

Abstract
Objective
To evaluate the impact of a nonstandard ventilation strategy on survival in congenital diaphragmatic hernia (CDH).
Background
Despite recent advances, including nitric oxide, CDH remains an unsolved problem with a mortality rate of 35% to 50%. Hyperventilation and alkalization remain common therapies.
Methods
In 1992, the authors prospectively abandoned hyperventilation and alkalization. Patients are lightly sedated and ventilated with the lowest pressure providing adequate chest movement, and the rate is set to patient comfort. Nitric oxide and extracorporeal membrane oxygenation (ECMO) are reserved for life-threatening instability. Surgical repair is delayed 1 to 5 days. Sixty consecutive patients are compared with 29 previous patients treated with hyperventilation and alkalization, 13 before and 16 after the availability of ECMO.
Results
Overall, 47 of 60 patients (78%) in study era 3 survived compared with 2 of 13 (15%) in the hyperventilation era and 7 of 16 (44%) in the hyperventilation/ECMO era (p < 0.0001). The disease severity and the incidence of associated anomalies did not differ between groups. To compare management strategies, patients who had treatment withheld because of lethal associated conditions were then removed from analysis. Peak inspiratory pressure and arterial pH were lower (p < 0.0001) and PaCO2 was higher (p < 0.05) in era 3 than in the previous eras. The rate of pneumothorax (1.9%) decreased (p < 0.0001). In era 3, survival was 47 of 53 (89%) treated patients, and 23 of 25 inborn patients with isolated CDH survived (92%).
Conclusions
Nonstandard ventilatory support of patients with CDH has led to significantly improved survival rates. This study sets a survival benchmark and strongly suggests the negative effects of hyperventilation and alkalization.
In 1963, the premature son of President John F. Kennedy died from relatively mild respiratory distress syndrome. In the ensuing 36 years, there have been dramatic changes in neonatal care, including the use of infant warmers, total parenteral nutrition, positive pressure ventilators, exogenous surfactant, extracorporeal membrane oxygenation (ECMO), nitric oxide, and refinements in both anesthetic and surgical techniques for newborns requiring surgical intervention. During this time, hyperventilation and alkalosis were shown to improve arterial oxygenation in many infants with hypoxemia resulting from persistent pulmonary hypertension, 1 and this became a deeply ingrained therapeutic strategy. With the advent of ECMO in the late 1970s, respiratory deaths in term infants with most forms of respiratory distress were nearly eliminated.
Congenital diaphragmatic hernia (CDH) proved to be an exception, however, and continued to carry a high mortality rate. Treatment of hypoxemia in these infants with methods accepted as the standard of care for other conditions frequently failed, even with the addition of ECMO. As reported in 1998 by the CDH study group, comprising participating centers that voluntarily report their treatment results, the survival rate remains low at 63%. 2
Fundamental physiologic problems confront infants with CDH. These include pulmonary hypoplasia, 3 pulmonary immaturity, 4,5 and pulmonary hypertension, 6–8,9 because the pulmonary vascular bed is both smaller and more muscularized than normal. 10 Treatments aimed at controlling pulmonary hypertension failed to improve results, leading to the commonly held opinion that pulmonary parenchymal mass is inadequate for survival in many infants with CDH. This conclusion stimulated experimentation with fetal surgery 11–15 and neonatal lung transplantation. 16 ECMO was used as a rescue therapy for patients in whom standard ventilatory techniques failed, when initial gas exchange suggested there was adequate lung parenchyma for long-term survival. 17–19
The impetus to change our treatment strategy was dissatisfaction with clinical results using hyperventilation, both with and without ECMO. Wung et al, in their landmark article in 1985, 20 showed the potential benefits of avoiding hyperventilation in babies without CDH who had pulmonary hypertension. One of us (DWK) trained in pediatric surgery at Columbia University from 1990 to 1992 and studied this ventilatory technique. Based on this experience, we postulated that hyperventilation and alkalosis were harmful to patients with CDH and that survival could be improved by eliminating those negative effects. We adopted and extended the approach of Wung et al and herein report the results of such treatment for patients with CDH at the University of Florida.
MATERIALS AND METHODS
Study Population
All newborns with CDH, symptomatic in the first 6 hours of life and cared for since December 1, 1983, were included, regardless of associated anomalies, degree of pulmonary hypoplasia, and medical condition on arrival. We searched the hospital medical record system, the Division of Pediatric Surgery records, the University of Florida autopsy database, the Neonatal Intensive Care Unit database, a prenatal database, and the neonatal transport log to confirm that no patients were omitted.
Type of Study
An historic cohort study was performed. The following data were used for this analysis: prenatal diagnosis, mode, date, and place of delivery, Apgar scores, gestational age, birth weight, side of defect, position of stomach and liver, presence of associated anomalies, surgical details, type of ventilator, adjuvant medications, use and type of ECMO, date of extubation, and date of death. Transport records and records from referring institutions were also reviewed. All ventilator settings and blood gas values for the first 120 hours (5 days) of life were analyzed. End points for analysis of ventilator settings and blood gas data were 120 hours of life, death, initiation of ECMO, or extubation.
Statistical Analysis
Categorical factors were analyzed by contingency tables. Statistical tests of these data were performed employing exact methods using StatXact 4 (Cytel Corp., Cambridge, MA). Quantitative outcomes were analyzed using general linear models, and serial assessments were analyzed as serial values of quantitative variables with respect to era of treatment by the inclusion of an interaction term. Statistical tests were all two-sided. Other statistical analyses were conducted using the Statistical Analysis System (SAS Institute, Cary, NC). Life-table analysis was used to estimate survival (SAS System). To evaluate differences in therapy and outcome, patients who had treatment withheld or prospectively withdrawn were removed from analysis (Table 1). Mean peak inspiratory pressure (PIP), mean PaCO2, and mean arterial pH were calculated for each patient and reported at 12-hour intervals. Mean values of PIP, PaCO2, and arterial pH for each era were compared at each interval and over time for 120 hours of life.
Table 1. PATIENTS EXCLUDED FROM SECONDARY (TREATMENT) ANALYSIS

Definition of Eras
Era 1
From December 1983 through May 1988, treatment consisted of paralysis, hyperventilation, and alkalization for pulmonary hypertension. When possible, postductal PaO2 was maintained well above physiologic levels (i.e., >200–300 mm Hg, hyperoxia) in an attempt to keep the pulmonary arteries dilated. Surgical repair was performed routinely within the first 24 hours of life, and ECMO was not available.
Era 2
ECMO became regionally available in 1988 and was inaugurated at our institution in 1989. Primary treatment during this period, from June 1988 through July 1992, remained paralysis with hyperventilation and alkalization, but ventilator pressures were moderated to minimize barotrauma. Hyperoxia was used less vigorously than in era 1, but ventilator support was frequently increased for PaO2 <100 mm Hg. The first two patients in this era underwent surgical repair at 12 hours, but surgery was delayed in all others from 1 to several days.
Era 3
In August 1992, we prospectively changed our ventilator management to avoid hyperventilation and alkalization. Specific details of our current treatment strategy follow.
All prenatally diagnosed patients are encouraged to deliver at our facility, and all potential transfers are accepted. Those with prenatal diagnosis undergo planned delivery by either induction or cesarean section at approximately 38 weeks of gestation. This timing allows advanced commitment of key personnel and equipment. Outborn patients are transferred as quickly as possible. Affected babies are intubated and ventilated with the lowest pressure that provides adequate chest movement (usually 20 to 24 cm H2O PIP and peak end-expiratory pressure of 4 to 5 cm H2O). Ventilation is delivered by a conventional pressure-limited neonatal ventilator (Infant Star, Infrasonics, San Diego, CA) capable of synchronized intermittent mandatory ventilation. High-frequency oscillatory ventilation is not used. The ventilation rate (usually 40 to 80 breaths per minute) is set to patient comfort, with a goal of maintaining postductal PaCO2 at 40 to 65 mm Hg. Patients are lightly sedated with midazolam (Versed; Roche, Somerville, NJ) given as a continuous infusion at 0.05 to 0.10 mg/kg/hr. Patients are not paralyzed except as needed for surgical procedures.
Maintenance fluid of 100 cc/kg/day is provided as total parenteral nutrition that has been premixed and frozen; it is thawed for infusion after birth or transfer. Dopamine is started at 3 μg/kg/min. A low-lying umbilical artery catheter is placed for monitoring and arterial blood gas analysis. Peripheral intravenous lines are used for venous access, or we percutaneously place a right internal jugular central line for resuscitation and rapid conversion to venovenous ECMO if needed. Crystalloid is given in boluses of 5 to 10 cc/kg, and the dose of dopamine is increased as needed to support mean arterial blood pressure at or above the patient’s gestational age in weeks.
Patients are continually monitored with both pre- and postductal oxygen saturation monitors (Nellcor, Inc., Pleasanton, CA). Inspired oxygen is started at 100%, and weaning commences at 6 hours of life if stable. Oxygen is weaned as tolerated with a goal of maintaining postductal saturations >97% (PaO2 80 to 100 mm Hg).
For critically ill patients who do not meet “ideal” goals, marginal pulmonary gas exchange is tolerated, with the absolute commitment of maintaining adequate cerebral oxygen delivery. Postductal saturations as low as 60% are accepted as long as preductal saturations remain ≥85% and perfusion is satisfactory. Postductal PaO2 as low as 30 mm Hg is common and by itself does not represent a need to escalate therapy. PaCO2 >65 is also tolerated if the patient is otherwise stable. NaHCO3 is given only as necessary to keep arterial pH >7.20, even if the baby is hypercarbic. Ventilator rates of up to 100 per minute are often used, and the inspiratory to expiratory time ratio is maintained at 1:2 for intermittent mandatory ventilation rates >30, with inspiratory time of 0.6 seconds for rates <30. PIP is maintained at ≤25 cm H2O. Exceptions are rare increases to 28 cm H2O for instability associated with decreased chest movement. PIP >28 cm H2O is used only as a bridge to ECMO.
Patients who cannot maintain preductal saturations >85% or postductal PaO2 >30 or who show evidence of inadequate oxygen delivery based on rising serum lactate levels are placed on ECMO or inhaled nitric oxide (NO). Since the availability of NO at our institution in 1993, it has been given at 20 to 40 ppm as the first choice for inadequate oxygenation in the hemodynamically stable baby. ECMO is started for the same indications, if NO therapy is unsuccessful.
ECMO is used before or after surgery as needed. Early in the series, all ECMO was venoarterial, but increasingly venovenous ECMO has been used. Patients whose condition fails to improve with venovenous ECMO are converted to venoarterial support. Ventilation is weaned as tolerated on ECMO to resting settings of synchronized intermittent mandatory ventilation 24, PIP 22, peak end-expiratory pressure 5 cm H2O, and inspiratory time 0.6 seconds. Patients placed on ECMO before surgery underwent repair 1 or 2 days before anticipated decannulation (n = 11) or 2 days after completion of the ECMO run (n = 1). Epsilon-aminocaproic acid is not used.
ECMO therapy is discontinued when patients can maintain postductal saturations of 100% with PaCO2 < 60 on PIP 22/5, rate of 40, inspiratory time of 0.5 seconds, and FIO2 of 0.50, off ECMO support.
Surgical repair is delayed at least 1 day. If patients are unstable or require significant ventilator support, surgery may be delayed further. Surgical repair is performed in the neonatal intensive care unit for all but the most stable patients. In this series, a subcostal incision was used in all but one patient, who underwent a thoracotomy. A primary repair is preferred when there is minimal tension; otherwise, a 1-mm-thick Gore-Tex (W.L. Gore and Associates, Charlotte, NC) soft tissue patch is used. Appendectomy is not performed, and malrotation is rarely addressed. The abdominal wall is stretched and closed without a patch. Chest tubes are inserted in a majority of patients but are not placed to suction and are removed at 7 to 10 days, based on output.
After successful resolution of instability, patients are slowly weaned from mechanical ventilation and extubated to an oxy-hood or nasal cannula, with the goal of maintaining postductal arterial saturation at 98% to 100%. Oxygen is used liberally to maintain this, and many patients are discharged home receiving 100 to 200 cc/min of oxygen through a nasal cannula.
RESULTS
A total of 89 infants with symptomatic CDH were identified. Survival rates at discharge from the hospital were 2 of 13 (15%) in era 1, 7 of 16 (44%) in era 2, and 47 of 60 (78%) in era 3 (p < 0.0001). All patients in era 3 were discharged home breathing spontaneously and on no ventilatory support other than oxygen. The Kaplan-Meier survival curve is shown as Figure 1 , and statistical analysis confirmed significant differences by era (Wilcoxon, p < 0.0001). Because of associated lethal conditions, treatment was withheld or prospectively withdrawn from 10 patients: 1 in era 1 (7.7%), 2 in era 2 (12.5%), and 7 in era 3 (11.7%). These patients are described in Table 1 and are not included in the following statistical comparison of the treatment outcomes between eras.
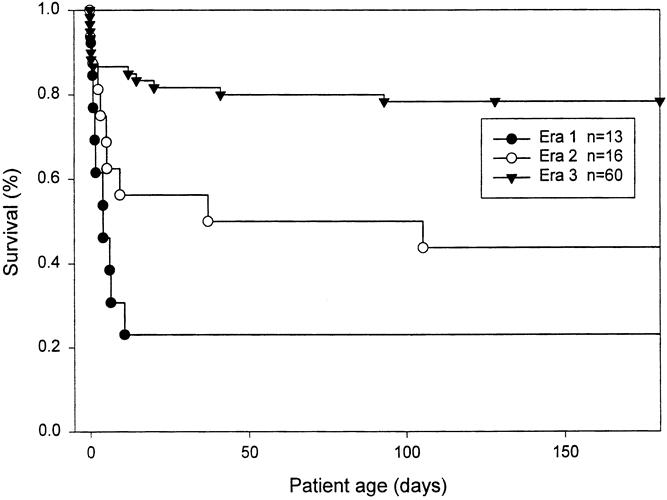
Figure 1. Life-table analysis of survival rate, all patients included, displayed over the first 180 days. Difference in the survival rate across eras is highly significant (p < 0.0001).
The remaining 79 infants were treated with strategies unique to each era. Fifty-three infants with symptomatic CDH treated without hyperventilation and alkalosis and with ECMO if needed (era 3) are compared with 14 patients treated with hyperventilation and ECMO if needed (era 2) and with 12 patients treated with hyperventilation alone (era 1). Two treated patients survived in era 1 (17%) compared with 7 in era 2 (50%) and 47 in era 3 (89%) (p < 0.0001).
Across eras, patient characteristics were similar with regard to birth weight, gestational age, Apgar score at 1 minute, and Apgar score at 5 minutes (Table 2). The incidence of serious but nonlethal anomalies was also similar across eras: 8% in era 1, 7% in era 2, and 6% in era 3 (Table 3). The incidence of right-sided defects was higher in era 3 (15/53, 28%) and era 1 (5/12, 42%) than in era 2 (1/14, 7%), but this difference was not significant (p = 0.14). There was one bilateral defect in era 3, and none in the other eras.
Table 2. DESCRIPTIVE VARIABLES OF TREATED PATIENTS BY ERA

Table 3. ASSOCIATED SEVERE BUT NONLETHAL ANOMALIES

Other measures of disease severity were also similar across eras. The stomach had herniated into the chest in 4 of 12 patients in era 1 (25%), in 7 of 14 in era 2 (50%), and in 26 of 53 in era 3 (49%) (p = 0.60). This finding was confined to left-sided defects. The liver had herniated into the chest in 7 of 12 patients (58%) in era 1, in 5 of 14 in era 2 (36%), and in 27 of 53 in era 3 (51%) (p = 0.56). This finding was more common in right-sided defects but also occurred in left-sided defects. Herniation of both stomach and liver into the chest occurred in 3 patients in era 1 (25%), 4 in era 2 (29%), and 12 in era 3 (23%) (p = 0.92). To compare severity across groups with varying ratios of left- and right-sided defects, we looked at the incidence of stomach and/or liver herniation into the chest. In era 1, 8 of 12 patients (67%) had liver and/or stomach herniation into the chest, in era 2 there were 8 of 14 (57%), and in era 3 there were 41 of 53 (77%) (p = 0.29).
The number of patients with a prenatal diagnosis of CDH rose significantly from 0 in era 1, to 5 of 14 (36%) in era 2, and to 22 of 53 (41%) in era 3 (p = 0.02) (see Table 2). The proportion born at our hospital also increased significantly, from 1 of 12 (8%) in era 1, to 5 of 14 (36%) in era 2, and to 25 of 53 (47%) in era 3 (p = 0.045).
The change in ventilator strategy resulted in predictable changes in ventilator settings and blood gas results. Peak airway pressures dropped dramatically (Fig. 2), PaCO2 was greater (Fig. 3), and arterial pH was lower (Fig. 4) in era 3 compared with previous eras. For example, between 24 and 36 hours of age, mean PIP was 32.8 ± 11.5 cm H2O (mean ± standard deviation) in era 1, 27.3 ± 6.3 in era 2, and 21.4 ± 3.6 in era 3 (p = 0.0001). During the same interval, mean PaCO2 was 31.3 ± 4.9 mm Hg in era 1, 34.0 ± 15.3 in era 2, and 44.6 ± 15.3 in era 3 (p = 0.0158). This was associated with a higher pH in both era 1 and era 2—7.53 ± 0.03 for era 1 and 7.49 ± 0.12 for era 2—compared with 7.33 ± 0.06 for era 3 (p = 0.0001). PIP, PaCO2, and pH were significantly different across eras, and not only at individual time points: the difference was maintained or increased over time (time*era effect).
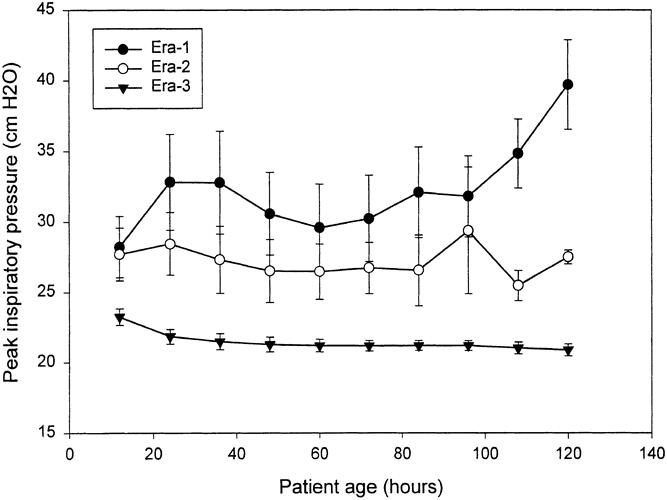
Figure 2. PIP over the first 120 hours of life, expressed as mean ± SEM at 12-hour intervals. Significantly higher peak ventilation pressures were delivered during eras 1 and 2 compared with era 3. The differences between peak ventilation pressure varied significantly across eras, and the difference increased over time (time*era effect, p = 0.00001).
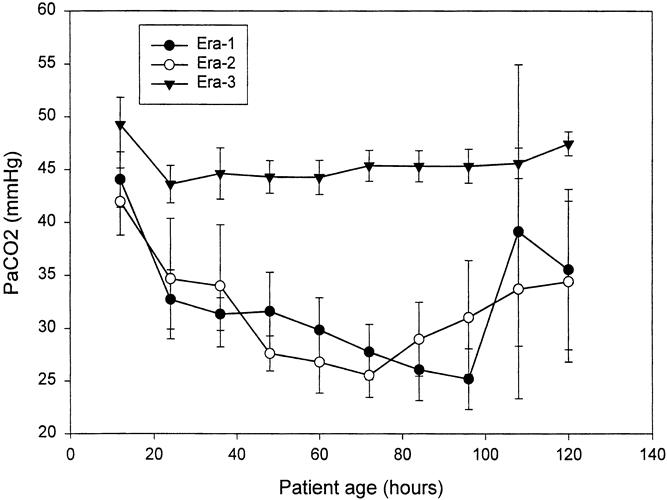
Figure 3. PaCO2 over the first 120 hours of life, expressed as mean ± SEM at 12-hour intervals. The effort to hyperventilate in eras 1 and 2 created nearly identical decrements in arterial PCO2 over time, which were difficult to sustain beyond 96 hours. PaCO2 means were statistically different at all time points except 12 and 108 hours. This difference across eras was maintained across time as well (time*era effect, p < 0.05).
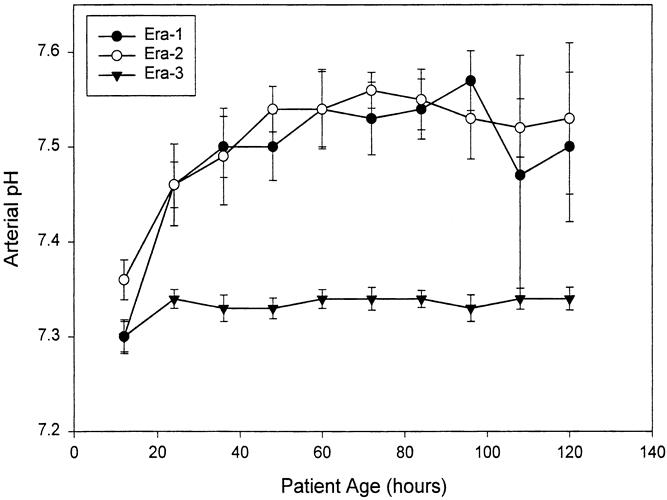
Figure 4. Arterial pH over the first 120 hours of life, expressed as mean ± SEM at 12-hour intervals. Alkalosis was achieved rapidly and maintained in eras 1 and 2. No attempt to alkalinize was made in era 3. Arterial pH was significantly different across the eras at all time points except 12 hours. This effect was maintained across eras and increased over time (time*era effect, p = 0.0003).
PIP increased over time in era 1 and to a lesser extent in era 2, as therapy was escalated in an effort to maintain hypocapnia (see Fig. 2). In era 3, however, mean PIP started significantly lower and declined over time as ventilation pressures were actively decreased (p = 0.0001, time*era effect). Because data collection was terminated when patients started ECMO, the decline in eras 2 and 3 may in part represent a selection effect, because the sickest patients were started on ECMO. The low standard deviation (previous paragraph) and standard error demonstrate, however, that even the sickest patients in era 3 were ventilated within the same narrow range of acceptable PIP.
Efforts to hyperventilate in eras 1 and 2 were successful, and PaCO2 dropped dramatically in the first few days of life (see Fig. 3). By 3 to 4 days, mean PaCO2 approached 25 mm Hg in both eras 2 and 3. This effect was not sustainable, however, and PaCO2 rose after this. In era 1, when ECMO was not available for salvage, these patients became acutely unstable, their PaCO2 rose uncontrollably, and they died quickly. In era 2, similar patients were treated with ECMO. Conversely, in era 3 there was no effort to hyperventilate, and mean PaCO2 for the group varied between 44 and 48 mm Hg throughout the 120-hour period of recording.
The therapeutic goal of alkalosis was achieved in eras 1 and 2 within the first 24 hours and was maintained at these levels (see Fig. 4). As patients became more difficult to ventilate and PaCO2 rose (see Fig. 3), increasing doses of exogenous alkali were given to maintain alkalosis. The standard error of these data increased beyond 72 hours, reflecting both the increasing instability of the patients and their decreasing number. By 96 hours, many patients in eras 1 and 2 had either died or were treated with ECMO. In contrast, mean arterial pH in era 3 remained constant at 7.30 to 7.35 throughout the 120-hour range, and the standard error of the mean did not increase with time.
Associated with the change in ventilator strategy, the rate of pneumothorax dramatically decreased in era 3 (1/53, 1.9%), compared with 6/14 (43%) in era 2 and 10/12 (83%) in era 1 (p < 0.0001). In fact, the only instance of pneumothorax encountered in era 3 occurred in an outborn patient with bilateral diaphragmatic hernias and was already evident on arrival from the referring institution.
The addition of NO to our armamentarium was not associated with either improved survival or decreased ECMO use. Eighteen patients in era 3 received NO, and 14 went on to ECMO. Survival of patients before NO availability was 10/10 (100%), and survival after NO was 37/43 (86%, p = 0.339). Need for ECMO was 3/10 (30%) before NO and 20/43 after NO availability (47%, p = 0.484).
No significant difference existed between need for ECMO in eras 3 and 2. In era 3, 23/53 patients were treated with ECMO (43%); in era 2, 7/14 (50%) were treated with ECMO (p = 0.77). Two patients in era 3 were treated with ECMO a second time, and one survived.
ECMO survival, however, was significantly improved in era 3. In era 2, 2/7 (29%) ECMO-treated patients survived; in era 3, 19/23 (83%) ECMO-treated patients survived to discharge (p = 0.014). Survival in patients not treated with ECMO was also improved in era 3—28/30 (93%) compared with 5/7 (71%)—but this difference was not statistically significant (p = 0.155). To assess whether general ECMO techniques had improved during this interval, we reviewed a parallel group of ECMO patients treated at our institution for meconium aspiration during the same time periods as era 2 and era 3. Survival in these reference patients was 8/9 (89%) from 1989 to 1992 and 35/35 (100%) from 1992 to 1998 (p = 0.2045). Overall survival of patients with meconium aspiration who required ECMO from 1989 to 1998 was 43/44 (98%).
We found no negative effect associated with prenatal diagnosis in era 3. Of the 60 patients in this group, 27 were prenatally diagnosed, and 4 of these had lethal associated anomalies (see Table 1). Another was born at home without medical assistance and is also included in Table 1. The survival rate of prenatally diagnosed patients (20/27, 74%) was no different from the overall survival rate in this series (47/60, 78%). The survival rate of inborn, prenatally diagnosed patients with isolated CDH was 20/22 (91%, Table 4). The survival rate of all inborn patients with isolated CDH, regardless of degree of pulmonary hypoplasia, was 23/25 (92%).
Table 4. SURVIVAL TO DISCHARGE BY ERA

In era 3, 13/60 patients died (22%). Seven patients had treatment withheld (see Table 1), and the six remaining patients were considered as treated. Four of these were outborn. One arrived profoundly asphyxiated with a pH of 6.9, received ECMO, and died with a grade IV intracranial hemorrhage. One was a boy with a gestational age of 27 weeks; he had an intrathoracic stomach and did not meet our criteria for ECMO because of profound prematurity. Two outborn, critically ill patients were overventilated before transport. One had stomach and liver in his chest, and the other had bilateral CDH and arrived with a pneumothorax. Both were treated with long ECMO runs without pulmonary recovery. Two inborn patients with isolated CDH died. One was prenatally diagnosed with a chromosomal inversion that was thought to be a normal variant. She was born with severe, bilateral thoracic hypoplasia and was unable to achieve a pH >6.7 or a PaCO2 <120 mm Hg. She was not offered ECMO and died. At postmortem examination, only 5 g of lung tissue was present. The other inborn patient had a left CDH with stomach and liver in his chest, could not be stabilized on ECMO, and died without repair.
We looked for anatomic markers in era 3 that predicted poor survival. Of patients with liver and/or stomach in the chest, the survival rate was 35/41 (85%) and 20/22 (91%) in the inborn subset of these patients. We also visually estimated the percentage of liver mass present in the chest. Nineteen of 53 had 50% or more of the liver in the chest; the survival rate was 15/19 (79%). Although subjectively we believed that patients with right-sided CDH and a large mass of liver in the chest were among our sickest, the survival rate was 14/14 (100%). Seven (50%) of these patients required ECMO. Twelve patients in era 3 had liver and stomach in the chest (left-sided defects), and eight patients (67%) survived to discharge. The highest-risk group in our series was patients with left CDH and with ≥50% of the liver in the chest: only one of five survived, including one of three inborn.
DISCUSSION
Survival in patients with CDH is ultimately dependent on both satisfactory function of available lung parenchyma and resolution of pulmonary hypertension. Because of hypoplasia and immaturity, CDH lungs may be particularly sensitive to ventilation injury. Pneumothorax, as shown by Kolobow 21–23 in an elegant series of experiments, represents an advanced level of ventilator-induced lung injury. By strictly limiting PIP in our patients, we encountered only one pneumothorax, which was already present on transfer. The reduction in the rate of pneumothorax from 83% in era 1 to 1.9% in era 3 parallels the decline we observed in the mortality rate.
Serious but less dramatic degrees of lung injury, including ruptured alveoli, torn alveolar basement membranes, alveolar hemorrhage and edema, with resultant atelectasis, decreased compliance, and seriously impaired gas exchange, occur well before pneumothorax. 21,23 These findings were present in postmortem analyses of babies with CDH. 24,25 Elimination of such injury, by whatever means, should allow an improved survival rate in these patients. The survival rate improved dramatically in our institution after limiting ventilator support and accepting both higher PaCO2 and lower PaO2.
One could argue that the improved survival rate in this series is due only to the removal of barotrauma, and benefit could still be conferred to these patients by correcting acidosis and alkalizing with exogenous buffers. Indeed, alkalosis has been shown to decrease pulmonary vascular resistance independently of hyperventilation. 26,27 Unfortunately, the effect of alkalosis on critically ill infants is often short-lived and difficult to maintain, and it may serve to increase rather than decrease the clinical instability of these patients. Our experience in era 3 confirms that pulmonary hypertension can resolve, as reflected by improved oxygenation, eventual extubation, and survival to discharge, in the presence of hypercarbia and acidosis (see Figs. 3 and 4).
Although it does not appear that the ventilatory changes we implemented decreased ECMO requirement in any significant way, the survival rate in patients treated with ECMO was dramatically improved. This result was not attributable to any changes in our indications for ECMO, nor to any technical improvements in ECMO delivery. Indeed, the survival rate of patients treated with ECMO for meconium aspiration at Shands Hospital did not change significantly from era 2 to era 3, and the ECMO techniques used to support infants with CDH are similar to those applied to other populations of hypoxic newborns. Accordingly, it is likely that the improved survival in patients with CDH receiving ECMO during era 3 reflects the changes in ventilatory technique, which are maintained before, during, and after the ECMO run.
Compared with national series, 25,26 our ECMO use in era 3 was lower than average and the survival rate was higher than average. Because these published series have excluded stillborns, we removed the single stillborn from our overall total of 60 era 3 patients to facilitate direct comparisons. Twenty-three of these 59 patients (39%) were treated with ECMO, and the survival rate was 19/23 (83%). Reickert et al 25 reported ECMO use of 46% ± 2% (mean ± SEM) in 411 patients collected from 16 level III nurseries interested in CDH management, with a survival rate of 55% ± 4%. ECMO use in institutions reporting to the CDH study group was 57%, with a survival rate of 54%. The current survival rate of 2899 patients with CDH treated with ECMO and reported to the Extra-Corporeal Life Support Organization stands at 58%. 28
We first used NO in a patient with CDH in December 1993. Since then, we have not experienced any overall improvement in CDH survival or any decreased requirement for ECMO. Therefore, the advent of NO has not appeared to contribute to our improved results. Although others have reported an apparent efficacy of NO in individual patients with CDH, 29–31 a prospective, randomized, masked study of 53 patients with CDH identified no decrease in either mortality rate or need for ECMO in those who received NO. 32
Surgery was delayed beyond 24 hours in both eras 2 and 3. Although delay in repair is becoming more widespread, reports pertaining to the impact on outcome have been inconsistent, and data analysis has been obscured by concomitant variations in ventilator management. Retrospective reviews by Breaux et al 33 and West et al 34 suggest that a delay in surgery beyond the first 24 hours improves the survival rate, whereas reports by Wilson et al 35 and a prospective, randomized trial by de la Hunt et al 36 show no survival benefit. The very prolonged delay in surgical intervention reported by Reickert et al was associated with both improved survival rate and decreased need for ECMO in a group of 33 patients treated from 1992 to 1995. 37 Given the mixed results in these reported series and the fact that surgery was delayed well beyond 24 hours in both eras 2 and 3, it seems unlikely that this factor significantly affected the outcome difference between the two eras.
This is not the first report documenting an improved survival rate by elimination of hyperventilation. Wung et al have treated patients with CDH without hyperventilation since 1983, and their experience provided our initial inspiration. These authors reported an overall survival rate of 52/71 patients (73%), with 17 of 19 (89%) surviving in their most recent group. 38 Wilson et al 24 reported an improved survival rate of 84% in isolated CDH when hyperventilation was avoided. Frenckner et al 39 stressed the importance of gentle ventilation in a report from Sweden on improved survival rates in CDH, but this series is otherwise not comparable to the others because patients were entirely outborn and <20% were ill enough to require intubation at birth.
Despite the seeming persuasiveness of these recent reports, many physicians remain unconvinced, and hyperventilation continues to be widely used in the management of CDH. In the Neonatal Inhaled Nitric Oxide Study Group trial of NO therapy in CDH patients, published in 1997, maximal therapy was allowed but not required. 32 This represented a state-of-the-art, multicenter trial enrolling many major university intensive care nurseries. Results showed that 91% of patients were treated with muscle paralysis and 70% were treated with hyperventilation and/or alkalosis. Even though 79% of patients were treated at some point with high-frequency oscillatory ventilation, air leaks from overly vigorous ventilation developed in 29%.
We have shown a dramatic increase in the survival rate of patients with CDH when treated without hyperventilation and alkalosis, compared with similar patients treated with this strategy. The improved survival rate cannot be attributed to differences in disease severity, to the use of NO, or to delayed surgery. Nor has there been any identifiable selection bias: in fact, many of the patients in era 3 were either prenatally diagnosed or inborn (42% and 47%, respectively). Both prenatal diagnosis and inborn status minimize selection bias and have been previously demonstrated to be potentially associated with decreased survival rates. 40–42 Our report of 23 survivors of 25 unselected, consecutively treated, inborn patients with isolated CDH, 22 of whom were prenatally diagnosed, is unsurpassed in the medical literature.
We therefore conclude that hyperventilation and alkalization, as a treatment strategy for patients with CDH, has prohibitive detrimental effects, and the approach should be abandoned. By eliminating such therapy, we significantly decreased the incidence of barotrauma and improved the survival rate, supporting our original hypothesis. Also, the survival rate of patients requiring ECMO improved significantly, supporting the conclusion that preexisting pulmonary hypoplasia complicated by iatrogenic lung injury represents a lethal combination, and elimination of such injury allows survival of the majority of patients with CDH who require ECMO.
Finally, the survival rate documented in this study using conventional ventilators represents a benchmark against which other therapies, such as high-frequency ventilation, liquid ventilation, and fetal intervention, must be measured.
Acknowledgments
The authors thank Brad H. Pollock, MPH, PhD, and his research assistant, Linda Garzarella, for their invaluable work with the statistical analysis of these data, and Willa H. Drummond, MD, for her longstanding support of this project.
Discussion
Dr. Kathryn D. Anderson (Los Angeles, California): This is an excellent paper, Dr. Kays, beautifully presented, and I appreciate the opportunity to review your manuscript and comment here.
The difficulty with developing a best practice for treating complex conditions such as congenital diaphragmatic hernia is that in comparing different treatment regimens in different institutions with different personnel and using historical eras such as Dr. Kays describes, it is impossible to control all the variables.
The term “congenital diaphragmatic hernia” encompasses a huge hodgepodge of conditions—degrees of hypoplasia, pulmonary hypertension, and associated anomalies, to name a few. Conventional ventilation, hyperventilation, and even high-frequency oscillatory ventilation techniques vary from institution to institution and even among personnel in the same institution. Nonetheless, with a large population of infants and a consistent technique, the authors have developed their best practice for their institution, and probably for many others as well.
I believe they have given us two lessons in this paper. The first is the one which Dr. Kays has concentrated on—that is, keeping the peak inspiratory pressure to the lowest possible value compatible with maintaining oxygenation, and ignoring, relatively speaking, rising carbon dioxide levels, avoiding damage to the already hypoplastic lungs, and reducing morbidity and mortality associated with the air leaks themselves. This has resulted in better survival.
The late Dr. Robert DeLemos showed us that these same principles apply to high-frequency oscillatory ventilation, decreasing the need for ECMO and resulting in improved outcome for these infants. And a large series reported by Jay Wilson from Boston confirms this also.
But I believe that the authors have demonstrated a second principle which they mention almost incidentally but in my opinion is extremely important. Dr. Robert Gross reported an 85%-plus survival for babies with congenital diaphragmatic hernia in the 1950s and was very critical of his pupils who could not achieve the same outcome. The dictum has always been that with better neonatal care, infants are now surviving to be transferred, but are dying later on of pulmonary problems, including hypoplasia. I don’t believe this is true any longer. The authors’ survival of outborn babies who needed ECMO in the last year of the series presented today is 68%. Inborn babies who needed ECMO had 92% survival.
These data are very difficult to extract from the National ECMO Registry. But an informal look at our own database of exclusively outborn infants and that of others shows a survival in the range of 65% to 75%, similar to the authors’ outborn survival.
The ability of one team to treat an infant with congenital diaphragmatic hernia immediately after birth and carry that treatment through ventilation, ECMO, surgery, and postoperative care with the same personnel is responsible for the vastly improved survival.
I would ask Dr. Kays to comment on this aspect of his work.
Presenter Dr. David W. Kays (Gainesville, Florida): Thank you, Dr. Anderson, for your very kind comments. Indeed, it has been very gratifying to see survival improve with this change in therapy. Additionally, survival of inborn patients has been improved over outborn patients, adding further support to the concept of improved therapy. Because of selection bias, survival of outborn patients has exceeded that of inborn patients in major reported series, and the reversal in this trend, reported here, may indeed represent a new observation.
To optimize outcome, it is important that the therapeutic style and goals be strictly maintained over both the short and long term. Although many physicians, nurses, and therapists contribute to the care of these patients, consistency of medical care and decision-making is maintained in our center by the managing surgeon, who actively oversees all aspects of ventilation, resuscitation, surgery, and ECMO if needed. We agree that this therapeutic consistency has contributed to the high survival reported in this series, and thank you for recognizing this.
Dr. Marshall Z. Schwartz (Wilmington, Delaware): I have a few comments myself, and then I will present Dr. Tapper’s comments as well.
Because I have been an advocate of the concept that less is better in the pulmonary management of infants with congenital diaphragmatic hernia, I applaud Dr. Kays’ approach and excellent results.
We have known that the two most significant iatrogenically induced insults to the lung in infants with diaphragmatic hernia are barotrauma and oxygen toxicity. However, our management has routinely led us in this direction. Every pediatric surgeon has cared for babies with diaphragmatic hernia that had adequate gases in the “honeymoon” period in the first 6 to 12 hours after birth, only to die later of respiratory failure. We attributed this course to pulmonary hypoplasia and pulmonary hypertension. Certainly these factors play a significant role. This presentation has demonstrated that minimizing barotrauma and oxygen toxicity by accepting marginal to adequate blood gases instead of attempting to achieve optimum blood gases can lead to a better outcome. This is not a new approach. However, your corroboration of previous work done at Columbia-Babies Hospital by Wung and Stolar will increase the awareness and potential benefit of this approach.
I have several questions that I would like to address to you.
I have found that the group most resistant to applying this approach is our neonatal colleagues who control the dials on the ventilator, and it is difficult to convince them that achieving optimum gases is not necessarily optimum management for these infants.
With your experience have you been able to identify early predictors of outcome, especially those infants that did not survive? In other words, can you tell on the first day of life based on ventilation requirements who is going to survive and who is not?
Finally, do you have long-term data on lung function and other organ functions? Clearly you have accomplished a tremendous first hurdle by significantly improving survival. The next hurdle is that the long-term follow-up data demonstrates that the infants are better off with this approach.
Dr. Kays: Thank you, Dr. Schwartz. Regarding early identification of patients who cannot survive, this can be difficult. We believe that the vast majority of patients with this disease can survive, and unless there is strong evidence to the contrary, such as a severe associated cardiac malformation, we treat virtually all patients as potential survivors.
In our series, several critically patients were not treated with ECMO due to severe associated cardiac malformations. At this juncture, we feel such patients have very little reasonable chance of survival, and counsel the families accordingly. Many other patients will look quite ill at birth, but will slowly improve in the first few hours, even if they eventually require ECMO. Of our 53 patients, we treated only four with ECMO in the first 12 hours of life.
Rarely, a newborn without associated anomalies will have such poor gas exchange that survival seems extremely unlikely, and we will not offer ECMO in this situation. We had one such patient in our series, with best pH less than 7.0 and best PaCO2 greater than 100, despite what we considered optimal therapy. Overall, however, it has been gratifying that most babies we have treated have survived, including several with very severe defects and poor gas exchange. Based on these experiences, we will continue to view the majority of patients as potential survivors, and treat them accordingly.
This series provides both the opportunity and the obligation to report the long-term outcome of these patients, many of whom previously would have died. Briefly, the morbidity in these patients is significant, including a spectrum of feeding problems, gastroesophageal reflux, and secondary growth issues. None of our patients requires assisted ventilation of any type. Long-term pulmonary function is good to excellent, and the feeding problems are manageable. The majority of these patients, because of compromised oxygen delivery and ECMO support, are at risk for neurologic injury, but we are very pleased, overall, with neurodevelopmental outcome in the survivors. Detailed analysis of these patients is ongoing, and will be subsequently reported.
Dr. Schwartz: Dr. David Tapper was also an invited discussant and unfortunately at the last minute was not able to come. He asked me if I wouldn’t mind reading his discussion, which I am happy to do.
Dr. David Tapper (Seattle, Washington): The field of pediatric surgery has been beset with the problem of improving the survival in babies born with diaphragmatic hernia. For over 50 years, the mortality from this congenital anomaly has been around 50%.
Great excitement heralded the introduction of extracorporeal membrane oxygenation as a technology to sustain the sickest infants and improve survival. Unfortunately, the survival of congenital diaphragmatic hernia patients treated with ECMO as reported by the Extracorporeal Life Support Organization stands at 58% of 2,899 patients.
In utero fetal surgery to correct the diaphragmatic defect has likewise been disappointing. A prospective randomized masked study of nitric oxide in 53 congenital diaphragmatic hernia patients revealed no decrease in mortality, nor any decrease in the need for ECMO in congenital diaphragmatic hernia patients treated with nitric oxide.
This presentation by Dr. Kays and associates has challenged current dogma. As you have seen, their results have been spectacular. Gregory, Phibbs, et al developed the hypothesis adopted by most pediatric surgeons that to treat the significant pulmonary hypertension that these infants all have, all babies should be maximally hyperventilated and made significantly alkalotic. This work by Kays et al, which supports previously published material by Wung and Stolar, significantly challenges previous recommendations. Their management can be characterized as gentle ventilation, permissive hypercapnia, and tolerance of metabolic acidosis. With these changes in patient management, the survival in treated patients was 89%. The number of infants requiring ECMO in their series was 47%, implying that their patients were severely ill, yet the survival in their patients after ECMO was an enviable 83%. Clearly, the rest of the pediatric surgical world must pay attention to the reports from Wung and Stolar from Babies Hospital in New York, and Kays et al, from Gainesville.
What do you believe is fundamentally wrong with the concept of hyperventilation and metabolic alkalosis?
How significant is the timing of the operative repair of diaphragmatic hernia?
When would you prefer to do the operation and what are the guidelines you use to determine the optimal time for surgical repair?
Based on your results, is there any indication for fetal intervention?
Dr. Kays: Thank you, Dr. Tapper. The main problem with hyperventilation and alkalosis is the risk of barotrauma, primarily related to overinflation of alveoli. Our best ventilation correlate of this is peak inspiratory pressure, or PIP. However, I do believe there is a secondary deleterious effect from reliance on alkalosis to cause pulmonary vasodilation. This effect, which may be very dramatic, is often transient, and clearly exhibits tachyphylaxis. Reliance on alkalosis to facilitate vasodilation introduces a new, unstable variable to what is already an unstable physiologic system, i.e., the neonate with congenital diaphragmatic hernia and pulmonary hypertension. It is difficult in a clinical situation, however, to separate the negative effects of barotrauma from the negative effects of alkalosis. In an otherwise equivalent situation, however, I would rather see a PCO2 of 50 and pH of 7.30, than a PCO2 of 30 and a pH of 7.50.
Regarding the timing of surgery, both the available literature and popular opinion has swung in favor of delayed surgical repair of CDH. Recent literature includes reports of repair delayed 6 to 10 days. We delay repair a minimum of 24 hours, but I do not believe that delayed repair is primarily responsible, nor even essential, for the improved survival we experienced. Meticulous ventilator care is the secret to good survival, not timing of surgery. In fact, I would rather operate at 24 hours of life in a baby destined for ECMO, and manage ECMO postoperatively, than to further delay surgery and be forced to manage a newborn with unrepaired CDH on ECMO. Therefore, as long as patients are improving, we will delay repair for 3 to 5 days. If a patient’s clinical state plateaus, then we usually repair at that time. If a patient is marginal, and appears to worsen, we may repair the defect quickly, try to stabilize for a few hours postoperatively, then go on to ECMO. As improved survival becomes more commonplace, then we will be able to look at quality, not quantity of outcome, and this may shed further light on the subject.
Fetal intervention for prenatally diagnosed CDH is both a scientific and emotional issue. Parents of a baby with prenatally diagnosed CDH are justifiably scared, and are frequently emotionally drawn to consider fetal intervention, as it is proactive in the fetal period and holds the vague potential of a better outcome at birth. Although the results with fetal intervention in highly selected patients may be improving, there is both insufficient size and maturity to these data to compare results to our larger, unselected experience. With our reported survival rate of 92% in unselected patients with CDH who deliver at our institution, I cannot personally recommend fetal intervention to any family that compares our results to those with fetal intervention.
Dr. Thomas C. Moore (Torrance, California): The authors are to be complimented for taking a critical look at so-called standard medical therapies employed in the management of congenital diaphragmatic hernia (CDH) in the newborn—a major surgical disorder which long has resisted a great variety of often ingenious efforts at successful management. Their central message clearly is to do no harm. CDH, at one occurrence in each 1,200 births, is one of the most frequently occurring of major malformations in the newborn as well as one of the most relentlessly fatal.
The principal cause of the fatality is the severely hypoplastic lung problem, identified so clearly in the classic paper of Campanale and Rowland from an Air Force hospital obstetrical unit, which appeared in the August 1955 issue of Annals of Surgery (142:176–189). They reported five cases of CDH (three operated upon) with 100% mortality and 100% hypoplastic lung (slides also checked by the illustrious Dr. Edith Potter of Chicago). Their published report also contained a most interesting personal communication from Dr. Robert Gross of Boston in which he did not recall ever seeing hypoplastic lung at operation for CDH and that he thought that it must be extremely rare, if it existed at all! Clearly two different literatures relating to CDH in the newborn were developing at this time—one from obstetrical units and one from children’s hospitals, where all CDH newborns arrive on referral from birth elsewhere.
Also in 1955, the late John Dorsey of this Association and associates reported in Obstetrics and Gynecology (6:262) an experience with 13 cases of CDH in the newborn. Nine of the 13 died within the first two hours of life (69.2%) and were diagnosed at autopsy. All four cases diagnosed clinically were operated upon and only one survived (for an overall survival of 7.7%). These 13 cases occurred during a 10-year period when there were 15,000 deliveries at their institution (Evanston Hospital).
Two years later (1957), I and associates reported in Surgery, Gynecology and Obstetrics (104:675–689) an experience with 16 cases of CDH (three of the 16 were autopsy diagnosed). Eight of the 13 operated-upon cases recovered (two of the eight were operated upon at 12 and 14 hours of age, with six operated upon at from 28 to 90 hours of age). An interesting and important finding was the progressive expansion of the small lung buds in the early hours and days after operation if baro damage could be avoided. Photomicrographs of a fatal case in this report showed alveolar distention and rupture, consistent with overly zealous baro resuscitative efforts and this was strongly warned against in the conclusions to this report, as the authors have emphasized here today. I and associates have a paper recently published in Pediatric Surgery International (1999; 15:97–104) suggesting, at the end of the discussion, a novel way of increasing in utero CDH fetal tracheobronchial fluid pressure, with the possibility of enhancing prebirth lung growth.
In two questions for the authors, how many of your inborn cases were diagnosed in the critical first 2 hours of life, and have you considered or initiated any prebirth manipulations of the fetus with known CDH in view of the virtually intractable difficulty with the hypoplastic lung postbirth?
DR. KAYS: Thank you, Dr. Moore. All of our patients were symptomatic in the first 6 hours of life, a common reference point in the literature. Fifty-nine of 60 patients in era 3 were symptomatic in the first 2 hours of life, and the vast majority, if not all, of these were symptomatic immediately at birth.
Footnotes
Correspondence: David W. Kays, MD, Dept. of Surgery, University of Florida, Box 100286, Gainesville, FL 32610-0286.
Presented at the 119th Annual Meeting of the American Surgical Association, April 15–17, 1999, Hyatt Regency Hotel, San Diego, California.
Accepted for publication April 1999.
References
- 1.Drummond WH, Gregory GA, Heymann MA, Phibbs RA. The independent effects of hyperventilation, tolazoline, and dopamine on infants with persistent pulmonary hypertension. J Pediatr 1981; 98 (4): 603–611. [DOI] [PubMed] [Google Scholar]
- 2.Clark RH, Hardin WD Jr, Hirschl RB, et al. Current surgical management of congenital diaphragmatic hernia: a report from the Congenital Diaphragmatic Hernia Study Group. J Pediatr Surg 1998; 33 (7): 1004–1009. [DOI] [PubMed] [Google Scholar]
- 3.Kitagawa M, Hislop A, Boyden EA, Reid L. Lung hypoplasia in congenital diaphragmatic hernia. A quantitative study of airway, artery, and alveolar development. Br J Surg 1971; 58 (5): 342–346. [DOI] [PubMed] [Google Scholar]
- 4.Suen HC, Catlin EA, Ryan DP, et al. Biochemical immaturity of lungs in congenital diaphragmatic hernia. J Pediatr Surg 1993; 28 (3): 471–477. [DOI] [PubMed] [Google Scholar]
- 5.George DK, Cooney TP, Chiu BK, Thurlbeck WM. Hypoplasia and immaturity of the terminal lung unit (acinus) in congenital diaphragmatic hernia. Am Rev Respir Dis 1987; 136 (4): 947–950. [DOI] [PubMed] [Google Scholar]
- 6.Ehrlich FE, Salzberg AM. Pathophysiology and management of congenital posterolateral diaphragmatic hernias. Am Surg 1978; 44 (1): 26–30. [PubMed] [Google Scholar]
- 7.Mishalany HG, Nakada K, Woolley MM. Congenital diaphragmatic hernias: eleven years’ experience. Arch Surg 1979; 114 (10): 1118–1123. [DOI] [PubMed] [Google Scholar]
- 8.Shochat SJ, Naeye RL, Ford WD, et al. Congenital diaphragmatic hernia. New concept in management. Ann Surg 1979; 190 (3): 332–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Geggel RL, Murphy JD, Langleben D, et al. Congenital diaphragmatic hernia: arterial structural changes and persistent pulmonary hypertension after surgical repair. J Pediatr 1985; 107 (3): 457–464. [DOI] [PubMed] [Google Scholar]
- 10.Reid LM. Structure and function in pulmonary hypertension. New perceptions. Chest 1986; 89 (2): 279–288. [DOI] [PubMed] [Google Scholar]
- 11.Harrison MR, Langer JC, Adzick NS, et al. Correction of congenital diaphragmatic hernia in utero, V. Initial clinical experience. J Pediatr Surg 1990; 25 (1): 47–57. [DOI] [PubMed] [Google Scholar]
- 12.Harrison MR, Adzick NS, Flake AW, et al. Correction of congenital diaphragmatic hernia in utero: VI. Hard-earned lessons. J Pediatr Surg 1993; 28 (10): 1411–1418. [DOI] [PubMed] [Google Scholar]
- 13.Hedrick MH, Estes JM, Sullivan KM, et al. Plug the lung until it grows (PLUG): a new method to treat congenital diaphragmatic hernia in utero. J Pediatr Surg 1994; 29 (5): 612–617. [DOI] [PubMed] [Google Scholar]
- 14.Skarsgard ED, Meuli M, VanderWall KJ, et al. Fetal endoscopic tracheal occlusion (Fetendo-PLUG) for congenital diaphragmatic hernia. J Pediatr Surg 1996; 31 (10): 1335–1338. [DOI] [PubMed] [Google Scholar]
- 15.Harrison MR, Mychaliska GB, Albanese CT, et al. Correction of congenital diaphragmatic hernia in utero IX: fetuses with poor prognosis (liver herniation and low lung-to-head ratio) can be saved by fetoscopic temporary tracheal occlusion. J Pediatr Surg 1998; 33 (7): 1017–1023. [DOI] [PubMed] [Google Scholar]
- 16.Van Meurs KP, Rhine WD, Benitz WE, et al. Lobar lung transplantation as a treatment for congenital diaphragmatic hernia. J Pediatr Surg 1994; 29 (12): 1557–1560. [DOI] [PubMed] [Google Scholar]
- 17.Bartlett RH, Gazzaniga AB, Toomasian J, et al. Extracorporeal membrane oxygenation (ECMO) in neonatal respiratory failure. 100 cases [published erratum appears in Ann Surg 1987 Jan;205(1): 11A]. Ann Surg 1986; 204 (3): 236–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Langham MR Jr, Krummel TM, Bartlett RH, et al. Mortality with extracorporeal membrane oxygenation following repair of congenital diaphragmatic hernia in 93 infants. J Pediatr Surg 1987; 22 (12): 1150–1154. [DOI] [PubMed] [Google Scholar]
- 19.Redmond C, Heaton J, Calix J, et al. A correlation of pulmonary hypoplasia, mean airway pressure, and survival in congenital diaphragmatic hernia treated with extracorporeal membrane oxygenation. J Pediatr Surg 1987; 22 (12): 1143–1149. [DOI] [PubMed] [Google Scholar]
- 20.Wung JT, James LS, Kilchevsky E, James E. Management of infants with severe respiratory failure and persistence of the fetal circulation, without hyperventilation. Pediatrics 1985; 76 (4): 488–494. [PubMed] [Google Scholar]
- 21.Kolobow T, Moretti MP, Fumagalli R, et al. Severe impairment in lung function induced by high peak airway pressure during mechanical ventilation. An experimental study. Am Rev Respir Dis 1987; 135 (2): 312–315. [DOI] [PubMed] [Google Scholar]
- 22.Mascheroni D, Kolobow T, Fumagalli R, et al. Acute respiratory failure following pharmacologically induced hyperventilation: an experimental animal study. Intensive Care Med 1988; 15 (1): 8–14. [DOI] [PubMed] [Google Scholar]
- 23.Tsuno K, Prato P, Kolobow T. Acute lung injury from mechanical ventilation at moderately high airway pressures. J Appl Physiol 1990; 69 (3): 956–961. [DOI] [PubMed] [Google Scholar]
- 24.Wilson JM, Lund DP, Lillehei CW, Vacanti JP. Congenital diaphragmatic hernia–a tale of two cities: the Boston experience. J Pediatr Surg 1997; 32 (3): 401–405. [DOI] [PubMed] [Google Scholar]
- 25.Reickert CA, Hirschl RB, Atkinson JB, et al. Congenital diaphragmatic hernia survival and use of extracorporeal life support at selected level III nurseries with multimodality support. Surgery 1998; 123 (3): 305–310. [PubMed] [Google Scholar]
- 26.Schreiber MD, Heymann MA, Soifer SJ. Increased arterial pH, not decreased Pa CO2, attenuates hypoxia-induced pulmonary vasoconstriction in newborn lambs. Pediatr Res 1986; 20 (2): 113–117. [DOI] [PubMed] [Google Scholar]
- 27.Morray JP, Lynn AM, Mansfield PB. Effect of pH and Pa CO2 on pulmonary and systemic hemodynamics after surgery in children with congenital heart disease and pulmonary hypertension. J Pediatr 1988; 113 (3): 474–479. [DOI] [PubMed] [Google Scholar]
- 28.ECMO Registry Report, International Summary. Extracorporeal Life Support Organization 1998.
- 29.Frostell CG, Lonnqvist PA, Sonesson SE, et al. Near-fatal pulmonary hypertension after surgical repair of congenital diaphragmatic hernia. Successful use of inhaled nitric oxide. Anaesthesia 1993; 48 (8): 679–683. [DOI] [PubMed] [Google Scholar]
- 30.Henneberg SW, Jepsen S, Andersen PK, Pedersen SA. Inhalation of nitric oxide as a treatment of pulmonary hypertension in congenital diaphragmatic hernia. J Pediatr Surg 1995; 30 (6): 853–855. [DOI] [PubMed] [Google Scholar]
- 31.Dillon PW, Cilley RE, Hudome SM, et al. Nitric oxide reversal of recurrent pulmonary hypertension and respiratory failure in an infant with CDH after successful ECMO therapy [see comments]. J Pediatr Surg 1995; 30 (5): 743–744. [DOI] [PubMed] [Google Scholar]
- 32.Inhaled nitric oxide and hypoxic respiratory failure in infants with congenital diaphragmatic hernia. The Neonatal Inhaled Nitric Oxide Study Group (NINOS). Pediatrics 1997; 99(6):838–845. [DOI] [PubMed]
- 33.Breaux CW Jr, Rouse TM, Cain WAS, Georgeson KE. Improvement in survival of patients with congenital diaphragmatic hernia utilizing a strategy of delayed repair after medical and/or extracorporeal membrane oxygenation stabilization. J Pediatr Surg 1991; 26 (3): 333–338. [DOI] [PubMed] [Google Scholar]
- 34.West KW, Bengston K, Rescorla FJ, et al. Delayed surgical repair and ECMO improves survival in congenital diaphragmatic hernia. Ann Surg 1992; 216 (4): 454–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wilson JM, Lund DP, Lillehei CW, et al. Delayed repair and preoperative ECMO does not improve survival in high-risk congenital diaphragmatic hernia. J Pediatr Surg 1992; 27 (3): 368–375. [DOI] [PubMed] [Google Scholar]
- 36.de la Hunt MN, Madden N, Scott JE, et al. Is delayed surgery really better for congenital diaphragmatic hernia? A prospective randomized clinical trial [see comments]. J Pediatr Surg 1996; 31 (11): 1554–1556. [DOI] [PubMed] [Google Scholar]
- 37.Reickert CA, Hirschl RB, Schumacher R, et al. Effect of very delayed repair of congenital diaphragmatic hernia on survival and extracorporeal life support use. Surgery 1996; 120 (4): 766–773. [DOI] [PubMed] [Google Scholar]
- 38.Wung JT, Sahni R, Moffitt ST, et al. Congenital diaphragmatic hernia: survival treated with very delayed surgery, spontaneous respiration, and no chest tube. J Pediatr Surg 1995; 30 (3): 406–409. [DOI] [PubMed] [Google Scholar]
- 39.Frenckner B, Ehren H, Granholm T, et al. Improved results in patients who have congenital diaphragmatic hernia using preoperative stabilization, extracorporeal membrane oxygenation, and delayed surgery. J Pediatr Surg 1997; 32 (8): 1185–1189. [DOI] [PubMed] [Google Scholar]
- 40.Nakayama DK, Harrison MR, Chinn DH, et al. Prenatal diagnosis and natural history of the fetus with a congenital diaphragmatic hernia: initial clinical experience. J Pediatr Surg 1985; 20 (2): 118–124. [DOI] [PubMed] [Google Scholar]
- 41.Manni M, Heydanus R, Den Hollander NS, et al. Prenatal diagnosis of congenital diaphragmatic hernia: a retrospective analysis of 28 cases. Prenat Diagn 1994; 14 (3): 187–190. [DOI] [PubMed] [Google Scholar]
- 42.Boedy RF, Howell CG, Kanto WP Jr. Hidden mortality rate associated with extracorporeal membrane oxygenation. J Pediatr 1990; 117 (3): 462–464. [DOI] [PubMed] [Google Scholar]