

Abstract
Objective
Specific and efficient tumor-targeted gene delivery is the major goal for successful cancer gene therapy.
Summary Background Data
A recombinant thymidine kinase-deleted vaccinia virus (vv) encoding the firefly luciferase (luc) reporter gene or the prodrug converter gene cytosine deaminase (CD) was constructed. The authors compared the extent, duration, and pattern of transgene (luc) expression in vivo after portal venous, intraperitoneal, or intravenous virus administration and survival after treatment with the vv containing CD followed by the prodrug 5-fluorocytosine (5-FC) in a murine model of disseminated liver metastases from colon cancer.
Methods
Recombinant vv containing the luc transgene within the thymidine kinase locus was administered to mice with isolated liver metastases from an MC38 adenocarcinoma. Transgene expression was determined in tumor and organs at various time points. Tumor-bearing mice were treated with recombinant vv containing CD and 5-FC or with appropriate controls and followed for survival.
Results
Tumor-specific gene delivery was achieved irrespective of administration route, with gene expression in tumors increased by up to 100,000-fold compared with normal tissues. There was significantly increased transgene expression in tumor after portal venous or intraperitoneal virus administration (p = 0.001 vs. systemic). Treatment using a CD-expressing vv and systemic 5-FC resulted in a significant survival benefit in all treatment groups compared with controls (p < 0.007); there was no additional benefit for portal venous or intraperitoneal virus administration.
Conclusions
Suicide gene therapy using vv with the CD/5-FC system leads to tumor-specific gene expression and improved survival and can result in cure of established liver metastases.
Colorectal cancer is one of the most common causes of cancer death worldwide: ≥500,000 people die from colorectal carcinoma each year. 1 Approximately 20% of patients have liver metastases at the time of diagnosis, 2 and even after curative resection of the primary tumor the liver is the most likely site of recurrence. 3,4 Cytotoxic chemotherapy has shown some palliative benefit, particularly regimens based on 5-fluorouracil (5-FU). 5 Overall, however, the outlook for patients with liver metastases from colon cancer is bleak, and alternative treatment methods are needed. 6
Cancer gene therapy approaches include expression of transgenes encoding cytokines, tumor antigens, and enzymes for enzyme/prodrug gene therapy. 7 Potentially attractive for liver metastases from colorectal cancer is regional enzyme/prodrug therapy. The “suicide gene therapy” concept involves a combination of a nonmammalian enzyme, cytosine deaminase (CD), and the nontoxic prodrug 5-fluorocytosine (5-FC), which is catalyzed by CD to the cytotoxic 5-FU. 8,9 When the CD gene is transferred to tumor cells, administered prodrug is converted selectively in tumors, thus eliminating toxicity to other organs not expressing the converter gene. 10,11 Therefore, tumor-specific gene delivery is of utmost importance for the effectiveness of this approach. 12
Recombinant vaccinia viruses are attractive candidates for cancer gene therapy vectors for a number of reasons. 13 Their safety profile in clinical use is well established, 14 they can carry large amounts of foreign DNA, 15 and they have shown tropism for tumor cells. 16,17 Recombinant thymidine kinase-deleted (TK−) vaccinia viruses have recently been shown to be efficient gene delivery vectors in vitro and in vivo. 18–20 However, it is not known whether selective tumor targeting by vaccinia virus can be substantially improved with regional versus systemic administration.
In this article, we describe the characteristics of the vaccinia vector system in which the transgene is inserted into the thymidine kinase (TK) locus and the CD/5-FC suicide gene therapy system is used for liver tumors, with particular emphasis on the impact of different routes of viral administration. Disseminated metastatic liver tumors of syngeneic colon carcinoma cells of C57BL/6 mice were established. We constructed recombinant vaccinia viruses expressing the firefly luciferase reporter gene and the Escherichia coli cytosine deaminase gene, respectively, and administered those viruses in vivo using different routes. The results show the feasibility of tumor-specific gene delivery and increased survival rates, with some permanent cures, in vivo using this vector system.
MATERIALS AND METHODS
Plasmids and Recombinant Viruses
The recombinant vaccinia viruses used are based on the previously described vaccinia shuttle plasmid pCB023II. 18 In short, the multiple cloning site of pBluescript KS II (Stratagene, La Jolla, CA) was inserted in the TK flanking region of pSC65 (kindly provided by B. Moss, Laboratory of Viral Diseases, NIAID, Bethesda, MD). An early termination signal was designed for all possible open reading frames and inserted just downstream of the left vaccinia virus TK flanking region to prevent antisense transcription from native vaccinia promoters. To facilitate selection of recombinant viruses, a gpt gene under the control of the p7.5 vaccinia early/late promoter was added. 21 The E. coli CD gene was amplified from bacterial genomic DNA and inserted into pCB023II using the pCRII cloning vector (Stratagene), resulting in pMP-CD. 18 A synthetic early/late promoter 22 controls the CD gene in this construct. All plasmid sequences were confirmed using automated sequencing (ABI Prism, Perkin Elmer Applied Biosystems, Norwalk, CT).
Recombination of vaccinia viruses (strain Western Reserve) was performed using CaCl precipitation. 23 Recombinant vaccinia viruses were subsequently isolated by mycophenolic acid selection, and the virus containing the CD gene was designated vvCD (Fig. 1A).

Figure 1. Recombinant vaccinia viruses. (A) vvCD recombinant vaccinia virus containing the E. coli CD gene. (B) vvLuc recombinant vaccinia virus containing the firefly luciferase gene. TK, vaccinia virus thymidine kinase gene locus; TF, termination fragment. The CD and luciferase genes are under the control of synthetic early/late promoter. The gpt gene for selection with mycophenolic acid is driven by the vaccinia virus p7.5 early/late promoter.
A previously described recombinant vaccinia virus expressing the firefly luciferase gene as reporter gene (vvLuc, Fig. 1B) was used for reporter gene experiments. 18
All recombinant viruses were amplified in HeLa S3 cells using spinner flasks. Virus titers were determined in standard plaque-forming assays using CV-1 cells. 23
Cell Lines
MC38 is a murine colon cancer line originally induced by the injection of dimethylhydrazine in C57BL/6 mice. 24 For the generation, amplification, and titration of recombinant vaccinia viruses, ovarian cancer HeLa S3 and monkey kidney cells CV-1 were purchased from the American Tissue Type Collection (ATTC, Manassas, VA). All cell lines were cultured in Dulbecco’s modified Eagle medium supplemented with 10% heat-inactivated fetal bovine serum, 2 mM L-glutamine, 1% penicillin/streptomycin, 1% gentamicin, and 0.2% amphotericin (Fungizone) (all from Biofluids, Rockville, MD) at 37°C in a 5% CO2 incubator.
Animal Care
Female C57BL/6 mice, 8 to 12 weeks old, were obtained from the NIH small animal facility. Animals had unlimited access to food and water and were housed at a maximum of five mice per cage. All animal protocols were approved by the Animal Care and Use Committee and conducted in strict compliance with the guidelines established by the NIH Animal Research Advisory Committee.
In Vivo Tumor Model of Liver Metastases
Liver metastases of MC38 adenocarcinoma were established using a modification of a previously described method. 25 Under sterile conditions, mice were anesthetized with ketamine (40 mg/kg; Fort Dodge, Fort Dodge, IA) and xylazine (12 mg/kg; Butler, Columbus, OH) given intraperitoneally. Animals were positioned in a right lateral position, and a left subcostal incision, approximately 6 mm long, was made. The lower pole of the spleen was exposed and completely exteriorized after division of the short gastric vessels and gastrosplenic ligament. The spleen was then positioned on a 3 × 3-cm piece of sterile gauze moisturized with phosphate-buffered saline. MC38 cells (2.5 × 105) were then slowly injected into the spleen in 200 μl HBSS with a 30-gauge needle (Becton Dickinson, Franklin Lakes, NJ). After removal of the needle, hemostasis was achieved by application of pressure to the spleen. After a delay of approximately 5 minutes to allow the tumor cells to enter the portal circulation, splenectomy was performed after ligature of the splenic pedicle with 4-0 silk (Ethicon, Somerville, NJ). Finally, the abdominal cavity was closed in one layer with 9-mm wound autoclips (Roboz Surgical, Rockville, MD). Animals were allowed to regain consciousness and were closely monitored after surgery.
The tumor model used is characterized by highly reliable tumor take (>99%). Untreated animals die from progressive liver metastases approximately 4 to 5 weeks after tumor inoculation.
Treatment Schedule and Administration of Recombinant Viruses and Drugs
In all experiments described, injection of the viral vector was performed 12 days after tumor inoculation. For the systemic treatment group, virus was injected into a lateral tail vein using a 30-gauge needle after vasodilatation using a heat lamp. The virus dose was adjusted to 500 μl of injection volume.
Vector application in the regional treatment groups was done by intraperitoneal injection with a 30-gauge needle. To ensure optimal distribution of the vector in the peritoneal cavity, the viral dose was adjusted to 2 ml of fluid volume.
For the local administration group, the vector was injected directly into the portal vein. For this purpose, animals were again anesthetized with ketamine (40 mg/kg) and xylazine (12 mg/kg) and placed in a supine position. A midline incision was made in the epigastrium, and the gastroduodenal ligament was exposed by application of slight sinistrodistal traction to the pylorus. The portal vein was identified using an operating microscope at sixfold magnification (Leica Microsystems, Deerfield, IL). The appropriate virus dose was injected into the portal vein under microscopic vision in 200 μl fluid using a 33-gauge microneedle (Roboz). Hemostasis was achieved by application of slight pressure for ≥1 minute. The abdominal wall was closed in one layer with 9-mm clips.
For the reporter gene experiments, sham laparotomies were performed in the systemic and regional administration groups.
Reporter Gene Experiments
For the reporter gene experiments, animals were injected with 108 pfu vvLuc using the different administration routes 12 days after tumor inoculation. Cohorts of animals were killed at days 2, 4, 6, 8, and 10 thereafter. Tumors and normal tissues (liver, kidneys, pancreas, uterus, ovary, bowel, heart, lung, muscle, skin, brain, and scar tissue) were harvested, immediately frozen, and stored at −70°C.
Firefly Luciferase Assay
Luciferase activity was assayed using a commercially available assay system (Promega, Madison, WI). Frozen tissue samples were briefly thawed, homogenized, and lysed in 750 μl of 1× reporter gene lysis buffer. Samples were centrifuged for 5 minutes at 13,000 rpm to pellet the cellular debris. A total of 10 μl of supernatant was added to 100 μl of luciferase assay reagent in an 8 × 50-mm disposable cuvette and immediately placed into a luminometer (TD-20/20, Turner, Sunnyvale, CA). After a 2-second delay, light emission was measured for 10 seconds. Each sample was measured in duplicate and mean values were recorded. The concentration of total protein in each sample was then determined using a bicinchoninic acid protein assay kit (Pierce, Rockford, IL) using a bovine serum albumin standard. All measurements were made in triplicate. The final firefly luciferase activity is expressed in relative light units per milligram protein.
In Vivo Treatment Experiments
For the in vivo treatment experiments, groups of animals were injected with 108 pfu vvCD or control vector vvLuc using the different administration routes 12 days after tumor inoculation. Beginning 2 days after virus administration, 5-FC was administered for a total of 7 days (daily dose 1180 mg/kg, administered intraperitoneally in 2 ml HBSS). Appropriate control groups (vector alone/no treatment, irrelevant vector/prodrug, no vector/prodrug, and no vector/no prodrug) were used in all experiments. Survival was the end point for all experiments; animals dying from other causes than tumor were considered censored for analysis. All animal experiments were repeated for confirmation of results.
Statistical Analysis
Data were calculated as mean ± standard deviation, and Student’s paired t test (two-sided) was used when appropriate. Survival analysis was performed using the Kaplan-Meier method. 26 Differences between subgroups were assessed using the log-rank test. 27
RESULTS
Validation of Tumor Model and Administration Techniques
Portal vein injections were performed in 92 mice for all experiments using the techniques described; of these, 80 (87%) were considered technically successful (complete injection of the intended amount of fluid [200 μl], no spillage of virus, no or minimal bleeding). All experiments were started with an excess number of animals to provide the intended group size even in case of technical problems.
Pattern of Reporter Gene Expression In Vivo
Groups of mice (n = 15 per group per time point) were killed at days 2, 4, 6, 8, and 10 after virus injection. Maximum gene expression in tumors was observed on day 4, irrespective of administration route (Tables 1, 2, and 3). The duration of transgene expression was limited to 1 week, presumably as a result of immune clearance of the vector. In general, tumor samples had a gene expression of at least three orders of magnitude greater than those of other tissues, providing evidence for the tumor-specific delivery of the transgene.
Table 1. LUCIFERASE GENE EXPRESSION AFTER INTRAVENOUS ADMINISTRATION OF 108 pfu vvLuc

* Accurate separation from tumor tissue is impossible in this model.
C57BL/6 mice (n = 15 per group time point) with 12-day-old liver metastases were injected intravenously with 108 pfu vvLuc on day 0. On days 2, 4, 6, 8, and 10 after virus administration, tumor and other tissues were harvested as described. Luciferase activity was determined and normalized to protein concentration. Data are given as mean of n = 15 in relative light units per milligram protein.
Table 2. LUCIFERASE GENE EXPRESSION AFTER INTRAPERITONEAL ADMINISTRATION OF 108 pfu vvLuc

* Accurate separation from tumor tissue is impossible in this model.
C57BL/6 mice (n = 15 per group time point) with 12-day-old liver metastases were injected intraperitoneally with 108 pfu vvLuc on day 0. On days 2, 4, 6, 8, and 10 after virus administration, tumor and other tissues were harvested as described. Luciferase activity was determined and normalized to protein concentration. Data are given as mean of n = 15 in relative light units per milligram protein.
Table 3. LUCIFERASE GENE EXPRESSION AFTER PORTAL VENOUS ADMINISTRATION OF 108 pfu vvLuc

* Accurate separation from tumor tissue is impossible in this model.
C57BL/6 mice (n = 15 per group time point) with 12-day-old liver metastases were injected with 108 pfu vvLuc administered directly into the portal vein on day 0. On days 2, 4, 6, 8, and 10 after virus administration, tumor and other tissues were harvested as described. Luciferase activity was determined and normalized to protein concentration. Data are given as mean of n = 15 in relative light units per milligram protein.
As shown in Table 1, gene expression in tumors was markedly increased over the entire duration of the experiment. The magnitude of expression differences between tumor and the next-highest expressing organ ranged from 40-fold (day 2) to 1700-fold (day 4). Although lung showed notable gene expression on day 2, the ovaries demonstrated the highest gene expression thereafter. Liver readings were disregarded because it is impossible in this model of diffuse metastases to harvest nontumor-bearing liver tissue reliably.
In general, gene expression in tumors was at least twice as high at all time points after intraperitoneal injection than in the intravenous group (see Table 2). Gene expression in other organs was similar. The minimum difference between tumor and other organs and tissues ranged from 330-fold (day 2) to 3700-fold (day 4).
Transgene expression in tumor was further increased after portal venous vector administration (see Table 3). Gene expression in other organs was almost negligible: differences between tumor and the highest normal organ were 32,600-fold (day 2), 120,000-fold (day 4), and 750-fold (day 6).
Transgene Expression in the Different Vector Administration Routes
As shown in Figure 2, portal venous and intraperitoneal delivery of the virus yielded the greatest maximal gene expression in tumors (day 4, p = 0.001 vs. systemic). There was no significant difference between the group with intraperitoneal administration versus direct portal venous injections (p NS, day 4), but an overall trend toward higher maximal luciferase expression in tumor in the portal venous treatment group was observed. Further, gene expression in other organs was lower after portal venous virus delivery, indicating a potentially beneficial widening of the therapeutic window. For example, gene expression in ovary on day 4 was 108 pfu local versus 2544 regional versus 2765 for systemic virus administration (p < 0.01).
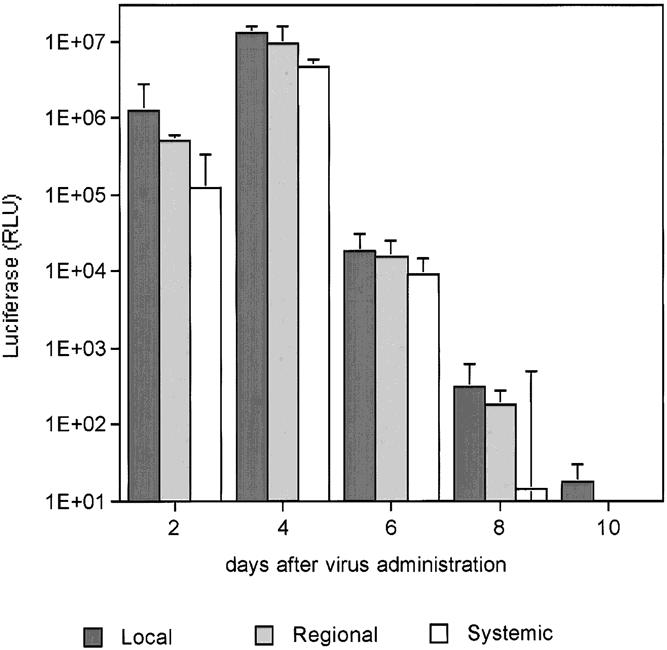
Figure 2. Luciferase expression in liver tumors after portal venous, intraperitoneal, and intravenous delivery of vvLuc. C57BL/6 mice with MC38 liver metastases were treated with 108 vvLuc. Vector was administered intravenously (systemic group), intraperitoneally (regional), or directly into the portal vein (local). Groups of 15 animals were killed at the time points given, and tumor and selected tissues were harvested and processed as described. Luciferase content is given in relative light units per milligram total protein. All data are presented as mean and SD of n = 15 per time point.
Impact on Survival
The liver metastases model was used for in vivo treatment experiments. In an attempt to make optimal use of the expression pattern of transgenes observed in the reporter gene experiments, prodrug treatment was given on days 2 to 8 after virus delivery. The maximum tolerated dose of 5-FC, 1180 mg/kg, was administered daily intraperitoneally. As shown in Figure 3, the treatment groups (vvCD/5-FC) had significantly better survival than the control groups (p < 0.001). The median survival was increased by 40% in treatment animals (47 vs. 33 days for controls). There was, however, no difference in survival when vv was administered using the intravenous, intraperitoneal, or portal venous route (p = 0.8). There was also no difference in survival between the multiple control groups. In all treatment groups, a proportion of animals survived >100 days and were considered cured. The cure rate was 17% in the local group (5/30 animals), 14% in the systemic group (3/22), and 13% in the regional group (4/32). There were no cures in any control group (Fig. 4).
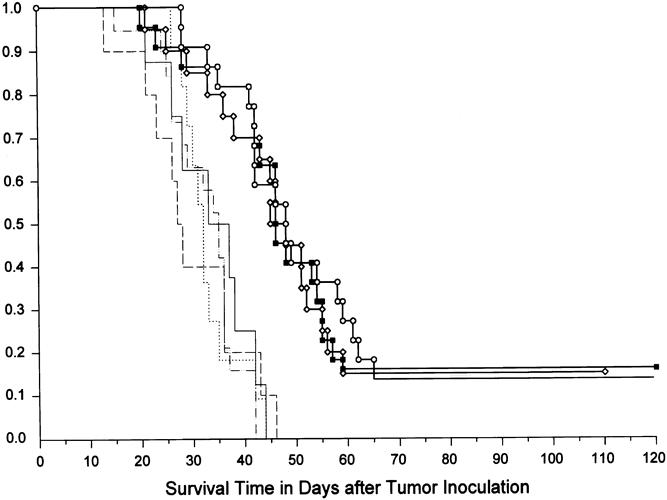
Figure 3. Survival after virus delivery using the different administration routes. At day 0, tumor-bearing mice were challenged with 108 pfu vvCD. The virus was administered intravenously, intraperitoneally, or directly into the portal vein. Prodrug treatment was given on days 2 to 8 (5-FC 1180 mg/kg/day intraperitoneally). Control groups included animals treated with irrelevant vector vvLuc and 5-FC, 5-FC alone, vvCD alone, and completely untreated controls. The control groups using regional virus administration are shown; there was no difference between control with local or systemic injection. Autopsies were performed on all mice to assess the cause of death. Mice surviving 120 days were considered cured; they were killed and underwent autopsy to confirm complete tumor regression. The three treatment groups yielded significantly better survival (p > 0.001) than the controls. There was no significant difference between the different virus administration routes. Squares, vvCD-local/5-FC treatment group (n = 30); circles, vvCD-regional/5-FC treatment group (n = 32); diamonds, vvCD-systemic/5-FC treatment group (n = 22); dots, vvCD-109/no prodrug vector alone control (n = 15); solid line, no vector/5-FC prodrug alone control (n = 12); dashed line, vvLuc/5-FC irrelevant vector control (n = 10); dots and dashes, no vector/no treatment negative control (n = 13).
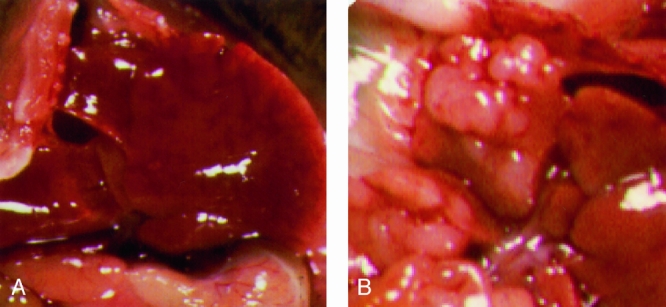
Figure 4. Representative photographs of livers from a treatment animal after virus injection versus control animal. Two representative animals are shown 14 days after intraperitoneal virus administration and subsequent 7-day treatment with 5-FC. Animal A is a treatment animal (vvCD given intraperitoneally) without visible liver metastases. The control animal, B (vvLuc), shows multiple tumor nodules.
DISCUSSION
In this study, data characterizing the properties of a recombinant vaccinia virus vector for suicide gene therapy of liver metastases were presented. We performed a series of experiments evaluating the effect of different routes of administration on gene expression and treatment efficacy in an in vivo tumor model setting using a TK− viral construct as vector. It has been shown in experimental gene therapy that tumor-specific gene delivery is the most important factor for success of in vivo treatments. 7,12,28 The presence of an artificially introduced gene within tumor cells and its absence in normal tissues provide the theoretical and practical basis for the validity of the suicide gene therapy approach. 29–31
Localized vector delivery has been used to achieve or increase transgene expression in tumor in different gene therapy settings. 32–35 Although systemic vector delivery may be the best option in many clinical settings, the unique anatomic features of the liver facilitate regional treatment approaches for unresectable hepatic metastases; thus, the liver has been proposed for the use of experimental gene therapy. 34 It has recently been shown that recombinant TK− vaccinia virus will traffic to tumor in rabbits 20 and mice. 19 However, there is also substantial gene expression in other tissues; this raises the possibility of treatment toxicity in the clinical setting. There is, however, no information yet available about the characteristics of recombinant vaccinia viral vectors in regionally administered gene therapy. The data from this study show that transgene expression in tumor can be significantly augmented by intraperitoneal or portal venous administration compared with systemic. However, transgene expression was so much greater in tumor than in other tissues after any route of administration that there was no additional therapeutic benefit with regional treatment using this construct under the conditions tested. Because gene expression in other organs is very low after regional administration of virus, refinements in treatment, such as prolonged prodrug administration or retreatment with regional administration of virus, may improve efficacy without additional toxicity.
Vector delivery of virus directly into the portal vein and intraperitoneally markedly increased maximum gene expression within tumors compared with intravenous delivery, suggesting an additional mechanism for the uptake of vaccinia into tumor cells, such as the existence of specific receptors on tumor cell surfaces. Although gene expression in other organs was similar after intravenous and intraperitoneal virus administration, it was markedly decreased after portal venous virus injection, supporting a first-pass uptake mechanism within tumors. The ovaries showed relatively high transgene expression compared with other organs; expression was lowest after portal venous delivery. Although accurate determination of gene expression in normal liver tissue is impossible in a liver metastasis model, there is no indication for hepatotropism of vaccinia virus, unlike adenovirus. 36
The mechanism of specific vaccinia infection of tumor cells is not fully understood. 16 There are several possible explanations for the observed tumor specificity, some of which can potentially be exploited by locoregional delivery strategies. The large size of the vaccinia virus may increase its uptake through tumor vasculature, which is generally considered more permeable than normal blood vessels. 15 In our view, however, the deletion of the vaccinia virus TK gene leads to the dependence of viral replication on host cell nucleotides and thus to predominant amplification in tumor cells. Also, viral binding to cell surfaces is highly increased by the presence of heparin sulfate proteoglycans, 37 which are present in high concentration within aggressively growing tumors. 38
In our in vivo treatment experiments, we used a recombinant TK− vaccinia virus expressing the E. coli CD gene and systemic treatment with 5-FC. This converter enzyme-prodrug system has been studied intensively in cancer gene therapy 7,12,36,39–44 and has a marked bystander effect. 9 This means that even with a limited proportion of tumor cells actually expressing the transgene, 12,45 a sufficient amount of 5-FC will be converted to 5-FU to kill nontransduced neighboring tumor cells. 46
It is difficult to predict whether the impressive tumor specificity of TK− vaccinia viruses will also be seen with the vector system in the clinical setting. Recombinant vaccinia viruses have been used in phase I clinical trials to induce specific immune responses against generalized tumors 47 and in other gene therapy fields. 48 Vaccinia virus induces a considerable immune response that induces long-lasting immunity. 15 Although this may preclude repeated application of the same viral vector in a given patient, 49 it also provides a safety benefit by virtually eliminating the likelihood of systemic infections with vaccinia virus in immunocompetent human hosts. In fact, there was no vector-associated clinical toxicity reported in several phase I gene therapy trials using recombinant vaccinia viruses. 50,51
In summary, we have demonstrated tumor-specific in vivo gene delivery using the TK− vaccinia virus system. Portal venous application of the viral vector led to an optimal transgene expression pattern and tumor specificity, compared with intraperitoneal and intravenous gene delivery, but did not translate into additional survival benefit. Overall, tumor-specific gene expression led to a significant survival benefit in an in vivo suicide gene/prodrug setting, including permanent cure of diffuse liver metastases from colon cancer in approximately one sixth of the animals. These results indicate that the TK− vaccinia virus system may be capable of sufficient tumor-selective gene transfer even without local vector application strategies. Modification of the treatment modalities, particularly prolonging transgene expression, may lead to further improvement of survival results and encourage the use of this system in clinical trials.
Acknowledgments
The authors thank Dr. Steven Rosenberg for helpful comments and Barbara Owen for technical assistance with the manuscript.
Discussion
Dr. Larry R. Kaiser (Philadelphia, Pennsylvania): I congratulate Dr. Alexander and his colleagues on this elegant and carefully performed study utilizing a recombinant vaccinia vector. This work, though preliminary, further expands our knowledge of regional and local gene therapy for malignant disease and suggests the potential utility of systemic administration of a recombinant vector that has tropism for tumors.
Indeed the holy grail of the prodrug approach to gene therapy for malignant disease is the possibility of systemic delivery of a vector carrying a transgene of choice that will home to all sites where tumor is present, and efficiently transduce the tumor, with resultant tumor cell killing. This approach requires that the vector be easily produced, cause little or no toxicity to normal tissue, and be targeted to tumor specifically.
Certain malignancies or manifestations of metastatic disease may lend themselves to a local or regional delivery approach. The experimental work and clinical trials of gene therapy for cancer to date mostly have relied on local or regional gene delivery to establish proof of principle. In our own work, we chose to use malignant mesothelioma, a tumor that for the most part remains confined to the pleural space, for our initial clinical trial after extensive experimental work that demonstrated the feasibility of intrapleural gene delivery.
Hepatic metastatic disease from colon cancer also lends itself to a local or regional gene delivery approach while we continue to make progress toward systemic delivery. Dr. Alexander and his colleagues have nicely demonstrated a large therapeutic ratio in gene expression between tumors and normal tissue no matter the route of administration. It seems that a prodrug model system using the vaccinia virus vector construct detailed for us today may be a significant improvement over an adenoviral construct.
I have several questions for you. First, have you performed any experiments to characterize the effect of the immune response directed toward the vaccinia vector? This likely will be important in the clinical situation.
Is there enhanced efficacy in immune-deficient animals or is the immune system important for the responses seen? Have you tried other colon tumor cell lines, or for that matter, other tumors, to assure that this vector construct has wide applicability?
Could you also comment for us on the so-called bystander effect, which is so important to efficacy when using an adenoviral construct? Do you think the vector itself plays a role in any observed bystander effect?
I was struck by the fact that your long-term cure rate remains less than 15% in your treated animals despite generous tumor transgene expression. To what do you attribute this disparity?—lack of adequate substrate, perhaps, because of toxicity? An antiapoptotic mechanism? Please comment.
Finally, would you care to elaborate on the possible mechanism by which this recombinant vaccinia vector targets tumor?
As with any excellent study, this one provokes lots of questions that should lead to further work, and I expect we will be hearing more from Dr. Alexander and his colleagues.
Presenter Dr. H. Richard Alexander (Bethesda, Maryland): Thank you, Dr. Kaiser, for your comments and your questions.
With respect to the role of immunization, that is a very important issue in the use of a vaccinia viral vector. As you know, the world’s population was subjected to widespread immunization for smallpox up until 1980, so that adults less than the age of 25 have not been routinely immunized but those that are older have. This poses a problem with the feasibility of using a vaccina virus in previously immunized individuals.
We have found that in immunocompromised mice, the results with this type of treatment are enhanced, suggesting that prolonged infection within tumor exists, although we still have to expand on the initial observations. We are also working on a model of immunizing immunocompetent mice and determining whether or not this kind of a vector can still work, perhaps inducing a brief period when the host is immunosuppressed prior to vaccinia viral therapy.
With respect to the bystander effect in vitro, the suicide gene system that we are using can induce significant cytotoxicity when only 1% to 5% of cells have been infected. We also believe that using a replication-competent vector allows for progressive infection of tumor cells in vivo, and that is a real advantage of this vector system.
We have tried this vector in vitro against a variety of tumor cells. It does work against a broad spectrum of histologies, it is not specific for the one cell type that we used in these experiments.
The vaccinia virus alone appears to have minimal cytopathic effect. Although others have shown that vaccinia virus can cause a significant cytopathic effect and reduction in tumor size, we have not seen that. To address your question about tumor targeting, we are inserting DNA into the thymidine kinase locus of the vaccinia virus, which markedly attenuates its ability to infect normal tissues. For reasons that we don’t fully understand, it will continue to proliferate and infect neoplastic tissue. We are evaluating the mechanism for that, but I don’t have an explanation for you today.
With respect to the low cure rates, I agree with you that further refinements in this treatment are necessary. I believe that modification in the treatment strategy, including using a second treatment or a more prolonged prodrug administration, will potentially improve survival.
Dr. Patricia K. Donahoe (Boston, Massachusetts): Dr. Alexander, since your vector includes the thymidine kinase, would you consider combining gene therapy with ganciclovir? Coexpression of two viral vectors often diminishes the transfection efficiency of the gene of interest. Have you seen that in these experiments? What do you propose will be your first clinical application?
Dr. Alexander: With respect to your first question, there are limited data available using a viral vector to deliver two therapeutic suicide genes in vivo. The precedent has been set in vitro and I believe that it invites one to consider doing that in this setting. A two suicide gene construct using systems that have different mechanisms of action would potentially result in an additive or synergistic effect in vivo and it is something that we are exploring. The vaccinia viral vectors will accommodate a large amount of foreign DNA without requiring any viral deletions and are ideally suited for two genes to be inserted and work in tandem.
With respect to your last question regarding the clinical applicability, that is a much more difficult one. We are interested in applying this therapy in the clinical setting, perhaps using a regional approach for peritoneal disease or isolated liver metastases. But there will be a number of safety issues that we have to satisfy before we are able to move this into the clinical setting. In addition, we are designing and using new constructs that may have improved tumor targeting characteristics.
Footnotes
Correspondence: H. Richard Alexander, Jr., MD, FACS, Surgical Metabolism Section, Surgery Branch, National Cancer Institute, National Institutes of Health, Bldg. 10 Rm. 2B07, 9000 Rockville Pike, Bethesda, MD 20892.
Presented at the 119th Annual Meeting of the American Surgical Association, April 15–17, 1999, Hyatt Regency Hotel, San Diego, California.
Dr. Gnant is the recipient of a research grant by the Max Kade Foundation, New York.
Accepted for publication April 1999.
References
- 1.Pisani P, Parkin DM, Ferlay J. Estimates of the worldwide mortality from eighteen major cancers in 1985. Implications for prevention and projections of future burden. Int J Cancer 1993; 55: 891–903. [DOI] [PubMed] [Google Scholar]
- 2.Bengmark S, Hafstrom L. The natural history of primary and secondary malignant tumors of the liver. I. The prognosis for patients with hepatic metastases from colonic and rectal carcinoma by laparotomy. Cancer 1969; 23: 198–202. [DOI] [PubMed] [Google Scholar]
- 3.Zavadsky KE, Lee YT. Liver metastases from colorectal carcinoma: incidence, resectability, and survival results. Am Surg 1994; 60: 929–933. [PubMed] [Google Scholar]
- 4.Bismuth H, Adam R, Navarro F, Castaing D, Engerran L, Abascal A. Re-resection for colorectal liver metastasis. Surg Oncol Clin North Am 1996; 5: 353–364. [PubMed] [Google Scholar]
- 5.van der Vilt CL, Smid K, Aherne GW, Noordhuis P, Peters GJ. Biochemical mechanisms of interferon modulation of 5-fluorouracil activity in colon cancer cells. Eur J Cancer 1997; 33: 471–478. [DOI] [PubMed] [Google Scholar]
- 6.Tannock IA. Conventional cancer therapy: promise broken or promise delayed? Lancet 1998; 351: SII9–SII16 [DOI] [PubMed] [Google Scholar]
- 7.Zwacka RM, Dunlop MG. Gene therapy for colon cancer. Gene Ther 1998; 12: 595–615. [DOI] [PubMed] [Google Scholar]
- 8.Rowley S, Lindauer M, Gebert JF, et al. Cytosine deaminase gene as a potential tool for the genetic therapy of colorectal cancer. J Surg Oncol 1996; 61: 42–48. [DOI] [PubMed] [Google Scholar]
- 9.Huber BE, Richards CA, Austin EA. Virus-directed enzyme/prodrug therapy (VDEPT). Ann NY Acad Sci 1994; 716: 104–114. [DOI] [PubMed] [Google Scholar]
- 10.Gabel M, Kim HJ, Kolozsvary A, Khil M, Freytag S. Selective in vivo radiosensitization by 5-fluorocytosine of human colorectal carcinoma cells transduced with the E. coli cytosine deaminase (CD) gene. Int J Radiat Oncol Biol Phys 1998; 41: 883–887. [DOI] [PubMed] [Google Scholar]
- 11.Huber BE, Austin EA, Good SS, Knick VC, Tibbels S, Richards CA. In vivo antitumor activity of 5-fluorocytosine on human colorectal carcinoma cells genetically modified to express cytosine deaminase. Cancer Res 1993; 1: 4619–4626. [PubMed] [Google Scholar]
- 12.Singhal S, Kaiser LR. Cancer chemotherapy using suicide genes. Surg Oncol Clin North Am 1998; 7: 505–536. [PubMed] [Google Scholar]
- 13.Peplinski GR, Tsung K, Norton JA. Vaccinia virus for human gene therapy. Surg Oncol Clin North Am 1998; 7: 575–588. [PubMed] [Google Scholar]
- 14.Fenner F, Henderson DA, Aritz I, Jezek Z, Ladnyi ID. Smallpox and its eradication. Geneva: World Health Organization; 1988.
- 15.Moss B. Genetically engineered poxviruses for recombinant gene expression, vaccination, and safety. Proc Natl Acad Sci USA 1996; 93: 11341–11348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Peplinski GR, Tsung K, Whitman ED, Meko JB, Norton JA. Construction and expression in tumor cells of a recombinant vaccinia virus encoding human interleukin-1β. Ann Surg Oncol 1995; 2: 151–159. [DOI] [PubMed] [Google Scholar]
- 17.Peplinski GR, Tsung K, Meko JB, Norton JA. Prevention of murine breast cancer by vaccination with tumor cells modified by cytokine-producing recombinant vaccinia viruses. Ann Surg Oncol 1996; 31: 15–23. [DOI] [PubMed] [Google Scholar]
- 18.Puhlmann M, Brown CK, Gnant M, et al. Vaccinia as a vector for tumor directed gene therapy: biodistribution of a thymidine kinase deleted mutant. Cancer Gene Ther 1999 (in press). [DOI] [PubMed]
- 19.Gnant MFX, Puhlmann M, Alexander HR Jr, Bartlett DL. Systemic administration of a recombinant vaccinia virus expressing the cytosine deaminase gene and subsequent treatment with 5-fluorocytosine leads to tumor specific gene expression and cures of established liver metastases. Cancer Res 1999 (in press). [PubMed]
- 20.Gnant M, Noll L, Irvine KR, et al. Tumor-specific gene delivery using recombinant vaccinia virus in a rabbit model of unresectable liver metastases: pattern of gene expression, vector elimination and immune response. JNCI 1999 (in press). [DOI] [PubMed]
- 21.Davison AJ, Moss B. Structure of vaccinia virus early promoters. J Mol Biol 1989; 210: 749–769. [DOI] [PubMed] [Google Scholar]
- 22.Davison AJ, Moss B. Structure of vaccinia virus late promoters. J Mol Biol 1989; 210: 771–784. [DOI] [PubMed] [Google Scholar]
- 23.Earl PL, Moss B. Current protocols in molecular biology. In: Ausubel FM, Kinston R, Kingston RE, et al., eds. Current protocols in molecular biology. New York: Greene/Wiley Interscience; 1991: 16.15.1–16.18.10.
- 24.Corbett TH, Griswold DP, Roberts BJ. Tumor induction relationships in development of transplantable cancers of the colon in mice for chemotherapy assays, with a note on carcinogen structure. Cancer Res 1975; 35: 2434–2439. [PubMed] [Google Scholar]
- 25.Lafreniere G, Rosenberg SA. A novel approach to the generation and identification of experimental hepatic metastases in a murine model. JNCI 1986; 76: 309–322. [PubMed] [Google Scholar]
- 26.Kaplan EL, Meier P. Nonparametric estimation from incomplete observation. J Am Stat Assoc 1958; 53: 457–481. [Google Scholar]
- 27.Mantel N. Evaluation of survival data and two new rank-order statistics arising in its consideration. Cancer Chem Rep 1966; 50: 163–170. [PubMed] [Google Scholar]
- 28.Dachs GU, Dougherty GJ, Stratford IJ, Chaplin DJ. Targeting gene therapy to cancer: a review. Oncol Res 1997; 9: 313–325. [PubMed] [Google Scholar]
- 29.Moolten FL. An alternative to the magic bullet paradigm for specific cancer therapy. Med Hypotheses 1987; 24: 43–51. [DOI] [PubMed] [Google Scholar]
- 30.Moolten FL, Wells JM, Heyman RA, Evans RM. Lymphoma regression induced by ganciclovir in mice bearing a herpes thymidine kinase transgene. Hum Gene Ther 1990; 1: 125–134. [DOI] [PubMed] [Google Scholar]
- 31.Huber BE, Richards CA, Krenitsky TA. Retroviral-mediated gene therapy for the treatment of hepatocellular carcinoma: an innovative approach for cancer therapy. Proc Natl Acad Sci USA 1991; 15: 8039–8043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ju DW, Cao X, Acres B. Intratumoral injection of GM-CSF gene encoded recombinant vaccinia virus elicits potent antitumor response in a murine melanoma model. Cancer Gene Ther 1997; 4: 139–144. [PubMed] [Google Scholar]
- 33.Bass C, Cabrera G, Elgavish A, et al. Recombinant adenovirus-mediated gene transfer to genitourinary epithelium in vitro and in vivo. Cancer Gene Ther 1995; 2: 97–104. [PubMed] [Google Scholar]
- 34.de Roos WK, Fallaux FJ, Marinelli AWKS, et al. Isolated-organ perfusion for local gene delivery: efficient adenovirus-mediated gene transfer into the liver. Gene Ther 1997; 4: 55–62. [DOI] [PubMed] [Google Scholar]
- 35.Lee SS, Einsenlohr LC, McCue PA, Mastrangelo MJ, Lattime EC. Intravesical gene therapy: in vivo gene transfer using recombinant vaccinia virus vectors. Cancer Res 1994; 54: 3325–3328. [PubMed] [Google Scholar]
- 36.Topf N, Worgall S, Hackett NR, Crystal RG. Regional pro-drug gene therapy: intravenous administration of an adenoviral vector expressing the E coli cytosine deaminase gene and systemic administration of 5-fluorocytosine suppresses growth of hepatic metastasis of colon carcinoma. Gene Ther 1998; 5: 507–513. [DOI] [PubMed] [Google Scholar]
- 37.Chung CS, Hsiao JC, Chang YS, Chang W. A27L protein mediates vaccinia virus interaction with cell surface heparin sulfate. J Virol 1998; 72: 1577–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brunner G, Reimbold K, Meissauer A, Schirrmacher V, Erkell LJ. Sulfated glycosaminoglycans enhance tumor cell invasion in vitro by stimulating plasminogen activation. Exp Cell Res 1998; 239: 301–310. [DOI] [PubMed] [Google Scholar]
- 39.Martin LA, Vile R, Lemoine NR, Sikora K, Pandha HS. Genetic prodrug activation therapy. Lancet 1997; 350: 1793–1794. [DOI] [PubMed] [Google Scholar]
- 40.Trinh QT, Austin EA, Murray DM, Knick VC, Huber BE. Enzyme/prodrug gene therapy: comparison of cytosine deaminase/5-fluorocytosine versus thymidine kinase/ganciclovir enzyme/prodrug systems in a human colorectal carcinoma cell line. Cancer Res 1995; 55: 4808–4812. [PubMed] [Google Scholar]
- 41.Richards CA, Austin EA, Huber BE. Transcriptional regulatory sequences of carcinoembryonic antigen: identification and use with cytosine deaminase for tumor-specific gene therapy. Hum Gene Ther 1995; 6: 881–893. [DOI] [PubMed] [Google Scholar]
- 42.Moss B. Replicating and host-restricted non-replicating vaccinia virus vectors for vaccine development. Dev Biol Stand 1994; 82: 55–63. [PubMed] [Google Scholar]
- 43.Hirschowitz EA, Ohwada A, Pascal WR, Russi TJ, Crystal RG. In vivo adenovirus-mediated gene transfer of the Escherichia coli cytosine deaminase gene to human colon carcinoma-derived tumors induces chemosensitivity to 5-fluorocytosine. Hum Gene Ther 1995; 6: 1055–1063. [DOI] [PubMed] [Google Scholar]
- 44.Mullen CA, Petropoulos D, Lowe RM. Treatment of microscopic pulmonary metastases with recombinant autologous tumor vaccine expressing interleukin 6 and Escherichia coli cytosine deaminase suicide genes. Cancer Res 1996; 56: 1361–1366. [PubMed] [Google Scholar]
- 45.Zhang J, Russell SJ. Vectors for cancer gene therapy. Cancer Metastasis Rev 1996; 15: 385–401. [DOI] [PubMed] [Google Scholar]
- 46.Caruso M, Panis Y, Gagandeep S, Houssin D. Regression of established macroscopic liver metastases after in situ transduction of a suicide gene. Proc Natl Acad Sci USA 1993; 90: 7024–7028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim CJ, Cormier J, Roden M, et al. Use of recombinant poxviruses to stimulate anti-melanoma T cell reactivity. Ann Surg Oncol 1998; 5: 64–76. [DOI] [PubMed] [Google Scholar]
- 48.Dolin R. Human studies in the development of human immunodeficiency virus vaccines. J Infect Dis 1995; 172: 1175–1183. [DOI] [PubMed] [Google Scholar]
- 49.Paoletti E. Applications of pox virus vectors to vaccination: an update. Proc Natl Acad Sci USA 1996; 93: 11349–11353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McAneny D, Ryan CA, Beazley RM, Kaufman HL. Results of a phase I trial of a recombinant vaccinia virus that expresses carcinoembryonic antigen in patients with advanced colorectal cancer. Ann Surg Oncol 1996; 3: 495–500. [DOI] [PubMed] [Google Scholar]
- 51.Cooney EL, Collier AC, Greenberg PD, et al. Safety of an immunological response to a recombinant vaccinia virus vaccine expressing HIV envelope glycoprotein. Lancet 1991; 337: 567–572. [DOI] [PubMed] [Google Scholar]