

Abstract
Objective
Pancreatic mucinous cystic neoplasms (MCNs) provide a spectrum of neoplastic changes ranging from benign to malignant. The authors have correlated K-ras mutations and p53 overexpression with the evolution of these tumors.
Methods
Areas of mild, moderate, or severe dysplasia were microdissected from paraffin-embedded tissue sections of 28 different MCNs (10 benign, 9 borderline, 9 malignant). Nonneoplastic pancreatic ducts were also microdissected from tissues adjacent to the tumors. Ten serous cystadenomas served as negative controls. K-ras codon 12 mutations were identified by a mutant-enriched nested polymerase chain reaction-restriction fragment length polymorphism assay and confirmed by sequencing. p53 overexpression was demonstrated by immunohistochemistry.
Results
K-ras mutations were detected in 20% of benign, 33% of borderline, and 89% of malignant MCNs. Histologically, mutations were found in 26% (7/27) of MCN epithelia with mild dysplasia, 38% (5/13) of MCN epithelia with moderate dysplasia, and 89% (8/9) of MCN epithelia with severe dysplasia or carcinoma. Ten percent (4/39) of nonneoplastic pancreatic ducts at the margins of MCN harbored mutations, all associated with borderline or malignant tumors. Overexpression of p53 occurred in none of the benign or borderline MCNs but in 44% (4/9) of the malignant tumors (p = 0.006 benign/borderline vs. malignant). p53 immunoreactivity was concentrated in areas of severe dysplasia/carcinoma or invasion, where K-ras mutation had been detected.
Conclusion
These findings demonstrate a sequential accumulation of genetic changes in the carcinogenesis of MCN. K-ras mutations appear early and increase in proportion with increasing dysplasia. Overexpression of p53 is a late finding observed only in carcinomas, and in combination with mutated K-ras genes. The presence of K-ras mutations in nonneoplastic ducts supports formal pancreatic resection over enucleation for treatment. Mucinous cystic neoplasms may be a useful model to study the evolution of pancreatic ductal adenocarcinomas, in which precursor lesions remain unknown.
Since the landmark paper of Compagno and Oertel, 1 cystic neoplasms of the pancreas have been increasingly reported in the medical literature. 2–4 Pancreatic cystic neoplasms are relatively uncommon, accounting for 4% to 6% of exocrine pancreatic tumors. Although there is a wide variety of cystic neoplasms, the majority (>90%) are accounted for by mucinous cystic neoplasms (MCNs) and serous cystadenomas. 5 Other tumors with a cystic appearance, such as intraductal papillary mucinous tumors (IPMTs), are now identified as separate pathologic entities. 6 Intraductal papillary mucinous tumors cause cystic dilatation at ducts but do not form true cysts. Among the truly cystic neoplasms, MCNs and serous cystadenomas differ markedly in their malignant potential. 1 Serous tumors almost always remain benign and may be managed by observation when asymptomatic. In contrast, MCNs are regarded as premalignant lesions with significant potential for invasion and metastasis; therefore, surgical resection is advocated as the primary treatment modality.
As opposed to the uniformly aggressive nature of ductal adenocarcinomas, MCNs display a clinical and histologic spectrum ranging from clearly benign to frankly malignant. 3 Accordingly, the World Health Organization subdivides MCNs into benign, borderline, or malignant (mucinous cystadenocarcinomas) variants based on increasing levels of dysplasia within tumors. 7 The presence of a continuum of premalignant lesions in MCNs differentiates them from ductal adenocarcinoma, where precursor lesions remain poorly defined. This property makes MCNs a potential model in which to study factors involved in tumor progression and malignant transformation. 8
Carcinogenesis involves the accumulation of genetic defects over time. 9 Genetic events described in the pathogenesis of pancreatic neoplasms include mutations in the K-ras, p16, p53, and DPC4 genes, among others. 10–12 The K-ras gene encodes a 21-kd protein that functions as a second messenger in signal transduction. 13 Point mutations, particularly within codon 12, can result in malignant transformation. 14,15 Activating K-ras oncogene mutations are present in 70% to 90% of pancreatic adenocarcinomas. 16 Tumors with a more favorable prognosis, such as IPMTs, have also been shown to be mutated at this gene. 17 Sporadic reports have documented the presence of K-ras mutations in a percentage of malignant MCNs, 18 but no data are available with regard to benign or borderline tumors.
The most commonly mutated tumor suppressor gene in human cancer is p53. 19 The normal p53 product plays a role in a variety of cellular functions, including regulation of transcription, DNA repair, and apoptosis. 20 Studies on pancreatic ductal adenocarcinoma and IPMT suggest that up to 50% of tumors contain inactivating p53 mutations. 21,22 Overexpression of the p53 protein has been shown to correlate with point mutation in the p53 gene in various neoplasms. 23–26 Wild-type p53 protein has a short half-life (5 to 45 minutes), but mutation leads to protein stabilization, resulting in intranuclear accumulation. 19,27 Immunohistochemical assays of mutant p53 are based on the ability to detect the stable mutated product but not the wild-type protein.
In this study, we have analyzed the presence of K-ras mutations and p53 overexpression in MCNs of the pancreas. Our principal goal was to investigate whether these genetic events correlate with the progression of these tumors to malignancy. We hope our data will provide some insights into some of the early events in pancreatic carcinogenesis.
MATERIALS AND METHODS
Paraffin-embedded tissue sections from 28 MCNs of the pancreas resected between 1988 and 1998 were collected from the surgical pathology files at the Massachusetts General Hospital. Tissue sections were reviewed by two pathologists (CCC, DZT), and each tumor was classified as benign, borderline, or malignant according to the criteria of the World Health Organization. 7 This scheme classifies tumors according to their maximal grade of dysplasia. Benign MCNs show no dysplasia or at most mild dysplasia, with slightly enlarged basal nuclei and no mitotic figures (Fig. 1A, B). Borderline MCNs exhibit moderate dysplasia, characterized by epithelial pseudostratification, nuclear crowding with atypia, and rare mitotic figures. Malignant noninvasive tumors (noninvasive mucinous cystadenocarcinomas) have severe dysplasia or carcinoma in situ, changes that can be focal and require careful examination of multiple tumor areas. Malignant epithelial changes include frequent mitoses, nuclear stratification into cell apices, nuclear pleomorphism, decreased mucin production, loss of cell polarization, and/or loss of epithelial–stromal contact (Fig. 1C). If stromal invasion is present, the tumor is classified as an invasive mucinous cystadenocarcinoma (Fig. 1D). The study sample included 10 benign, 9 borderline, and 9 malignant MCNs (4 invasive and 5 noninvasive).
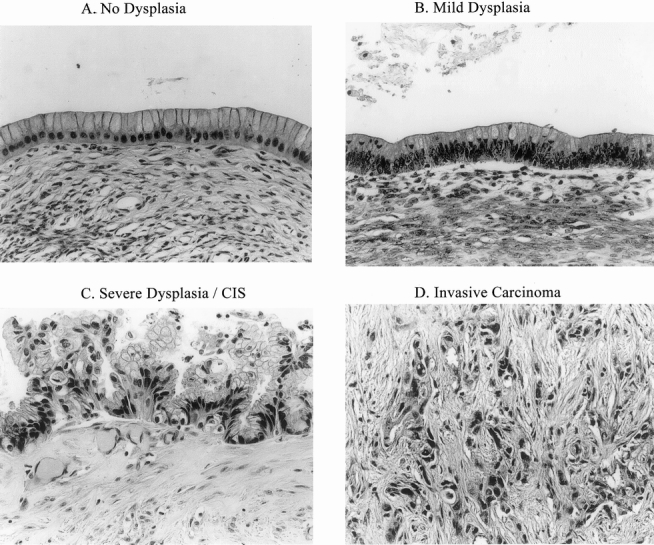
Figure 1. Spectrum of lesions microdissected from mucinous cystic neoplasms. (A) Cyst epithelium without dysplasia. (B) Epithelium with mild dysplasia. (C) Epithelium with severe dysplasia. (D) Stromal invasion by malignant epithelial islands.
Negative control specimens (wild-type gene) for the K-ras assay included white blood cells from the peripheral blood of a healthy person and paraffin-embedded tissue from 10 serous cystadenomas. The absence of K-ras mutations in serous cystadenomas has been documented in previous reports. 28 Positive control tissue for the K-ras assay was obtained from AspC-1 cells (American Tissue Culture Collection, Rockville, MD), which are known to have a mutated K-ras gene. 29 Paraffin-embedded tissue sections from a poorly differentiated human colon carcinoma were used as positive controls in the p53 immunohistochemistry protocol.
Specimen Microdissection
Three consecutive 10-μm sections were prepared from each paraffin-embedded tissue block and labeled by the study pathologists for neoplastic epithelium with or without dysplasia. Additionally, nonneoplastic pancreatic ducts surrounding MCN were identified and marked for the presence or absence of dysplasia. The unstained 10-μm sections were microdissected under an operating microscope at 30× magnification. Designated epithelium was carefully selected with minimal inclusion of associated fibrous reactive tissue. Disposable fine-tipped scalpels were used for microdissection to prevent contamination. At the conclusion of the procedure, the 10-μm slides were stained with hematoxylin and eosin to confirm the correctness of the dissection.
DNA Extraction
Microdissected samples were deparaffinized in xylene and gradually rehydrated. Tissues were then digested with proteinase K (200 mg/ml) for 16 to 18 hours at 50°C. Genomic DNA was extracted sequentially with phenol, phenol:chloroform (1:1), and chloroform. Subsequently, DNA was precipitated with 100% ethanol and washed with 70% ethanol. After centrifugation, each sample was allowed to dry for ≥1 hour at room temperature and was then resuspended in 25 μl of Tris-EDTA buffer. DNA concentration and purity were measured by optical density.
K-ras Mutational Assay
K-ras codon 12 mutations were detected by the mutant-enriched, nested polymerase chain reaction (PCR) technique described by Banerjee et al. 30 In brief, an initial PCR reaction using mismatched 5′ and 3′ primers was performed, yielding a 157 base pair (bp) product corresponding to exon 1 of the K-ras gene. These mismatched primers were designed to introduce two BstN1 restriction sites in wild-type codon 12 PCR products but one BstN1 site (at the 3′ end) in mutated products. Accordingly, the 157 bp product was digested with BstN1, thereby enriching the DNA in mutated sequences. A second PCR amplification followed, using the same mismatched 5′ primer but a normal 3′ primer upstream of the one previously used. This second PCR reaction yielded a 115 bp product, which was subsequently digested with BstN1. BstN1 digests wild-type products to an 86 bp fragment but leaves mutated products intact (115 bp). Detection of these restriction fragment length polymorphisms (RFLP) by gel electrophoresis allowed discrimination between normal and mutated K-ras genes.
All PCR reactions were 50 μl in volume and included 5 μl DNA (10 to 50 μg/ml), 1× PCR buffer A, 200 μM of deoxynucleotide triphosphate (dNTP), 1 μM concentration of each primer, and 0.5 U Taq polymerase. Primers used were identical to those described by Banerjee et al. 30 Each PCR run included a sample with no template DNA to control for carryover contamination. Samples were subjected to 25 cycles of amplification in a DNA thermal cycler under the following parameters: 45 seconds at 94°C, 45 seconds at 55°C, and 1 minute at 72°C. After amplification, 20% of the PCR reaction mixture was digested with BstN1. Each BstN1 digestion totaled 50 μl in volume, containing 10 μl PCR product and 50 U BstN1 in 1× NE Buffer2 supplemented with 100 g/ml bovine serum albumin. Incubation was at 60°C for 3 hours. PCR fragments and BstN1 digestion products were electrophoresed on ethidium bromide-stained, 2% low-melt agarose gels. DNA was viewed under an ultraviolet light, and gels were photographed using a video system.
Results of a sample assay are illustrated in Figure 2. The first PCR amplification product is included in lane 2 (157 bp). Lanes 3 and 4 already show differentiation between wild-type and mutant genes by RFLP (114 vs. 143 bp, respectively). Lane 5 represents the second PCR amplification (115 bp), whereas lanes 6 and 7 show the RFLP used to identify mutations in this study (86 vs. 115 bp for wild-type and mutant genes, respectively). Notice the signal enhancement, particularly in lane 7, which results from the nested, mutant-enriched protocol used. The mutant-enriched technique (lanes 5 to 7) allowed detection of mutated sequences in small amounts of DNA, improving the sensitivity of the traditional assay using single DNA amplification and RFLP (lanes 2 to 4).
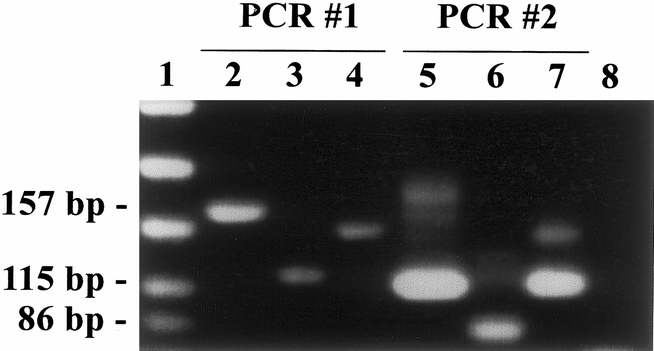
Figure 2. Detection of K-ras gene mutation by restriction fragment length polymorphism. Lane 1: Size standards. Lane 2: Example of 157 bp product resulting from first PCR. Lane 3: 114 bp fragment after BstN1 digestion of wild-type 157 bp product. Lane 4: 143 bp fragment after BstN1 digestion of mutated 157 bp product. Lane 5: Example of 115 bp product resulting from second (nested) PCR. Lane 6: 86 bp fragment after BstN1 digestion of wild-type 115 bp product. Lane 7: Mutated 115 bp product resists digestion by BstN1. Lane 8: No DNA (H2O) control.
DNA Sequencing
Mutations detected by RFLP were confirmed by DNA sequencing. Selected bands were cut from 2% low-melt agarose gels and subjected to DNA extraction. Products obtained were sequenced by the dye terminator method using an ABI 373a automatic sequencer.
Immunohistochemistry
A mouse monoclonal antibody against p53 (Dako, Carpinteria, CA; clone DO-7) was used for immunolocalization. Paraffin sections cut at 5 μm were initially deparaffinized, rehydrated, incubated in H2O2, and subjected to antigen retrieval by heating in 0.1 mM citrate buffer. Alternatively, cryostat sections were prepared by fixing in acetone and blocking endogenous peroxidase activity with H2O2. Both paraffin and cryostat sections were then processed in identical fashion. To block nonspecific antibody binding, blocking (horse) serum (Vector ABC Kit, Vector Lab, Burlingame, CA) was applied for 20 minutes, then washed off. Primary antibodies diluted 1:50 were applied, and slides were incubated overnight in a moist chamber at 4°C. Controls were incubated overnight with either PBS buffer or purified mouse IgG standards in the same dilution as the primary antibodies. The next day, unbound antibody was washed off, and slides were incubated consecutively in secondary biotinylated antimouse antibodies and ABC (avidin-biotin-peroxidase complex) reagent (Vector ABC Kit). Slides were then developed in AEC solution (80 mg 3-amino-9-ethylcarbazole in 10 ml NN-dimethyl formamide diluted in 29.6 ml 0.2 M acetic acid, 70.4 ml 0.2 M sodium acetate, 0.4 ml H2O2, and 100 ml distilled water). After fixation in acetate/formaldehyde, slides were counterstained with hematoxylin and mounted on Dako glycergel (Dako) for light microscopy.
Statistical Analysis
The two-tailed Fisher’s exact test was used for all statistical comparisons. A probability value ≤0.05 was accepted as significant.
RESULTS
K-ras Mutational Analysis
Eighty-eight different areas were identified within the 28 tumor specimens for microdissection and extraction of genomic DNA. These included 23 normal ducts, 16 ducts with dysplasia, 21 MCN epithelia without dysplasia, 6 MCN epithelia with mild dysplasia, 13 MCN epithelia with moderate dysplasia, and 9 MCN epithelia with severe dysplasia or carcinoma in situ. One to five different lesions were obtained per patient, depending on the tissue blocks available. Representative examples of these lesions can be seen in Figure 1.
K-ras codon 12 mutations were found in 27% (24/88) of successful PCR-RFLP reactions, corresponding to 46% (13/28) of examined tumors. Mutations were found in 20% (2/10) of benign, 33% (3/9) of borderline, and 89% (8/9) of malignant MCNs (p < 0.05, benign or borderline vs. malignant) (Fig. 3). The only malignant MCN lacking mutation within tumor DNA was found to harbor mutation in adjacent, dysplastic pancreatic ducts. No mutations were found in any of the 10 serous cystadenomas analyzed as negative controls. The presence of mutations did not correlate with tumor size. Examination of pancreatic ducts surrounding benign MCN revealed no abnormal K-ras genes. In contrast, mutations in adjacent pancreatic tissue were detected in 11% (1/9) of borderline and 38% (3/8) of malignant MCNs.
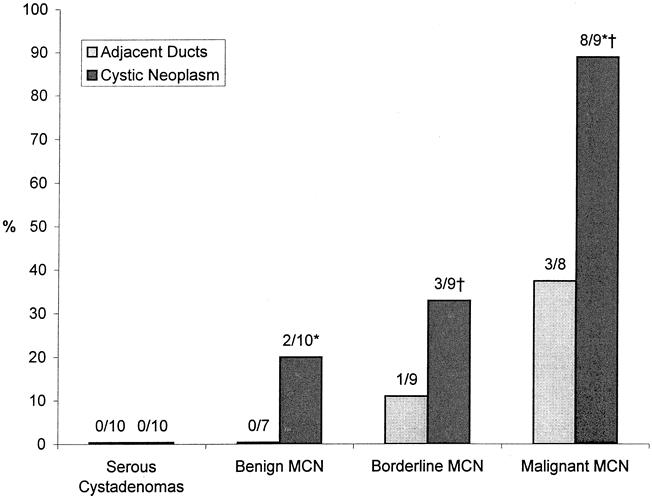
Figure 3. Frequency of K-ras mutations within mucinous cystic neoplasm (MCN) epithelia and in adjacent pancreatic ducts. *, p < 0.006; †, p < 0.05.
Histologically, the prevalence of K-ras mutations in MCNs was 26% (7/27) in areas without dysplasia or with mild dysplasia, 38% (5/13) in areas with moderate dysplasia, and 89% (8/9) in areas with severe dysplasia or carcinoma (p = 0.002 mild vs. severe; p = 0.04 moderate vs. severe; Tables 1, 2, and 3). Most of the mutations detected (14/20) were found in association with malignant MCNs, where even tumor areas without dysplasia showed genetic changes. Ten percent (4/39) of pancreatic ducts at the margins of MCN showed K-ras mutations. Mutations occurred in none (0/23) of the pancreatic ducts without dysplasia but in 33% (4/16) of the ducts with dysplastic changes (p = 0.03). Mutated pancreatic ducts were found only in association with borderline or malignant tumors.
Table 1. BENIGN MUCINOUS CYSTIC NEOPLASMS

* 0 stands for wild-type codon 12, which is GGT (glycine). Mutations include GAT (aspartic acid), GTT (valine), and CGT (arginine).
† p53 data summarized as negative (−) or positive (+).
Table 2. BORDERLINE MUCINOUS CYSTIC NEOPLASMS

* 0 stands for wild-type codon 12, which is GGT (glycine). Mutations include GAT (aspartic acid), GTT (valine), and CGT (arginine).
† p53 data summarized as negative (−) or positive (+).
Table 3. MALIGNANT MUCINOUS CYSTIC NEOPLASMS

† 0 stands for wild-type codon 12, which is GGT (glycine). Mutations include GAT (aspartic acid), GTT (valine), and CGT (arginine).
‡ p53 data summarized as negative (−) or positive (+).
§ Cases 6–9 were invasive tumors. All others had only severe dysplasia or carcinoma in situ.
DNA Sequencing
All mutations detected by PCR-RFLP were successfully confirmed by DNA sequencing. When multiple areas were mutated within a tumor, all were found to have the same sequence abnormality. The most frequent mutation found was a GGT→GAT transition, which occurred in 38% (5/13) of mutated samples. Other mutations included GGT→CGT transversions (31%, 4/13) and GGT→GTT transversions (31%, 4/13) (see Tables 1 through 3). All mutations detected were of the missense category, encoding an amino acid change in the translated polypeptide.
p53 Immunohistochemistry
Immunohistochemistry was performed in the 28 MCNs and 10 serous cystadenomas studied. Positive controls consisted of poorly differentiated colon adenocarcinoma, which showed intense nuclear staining in neoplastic areas. Serous cystadenomas showed no immunoreactivity to p53 protein. In contrast, p53 overexpression was observed in 14% (4/28) of MCNs. Overexpression occurred in none of the benign and borderline tumors but in four of nine (44%) of the malignant MCNs (p = 0.006, benign/borderline vs. malignant). When present, p53 immunoreactivity localized to the cell nucleus in areas of severe dysplasia or invasion (Fig. 4). Two of four invasive tumors displayed p53 overexpression.
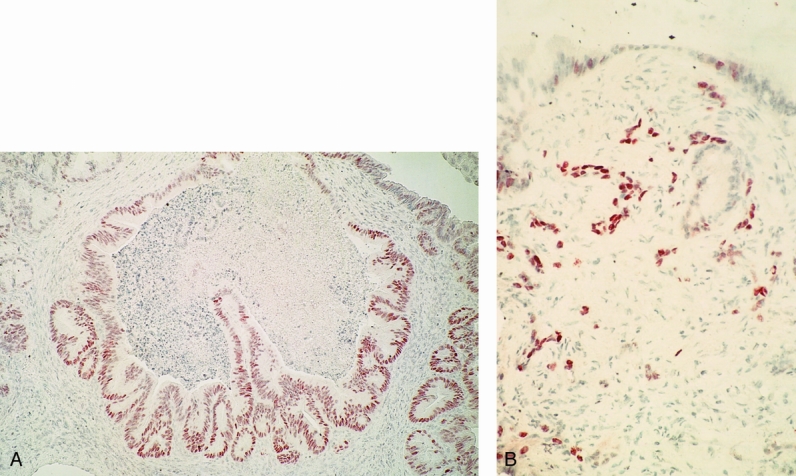
Figure 4. p53 immunohistochemistry in MCN. (A) p53 overexpression in cystic lesion with severe dysplasia (paraffin section, ABC reaction, 100×). (B) Positive staining within invasive cells (paraffin section, ABC reaction, 150×).
DISCUSSION
Most of the current knowledge of genetic events involved in the process of neoplasia has emerged from studies in colorectal carcinoma. These studies exploited the well-established sequence of adenoma to carcinoma to analyze genetic mutations at different stages of carcinogenesis. 9 The study of genetic events involved in pancreatic ductal adenocarcinoma has been complicated by the absence of a progressive array of precursor lesions comparable to colorectal tumors. In this setting, tumors of attenuated malignant potential such as MCNs may serve as a good model to study the genetic changes involved in pancreatic carcinogenesis.
Mutations in the K-ras gene are considered the hallmark of pancreatic neoplasms. 16 Initial reports on the prevalence of this mutation in pancreatic adenocarcinoma varied from 56% to 95%. With recent improvements in the sensitivity of detection assays for K-ras codon 12 mutations, >95% of ductal cell carcinomas can be shown to carry this specific genetic alteration. 30,31 In the present study, tissue microdissection combined with a sensitive PCR-RFLP assay was used to assess the presence of K-ras mutations in cystic tumors of the pancreas. Use of other, less sensitive techniques has previously failed to detect mutations in MCNs. 32
Activating K-ras codon 12 mutations were found in 46% of MCNs but in none of the serous cystadenomas studied. The presence of these mutations, therefore, correlates with the malignant potential inherent in MCNs but absent in serous tumors. Virtually all of the malignant MCNs studied harbored mutations; this is comparable to ductal adenocarcinoma. Also, the spectrum of mutations found (69% GAT or GTT) agrees strongly with previously reported data on ductal adenocarcinoma. 16 Therefore, mutations in the K-ras gene suggest similar neoplastic pathways in MCN and ductal adenocarcinoma but not in serous cystadenomas. Similarities between MCN and ductal adenocarcinoma are consistent with a common origin from ductal cells, as opposed to serous tumors, which are thought to originate from centroacinar cells. 33,34
The frequency of K-ras mutations correlated with the degree of dysplasia in the MCNs studied, with increasing histologic grade predicting acquisition of mutations. We made similar observations for pancreatic IPMT, another mucin-producing tumor of the pancreas with low malignant potential. 17 Mutation of the K-ras gene appears to be an early event in the pathogenesis of MCN, appearing at low frequencies in lesions with mild dysplasia and at maximal frequency in severe dysplasia or carcinoma. Interestingly, one of our malignant tumors had no mutations found within neoplastic areas of severe dysplasia or carcinoma, but only in adjacent dysplastic pancreatic ducts. However, this particular tumor had an associated intense desmoplastic reaction, compromising the quality of microdissection. Within the malignant MCN, the presence of K-ras mutations did not correlate with aggressiveness: both noninvasive and invasive tumors were found to be mutated.
Evaluation of pancreatic tissue at the margins of cystic neoplasms revealed K-ras mutations only in association with mutated borderline or malignant MCNs. The detection of K-ras mutations in nonneoplastic ducts was not surprising, because these are known to occur in the setting of benign conditions such as chronic pancreatitis. 35 Our results suggest that neoplastic transformation is ongoing at the periphery of MCN, particularly in association with malignant MCN. The presence of K-ras mutations in grossly normal pancreatic tissue supports formal pancreatic resection for the treatment of MCN and raises the question of incomplete resection when simple enucleation is performed. 36
Abnormal expression of the p53 gene was assessed by immunohistochemistry in this study. In most instances, accumulation of p53 protein to levels detectable by immunohistochemistry indicates inactivating point mutations in that gene. 27 Studies of colorectal carcinomas with known mutated p53 genes confirm a close correlation between p53 overexpression and gene mutation. 23 However, false-negative or false-positive results can occur. The assay will fail to detect mutations resulting in gross deletions or stop codons (nonsense mutations), where protein accumulation does not occur. 37 Likewise, some tumors with strong p53 immunoreactivity have been shown to have wild-type genes. 38 In particular, this phenomenon has been described in association with the large T antigen of SV40 or with the product of the mdm2 gene, both of which bind and stabilize p53, resulting in a functional inactivation. 19,39 These cases, however, are considered the exception rather than the rule.
Overexpression of the p53 gene product was found in 14% of MCNs, all within malignant variants. We are aware of only two other studies evaluating p53 mutations in MCNs that demonstrated none or minimal overexpression in benign or borderline tumors but strong immunoreactivity in approximately 50% of invasive, malignant neoplasms. 32,40 In our study, 44% of malignant MCNs showed immunohistochemically detectable p53 protein, approaching the 50% incidence of p53 overexpression in ductal adenocarcinomas and IPMTs. 22,41 Differences in the rate of p53 overexpression between benign or borderline MCNs and ductal adenocarcinoma may explain their contrasting biologic behaviors. Recent studies in transgenic mice demonstrate accelerated tumor growth as a consequence of p53 inactivation in pancreatic carcinogenesis. 42
Two observations suggest that p53 overexpression is a later event than K-ras mutation. First, p53 overexpression was not seen in benign or borderline tumors, and within malignant MCNs, detection was observed only in areas of severe dysplasia. Second, p53 overexpression occurred only in tumors with mutated K-ras genes. Conversely, p53 protein was never detected in the absence of a K-ras gene mutation. Similar findings are reported for ductal adenocarcinoma, where mutations in the p53 gene are almost always noted to accompany K-ras mutations. 21,43
Our data suggest that a sequential accumulation of genetic events may explain part of the progression of benign MCN to malignancy. Although abnormal activation of the K-ras gene appears important in the early progression of mild to severe dysplasia, disruption of the function of p53 may confer a more important change into an aggressive phenotype. This type of cooperation between mutated K-ras and p53 genes in the development of carcinoma is supported by results from animal models. 44–46 Our results, however, do not support that p53 mutation is sufficient for invasion and metastasis, because two of four tumors with p53 overexpression showed only carcinoma in situ.
The findings described in this study may have clinical applications. Previous studies from our institution have shown the value of cyst fluid analysis and cytologic examination in the diagnosis of cystic tumors of the pancreas. 47,48 Recent reports confirm that fluid from pancreatic cystic tumors obtained by fine-needle aspiration may be used for K-ras mutation assays. 49 At this time, we have not used K-ras data from cyst fluid to guide diagnosis or treatment, and further studies will be needed to evaluate the relevance of this information to clinical management. The significance and prognostic value of p53 overexpression have been evaluated for ductal adenocarcinoma, where it seems to correlate with enhanced tumor aggressiveness. 50 Larger studies on malignant MCNs with adequate patient follow-up will be needed to determine the significance of p53 overexpression in these tumors.
Discussion
Dr. L. William Traverso (Seattle, Washington): I rise to confirm the MGH data and to ask one question, but with first a little verbosity in between.
In recent years, we have turned to molecular biology with these tumors, first to define the neoplastic process, and second to help clinical decision-making among surgeons who must make decisions to remove tumor and how much. The MGH group has provided a number of vital portions to this puzzle that perplexes many of us.
Normally, this ductal epithelial lining does not ordinarily make mucus and when the mucous-secreting epithelium is seen in these cystic lesions of the pancreas, it occurs in one of two types of tumors, depending on whether or not the cystic lesion connects to the pancreatic ductal system. The most common cystic lesion is mucinous cystadenoma, and this is not connected to the pancreatic ductal system. The modern term is the one used in this talk, and that is mucinous cystic lesion. The other but more uncommon cystic lesion is connected to the ductal system, and that is the so-called intraductal papillary mucinous tumor. Since the lining of both types of these cystic lesions of the pancreas are identical, it is not surprising that the MGH group has found an increasing frequency of K-ras mutations as the dysplasia increases in both of these lesions. They presented the intraductal data at this meeting in Quebec City 2 years ago and are presenting the mucinous cystadenoma type data today.
How can this information help the surgeon make clinical decisions, particularly when many of these patients are asymptomatic and these lesions are premalignant? I believe assistance can be obtained by looking at the p53 overexpression data.
At the Mason Clinic, we have analyzed our last 30 intraductal tumors for K-ras, p53, HER-2/NEU, and cyclic D-1. These analyses were done in collaboration with the University of Washington Department of Pathology. These data confirm the frequent finding of K-ras mutations in either nondysplastic, dysplastic, or malignant epithelium. HER-2/NEU and cyclic D-1 was also observed frequently, just like K-ras. However, p53 was only seen in invasive tumors and is 100% specific for invasion, while occurring in 64% of the cases. Therefore, p53 might be measured in the pancreatic juice, which contains epithelial DNA, or from brushings of the ductal system. The likelihood of invasion would be very high if p53 were found and indicate the need for resective surgery.
My question to the MGH group is, have you tried this? Are you looking at pancreatic juice or brushings in applying this very informative data which you have provided us today and also at the Quebec City meeting 2 years ago?
Presenter Dr. Ramon E. Jimenez (Boston, Massachusetts): As mentioned by Dr. Traverso, we have published similar findings pertaining to K-ras mutations in the intraductal papillary mucinous tumors. Likewise, we agree that intraductal papillary mucinous tumors almost always connect with the main pancreatic duct, while mucinous cystic tumors do not. We have not looked for K-ras and p53 mutations in pancreatic juice from patients with mucinous cystic neoplasms because these tumors rarely connect with the main pancreatic duct. Although we have extensive experience with cyst fluid analysis in these tumors obtained by fine-needle aspiration, mutations in the K-ras gene have not been evaluated in such aspirates. In terms of the intraductal papillary mucinous tumors, we have not used this K-ras data for clinical decision-making yet either.
Regarding clinical applications of the p53 data, some investigators have found that p53 overexpression correlates to poor prognosis in patients with mucinous cystic neoplasms. A recent paper from the Armed Forces Institute published in January 1999 (Am J Surg Pathol 1999; 23:1–16) analyzed 130 mucinous cystic neoplasms and demonstrated that positive p53 immunoreactivity correlated to poor prognosis and disease recurrence. At our institution, however, we have not used p53 immunohistochemistry to guide postoperative patient management.
Dr. Richard H. Bell, Jr. (Seattle, Washington): Dr. Jimenez, I enjoyed your paper very much. In your manuscript, you suggested that the relatively slow-growing nature of MCNs might give us an opportunity to observe, in a single lesion, sequential genetic changes that may parallel those that occur in the more common form of pancreatic cancer, where genetic analysis is difficult because of the advanced nature of the lesions when they are diagnosed.
In that regard, I would like to ask for your thoughts about the cell of origin of MCNs as compared to ductal carcinoma of the pancreas. Do you think that MCNs arise from the same cells as pancreatic duct adenocarcinoma, but that the rate of progression is different because there is a different sequence of genetic changes? Or do you think that MCNs arise from a different population of cells in the pancreatic duct?
Dr. Jimenez: Some information is available on the cell of origin of the cystic tumors of the pancreas. Most investigators agree that serous cystadenomas arise from centroacinar cells. In contrast, mucinous cystic neoplasms, intraductal papillary mucinous tumors, and ductal adenocarcinoma appear to arise from ductal cells. In general, intraductal papillary mucinous tumors and ductal adenocarcinoma are thought to arise from cells in or near the main pancreatic duct, while mucinous cystic neoplasms are thought to arise from the cellular components of peripheral ducts.
It is not known why mucinous cystic neoplasms and ductal adenocarcinoma differ in biologic behavior despite a similar cell of origin. It is clear that the cystic neoplasms progress slower and less aggressively than ductal adenocarcinomas. This may be due to different amounts or combinations of genetic mutations, but this remains to be proven.
Some newer and more unusual theories regarding the origin of mucinous cystic neoplasms are beginning to surface. One such theory postulates that these tumors arise from remnants of ovarian tissue left behind in the area of the pancreas during organogenesis. This may explain in part why mucinous cystic tumors occur almost exclusively in females. The validity of this theory, however, has not been demonstrated (Am J Surg Pathol 1999; 23:410–422).
Dr. Keith D. Lillemoe (Baltimore, Maryland): I would like to congratulate Dr. Jimenez and his colleagues for a very nice paper. In his first slide, he said that these lesions were uncommon. But with the increasing use of abdominal imaging, we are seeing more and more of these lesions that are being detected in asymptomatic patients. This leaves us with the difficult clinical decision for whom to perform aggressive surgery versus observation.
The group at the MGH for years has been trying to help us with this decision-making process by various techniques of cyst fluid analysis. Now they go on to the molecular basis for analysis. And I think these results are both encouraging and exciting in terms of providing information on the pathogenesis of these tumors.
I would like to just ask a couple of questions. First of all, you looked at the molecular alteration in the K-ras, but only looked at the gene product expression of the p53 alteration. Have you looked at the p53 molecular alteration itself? And do you have those results? Along that same line, have you done analysis of some of the other molecular alterations that take place in pancreatic cancer such as p16 and DPC-4?
Finally, the most important question, which you have been asked before, concerning the other analyses that you have ever done for pancreatic cystic neoplasms: What is your level of confidence—and I accept the fact you have not applied this clinically—that you will be able to stick a needle into one of these lesions either endoscopically or percutaneously and get an adequate piece of tissue to allow you to make a clinical decision? Because that is clearly where these techniques will have their most important role in predicting pathologic diagnosis and dictating surgical management.
Dr. Jimenez: As mentioned by Dr. Lillemoe, the p53 assay was based on immunohistochemistry and not on direct DNA analysis. We have not done a DNA-based analysis on the p53 gene comparable to that done for K-ras. This is a difficult endeavor because mutations in p53 do not localize to a “hot spot” as occurs in the K-ras gene and therefore mutation enrichment is not possible. The group at Johns Hopkins has ingeniously circumvented many of these shortcomings by growing human pancreatic tumors in nude mice. This technique facilitates detection of gene deletions and minimizes contamination with normal human DNA. We do not have such a tissue bank and consequently have not evaluated other tumor suppressor genes such as p16 or DPC-4.
Finally, K-ras and p53 mutations could be analyzed in biopsy samples of mucinous cystic tumors. Most of our experience in the evaluation of these tumors has been with cyst fluid analysis, which is very acellular and scarce in DNA. Obtaining a biopsy of the cyst epithelium may not be straightforward because the cyst lining is often discontinuous. Therefore, obtaining tissue from the cyst wall will not guarantee the presence of neoplastic epithelium. Again, we have not used K-ras or p53 data to evaluate our patients.
Dr. Michael G. Sarr (Rochester, Minnesota): You have unraveled another piece of the puzzle. And this does look to be a reasonable model in the human for carcinogenesis.
My colleagues and I at the Mayo Clinic have just reviewed our 50-year experience with 84 of these mucinous cystic neoplasms, but we hesitate to call them malignant unless there is actual invasion (Ann Surg; in press). We have developed a new classification that we will use to call them malignant only if they have had tissue invasion. Of the 84 patients, only seven had frank tissue invasion. This leads to my question to you.
You have microdissected out ducts at the periphery of the tumor. Do you mean that this represents “normal” pancreas? Or is this periphery still part of the neoplasm? The importance of this is, if it is part of the neoplasm, we can understand this to be a type of clonal proliferation or some similar concept. But if this is outside the neoplasm and in the “normal” nonneoplastic parenchyma, this suggests that with these isolated mucinous cystic neoplasms that it is a segmental or a field defect and not a global defect. Is that what you really mean to say? In other words, is this a point mutation that has proliferated or is this a generalized defect in the epithelial lining?
Dr. Jimenez: The pancreatic ducts analyzed in the study were located outside the mucinous cystic neoplasms. The presence of K-ras mutations in some of these ducts provides evidence for a field defect in the parenchyma surrounding the tumors. We have used this information to advocate formal pancreatic resection in the treatment of these tumors.
Recent studies have shown that enucleation may be sufficient in the treatment of mucinous cystic neoplasms. When dealing with a benign tumor, enucleation may be adequate. However, more often than not, it is not possible to predict if a tumor is benign, borderline, or malignant until an extensive amount of cyst epithelium is examined by the pathologist. In view of this uncertainty, we do not recommend enucleation in the treatment of mucinous cystic neoplasms.
Dr. Andrew L. Warshaw (Boston, Massachusetts): Since a number of the discussants have geared their questions toward the utilization of these observations for clinical decision-making, I would like to disavow that intent in this work. The changes that we have seen, as Dr. Jimenez has so carefully shown, are not only discontinuous but very much depend on sampling and other issues. Our approach to these tumors is that they are premalignant and should be resected unless the patient has too many comorbidities to be a surgical candidate. We don’t rely on any of this information for clinical decision-making.
Footnotes
Correspondence: Carlos Fernández-del Castillo, MD, Dept. of Surgery, WACC 336, Massachusetts General Hospital, Boston, MA 02114.
Presented at the 119th Annual Meeting of the American Surgical Association, April 15–17, 1999, Hyatt Regency Hotel, San Diego, California.
Supported by the Marshall K. Bartlett, MD, Resident Research Fellowship, Harvard Medical School (REJ), and grants from the Swiss National Foundation and the Swiss Cancer Society (KZ).
Accepted for publication April 1999.
References
- 1.Compagno J, Oertel JE. Mucinous cystic neoplasms of the pancreas with overt and latent malignancy (cystadenocarcinoma and cystadenoma). A clinicopathologic study of 41 cases. Am J Clin Pathol 1978; 69: 573–580. [DOI] [PubMed] [Google Scholar]
- 2.Kerlin DL, Frey CF, Bodai BI, Twomey PL, Ruebner B. Cystic neoplasms of the pancreas. Surg Gynecol Obstet 1987; 165: 475–478. [PubMed] [Google Scholar]
- 3.Warshaw AL, Compton CC, Lewandrowski K, Cardenosa G, Mueller PR. Cystic tumors of the pancreas. New clinical, radiologic, and pathologic observations in 67 patients. Ann Surg 1990; 212: 432–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yamaguchi K, Enjoji M. Cystic neoplasms of the pancreas. Gastroenterology 1987; 92: 1934–1943. [DOI] [PubMed] [Google Scholar]
- 5.Fernandez-del Castillo C, Warshaw AL. Cystic tumors of the pancreas. Surg Clin North Am 1995; 75: 1001–1016. [DOI] [PubMed] [Google Scholar]
- 6.Rivera JA, Fernandez-del Castillo C, Pins M, et al. Pancreatic mucinous ductal ectasia and intraductal papillary neoplasms. A single malignant clinicopathologic entity. Ann Surg 1997; 225: 637–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kloppel G, Solcia E, Longnecker DS. World Health Organization international histological classification of tumours. Histological typing of tumours of the exocrine pancreas. New York: Springer-Verlag; 1996.
- 8.Kirby RE, Lewandrowski KB, Southern JF, Compton CC, Warshaw AL. Relation of epidermal growth factor receptor and estrogen receptor-independent pS2 protein to the malignant transformation of mucinous cystic neoplasms of the pancreas. Arch Surg 1995; 130: 69–72. [DOI] [PubMed] [Google Scholar]
- 9.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell 1990; 61: 759–767. [DOI] [PubMed] [Google Scholar]
- 10.Caldas C, Hahn SA, da Costa LT, et al. Frequent somatic mutations and homozygous deletions of the p16 (MTS1) gene in pancreatic adenocarcinoma. Nature Genet 1994; 8: 27–32. [DOI] [PubMed] [Google Scholar]
- 11.Hahn SA, Schutte M, Hoque AT, et al. DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science 1996; 271: 350–353. [DOI] [PubMed] [Google Scholar]
- 12.Redston MS, Caldas C, Seymour AB, et al. p53 mutations in pancreatic carcinoma and evidence of common involvement of homocopolymer tracts in DNA microdeletions. Cancer Res 1994; 54: 3025–3033. [PubMed] [Google Scholar]
- 13.Santos E, Nebreda AR. Structural and functional properties of ras proteins. FASEB J 1989; 3: 2151–2163. [DOI] [PubMed] [Google Scholar]
- 14.Barbacid M. ras genes. Annu Rev Biochem 1987; 56: 779–827. [DOI] [PubMed] [Google Scholar]
- 15.Quaife CJ, Pinkert CA, Ornitz DM, Palmiter RD, Brinster RL. Pancreatic neoplasia induced by ras expression in acinar cells of transgenic mice. Cell 1987; 48: 1023–1034. [DOI] [PubMed] [Google Scholar]
- 16.Hruban RH, van Mansfeld AD, Offerhaus GJ, et al. K-ras oncogene activation in adenocarcinoma of the human pancreas. A study of 82 carcinomas using a combination of mutant-enriched polymerase chain reaction analysis and allele-specific oligonucleotide hybridization. Am J Pathol 1993; 143: 545–554. [PMC free article] [PubMed] [Google Scholar]
- 17.Z’graggen K, Rivera JA, Compton CC, et al. Prevalence of activating K-ras mutations in the evolutionary stages of neoplasia in intraductal papillary mucinous tumors of the pancreas. Ann Surg 1997; 226: 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bartsch D, Bastian D, Barth P, et al. K-ras oncogene mutations indicate malignancy in cystic tumors of the pancreas. Ann Surg 1998; 228: 79–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Levine AJ, Momand J, Finlay CA. The p53 tumour suppressor gene. Nature 1991; 351: 453–456. [DOI] [PubMed] [Google Scholar]
- 20.Steele RJ, Thompson AM, Hall PA, Lane DP. The p53 tumour suppressor gene. Br J Surg 1998; 85: 1460–1467. [DOI] [PubMed] [Google Scholar]
- 21.Rall CJ, Yan YX, Graeme-Cook F, et al. Ki-ras and p53 mutations in pancreatic ductal adenocarcinoma. Pancreas 1996; 12: 10–17. [DOI] [PubMed] [Google Scholar]
- 22.Satoh K, Shimosegawa T, Moriizumi S, Koizumi M, Toyota T. K-ras mutation and p53 protein accumulation in intraductal mucin- hypersecreting neoplasms of the pancreas. Pancreas 1996; 12: 362–368. [DOI] [PubMed] [Google Scholar]
- 23.Baas IO, Mulder JW, Offerhaus GJ, Vogelstein B, Hamilton SR. An evaluation of six antibodies for immunohistochemistry of mutant p53 gene product in archival colorectal neoplasms. J Pathol 1994; 172: 5–12. [DOI] [PubMed] [Google Scholar]
- 24.Bodner SM, Minna JD, Jensen SM, et al. Expression of mutant p53 proteins in lung cancer correlates with the class of p53 gene mutation. Oncogene 1992; 7: 743–749. [PubMed] [Google Scholar]
- 25.Davidoff AM, Humphrey PA, Iglehart JD, Marks JR. Genetic basis for p53 overexpression in human breast cancer. Proc Natl Acad Sci USA 1991; 88: 5006–5010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shin DM, Kim J, Ro JY, et al. Activation of p53 gene expression in premalignant lesions during head and neck tumorigenesis. Cancer Res 1994; 54: 321–326. [PubMed] [Google Scholar]
- 27.Finlay CA, Hinds PW, Tan TH, Eliyahu D, Oren M, Levine AJ. Activating mutations for transformation by p53 produce a gene product that forms an hsc70–p53 complex with an altered half-life. Mol Cell Biol 1988; 8: 531–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ishikawa T, Nakao A, Nomoto S, et al. Immunohistochemical and molecular biological studies of serous cystadenoma of the pancreas. Pancreas 1998; 16: 40–44. [DOI] [PubMed] [Google Scholar]
- 29.Kainuma O, Asano T, Hasegawa M, et al. Inhibition of growth and invasive activity of human pancreatic cancer cells by a farnesyltransferase inhibitor, manumycin. Pancreas 1997; 15: 379–383. [DOI] [PubMed] [Google Scholar]
- 30.Banerjee SK, Makdisi WF, Weston AP, Campbell DR. A two-step enriched-nested PCR technique enhances sensitivity for detection of codon 12 K-ras mutations in pancreatic adenocarcinoma. Pancreas 1997; 15: 16–24. [DOI] [PubMed] [Google Scholar]
- 31.Longnecker DS, Terhune PG. What is the true rate of K-ras mutation in carcinoma of the pancreas? Pancreas 1998; 17: 323–324. [DOI] [PubMed] [Google Scholar]
- 32.Thompson LD, Becker RC, Przygodzki RM, Adair CF, Heffess CS. Mucinous cystic neoplasm (mucinous cystadenocarcinoma of low-grade malignant potential) of the pancreas: : a clinicopathologic study of 130 cases. Am J Surg Pathol 1999; 23: 1–16. [DOI] [PubMed] [Google Scholar]
- 33.Alpert LC, Truong LD, Bossart MI, Spjut HJ. Microcystic adenoma (serous cystadenoma) of the pancreas. A study of 14 cases with immunohistochemical and electron-microscopic correlation. Am J Surg Pathol 1988; 12: 251–263. [DOI] [PubMed] [Google Scholar]
- 34.Shorten SD, Hart WR, Petras RE. Microcystic adenomas (serous cystadenomas) of pancreas. A clinicopathologic investigation of eight cases with immunohistochemical and ultrastructural studies. Am J Surg Pathol 1986; 10: 365–372. [DOI] [PubMed] [Google Scholar]
- 35.Rivera JA, Rall CJ, Graeme-Cook F, et al. Analysis of K-ras oncogene mutations in chronic pancreatitis with ductal hyperplasia. Surgery 1997; 121: 42–49. [DOI] [PubMed] [Google Scholar]
- 36.Talamini MA, Moesinger R, Yeo CJ, et al. Cystadenomas of the pancreas:: is enucleation an adequate operation? Ann Surg 1998; 227: 896–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wynford-Thomas D. P53 in tumour pathology:: can we trust immunocytochemistry? J Pathol 1992; 166: 329–330. [DOI] [PubMed] [Google Scholar]
- 38.Hall PA, Lane DP. p53 in tumour pathology:: can we trust immunohistochemistry?—Revisited! J Pathol 1994; 172: 1–4. [DOI] [PubMed] [Google Scholar]
- 39.Momand J, Zambetti GP, Olson DC, George D, Levine AJ. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell 1992; 69: 1237–1245. [DOI] [PubMed] [Google Scholar]
- 40.Flejou JF, Boulange B, Bernades P, Belghiti J, Henin D. p53 protein expression and DNA ploidy in cystic tumors of the pancreas. Pancreas 1996; 13: 247–252. [DOI] [PubMed] [Google Scholar]
- 41.DiGiuseppe JA, Hruban RH, Goodman SN, et al. Overexpression of p53 protein in adenocarcinoma of the pancreas. Am J Clin Pathol 1994; 101: 684–688. [DOI] [PubMed] [Google Scholar]
- 42.Wagner M, Deppert W, Stacke V, Kern H, Adler G, Schmid RM. Loss of p53 accelerates tumor formation in the pancreas of TGF transgenic mice. Pancreas 1999; 17: 458. [Google Scholar]
- 43.Pellegata NS, Sessa F, Renault B, et al. K-ras and p53 gene mutations in pancreatic cancer: : ductal and nonductal tumors progress through different genetic lesions. Cancer Res 1994; 54: 1556–1560. [PubMed] [Google Scholar]
- 44.Hicks GG, Egan SE, Greenberg AH, Mowat M. Mutant p53 tumor suppressor alleles release ras-induced cell cycle growth arrest. Mol Cell Biol 1991; 11: 1344–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Taylor WR, Egan SE, Mowat M, Greenberg AH, Wright JA. Evidence for synergistic interactions between ras, myc and a mutant form of p53 in cellular transformation and tumor dissemination. Oncogene 1992; 7: 1383–1390. [PubMed] [Google Scholar]
- 46.Zambetti GP, Olson D, Labow M, Levine AJ. A mutant p53 protein is required for maintenance of the transformed phenotype in cells transformed with p53 plus ras cDNAs. Proc Natl Acad Sci USA 1992; 89: 3952–3956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lewandrowski KB, Southern JF, Pins MR, Compton CC, Warshaw AL. Cyst fluid analysis in the differential diagnosis of pancreatic cysts. A comparison of pseudocysts, serous cystadenomas, mucinous cystic neoplasms, and mucinous cystadenocarcinoma. Ann Surg 1993; 217: 41–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Centeno BA, Warshaw AL, Mayo-Smith W, Southern JF, Lewandrowski K. Cytologic diagnosis of pancreatic cystic lesions. A prospective study of 28 percutaneous aspirates. Acta Cytol 1997; 41: 972–980. [DOI] [PubMed] [Google Scholar]
- 49.Hammel P, Bouisson M, Palazzo L, et al. Diagnostic interest in Ki-ras gene mutations in fluid from cystic lesions of the pancreas. Gastroenterology 1999; 116: A417. [Google Scholar]
- 50.Yokoyama M, Yamanaka Y, Friess H, Buchler M, Korc M. p53 expression in human pancreatic cancer correlates with enhanced biological aggressiveness. Anticancer Res 1994; 14: 2477–2483. [PubMed] [Google Scholar]