

Abstract
Objective
To determine whether expression of P- and E-selectin molecules is associated with the development of systemic organ manifestations in acute pancreatitis (AP).
Summary Background Data
Overproduction of inflammatory cytokines in AP induces expression of adhesion molecules, which may lead to increased leukocytic infiltration and tissue damage. Understanding the temporal expression of these molecules could afford better measures for therapeutic intervention.
Methods
Acute pancreatitis was induced in 30-day-old female C57/bl/6J mice by feeding a choline-deficient/ethionine-supplemented diet (n = 95). Mice were divided into three groups. Group I (n = 35) was used to study the biochemical and histologic manifestations of AP and to evaluate the neutrophilic infiltration by myeloperoxidase activity and immunofluorescence. Groups II (n = 35) and III (n = 25) were used to evaluate expression of P- and E-selectin by the dual radiolabeled monoclonal antibody technique.
Results
Biochemical and histologic evidence of AP developed in all mice. The inflammatory cytokine tumor necrosis factor-α gradually increased in serum as early as 18 hours, reaching more than 800-fold background levels by 72 hours. Biphasic P-selectin expression in the lung was seen with peaks at 24 and 48 hours; E-selectin expression peaked at 48 hours. CD18-positive leukocytes and increased myeloperoxidase activity in the lung were demonstrated at 24 hours, correlating with the onset of selectin upregulation. Histologic scoring of lung tissue demonstrated mild damage at 24 hours, with progressive injury occurring from 48 to 72 hours.
Conclusions
In AP, the production of inflammatory cytokines precedes upregulation of P- and E-selectin, whose expression coincided with the increased infiltration of CD18-positive cells and neutrophil sequestration in lung tissue. Temporally, these events correlate with evidence of histologic pulmonary injury and underscore the role of adhesion molecules as mediators of pathophysiologic events. This mechanistic pathway may afford novel therapeutic interventions in clinical disease by using blocking agents to ameliorate the systemic manifestations of AP.
Acute pancreatitis (AP) is a multiple system disorder that, in its severe state, has manifestations sometimes indistinguishable from those seen in septic shock. Like sepsis, the death and complications related to this disease are due to the respiratory distress syndrome and multiple organ failure that ensue in severe cases. 1,2 Data from our laboratory as well as others have proven that cytokines (tumor necrosis factor-α [TNFα, interleukin [IL]-1, IL-6) released during the early stage of the disease are mediators of the associated systemic manifestations 3,4 and that by blocking the cytokine cascade in its early stage, amelioration of the disease and its systemic complications occurs. 5,6 The difficulty in applying these findings to clinical practice is that most patients are seen after the systemic damage is manifested. Thus, there is a need to develop a therapeutic strategy that intervenes in later stages of the disease. To achieve this goal, we need to define the pathophysiologic mechanisms associated with disease progression.
The release of inflammatory mediators such as TNFα and IL-1β during AP propagates a complex cascade of events between the tissue vasculature and the inflammatory cell. Inflammatory cytokines have been shown to mediate organ damage by their action on vascular endothelia and leukocytes in part by upregulating the expression of adhesion molecules, which in turn promote rolling, adhesion, aggregation, and transmigration of leukocytes into the involved tissues. 7 Cytokines stimulate the endothelial cell membranes and induce upregulation of selectin molecules, which bind to the leukocytes through their respective receptors. This causes rolling of the leukocytes along the postcapillary venule wall. Activation of the intracellular adhesion molecules (ICAM) and vascular cell adhesion molecules (VCAM) on the endothelial cells promotes firm adhesion of leukocytes to the vascular endothelium and facilitates their transmigration into the tissue. 8 Inflammatory cells are then free to respond to the inciting event by releasing enzymes and reactive oxygen species that cause tissue injury. 9–11 Selectin molecules have been shown to be an important factor in the lung injury associated with inflammatory syndromes. 12,13
Our laboratory has been interested in defining the role of adhesion molecules in mediating the lung injury seen in AP. To prove this, we and others have demonstrated that lung injury could be ameliorated by preventing leukocyte migration using anti-TNFα and anti-CD18 antibodies, thus providing evidence that the cytokine/adhesion molecule axis is one possible mechanism for extrapancreatic organ injury in AP. 5,14
Until now, immunofluorescent staining techniques have been the major tool used to generate a semiquantitative demonstration of adhesion molecule upregulation in tissues during AP. The radiolabeled dual monoclonal antibody (mAb) method is a novel technique used to quantify the surface expression of endothelial cell adhesion molecules in inflamed tissue. 15 One of the goals of this study was to use this technique to quantify the expression of the selectin adhesion molecules on endothelial cells during progressive severe AP and to determine the temporal relation of adhesion molecule upregulation with activated leukocyte infiltration, neutrophil sequestration, and histologic lung injury.
MATERIALS AND METHODS
Antibodies and Radiolabeling
The mAbs RB40.34, a rat IgG1 directed against mouse P-selectin 16 (Pharmingen Inc., San Diego, CA), and 10E9.6, a rat IgG2 directed against mouse E-selectin (Pharmingen), 17 were used for the in vivo evaluation of adhesion molecule expression. Previous studies using immunohistochemical staining techniques have shown that the antibody RB40.34 binds to stimulated endothelial cells and platelets in the blood vessels of normal strain mice and does not adhere to the endothelia of P-selectin knockout mice. 18 The nonbinding murine IgG1 (P-23) is directed at the human P-selectin and was used as a control antibody to correct for nonspecific accumulation of immunoglobulins in the vasculature. 19
The binding and nonbinding mAbs were radiolabeled with I125 and I131, respectively (DuPont NEN, Boston, MA), using the iodogen method. 20 Iodogen was dissolved in chloroform at a concentration of 0.5 mg/mL. One hundred twenty-five micrograms of the iodogen was added to 250 μg of the mAb and then incubated with 250 μCi sodium I125 or I131 for 20 minutes at 4°C to obtain a 1 μCi/μg protein mixture. The radiolabeled mAbs were then gel-filtered through a Sephadex PD-10 column (Pharmacia, Uppsala, Sweden) to separate the radiolabeled protein from the free I125 or I131. After equilibration of the column, PBS containing 1% bovine serum albumin was used to elute the column. Four fractions were collected, and the second 2.5-mL fraction contained the radiolabeled antibody. This was confirmed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis analysis showing normal heavy- and light-chain moieties of expected molecular weight. Previous studies have shown that the iodogen method for labeling mAbs does not interfere with their functional activity. 21 Anti-CD18 mAbs (Clone WT3) were purchased from Seikagaku Corp. (Tokyo, Japan) for use in immunofluorescent staining.
Calculation of Endothelial Cell Adhesion Molecule Expression
The method used to determine the expression of P- and E-selectin has been described previously. 22 Briefly, the I125 binding and I131 nonbinding mAb activities are measured in different tissues and 50-μL samples of serum are counted in a 14800 Wizard 3 gamma counter (Wallace, Turku, Finland), with correction for background activity and spillover taken into account. The activity of the injected dose is measured before injection into the animal. The activity remaining in the tube used for mixing the mAbs as well as the syringe used for the injection is subtracted from the total injected activity. These average less than 1% of the total injected activity. The accumulated activity of each mAb in an organ is expressed as the percent of the injected dose (%ID) per gram of dry weight tissue. The amount of binding mAb in micrograms (μg) injected into each animal allows calculation of the expression of target molecules, which is expressed as μg mAb/gram dry tissue weight. The equation used to calculate the P- and E-selectin expression was as follows:
 |
Animals
Young female C57/bl/6J (15.7 ± 0.2 g) mice were purchased from Jackson Laboratory (Bar Harbor, MA). All animals were housed in wire-bottom cages and fed standard mouse chow and water ad libitum until the beginning of the experiment, at which time the animals were fasted for 12 hours. A choline-deficient/ethionine-supplemented (CDE) (Harlan Teklad Madison, WI) diet in powder form was fed to the fasted animals ad libitum. DL-Ethionine (Sigma, St. Louis, MO) was supplemented at a concentration of 0.5% and kept at 4°C until use. The animals were kept in isolation with strict biohazard precautions in a room maintained with 12-hour light/dark cycles at 21°C. Care of the animals was in accordance with NIH standards published in the “Guide for Care and Use of Laboratory Animals” (NIH 85-23, 1985).
The CDE diet model described by Lombardi et al 23,24 is an established model of AP that has been reported in more than 40 publications studying various aspects of the disease. Since then, the model has been tested in the C57/bl/6J mice, which results in comparable pancreatic and systemic manifestations. 25–28
Experimental Protocol
Mice were divided into three groups. Group I (n = 35) was used for biochemical measurements, serum cytokine determinations, and histology, immunofluorescent staining, and measurement of myeloperoxidase activity in the lung tissue. Groups II (n = 35) and III (n = 25) were used to evaluate the expression of P- and E-selectin by the dual radiolabeled mAb technique. 21 Five mice from each group were killed at time 0, having never been fed the CDE diet. The remaining mice were killed at 18, 24, 36, 48, and 72 hours after starting the CDE diet.
Anesthesia was induced by an intramuscular injection of combined ketamine (87 mg/mL) and xylazine (13 mg/mL) at a dose of 0.1 mL/100 g. Group I mice underwent a laparotomy and the aorta was cannulated with a 22g heparinized syringe. Serum was collected and stored at −80°C until assayed. After the animal was exsanguinated, the pancreas, liver, and lungs were excised, divided, and either stored in formalin fixative until stained with hematoxylin and eosin for histologic evaluation or snap-frozen at −80°C. After storage, lung tissue was used for myeloperoxidase extraction, and pancreas, lung, and liver tissues were subsequently stained by immunofluorescent techniques for CD18 receptors located on leukocytes.
For quantitative evaluation of P- and E-selectin adhesion molecules, the animals in groups II and III were anesthetized and their jugular vein and carotid artery were cannulated using PE10 and PE50 tubing, respectively. For assessment of selectin expression, 10 μg of the respective P- or E-selectin mAb labeled with I125 and 0.5 to 5 μg of the nonbinding mAb P-23 labeled with I131 were introduced into the venous catheter. A blood sample was obtained from the carotid artery 5 minutes after injection of the mAb mixture. The animals were then given 40 units heparin intravenously and exsanguinated through the carotid artery catheter while a bicarbonate-buffer saline solution was infused through the venous catheter. At this point the organs were excised and weighed. Wet weight was measured after harvesting the organs. Dry weight was obtained after the organs were heated in the oven until the weight remained unchanged.
Assays
A bioassay using the WEHI 164 subclone 13 cell line was used to detect TNFα activity. This assay has been shown to have high specificity and sensitivity in the measurement of TNFα. One hundred microliters of WEHI cells (5 × 104 cells/well) with 0.5 μg/mL actinomycin D (Calbiochem, LaJolla, CA) was added to 96-well flat-bottomed cell culture trays and incubated with serial dilutions of plasma samples for 20 hours at 37°C. The next day, 20 μL 3-(4,5-dimethylthazol-2-yl)-2,5-diphenyltetrazolium bromide (Sigma) at a concentration of 5 mg/mL in phosphate-buffered saline (PBS) was added to all wells and the plates were incubated for another 4 hours. Finally, 150 μL medium was removed and 100 μL 0.04N HCl in isopropanol was added to each well to dissolve the purple formazan crystals. The absorbance at 550 nm was read the next day using a Bio-Rad ELISA reader (Richmond, CA). TNFα values were calculated using a standard curve prepared from rat TNFα (R&D Systems, Minneapolis, MN) and expressed in picograms per milliliter (pg/mL).
Myeloperoxidase extraction was performed as previously described. 29,30 Frozen tissue was thawed, weighed, and placed in 20 mmol/L potassium phosphate buffer (pH 7.4), which was then homogenized. Centrifugation at 20,000 relative centrifugal force was performed at 4°C for 15 minutes. The supernatant was discarded and the pellet resuspended in 50 mmol/L phosphate buffer containing 0.5% hexadecyltrimethylammonium bromide (Sigma); this was followed by sonication for 40 seconds while on ice. The samples were recentrifuged and the supernatant was assayed for myeloperoxidase activity using a mixture of water, HETAB 0.5%, tetramethylbenzidine, sodium acetate buffer, and H2O2. The reaction was stopped with catalase. Activity was measured at 655 nm after 3 minutes of incubation. Activity units (AU) per gram of tissue were calculated, and to ensure the quality of the assay a standard curve was constructed using human myeloperoxidase.
Histology
Tissue was fixed and stained with hematoxylin and eosin for histologic examination by a pathologist unaware of the experimental assignment of the animals. Histologic sections of the pancreas were examined, recording the degree of edema, zymogen crowding, vacuolation, and cellular necrosis. Changes were assigned numeric scores based on the severity of the findings (normal = 1, mild = 2, moderate = 3, severe = 4). Lung sections were examined for the degree of edema, leukocyte infiltration, congestion, and alveolar hemorrhage, and the histologic changes were also assigned numeric scores based on the severity of the findings (normal lung morphology = 1, mild = 2, moderate = 3, severe = 4).
Biochemical Analysis
Serum hematocrit was measured using an Adams Micro-hematocrit Centrifuge (New York, NY) and was read from a Critocap micro-hematocrit capillary tube reader (Lancer, St Louis, MO). Serum glucose levels were measured using the Accu-check 111 kit (Boehringer Mannheim Biochemica, Indianapolis, IN) and reported in mg/dL. Amylase was measured by a quantitative enzymatic assay (Sigma, No. 575-UV). Ten microliters are required per determination and results are expressed as International Units per liter.
Immunofluorescent Staining
Snap-frozen organs (pancreas, lung, and liver) from animals at each time point were cut into 4-micron sections and mounted on Superfrost plus slides (Fisher Scientific, Pittsburgh, PA). Tissue samples were fluorescently stained using techniques already described. 31,32 The slides were fixed in acetone for 30 seconds and then bleached for 15 minutes using 1:4, 3% H2O2 and methanol. To block any nonspecific binding, the sections were incubated in 1% BSA/PBS Tween + 20% mouse serum for 20 minutes. Lung, pancreas, and liver were stained with fluorescein (FITC)-conjugated mouse antimouse LFA-1β chain (CD18) mAb (clone WT3) using 1:100 dilution. Isotypic controls stained with rat IgG and PBS were used as negative controls. Immunofluorescent sections were examined with the fluorescent microscope.
Statistical Analysis
Results are expressed as mean ± standard error of the mean. Data for response variables were analyzed using two-way analysis of variance and the Student unpaired t test. Nonparametric data (histologic grading) was analyzed with the Mann-Whitney U test. Statistical significance was set at P < .05.
RESULTS
Ingestion of the CDE diet was associated with the development of progressive AP with swelling of the pancreas, weight loss, decreased blood glucose, and increased amylase (Fig. 1). Serum amylase levels increased significantly from 2154 ± 270 U/L to 6400 ± 3209 U/L (P < .02) in animals with AP by 24 hours. Amylase levels continued to be elevated throughout the experiment. Significant hypoglycemia was also seen in AP animals as early as 18 hours after initiation of the CDE diet (130 ± 18 mg/dL vs. 204 ± 13 mg/dL at baseline, P < .0001) and progressed until the animals were killed or died. Hemoconcentration was manifested by an increased serum hematocrit throughout the experiment, from 39 ± 1% to 44 ± 0.5% (P < .007). Animals with AP also had progressive weight loss during the experiment.

Figure 1. Biochemical measurements in diet-induced acute pancreatitis. Amylase levels increased over the 72-hour feeding period (*, P < .0003). Significant progressive hypoglycemia (†, P < .0001) and severe hemoconcentration (‡, P < .008) occurred during the feeding period, consistent with diet-induced acute pancreatitis. Also, progressive weight loss (§, P < .002) occurred during the feeding period. All values are expressed as mean ± SEM.
Serum TNFα levels continuously increased throughout the experiment, in concert with the biochemical evidence of severe AP. The serum TNFα level increased to 32 ± 31 pg/mL by 18 hours and continued to increase to 888 ± 359 pg/mL at 72 hours (Fig. 2). AP was associated with significant P-selectin upregulation in the pancreas, spleen, lung, and liver (Fig. 3). Baseline (constitutive) expression of P-selectin in the pancreas was 0.038 ± 0.20 μg mAb/g tissue. Development of pancreatitis was associated with significant upregulation of P-selectin starting at 48 hours (0.064 ± 0.008 μg mAb/g tissue); it peaked at 0.153 ± 0.027 μg mAb/g tissue by 72 hours. Lung tissue also showed a progressive increase in P-selectin expression from 0.113 ± 0.032 μg mAb/g tissue at baseline to 0.362 ± 0.186 μg mAb/g tissue by 24 hours. P-selectin expression peaked at 48 hours in the lung (0.546 ± 0.228 μg mAb/g) and then significantly declined to 0.232 ± 0.060 μg mAb/g tissue by 72 hours. The liver revealed a gradual increase in P-selectin expression from 0.012 ± 0.015 μg mAb/g tissue at baseline to 0.213 ± 0.074 μg mAb/g tissue at 24 hours; it peaked at 0.498 ± 0.172 μg mAb/g tissue at 72 hours. Splenic tissue demonstrated a rise from baseline (0.00 μg mAb/g tissue), but significance was not seen until 48 hours after initiation of the diet (0.249 ± 0.162 μg mAb/g tissue). Peak levels in the spleen occurred at 72 hours at 0.319 ± 0.176 μg mAb/g tissue, again correlating with progression of disease. Heart, mesentery, and small bowel all showed significant increases in P-selectin, but there was no upregulation in the kidney.
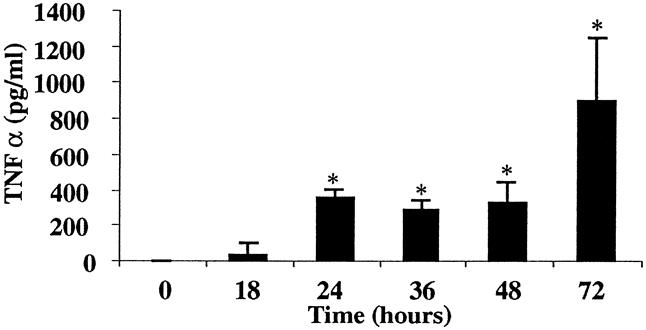
Figure 2. Serum levels of tumor necrosis factor-α in diet-induced pancreatitis. Inflammatory cytokine levels were elevated during the diet model of acute pancreatitis (*, P < .0001).

Figure 3. P-selectin expression during acute pancreatitis. P-selectin expression was upregulated in the pancreas, spleen, lung, and liver (*, P < .04; †, P < .0001; ‡, P < .0004; §, P < .0001).
In contrast to the widespread upregulation of P-selectin, E-selectin expression was demonstrated only in lung tissue of animals with AP. E-selectin increased from 0.12 ± 0.099 μg mAb/g tissue at baseline to a peak at 48 hours of 0.38 ± 0.18 μg mAb/g tissue (Fig. 4). The timing of maximum E-selectin upregulation coincided with the second peak of P-selectin expression in the lung.
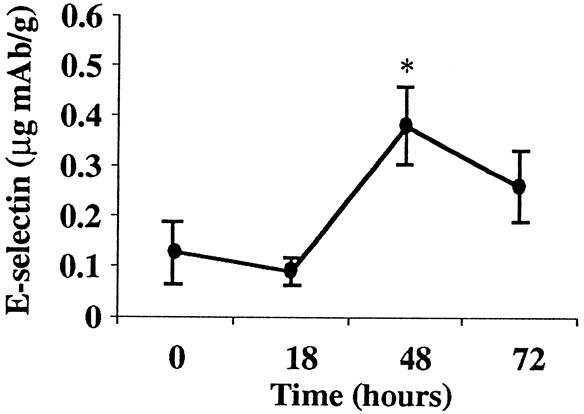
Figure 4. E-selectin expression during acute pancreatitis. E-selectin expression was upregulated in lung tissue at 48 hours (*, P < .005).
Immunofluorescent staining for the leukocyte CD18 receptor demonstrated increased intensity of CD18 staining in the pancreas at 18 hours. In the lung, CD18-positive leukocytic infiltrates appeared at 24 hours and persisted until the animal was sacrificed (Fig. 5). Concomitantly, there was an increase in myeloperoxidase activity in lung tissue from control animal levels of 1.44 ± 0.25 AU/g to 6.55 ± 1.26 AU/g (P < .001) in animals that had been fed the CDE diet for 24 hours (Fig. 6). This increased myeloperoxidase activity persisted throughout the experiment.
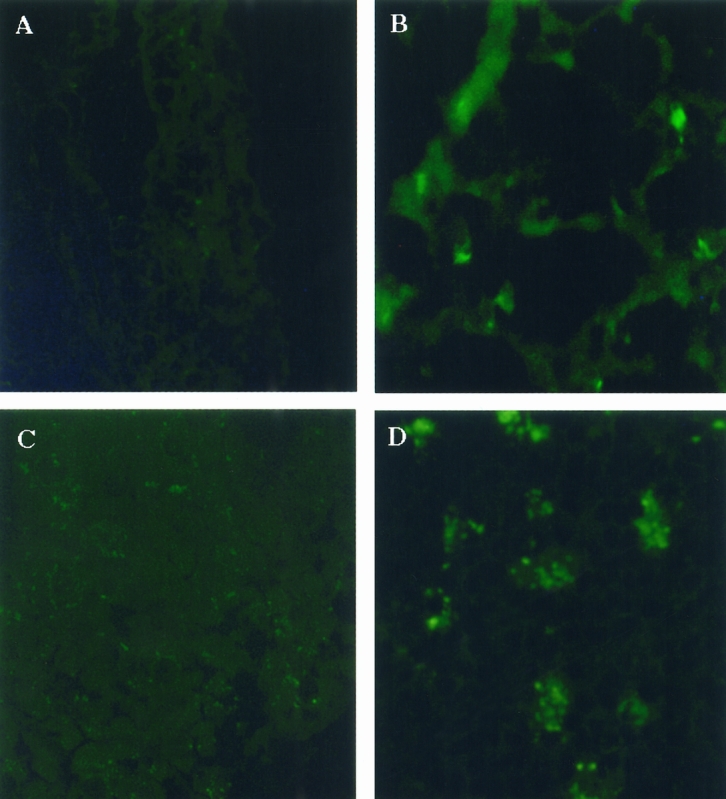
Figure 5. Immunofluorescent staining of CD18-positive leukocytes in lung and pancreas. (A) Absence of leukocyte infiltration in lung tissue of control mice versus (B) those fed the choline-deficient/ethionine-supplemented (CDE) diet at 24 hours, which correlated with upregulation of P-selectin expression in lung tissue. (C) Absence of leukocyte infiltration in pancreas tissue in control mice versus (D) CD18-positive infiltrating cells by 18 hours in those fed the CDE diet. Infiltration of leukocytes in the pancreas occurred before P-selectin upregulation.
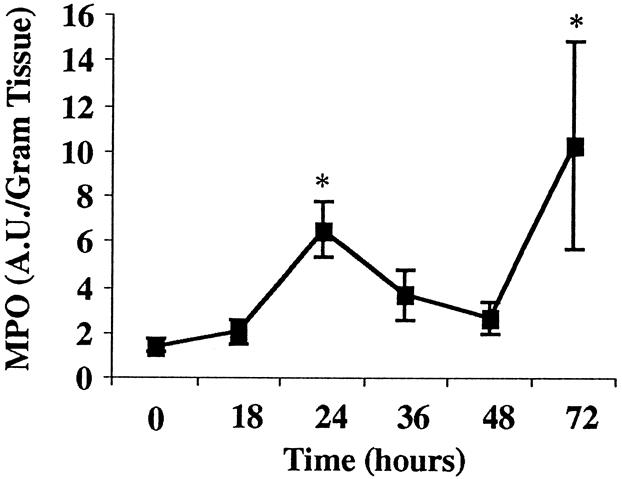
Figure 6. Pulmonary leukocyte infiltration in diet-induced acute pancreatitis. Lung myeloperoxidase activity in mice increased at 24 hours, which correlated with P- and E-selectin upregulation (*, P < .0001).
Histologically, the pancreas of the animals fed the CDE diet showed a progressive worsening of edema, congestion, and hemorrhage as well as vacuolization, pyknosis, and cellular necrosis. Changes in the pancreas were noted as early as 18 hours after the initiation of the CDE diet, with continued worsening to moderate/severe changes between 48 and 72 hours (Fig. 7). At 24 hours, the lung tissue revealed mild histologic changes with edema and early leukocyte infiltration. These mild changes continued through 48 hours and then progressed to moderate injury by 72 hours with significant edema, leukocyte infiltration, and hemorrhage into the alveoli. The liver also demonstrated congestion as well as cellular necrosis starting at 24 hours; this progressively worsened throughout the study.
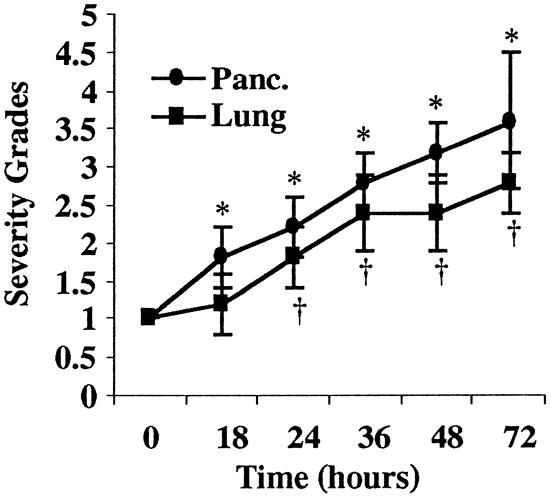
Figure 7. Scoring of histologic severity in diet-induced pancreatitis. Progressive pancreatic injury (closed circle) worsened throughout the diet model of AP (*P < .03). Lung injury (closed square) followed the progression of acute pancreatitis (†P < .03) and correlated with the upregulation of cytokines, selectins, and leukocyte infiltration. Histologic data was analyzed by the Mann-Whitney test. Statistical significance was set at P < .05.
DISCUSSION
This study is the first demonstration that AP is associated with a significant upregulation of the P- and E-selectin endothelial cell adhesion molecules in the pancreas and lung as well as other distant organs. We demonstrated how selectin expression is temporally related to cytokine upregulation, leukocyte infiltration, and the progressive histologic changes seen during the disease process. The quantitation of adhesion molecule expression provides some guide to the relative role of the selectin molecules in the differential injury occurring in various organs.
The CDE diet model was chosen for these experiments because of its reported clinical and biochemical similarities to human disease. 33 Ingestion of this diet is associated with gradual evolution of pancreatitis with increasing severity during several days, which is highly reproducible. 33,34 This allowed us to examine the correlation between changes in cytokines and adhesion molecules and the clinicopathologic development of lung disease. One of the traditional criticisms of the CDE diet model is that there is no defined starting point and that there are variable differences in disease severity between animals. Some of this disparity has been attributed to variable ingestion of the diet by the mice, leading to discrepancy in disease expression. In our laboratory, we observed that a 24-hour fast before feeding the animals, as well as constant filling of food jars and removal of old food, resulted in a steady progression of AP in all CDE-fed groups, and that group variability was minimal. 28 The success of this methodology is seen in our results: the standard deviations suggest that the disease severity in each group of animals was relatively similar. Clearly, like most models of AP, the CDE diet model has advantages and potential disadvantages, including the potential hepatotoxicity of the diet; however, the use of this model has been popular because of its reproducibility, noninvasiveness, and ease in manipulating mortality. 25
Previously, we and others demonstrated the important role of increased cytokine production in the development of systemic manifestations of AP. 35,36 Inflammatory cytokines have been shown in other disease models to mediate organ damage through upregulation of the expression of endothelial cell and leukocyte adhesion molecules. 37,38 Until now, however, there have been few data regarding the changes in these molecules during AP, despite their probable key role in initiating tissue injury associated with this disease. Demonstrating the temporal relation of selectin upregulation to the cytokine cascade and the histologic injury in AP is based on the recognition of the pivotal role of the selectin molecules as the first of the endothelial cell adhesion molecules to be expressed during the inflammatory response. 39
Because of the ability to sample lung tissue frequently, we were able to demonstrate some of the earliest pulmonary changes associated with the onset of AP. These pulmonary histologic changes correlated temporally with the kinetics of selectin expression. Because of the fact that P- and E-selectin are the earliest adhesion molecules expressed in response to cytokine expression, this suggests a role of these molecules in early pulmonary injury associated with AP. In the lung, we were able to determine that both the P- and E-selectin molecules are expressed during the evolution of pancreatitis, with the former molecule having biphasic peaks. The P-selectin is constitutively expressed at low levels in several organs and is stored in a preformed pool of the endothelial cell (Weibel-Palade bodies). 12 The P-selectin molecules can also be transcriptionally induced on the endothelial cell after stimulation with an inflammatory mediator. 40 Thus, the biphasic peaks represent the release of preformed molecules and de novo transcription in response to the cytokine stimulus generated by pancreatic inflammation. In contrast, E-selectin is transcriptionally induced, and thus the lack of a constitutive pool of E-selectin accounts for its later appearance on the endothelial cells after an inflammatory stimulus. 22,41 In our model, the second peak of P-selectin expression seen in the lung at 48 hours after AP coincided with the single peak of E-selectin, indicating that both molecules were transcribed in response to the severe inflammatory signal after the initiation of AP.
In this study, pulmonary leukocytic infiltration was detected by immunofluorescent staining for the CD18-positive cells (leukocytes) and by measuring neutrophil sequestration in lung parenchyma through measurement of myeloperoxidase activity. There is a large influx of CD18-positive inflammatory cells into the lungs. When considered in the context of the elevated myeloperoxidase activity, this probably represents an influx of neutrophils into the lung tissue, although the presence of other leukocytic populations cannot be ruled out. Many investigators have reported that the neutrophil is a key cell that mediates organ injury in AP, 42–44 and it has been shown that neutrophil depletion before the onset of AP reduces the associated lung injury. 42,44,45 The importance of other cell types involved in AP-associated pulmonary injury has been suggested in several studies. 45,46,47 Recently, Frossard et al 45 demonstrated that AP-associated lung injury may be partially caused by neutrophil-independent events, thus confirming that other inflammatory cells are involved in injury progression.
We observed that the initial peak of P-selectin expression coincided with the appearance of CD18-positive leukocytes in the lung as well as with the initial rise in myeloperoxidase activity. In addition, the progression to the second peak of P-selectin and the appearance of E-selectin was associated with a significant accumulation of leukocytes in the lung, as detected by immunofluorescence and myeloperoxidase activity. These changes in adhesion molecules and the associated increase in pulmonary leukocytic infiltration were accompanied by worsening of the lung injury severity score. Together, these changes strongly suggest that pulmonary injury in AP is mediated by the upregulation of adhesion molecules.
In contrast to the correlation between histologic injury and adhesion molecule upregulation in the lung, we have demonstrated that the initial infiltration of leukocytes and the early histologic changes in the pancreas occur before demonstrable expression of P- and E-selectin molecules in this organ. Beginning at 48 hours, there is an upregulation of P-selectin in the pancreas, correlating with the worsening of pancreatic injury between 48 and 72 hours. However, because leukocytic infiltration in the pancreas occurs before 48 hours, this contradicts the current hypothesis of inflammatory cell migration in that a requirement for leukocytic infiltration is the expression of endothelial cell adhesion molecules. 48,49 A possible explanation for this phenomenon is that the CDE diet itself causes pancreatic injury, which may induce the release of chemokines and cytokines by the acinar cells or interstitial leukocytes, which attracts leukocytes before adhesion molecule expression. 50,51 Another possible explanation is that the selectin molecules play a minor role in the pancreas during AP and that other cellular adhesion molecules are the major contributors to leukocyte infiltration. This remains to be investigated.
Another finding of our study is that the level and timing of adhesion molecule expression varies in different organs, a phenomenon seen in other inflammatory conditions. 22 For example, in the liver and spleen, the pattern of P-selectin upregulation occurred at 24 hours and 48 hours, respectively. We also observed an increased expression of P-selectin molecules in the heart, small bowel, mesentery, and spleen during AP, demonstrating that this is truly a systemic disease, although leukocyte infiltration was not evaluated in these organs.
In previous work, histologic injury could be ameliorated by blocking the inflammatory cytokines TNFα and IL1β. 5,6 However, despite the theoretical mechanistic significance of these observations, their practical utility in management of human disease is limited. Our data demonstrated that the triggering of the cytokine cascade occurs extremely early in AP, and that TNFα, once released, induces further cytokine production from a variety of cells, including macrophages, leukocytes, T lymphocytes, and mast cells. 52–54 Once the cytokine cascade is triggered, there is an activation of downstream events that leads to organ injury and is not amenable to cytokine blockade. Because of these factors, the cytokine cascade must be blocked either before the insult that causes AP or within a few hours after the onset of the disease. Difficulties in the timing of anticytokine interventions and in administering effective cytokine blocking have been demonstrated in systemic inflammatory response syndromes. 55,56 Because of these difficulties, we attempted to study more downstream events in the inflammatory response, particularly those of the adhesion molecules. Although our model shows a temporal correlation between the cytokines, selectin, leukocytes, and organ injury, we have not confirmed a direct mechanism by using inhibitors of the selectin molecules. The literature suggests that treatment against selectin expression in some models has a positive outcome with regard to leukocyte infiltration and organ injury. 48 Based on this evidence, we would expect that inhibitors of the selectin molecules would decrease leukocyte infiltration and would ultimately ameliorate organ injury associated with AP. In this regard, blocking of adhesion molecules has been attempted in several inflammatory diseases with promising results. Many researchers have demonstrated that treatment with antiselectin and anti-ICAM antibodies in ischemia/reperfusion and sepsis models is beneficial in the treatment of organ injury and death. 57,58 Also, treatment against leukocyte activation and migration using anti-CD18 and anti-ICAM-1 mAbs has been attempted in AP, resulting in a decrease in the inflammatory mediators released by the leukocytes and amelioration of distant organ injury. 14 The data suggest an important role for adhesion molecules in AP and suggest that they could be targeted for therapeutic intervention.
There is an inherent redundancy of cellular migration processes during inflammation, 7,40 and thus P- and E-selectins are not the only endothelial cell adhesion molecules involved in mediating inflammatory cells to the stimulated tissues. Evidence of the role played by other adhesion molecules has been demonstrated in other inflammatory models by blocking the different adhesion molecules with specific inhibitors. Blocking specific molecules results in decreased markers of injury, but none is associated with complete amelioration of the inflammatory response. 13,18,59 In AP-associated lung injury, it has been reported that other molecules, such as ICAM-1, play an important role in the injury pathogenesis. This was demonstrated by using ICAM-1 genetically deficient mice, thereby decreasing AP-associated lung injury. 45 However, in that report, the authors point out that ICAM-1 deficiency did not totally ameliorate lung injury, confirming that as in other models, lung injury in AP is multifactorial in origin.
Our data indicate that the selectin upregulation is the first of these adhesion molecules to be expressed, as is seen in other inflammatory models. 39 We believe that this closely correlates with the temporal onset of distant organ injury. In conclusion, we believe that the sequences of events associated with leukocyte infiltration are mediated initially by the P- and E-selectins.
Footnotes
Correspondence: A. Osama Gaber, MD, Dept. of Surgery, Division of Transplantation, University of Tennessee, Memphis, 956 Court Ave., Suite A202, Memphis, TN 38163.
Supported by the Transplant Surgical Immunology Laboratory at the University of Tennessee at Memphis and a grant (to DNG) from the National Institutes of Health (PO1 DK-43785).
Accepted for publication June 16, 1999.
References
- 1.Ranson JH, Turner JW, Roses DF, et al. Respiratory complications in acute pancreatitis. Ann Surg 1974; 179:557–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lankisch PG, Rahlf G, Koop H. Pulmonary complications in fatal acute hemorrhagic pancreatitis. Dig Dis Sci 1983; 28:110–116. [DOI] [PubMed] [Google Scholar]
- 3.Grewal HP, Kotb M, el Din AM, et al. Induction of tumor necrosis factor in severe acute pancreatitis and its subsequent reduction after hepatic passage. Surgery 1994; 115:213–221. [PubMed] [Google Scholar]
- 4.Guice KS, Oldham KT, Remick DG, et al. Anti-tumor necrosis factor antibody augments edema formation in caerulein-induced acute pancreatitis. J Surg Res 1991; 51:495–499. [DOI] [PubMed] [Google Scholar]
- 5.Hughes CB, Gaber LW, Mohey el-Din AB, et al. Inhibition of TNF alpha improves survival in an experimental model of acute pancreatitis. Am Surg 1996; 62:8–13. [PubMed] [Google Scholar]
- 6.Norman JG, Franz MG, Fink GS, et al. Decreased mortality of severe acute pancreatitis after proximal cytokine blockade. Ann Surg 1995; 221:625–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Springer TA. Traffic signals on endothelium for lymphocyte recirculation and leukocyte emigration. Annu Rev Physiol 1995; 57:827–872. [DOI] [PubMed] [Google Scholar]
- 8.Panes J, Granger DN. Leukocyte-endothelial cell interactions: molecular mechanisms and implications in gastrointestinal disease. Gastroenterology 1998; 114:1066–1090. [DOI] [PubMed] [Google Scholar]
- 9.Donnelly SC, Haslett C, Dransfield I, et al. Role of selectins in development of adult respiratory distress syndrome. Lancet 1994; 344 (8917):215–219. [DOI] [PubMed] [Google Scholar]
- 10.Mulligan MS, Polley MJ, Bayer RJ, et al. Neutrophil-dependent acute lung injury. Requirement for P-selectin (GMP- 140). J Clin Invest 1992; 90:1600–1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Till GO, Johnson KJ, Kunkel R, Ward PA. Intravascular activation of complement and acute lung injury. Dependency on neutrophils and toxic oxygen metabolites. J Clin Invest 1982; 69:1126–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Windsor AC, Mullen PG, Fowler AA, Sugerman HJ. Role of the neutrophil in adult respiratory distress syndrome. Br J Surg 1993; 80:10–17. [DOI] [PubMed] [Google Scholar]
- 13.Doerschuk CM, Quinlan WM, Doyle NA, et al. The role of P-selectin and ICAM-1 in acute lung injury as determined using blocking antibodies and mutant mice. J Immunol 1996; 157:4609–4614. [PubMed] [Google Scholar]
- 14.Inoue S, Nakao A, Kishimoto W, et al. LFA-1 (CD11a/CD18) and ICAM-1 (CD54) antibodies attenuate superoxide anion release from polymorphonuclear leukocytes in rats with experimental acute pancreatitis. Pancreas 1996; 12:183–188. [DOI] [PubMed] [Google Scholar]
- 15.Henninger DD, Panes J, Eppihimer M, et al. Cytokine-induced VCAM-1 and ICAM-1 expression in different organs of the mouse. J Immunol 1997; 158:1825–1832. [PubMed] [Google Scholar]
- 16.Patel KD, Moore KL, Nollert MU, McEver RP. Neutrophils use both shared and distinct mechanisms to adhere to selectins under static and flow conditions. J Clin Invest 1995; 96:1887–1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Norton CR, Rumberger JM, Burns DK, Wolitzky BA. Characterization of murine E-selectin expression in vitro using novel anti-mouse E-selectin monoclonal antibodies. Biochem Biophys Res Commun 1993; 195:250–258. [DOI] [PubMed] [Google Scholar]
- 18.Bullard DC, Qin L, Lorenzo I, et al. P-selectin/ICAM-1 double mutant mice: acute emigration of neutrophils into the peritoneum is completely absent but is normal into pulmonary alveoli. J Clin Invest 1995; 95:1782–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ma L, Raycroft L, Asa D, et al. A sialoglycoprotein from human leukocytes functions as a ligand for P-selectin. J Biol Chem 1994; 269:27739–27746. [PubMed] [Google Scholar]
- 20.Fraker PJ, Speck JC Jr. Protein and cell membrane iodinations with a sparingly soluble chloroamide, 1,3,4,6-tetrachloro-3a,6a-diphrenylglycoluril. Biochem Biophys Res Commun 1978; 80:849–857. [DOI] [PubMed] [Google Scholar]
- 21.Panes J, Perry MA, Anderson DC, et al. Regional differences in constitutive and induced ICAM-1 expression in vivo. Am J Physiol 1995; 269:H1955–H1964. [DOI] [PubMed] [Google Scholar]
- 22.Eppihimer MJ, Wolitzky B, Anderson DC, et al. Heterogeneity of expression of E- and P-selectins in vivo. Circ Res 1996; 79:560–569. [DOI] [PubMed] [Google Scholar]
- 23.Lombardi B, Estes LW, Longnecker DS. Acute hemorrhagic pancreatitis (massive necrosis) with fat necrosis induced in mice by DL-ethionine fed with a choline-deficient diet. Am J Pathol 1975; 79:465–480. [PMC free article] [PubMed] [Google Scholar]
- 24.Lombardi B, Rao NK. Acute hemorrhagic pancreatic necrosis in mice. Influence of the age and sex of the animals and of dietary ethionine, choline, methionine, and adenine sulfate. Am J Pathol 1975; 81:87–99. [PMC free article] [PubMed] [Google Scholar]
- 25.Niederau C, Luthen R, Niederau MC, et al. Acute experimental hemorrhagic-necrotizing pancreatitis induced by feeding a choline-deficient, ethionine-supplemented diet. Methodology and standards. Eur Surg Res 1992; 24(suppl 1):40–54. [DOI] [PubMed] [Google Scholar]
- 26.Norman JG, Fink G, Franz M, et al. Active interleukin-1 receptor required for maximal progression of acute pancreatitis. Ann Surg 1996; 223:163–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gloor B, Todd KE, Lane JS, et al. Mechanism of increased lung injury after acute pancreatitis in IL-10 knockout mice. J Surg Res 1998; 80:110–114. [DOI] [PubMed] [Google Scholar]
- 28.Eubanks JW 3rd, Sabek O, Kotb M, et al. Acute pancreatitis induces cytokine production in endotoxin-resistant mice. Ann Surg 1998; 227:904–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bradley PP, Priebat DA, Christensen RD, Rothstein G. Measurement of cutaneous inflammation: estimation of neutrophil content with an enzyme marker. J Invest Dermatol 1982; 78:206–209. [DOI] [PubMed] [Google Scholar]
- 30.Yamanaka K, Saluja AK, Brown GE, et al. Protective effects of prostaglandin E1 on acute lung injury of caerulein-induced acute pancreatitis in rats. Am J Physiol 1997; 272 (1 pt 1):G23–G30. [DOI] [PubMed] [Google Scholar]
- 31.Dickler HB, Kubicek MT. Interactions between lymphocyte membrane molecules. I. Interaction between B lymphocyte surface IgM and Fc IgG receptors requires ligand occupancy of both receptors. J Exp Med 1981; 153:1329–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ljungstrom I, Ruitenberg EJ. A comparative study of the immunohistological and serological response of intact and T cell-deprived mice to Trichinella spiralis. Clin Exp Immunol 1976; 24:146–156. [PMC free article] [PubMed] [Google Scholar]
- 33.Banerjee AK, Galloway SW, Kingsnorth AN. Experimental models of acute pancreatitis. Br J Surg 1994; 81:1096–1103. [DOI] [PubMed] [Google Scholar]
- 34.Bilchik AJ, Leach SD, Zucker KA, Modlin IM. Experimental models of acute pancreatitis. J Surg Res 1990; 48:639–647. [DOI] [PubMed] [Google Scholar]
- 35.Grewal HP, Mohey el Din A, Gaber L, et al. Amelioration of the physiologic and biochemical changes of acute pancreatitis using an anti-TNF-alpha polyclonal antibody. Am J Surg 1994; 167:214–219. [DOI] [PubMed] [Google Scholar]
- 36.Norman JG, Fink GW, Denham W, et al. Tissue-specific cytokine production during experimental acute pancreatitis. A probable mechanism for distant organ dysfunction. Dig Dis Sci 1997; 42:1783–1788. [DOI] [PubMed] [Google Scholar]
- 37.Mulligan MS, Vaporciyan AA, Miyasaka M, et al. Tumor necrosis factor alpha regulates in vivo intrapulmonary expression of ICAM-1. Am J Pathol 1993; 142:1739–1749. [PMC free article] [PubMed] [Google Scholar]
- 38.Fassbender K, Kaptur S, Becker P, et al. Adhesion molecules in tissue injury: kinetics of expression and shedding and association with cytokine release in humans. Clin Immunol Immunopathol 1998; 89:54–60. [DOI] [PubMed] [Google Scholar]
- 39.Granger DN, Kubes P. The microcirculation and inflammation: modulation of leukocyte-endothelial cell adhesion. J Leukocyte Biol 1994; 55:662–675. [PubMed] [Google Scholar]
- 40.Smith CW. Endothelial adhesion molecules and their role in inflammation. Can J Physiol Pharmacol 1993; 71:76–87. [DOI] [PubMed] [Google Scholar]
- 41.Malik AB, Lo SK. Vascular endothelial adhesion molecules and tissue inflammation. Pharmacol Rev 1996; 48:213–229. [PubMed] [Google Scholar]
- 42.Inoue S, Nakao A, Kishimoto W, et al. Anti-neutrophil antibody attenuates the severity of acute lung injury in rats with experimental acute pancreatitis. Arch Surg 1995; 130:93–98. [DOI] [PubMed] [Google Scholar]
- 43.Barie PS, Tahamont MV, Blumenstock FA, Malik AB. The role of neutrophils in the pathogenesis of pulmonary vascular injury after acute pancreatitis. Curr Surg 1982; 39:411–413. [PubMed] [Google Scholar]
- 44.Bhatia M, Saluja AK, Hofbauer B, et al. The effects of neutrophil depletion on a completely noninvasive model of acute pancreatitis-associated lung injury. Int J Pancreatol 1998; 24 (2):77–83. [DOI] [PubMed] [Google Scholar]
- 45.Frossard JL, Saluja A, Bhagat L, et al. The role of intercellular adhesion molecule 1 and neutrophils in acute pancreatitis and pancreatitis-associated lung injury. Gastroenterology 1999; 116:694–701. [DOI] [PubMed] [Google Scholar]
- 46.Closa D, Sabater L, Fernandez-Cruz L, et al. Activation of alveolar macrophages in lung injury associated with experimental acute pancreatitis is mediated by the liver. Ann Surg 1999; 229:230–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tsukahara Y, Morisaki T, Horita Y, et al. Phospholipase A2 mediates nitric oxide production by alveolar macrophages and acute lung injury in pancreatitis. Ann Surg 1999; 229:385–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Butcher EC. Cellular and molecular mechanisms that direct leukocyte traffic. Am J Pathol 1990; 136:3–11. [PMC free article] [PubMed] [Google Scholar]
- 49.Sakai A. Protection against septic shock in mice with SJC13, an azaindolidine derivative that is a cell adhesion molecule inhibitor. Inflamm Res 1996; 45:448–451. [DOI] [PubMed] [Google Scholar]
- 50.Grady T, Liang P, Ernst SA, Logsdon CD. Chemokine gene expression in rat pancreatic acinar cells is an early event associated with acute pancreatitis. Gastroenterology 1997; 113:1966–1975. [DOI] [PubMed] [Google Scholar]
- 51.Gerard C, Frossard JL, Bhatia M, et al. Targeted disruption of the beta-chemokine receptor CCR1 protects against pancreatitis-associated lung injury. J Clin Invest 1997; 100:2022–2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kingsnorth A. Role of cytokines and their inhibitors in acute pancreatitis. Gut 1997; 40:1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mannel DN. Biological aspects of tumor necrosis factor. Immunobiology 1986; 172:283–290. [DOI] [PubMed] [Google Scholar]
- 54.McKay CJ, Gallagher G, Brooks B, et al. Increased monocyte cytokine production in association with systemic complications in acute pancreatitis. Br J Surg 1996; 83:919–923. [DOI] [PubMed] [Google Scholar]
- 55.Norman JG, Fink GW, Messina J, et al. Timing of tumor necrosis factor antagonism is critical in determining outcome in murine lethal acute pancreatitis. Surgery 1996; 120:515–521. [DOI] [PubMed] [Google Scholar]
- 56.Dinarello CA, Gelfand JA, Wolff SM. Anticytokine strategies in the treatment of the systemic inflammatory response syndrome. JAMA 1993; 269:1829–1835. [PubMed] [Google Scholar]
- 57.Vedder NB, Fouty BW, Winn RK, et al. Role of neutrophils in generalized reperfusion injury associated with resuscitation from shock. Surgery 1989; 106:509–516. [PubMed] [Google Scholar]
- 58.Winn RK, Sharar SR, Vedder NB, Harlan JM. Leukocyte and endothelial adhesion molecules in ischaemia/reperfusion injuries. Ciba Found Symp 1995; 189:63–71. [DOI] [PubMed] [Google Scholar]
- 59.Bullard DC, Kunkel EJ, Kubo H, et al. Infectious susceptibility and severe deficiency of leukocyte rolling and recruitment in E-selectin and P-selectin double mutant mice. J Exp Med 1996; 183:2329–2336. [DOI] [PMC free article] [PubMed] [Google Scholar]