI] [PubMed] [Google Scholar]
- 34.Michon J, Hartmann O, Demeocq F, et al. Consolidation with busulfan and melphalan followed by blood progenitor cell graft in children and young adults with high-risk Ewing’s sarcoma: a report of the French Society of Pediatric Oncology. Proc Am Soc Clin Oncol 1994; 13: 414. [Google Scholar]
- 35.Ladenstein R, Lasset C, Pinkerton R, et al. Impact of megatherapy in children with high-risk Ewing’s tumours in complete remission: a report from the EBMT Solid Tumour Registry. Bone Mar Trans 1995; 15: 697–705. [PubMed] [Google Scholar]
- 36.Cangir A, Vietti TJ, Gehan EA, et al. Ewing’s sarcoma metastatic at diagnosis. Results and comparisons of two intergroup Ewing’s sarcoma studies. Cancer 1990; 66: 887–893. [DOI] [PubMed] [Google Scholar]
- 37.Gobel V, Jurgens H, Etspuler G, et al. Prognostic significance of tumor volume in localized Ewing’s sarcoma of bone in children and adolescents. J Cancer Res Clin Oncol 1987; 113: 187–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rosen G, Caparros B, Nirehberg A, et al. Ewing’s sarcoma: ten-year experience with adjuvant chemotherapy. Cancer 1981; 47: 2204–2213. [DOI] [PubMed] [Google Scholar]
- 39.Evans RG, Nesbit ME, Gehan EA, et al. Multimodal therapy for the management of localized Ewing’s sarcoma of pelvic and sacral bones: A report from the second Intergroup study. J Clin Oncol 1991; 9: 1173–1180. [DOI] [PubMed] [Google Scholar]
- 40.Sauer R, Jurgens H, Burgers J, et al. Prognostic factors in the treatment of Ewing’s sarcoma. The Ewing’s Sarcoma Study Group of the German Society of Paediatric Oncology CESS 81. Radiother Oncol 1987; 10: 101–110. [DOI] [PubMed] [Google Scholar]
- 41.Meyers PA, Wunder J, Healey JM, et al. Ewing’s sarcoma/primitive neuroectodermal tumor of bone: histological response to preoperative chemotherapy predicts event-free survival. Proc Am Soc Clin Oncol 1995; 14: 441. [Google Scholar]
- 42.Oberlin O, Patte C, Demeocq F, et al. The response to initial chemotherapy as a prognostic factor in localized Ewing’s sarcoma. Eur J Cancer Clin Oncol 1985; 21: 463–467. [DOI] [PubMed] [Google Scholar]
- 43.Pritchard DJ, Dahlin DC, Dauphine RT, et al. Ewing’s sarcoma: a clinicopathological and statistical analysis of patients surviving five years or longer. J Bone Joint Surg 1975; 57A: 10–16. [PubMed] [Google Scholar]
- 44.Tucker MA, D’Angio GJ, Boice JD, et al. Bone sarcomas linked to radiotherapy and chemotherapy in children. N Engl J Med 1987; 317: 588–593. [DOI] [PubMed] [Google Scholar]
- 45.Kuttesch JF, Wexler LH, Marcus RB, et al. Second malignancies after Ewing’s sarcoma: radiation dose-dependency of secondary sarcomas. J Clin Oncol 1996; 14: 2818–2825. [DOI] [PubMed] [Google Scholar]