

Abstract
Objective
To evaluate the putative relation of platelet activating factor (PAF), xanthine oxidase, reactive oxidants, and leukocytes in the pathogenesis of hepatic injury after shock/resuscitation (S/R) in vivo.
Background
Reactive oxygen metabolites generated by xanthine oxidase at reperfusion have been found to trigger postischemic injury in many organs, including the liver. However, the precise linear sequence of the mechanism of consequent hepatic injury after S/R remains to be characterized.
Methods
Unheparinized male rats were bled to a mean blood pressure of 45 ± 3 mmHg. After 2 hours of shock, they were resuscitated by reinfusion of shed blood (anticoagulated with citrate-phosphate-dextrose) and crystalloid and observed for the next 6 or 24 hours.
Results
S/R caused the oxidation of hepatic glutathione and generated centrolobular leukocyte accumulation at 6 hours, followed by predominantly centrolobular hepatocellular injury at 24 hours. Each of these components was attenuated by PAF inhibition with WEB 2170, xanthine oxidase inhibition with allopurinol, antioxidant treatment with N-acetylcysteine, or severe leukopenia induced by vinblastine. In each case, the degree of leukocyte accumulation at 6 hours correlated with the hepatocellular injury seen at 24 hours. However, xanthine oxidase inhibition with allopurinol failed to attenuate further the small level of residual hepatocellular injury seen in leukopenic rats.
Conclusion
These findings suggest that reactive oxidants generated by xanthine oxidase at reperfusion, stimulated by PAF, mediate hepatocellular injury by triggering leukocyte accumulation, primarily within the centrolobular sinusoids.
Postischemic hepatic injury can be seen after hypovolemic shock, major surgery, liver transplantation, trauma, and other forms of severe physiologic stress. 1–5 This condition, variously termed ischemic hepatitis, postshock hepatic insufficiency, or shock liver, is thought to be a consequence of hepatic ischemia followed by reperfusion. 2,6–8 Although the clinical consequences of hepatic insufficiency are frequently transient and self-limited, 3 more severe degrees of insult can lead to terminal hepatic failure or can contribute to the evolution of multiple organ dysfunction syndrome. 9 For example, we have found that hepatic reticuloendothelial function, normally accounting for approximately 80% of the clearance and killing of bacteria from the systemic circulation, 10 is profoundly influenced by hepatic ischemia/reperfusion in a complex and biphasic manner. 11
Much of the injury sustained by many organs, including the liver, as a consequence of ischemia is triggered by the generation of reactive oxygen metabolites by activated xanthine oxidase at reperfusion. 12 This paradigm of reperfusion injury, described first in the feline small intestine, 13,14 entails the initial activation of xanthine oxidase by ischemia, the generation of superoxide at the reintroduction of oxygen at reperfusion, and a characteristic free radical cascade as the unpaired electron is passed from molecule to molecule within the endothelial cell. The resultant upregulation of endothelial adhesion molecules, particularly within the venules, and the consequent trapping and activation of circulating leukocytes, results in microvascular injury and ultimately in parenchymal organ dysfunction. 15,16 More recent studies have indicated that platelet activating factor (PAF) may well play an important early role in this inflammatory sequence. 17 Although several studies have evaluated these individual components of this system with respect to the liver, 18,19 we are aware of no studies that have evaluated this entire paradigm sequentially and in vivo.
This deficiency has been exacerbated by the need for most hemorrhagic shock/resuscitation (S/R) models to use heparin to maintain the patency of intravascular catheters and to prevent thrombosis in the shed blood, thereby allowing it to be reinfused at reperfusion. 20,21 There are severe limitations of heparinized models of hemorrhagic shock, however, particularly because microvascular patency is one of the limiting factors in tissue recovery after ischemia, and the use of heparin alone has been found to improve recovery substantially. 21,22 Moreover, heparin has been reported to release xanthine oxidase from the surface of endothelial cells into the circulation 23 and therefore could profoundly affect the results of any experiments evaluating the role of xanthine oxidase in this mechanism.
We therefore modified a standardized hemorrhagic S/R model in rats, 20,24 based on the isobaric principle of Wiggers, 25 to be functional and reproducible without the use of heparin. Using this model, we evaluated the relative, potentially sequential roles of PAF, xanthine oxidase, the generation of reactive oxygen species, hepatic oxidant stress, and hepatic neutrophil accumulation in the evolution of postischemic hepatic injury after whole-body S/R.
MATERIALS AND METHODS
Experimental Animals
All studies were conducted in accordance with standard criteria for the humane care and treatment of animals, in accordance with protocols previously approved by the Animal Care and Use Committee of the Johns Hopkins University, School of Medicine. Male Sprague-Dawley rats weighing 250 to 300 g were fed standard laboratory chow (Labdiet, PMI Feeds, St. Louis, MO) and were kept under clean conditions in individual cages in a light-cycled room.
Model
We modified a standardized rat model 20,24 of hemorrhagic shock based on Wiggers’ isobaric model. 25 Rats were fasted of food but not water for 2 days and were then anesthetized with 150 mg/kg ketamine (Phoenix Scientific Inc., St. Joseph, MO) and 0.075 mg/kg acepromazine (Ayerst Laboratories Inc., Rouses Point, NY) by intramuscular injection. Core body temperature, monitored rectally, was maintained between 36° and 38°C with a heating lamp. Through bilateral inguinal incisions, each femoral artery was cannulated with a PE-50 catheter. To avoid clotting within the catheters during the hemorrhagic shock period in this unheparinized model, the catheters had been previously coated with heparin bonded to tridodecylmethylammonium chloride (TDMAC, Polyscience Inc, Warrington, PA). 26 Citrate-phosphate-dextrose (CPD, containing 0.14 mL/mL shed blood; Sigma Chemical Co., St. Louis, MO) was used as the anticoagulant in the shed-blood reservoir to avoid the need for heparinization. The rat’s systemic arterial pressure was monitored continuously via the right femoral artery catheter with a physiologic recorder (Grass Instrument Co., Quincy, MA) and a strain gauge arterial pressure transducer (Gould Statham, Valley, MA). After the surgical preparation, a 15-minute period of hemodynamic stabilization was allowed before starting the experiment.
Hypotension was then induced by withdrawing blood from the left femoral artery slowly during a 30-minute period until the mean arterial pressure fell to 45 ± 3 mmHg. The mean arterial pressure was then held constant at 45 ± 3 mmHg by withdrawing or reinfusing small (0.5-mL) aliquots of shed blood to effect an isobaric (not isovolemic) shock state (Fig. 1). After 2 hours of this hypotension, the shed blood (containing the CPD) plus 1% body weight lactated Ringer’s solution was reinfused during a 20-minute period. Serum calcium levels, obtained from the systemic circulation of the intact rat after reinfusion of the shed blood, were no lower than those measured before preshock, nor than normal levels (7.8 ± 0.9 mg/dL), indicating no systemic effect of the CPD at reinfusion. Both catheters were then removed, the vessels were ligated, and the inguinal incisions were sutured closed. The rats were then given free access to water and food for either 6 or 24 hours after S/R. Sham-shock rats were cannulated and so maintained without hemorrhage for 2.5 hours and then observed for 6 or 24 hours. After 6 or 24 hours of resuscitation, the rats were euthanized, under anesthesia, by exsanguination via cardiac puncture so that the blood could be collected for analysis.

Figure 1. Hemokinetics of rat model of hemorrhagic shock/resuscitation. Both femoral arteries were cannulated with PE-50 catheters. The arterial pressure was continuously monitored with one of the catheters and a Grass recorder. Blood was withdrawn or instilled through another catheter to maintain a constant level of hemorrhagic shock (at 45 ± 3 mmHg) within 30 minutes. After 120 minutes of hemorrhagic hypotension, the shed blood and 1% body weight lactated Ringer’s solution were reinfused during a 20-minute period to restore blood pressure.
The portal vein was then cannulated with an 18-gauge polyethylene catheter, the suprahepatic inferior vena cava was incised, and the infrahepatic inferior vena cava was ligated just below the liver, above the right renal vein. The liver was then flushed via the portal vein with 5 mL phosphate-buffered saline (PBS; pH 7.4) at 37°C for 20 seconds, after which the left lateral and median lobes of the liver were removed immediately. Half of each excised lobe was immediately frozen in liquid nitrogen and stored at −80°C for myeloperoxidase and reduced (GSH) and oxidized glutathione (GSSG) assays. The other half of each excised lobe was fixed in 10% buffered formalin for histologic and histochemical processing. The sections for histologic examination were stained with hematoxylin and eosin for conventional morphologic evaluation of hepatic injury.
The remaining liver was perfused in situ by a modification of the technique previously described 20,27 that involves trypan blue exclusion by the hepatocyte plasma membrane to assess hepatocyte viability in situ. The flushed liver was perfused via the portal vein with 150 mL 0.2 mmol/L trypan blue (Sigma) in PBS (pH 7.4) at 37°C at 3.0 mL/min/g liver for 10 minutes. Then 30 mL 4% paraformaldehyde (pH 7.4) (J.T. Baker Inc., Philipsburg, NJ) in PBS was perfused for 2 minutes. This liver was then excised and immersed in 10% buffered formalin and processed routinely for histologic examination. These particular sections were counterstained with only eosin to allow the visualization of trypan blue-stained nuclei as a more sensitive indicator of loss of hepatocyte viability (i.e., loss of plasma membrane permeability).
Assessment of Hepatic Oxidant Stress
Liver samples were stored at −80°C until measurement. Total and oxidized glutathione were measured as indices of net hepatic oxidant stress using an enzymatic recycling procedure whereby glutathione is sequentially oxidized by 5, 5′-dithiobis (2-nitrobenzoic acid) (DTNB) and then reduced by nicotinamide-adenine dinucleotide phosphate (NADPH) in the presence of glutathione reductase, as described of Tietze, 28 modified by Griffith. 29 Briefly, liver samples were homogenized in 5% 5-sulfosalicylic acid (Sigma) and then centrifuged at 15,000 g for 5 minutes at 4°C. 5-sulfosalicylic acid was used to prevent the postsampling oxidation of GSH to GSSG and thereby to maintain the experimental redox state. The assay medium contained 0.2 mmol/L NADPH, 0.6 mmol/L DTNB, 0.5 U glutathione reductase, and 1 μL sample in a final volume of 1 mL. The reaction was initiated by the addition of glutathione reductase to the assay medium. The rate of formation of reduced DTNB was measured spectrophotometrically at 412 nm for 3 minutes using the Beckman DU-64 spectrophotometer (Beckman Instruments, Fullerton, CA). For calculation of [GSH] from the liver samples, a standard curve was made with reduced glutathione (Sigma). GSSG was measured using the recycling assay described by Griffith, 29 after reduced glutathione had been derivatized by 2-vinylpyridine (Aldrich Chem. Co. Inc., Milwaukee, WI). With 2-vinylpyridine, the formation of glutathione-containing disulfides by autoxidation or thiol transfer is less likely to occur. Briefly, 100 μL sample contained 2 μL 2-vinylpyridine and 6 μL triethanolamine (Aldrich). After 60 minutes, the GSSG level was determined by the assay described in the DTNB-GSSG reductase assay above. For calculation of [GSSG] from the liver samples, a standard curve was made with oxidized glutathione (Sigma).
Assessment of Hepatic Neutrophil Accumulation
Assay of Total Hepatic Myeloperoxidase Activity Levels
Total hepatic myeloperoxidase activity was used to estimate overall hepatic leukocyte accumulation, without respect to intrahepatic localization. The liver samples were thawed and homogenized in 20 mmol/L PBS (pH 7.4), then centrifuged at 20,000 g for 15 minutes at 4°C. The supernatant was discarded, and the pellet was rehomogenized in 10 mmol/L EDTA and 0.5% hexadecyltrimethylammonium bromide (Sigma) in 50 mmol/L PBS (pH 6.0). The samples were then recentrifuged at 40,000 g for 30 minutes at 4°C. Myeloperoxidase activity was assayed by mixing 0.1 mL of the supernatant with 2.9 m of 50 mmol/L PBS (pH 6.0) containing 0.167 mg/mL O-dianisidine hydrochloride (Sigma) and 0.0005% hydrogen peroxide (J.T. Baker). The change in absorbance at 460 nm was measured for 2 minutes using the Beckman spectrophotometer. The change in absorbance between 30 and 90 seconds was used for comparison. One unit of myeloperoxidase activity was defined as that amount degrading one μmole of peroxide per minute at 25°C. 30,31 For calculation of myeloperoxidase activity from the liver samples, a standard curve was made with human myeloperoxidase (Sigma).
Histochemical Localization of Intrahepatic Leukocyte Accumulation
A modification of the technique described by Moloney et al 32 and Katayama and Mochino 33 involving esterase histochemistry was used to quantitate the localization of hepatic leukocyte accumulation. Briefly, after PBS perfusion, the liver tissue was fixed in 10% buffered formalin and sectioned at 5 μm. These sections were stained with naphthol ASD-chloroacetate esterase (Sigma), as previously described. 20 At microscopy, three regions of the liver lobule were designated as follows:
• Zone 1 (periportal region): within a five-hepatocyte circumference around the portal vascular bundle
• Zone 2 (midzone): beyond a 10-hepatocyte circumference from both the portal vascular bundle and the central vein
• Zone 3 (pericentral region): within a five-hepatocyte circumference around the central vein.
The leukocytes were identified by this bright-red esterase staining and were counted in 10 randomly selected high-power (H450) fields per zone, using a light microscope.
Assessment of Hepatocellular Injury
Plasma Aspartate Aminotransferase Levels
Levels of plasma aspartate aminotransferase (AST) were measured spectrophotometrically using a commercial kit (Sigma) as an index of nonlocalization-specific hepatocellular injury. AST catalyzes the transfer of α-amino groups from specific amino acids to α-ketoglutaric acid to yield glutamic acid and oxaloacetic acid. These keto acids are then determined colorimetrically after their reaction with 2,4-dinitrophenyl-hydrazine, and the keto acid present in the substrate and those formed as products are reacted with 2,4-dinitrophenyl-hydrazine. 34,35 The absorbance is then measured at 505 nm. For calculating AST levels from rat plasma, a standard curve was made using a commercial kit (Sigma).
Plasma Alcohol Dehydrogenase Levels
Plasma alcohol dehydrogenase (ADH) activity was measured as an index of hepatocellular injury that is more specifically localized within the centrolobular region. 36,37 The reaction mixture contained 250 μl 100 mmol/L glycine (J.T. Baker) buffer (pH 10.6), 50 μL 5 mmol/L NAD (Sigma), 30 μL sample, and 60 μL water. Samples were incubated at 37°C for 5 minutes. The reaction was initiated by addition of 10 μL 20 mmol/L ethanol. The absorbance was assayed spectrophotometrically at 340 nm at 37°C for 3 minutes. A blank reaction without ethanol was measured in each case and the rate of NAD reduction without ethanol was subtracted from the rate obtained with ethanol. For calculating ADH levels from rat plasma, a standard curve was made with equine liver ADH (Sigma).
Histologic Evidence of Hepatocellular Necrosis
The liver sections were stained with hematoxylin and eosin to allow assessment of overall tissue morphology and injury grade. The degree of hepatic injury was graded from 0 to 3:
• Grade 0: no morphologic evidence of injury
• Grade 1: scattered pericentral necrosis seen occasionally, only within a five-hepatocyte circumference around the central vein
• Grade 2: pericentral necrosis seen uniformly, but only within a five-hepatocyte circumference around the central vein, with the other areas undamaged
• Grade 3: widespread hepatocellular necrosis, extending beyond a five-hepatocyte circumference around the central vein, frequently reaching to the midzone.
Higher levels of hepatic injury were not seen in this model.
To obtain a more quantitative estimate of the degree of hepatic necrosis, sections from the same but trypan blue-perfused liver were counterstained with eosin alone and were used to determine the degree of loss of hepatocyte viability, indicated by the presence of trypan blue staining of the hepatocyte nuclei, as previously described. 20 This method detects cells with increased nonspecific permeability of the plasma membrane. Trypan blue staining was evaluated in 10 randomly selected high-power fields per zone. The percentage of trypan blue-positive nuclei in each zone was then calculated.
Experimental Protocols
The rats were divided into seven groups. The rats in groups 2 through 7 all underwent hemorrhagic S/R as described.
1. Sham shock (n = 8) as described
2. S/R alone (n = 10)
3. WEB 2170 + S/R (n = 8): Rats received 10 mg/kg WEB 2170, a specific PAF receptor antagonist, intraperitoneally 90 minutes after the induction of shock (i.e., 30 minutes before resuscitation).
4. Allopurinol + S/R (n = 8): Rats received 50 mg/kg allopurinol, a specific xanthine oxidase inhibitor, by gavage 36, 12, and 2 hours before shock.
5. N-acetylcysteine + S/R (n = 8): Rats received 200 mg/kg N-acetylcysteine, the cell permeable antioxidant, intraperitoneally 90 minutes after the induction of shock (i.e., 30 minutes before resuscitation).
6. Vinblastine + S/R (n = 8): Rats received 0.75 mg/kg vinblastine as an intravenous bolus 4 days before shock to induce leukopenia.
7. Vinblastine + allopurinol + S/R (n = 8): Rats received both allopurinol, as in group 3, and vinblastine, as in group 6.
The experiments in groups 3 through 7 were all performed with appropriate vehicle for WEB 2170 as in group 3, for allopurinol as in group 4, for N-acetylcysteine as in group 5, for vinblastine as in group 6, and for vinblastine plus allopurinol as in group 6. All experiments, except those in group 1 (sham), were conducted and all results analyzed with the investigator masked (by prior coding of animals and samples) to the treatment received. (All samples for group 1 rats were also evaluated in masked fashion, but it obviously was evident to the investigators conducting the shock portion of the experiment as to the identity of the sham-shocked rats.)
Statistical Analysis
All continuous data were expressed as means ± 1 SD. Apparent differences between the time course of disparate groups were evaluated by one-way analysis of variance. Discrete (not continuous) data (histologic grade of injury) were expressed as individual values, with the median value circled. Apparent differences between groups in these experiments were evaluated for significance by nonparametric Wilcoxon rank-sum analysis based on an overall masked ranking of the histologic specimens. P < .05 was considered to be significant.
RESULTS
Effect of Sham Shock
Fasted, sham-operated rats showed evidence of marked transient hepatic GSH depletion at 6 hours, but this had returned to normal levels by 24 hours (Fig. 2) . However, there was little indication of the generation of oxidized GSSG at either time. There was no evidence of hepatic leukocyte accumulation (Fig. 3) or hepatic injury at either time.
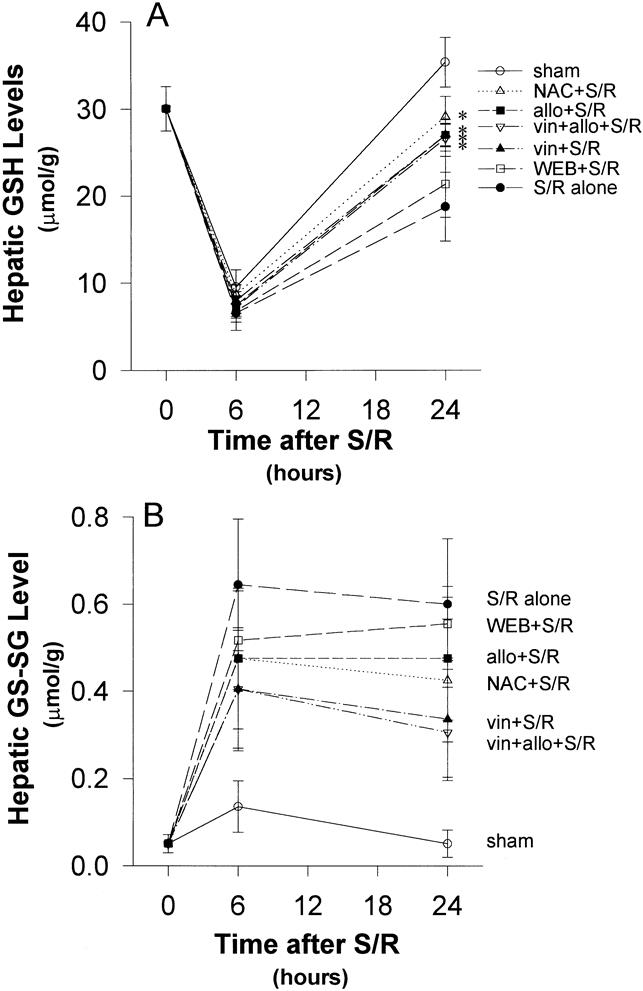
Figure 2. (A) Reduced glutathione (GSH). Starvation reduced hepatic glutathione levels markedly, even in the sham-shocked rats. Shock/resuscitation (S/R) slightly increased this marked depletion of GSH at 6 hours and significantly inhibited its recovery at 24 hours. Allopurinol, N-acetylcysteine, and vinblastine generated a more rapid return to baseline compared with S/R alone. *P < .05 versus S/R alone. (B) Oxidized glutathione (GSSG). All the rates showed a peak elevation of hepatic GSSG at 6 hours. Despite clear trends toward protection, none of the reductions in GSSG generation were statistically significant when compared with S/R alone. There were no significant differences in the trends between groups.
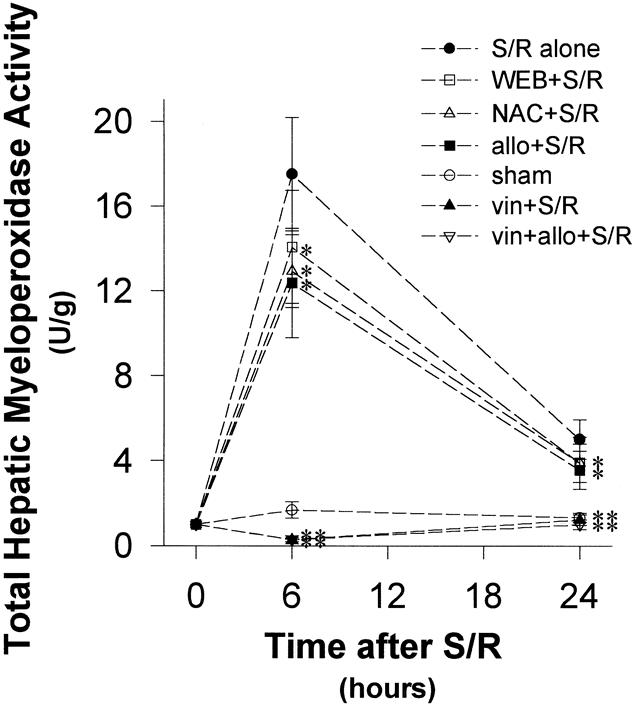
Figure 3. Hepatic myeloperoxidase (MPO) activity after shock/resuscitation (S/R). Leukocyte accumulation at 6 and 24 hours, as measured by MPO activity, was blunted by allopurinol, N-acetylcysteine, and WEB 2170 compared with S/R alone. The apparent protection produced by N-acetylcysteine was not significant due to a large standard deviation, but the mean value for the group was reduced compared with S/R alone at 24 hours. *P < .05 versus S/R alone; **P < .001 versus S/R alone.
Effect of S/R
S/R increased the depletion of GSH, but coupled with marked GSSG generation at 6 hours. This showed some recovery at 24 hours (Fig. 2). This was associated with leukocyte accumulation, localized predominantly within the periportal and centrolobular regions, at 6 hours, but with substantial resolution by 24 hours (Figs. 4 and 5). This, in turn, was associated with striking elevations of plasma AST and especially ADH levels (Fig. 6) and with histologic evidence of hepatocyte necrosis (Figs. 7–9). This morphologic indication of necrosis was localized predominantly within the centrolobular regions, and all of these indicators of hepatocellular injury were more marked at 24 hours than at 6 hours. There was a striking correlation between (centrolobular) neutrophil accumulation at 6 hours and (centrolobular) hepatocellular injury at 24 hours.
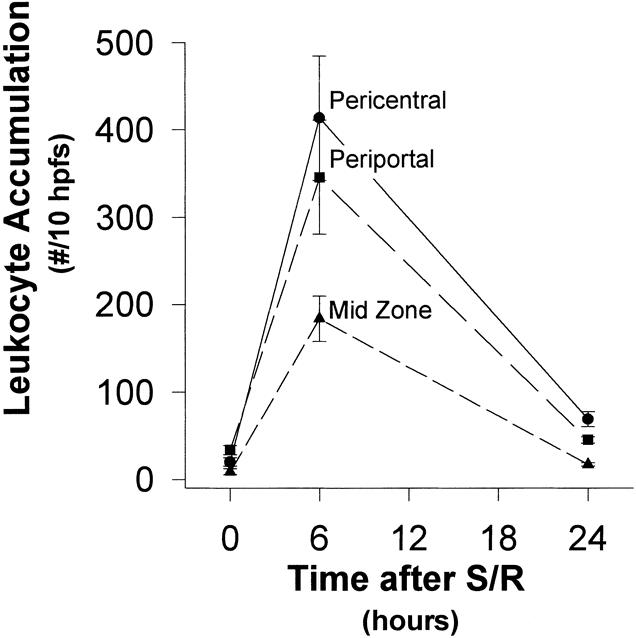
Figure 4. Localization of hepatic leukocyte accumulation (time course). Shock/resuscitation increased leukocyte accumulation, localized predominantly in the periportal and the pericentral regions, at 6 hours, but there was a substantial resolution by 24 hours.

Figure 5. Localization of hepatic leukocyte accumulation. (A) (Periportal region) Allopurinol and N-acetylcysteine significantly decreased leukocyte accumulation after shock/resuscitation (S/R). This accumulation peaked at 6 hours but was still significantly decreased at 24 hours in the allopurinol and N-acetylcysteine groups. The WEB 2170 group also showed a peak neutrophil accumulation at 6 hours, but the number of leukocytes was not significantly decreased compared with the S/R alone group at either 6 or 24 hours. (B) (Midzone) Leukocyte accumulation peaked at 6 hours but was not significantly reduced with allopurinol, N-acetylcysteine, or WEB 2170 treatment at either 6 or 24 hours. (C) (Pericentral region) Pericentral leukocyte accumulation was significantly decreased by allopurinol, N-acetylcysteine, and WEB 2170 treatment at both 6 and 24 hours compared with the S/R alone group. *P < .05 versus S/R alone; **P < .001 versus S/R alone.

Figure 6. (A) Plasma aspartate aminotransferase (AST) levels continued to rise for the first 24 hours after shock/resuscitation (S/R) for all groups with substantial elevation in the first 6 hours. However, this early elevation was significantly blunted in the allopurinol, N-acetylcysteine, and WEB 2170 groups. At 24 hours, allopurinol and vinblastine treatment blunted the elevated AST levels compared with the S/R alone group. (B) Plasma alcohol dehydrogenase (ADH) levels in the S/R rats rose rapidly in the first 6 hours and then started to decline but remained elevated at 24 hours. In contrast, the allopurinol and N-acetylcysteine groups showed a steady increase in ADH levels. At 6 hours, treatment with allopurinol, N-acetylcysteine, WEB 2170, and vinblastine significantly blunted the rise in ADH levels compared with the S/R alone group. At 24 hours, the allopurinol and vinblastine-treated groups had significantly lower ADH levels than the S/R alone group. *P < .05 versus S/R alone; **P < .001 versus S/R alone.
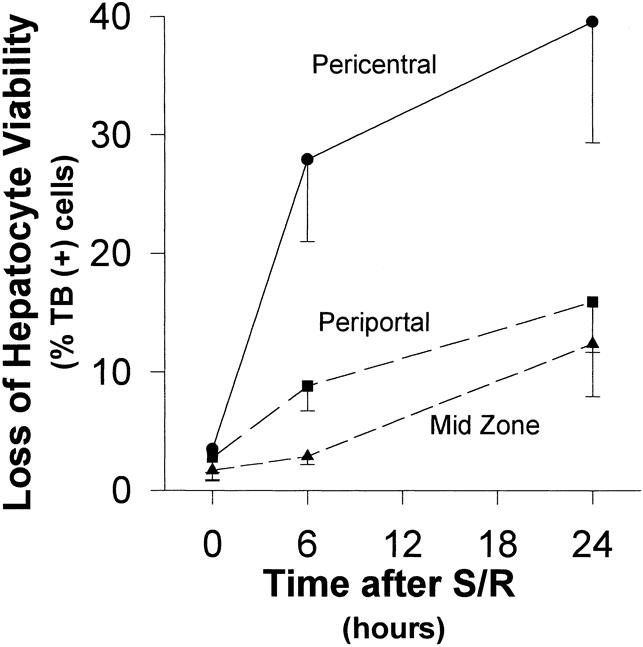
Figure 7. Loss of hepatocyte viability (time course). The pattern of hepatic injury, as assessed by histologic examination of trypan blue-stained liver section for loss of hepatocyte viability, was greatest in the pericentral region at both 6 and 24 hours. Although the greatest increase in the degree of injury was in the first 6 hours after shock/resuscitation, the injury continued to increase at 24 hours.

Figure 8. Loss of hepatocyte viability. (A) The mild degree of injury in the periportal region was not significantly affected by treatment, compared with the shock/resuscitation (S/R) alone group. (B) Similar to the periportal region, the midzone also did not show significant reduction in the mild degree of injury with treatment. (C) Although the pericentral region was most injured by S/R, these parameters did not reflect significant protection by treatment with allopurinol, N-acetylcysteine, or WEB 2170. **P < .05 versus S/R alone.

Figure 9. Masked ranking of histologic hepatic injury (A) 6 hours and (B) 24 hours after shock/resuscitation (S/R). S/R-induced centrolobular hepatocellular injury was decreased in the treatment groups and was more marked at 24 hours than at 6 hours. Each treatment protected significantly at 24 hours. Each point represents an individual rat. Median values are represented by circles. The Wilcoxon rank-sum analysis was used to evaluate these data. *P < .05 versus sham. †P < .05 versus S/R alone.
PAF Inhibition + S/R
Inhibition of PAF receptor binding with WEB 2170 significantly attenuated leukocyte accumulation at both 6 and 24 hours (Figs. 3 and 5). Despite the fact that there was no decrease in the generation of hepatic oxidant stress at both 6 and 24 hours after S/R with WEB 2170 (Fig. 2), there was significantly less hepatic injury at both time points (Figs. 6, 8, and 9).
Xanthine Oxidase Inhibition + S/R
Xanthine oxidase inhibition with allopurinol significantly attenuated the GSH depletion at 24 hours (Fig. 2), but not at 6 hours. This was associated with significantly less leukocyte accumulation at 6 hours (Figs. 3–5) and less evidence of hepatic oxidant stress and injury at both 6 and 24 hours after S/R (Figs. 6, 8, and 9).
Antioxidant + S/R
Antioxidant treatment with N-acetylcysteine had a similar effect to that of allopurinol on the response to S/R. GSH depletion was significantly attenuated at 24 hours, but the apparently small attenuation at 6 hours was not significant (Fig. 2). However, this was associated with significantly less leukocyte accumulation at 6 hours (Figs. 3 and 5) and significantly less evidence of hepatic oxidant stress (GSSG) and less injury at both 6 and 24 hours after S/R (Figs. 6, 8, and 9).
Leukopenia + S/R
Treatment with vinblastine induced severe peripheral leukopenia (<103/mL) and thereby virtually eliminated hepatic leukocyte accumulation in response to S/R (Figs. 3 and 5). This was associated with significant attenuation of the indicators of hepatic oxidant stress and injury at both 6 and 24 hours after S/R (Figs. 6, 8, and 9).
Vinblastine + Allopurinol + S/R
When the rats were pretreated with both allopurinol and vinblastine, there was no measurable improvement in the level of protection over that provided by vinblastine alone or allopurinol alone (Figs. 6, 8, and 9).
DISCUSSION
This model of hemorrhagic S/R evolved from earlier versions used in our laboratory 20 to one that consistently produces a characteristic severe hepatic injury, but without the death of the animal. Indeed, these animals routinely survive for the 24-hour period of observation (and thereafter, if allowed to do so). Due to reasonable limitations imposed by the ethical use of animals, as well as economic considerations, we focused on two time periods after this shock-induced injury. The first, at 6 hours, represents an optimal point to evaluate the early inflammatory response in the liver, based on our previous experiments. 20 Likewise, the 24-hour end point represents a maximal level of evidence of hepatocellular injury as the consequence of these inflammatory changes. Therefore, although we did not formally evaluate the further evolution of this injury in this study, the level seen at 24 hours represents a near-maximal injury end point. Likewise, this study did not evaluate any subsequent hepatic recovery or regeneration. Indeed, our focus was primarily on the initial pathogenesis of injury.
The most important change from our previous models was based on prior, 20,24,25 legitimate criticism of our models studying the xanthine oxidase system, which had involved the use of heparin to prevent coagulation of blood in the intravascular catheters and of the shed blood in the reservoir. A number of studies have indicated that heparin can release the free radical-generating enzyme xanthine oxidase from its constitutive location, bound to the surface of the endothelial cell. 21,23,38 Certainly most patients who are subjected to hemorrhagic S/R do not have the benefit of heparin before the insult, and therefore this model is more representative of the clinical situation in a theoretically important way.
Despite the validity of this theoretical argument, however, the results of this model of S/R were strikingly similar to those seen in the heparinized model that we had used earlier. 20 The characteristic lesion produced in the liver in both versions in this model is centrolobular necrosis, as indicated morphologically and by the preferential elevation of serum levels of ADH relative to the elevation of the levels of the enzyme AST. AST is distributed more uniformly among hepatocytes throughout the hepatic lobule; ADH is characteristically localized within centrolobular hepatocytes. 36,37 There is close correlation between the primarily centrolobular leukocyte accumulation at 6 hours and biochemical (ADH, AST) and histologic (conventional morphology and trypan blue exclusion) indications of hepatocellular injury at 24 hours. These features are very similar to those described in patients with ischemic hepatitis. Therefore, although no animal model can perfectly simulate the clinical situation, this model represents a highly reproducible and quantifiable model of the clinicopathologic picture of ischemic hepatitis.
The level of injury sustained by the liver as a consequence of ischemia/reperfusion is closely related to the fasting state of the animal before injury. In pilot studies, we also had found a substantially diminished level of injury if the rats were fed up until the time of surgery. Although this well-described phenomenon is undoubtedly due to a number of factors, an important component has been attributed to the depletion of hepatocyte stores of GSH, 39,40 a situation that we saw strikingly reproduced in this model, even in the sham-shock animals, which were subjected to the same period of starvation without undergoing shock. These animals, however, showed no evidence of the generation of oxidized glutathione (GSSG) and no evidence of hepatic inflammation or hepatocellular injury.
In the remaining experiments, all animals showed a remarkable correlation between the levels of oxidant stress, indicated more by the generation of GSSG than by the depletion of GSH, and leukocyte accumulation at 6 hours with subsequent hepatocellular injury at 24 hours. Although this sequence of events alone is strongly suggestive of an oxidant mechanism triggered by the generation of the reactive oxygen metabolites at reperfusion, it does not unequivocally establish such a cause-and-effect relationship. To this end, the following specific inhibitory studies were designed.
A blockade of leukocyte accumulation at 6 hours as well as hepatocellular injury at 24 hours by the inhibition of xanthine oxidase with allopurinol is strongly suggestive of the triggering of this mechanism by xanthine oxidase. We were careful to use a dose of allopurinol that had been definitively shown to inhibit xanthine oxidase activity in vivo without producing the (largely theoretical) direct hydroxyl scavenging effect that has been seen with high allopurinol concentrations in vitro. 41–43 Although xanthine oxidase inhibition has also been reported to be protective by virtue of the salvage of purines to potentiate the regeneration of ATP after ischemia, 44–46 the fact that allopurinol did not further improve the protection provided by neutropenia, and the fact that equivalent protection was provided by antioxidant therapy or leukopenia, argue against this mechanism, a mechanism for which there has never been any direct evidence.
PAF can produce a microvascular injury, particularly within the intestine 17,47–49 and also within the liver. 50–52 When we injected 5 μg/kg PAF intravascularly into these same rats without subjecting them to shock, we saw similar degrees of leukocyte infiltration (data not shown). However, as previously reported by Granger et al in the small intestine, 17,53 here PAF activity seems to be working in series with the xanthine oxidase-oxidant-leukocyte accumulation mechanism in that PAF inhibition with WEB 2170 not only inhibited hepatocellular injury but also blocked hepatic leukocyte accumulation.
Finally, when leukocyte accumulation was blocked directly by inducing systemic neutropenia, we saw striking protection from hepatocellular injury. This suggests that the xanthine oxidase-generated, oxidant-mediated injury mechanism works not only by triggering with PAF, but also through the activation of leukocyte accumulation. This is consistent with other studies in the feline small intestine 54 and in other organs. 55,56 Although many have equated antioxidant effects with antileukocyte effects, 57 the inhibition of xanthine oxidase does not directly inhibit the generation of oxidants from leukocytes. 58 The generation of reactive oxygen species by leukocytes is mediated by a distinctly different enzyme, the “leukocyte” NADPH oxidase. 58 Because xanthine oxidase inhibition with allopurinol and antioxidant therapy with N-acetylcysteine blocked both leukocyte accumulation and hepatocellular injury, this suggests that the oxidants that trigger this response are generated by endothelial cell xanthine oxidase, 12,38 not the leukocytes that accumulate subsequently. This suggests that leukocyte-generated reactive oxygen species are not likely to be the direct source of subsequent hepatocellular injury. This is consistent with the earlier findings of Kvietys et al, 59,60 based on in vitro studies, that suggest that leukocyte proteases in general, and elastases in particular, are more important for the microvascular injury generated by leukocytes than are the reactive oxidants generated by the leukocyte NADPH oxidase. Certainly the localization of this inflammation and injury in the distal (pericentral) hepatic sinusoids and central hepatic veins, which corresponds to the mesenteric and intestinal venules, and which also corresponds with the localization of endothelial xanthine oxidase, 38 strongly supports this concept.
Reactive oxidants mediate neutrophil accumulation in this injury, as they do in other injury models, by signaling the upregulation of vascular adhesion molecules, 61–63 as previously described in the feline small intestine 13,14,54 and in other organs. Unfortunately, due to the limited availability and the extraordinary expense of specific inhibitors to these molecules for the rat, we were unable to explore the specific adhesion molecules that might be involved in this model. Moreover, although we think it is likely that this mechanism operates in a manner similar to that previously described, we cannot distinguish the consequences of microvascular leukocyte accumulation due to nonspecific microvascular injury from those that might be due to the more specific upregulation of endothelial adhesion molecules, both of which could be mediated by reactive oxygen species generated from xanthine oxidase.
In summary, these studies confirm, in a highly reproducible and unheparinized model, the apparent linear etiologic relation between reactive oxygen metabolites generated by xanthine oxidase, oxidant stress, PAF receptor activation, and leukocyte accumulation in the first 6 hours with the consequent development of centrolobular hepatocellular injury characteristic of ischemic hepatitis over the next 24 hours. It appears likely that a similar mechanism might contribute substantially to the generation of ischemic hepatitis in humans.
Footnotes
Correspondence: Gregory B. Bulkley, MD, Dept. of Surgery, The Johns Hopkins University School of Medicine, Blalock 685, Johns Hopkins Hospital, 600 N. Wolfe St., Baltimore, MD 21287-4685.
Supported by National Institutes of Health grant DK-31764. WEB 2170 was a gift from Boehringer Ingelheim KG (Ingelheim, Germany).
Accepted for publication June 10, 1999.
References
- 1.Bulkley GB, Oshima A, Bailey RW. Pathophysiology of hepatic ischemia in cardiogenic shock. Am J Surg 1986; 151:87–97. [DOI] [PubMed] [Google Scholar]
- 2.Rawson JS, Achord JL. Shock liver. South Med J 1985; 78:1421–1425. [DOI] [PubMed] [Google Scholar]
- 3.Champion HR, Jones RT, Trump BF, et al. A clinicopathologic study of hepatic dysfunction following shock. Surg Gynecol Obstet 1976; 142:657–663. [PubMed] [Google Scholar]
- 4.Nunes G, Blaisdell FW, Margaretten W. Mechanism of hepatic dysfunction following shock and trauma. Arch Surg 1970; 100:546–556. [DOI] [PubMed] [Google Scholar]
- 5.Konishi Y, Shaked A, Egawa H, et al. Correlation of hepatic injury, synthetic function, and mitochondria energy level in orthotopic liver transplantation. J Surg Res 1992; 52:466–471. [DOI] [PubMed] [Google Scholar]
- 6.Gibson PR, Dudley FJ. Ischemic hepatitis: clinical features, diagnosis and prognosis. Aust NZ J Med 1984; 14:822–825. [DOI] [PubMed] [Google Scholar]
- 7.Bynum TE, Boitnott JK, Maddrey WC. Ischemic hepatitis. Dig Dis Sci 1979; 24:129–135. [DOI] [PubMed] [Google Scholar]
- 8.Birgens HS, Henriksen J, Matzen P, Poulsen H. The shock liver: clinical and biochemical findings in patients with centrilobular liver necrosis following cardiogenic shock. Acta Med Scand 1978; 204:417–421. [PubMed] [Google Scholar]
- 9.Schiller HJ, Buchman TG, Bulkley GB. Multiple system organ failure. In: Cameron JL, ed. Current Surgical Therapy. Philadelphia: BC Decker; 1992: 1067–1072.
- 10.Klein A, Zhadkevich M, Margolick J, et al. Quantitative discrimination of hepatic reticuloendothelial clearance and phagocytic killing. J Leukoc Biol 1994; 55:248–252. [DOI] [PubMed] [Google Scholar]
- 11.Kondo S, Wang D, Mayumi T, et al. Effect of hemorrhagic shock and resuscitation upon hepatic phagocytic clearance and killing of circulating microorganisms. Shock 1996; 5:106–111. [DOI] [PubMed] [Google Scholar]
- 12.Ratych RE, Chuknyiska RS, Bulkley GB. The primary localization of free radical generation after anoxia/reoxygenation in isolated endothelial cells. Surgery 1987; 102:122–131. [PubMed] [Google Scholar]
- 13.Granger DN, Rutili G, McCord JM. Superoxide radicals in feline intestinal ischemia. Gastroenterology 1981; 81:22–29. [PubMed] [Google Scholar]
- 14.Parks DA, Bulkley GB, Granger DN, et al. Ischemic injury to the cat small intestine: role of superoxide radicals. Gastroenterology 1982; 82:9–15. [PubMed] [Google Scholar]
- 15.Bulkley GB. Endothelial xanthine oxidase: a radical transducer of inflammatory signals for reticuloendothelial activation. Br J Surg 1993; 80:684–686. [DOI] [PubMed] [Google Scholar]
- 16.Bulkley GB. Reactive oxygen metabolites and reperfusion injury: aberrant triggering of reticuloendothelial function. The Lancet 1994; 344:934–936. [DOI] [PubMed] [Google Scholar]
- 17.Kubes P, Suzuki M, Granger DN. Platelet-activating factor-induced microvascular dysfunction: role of adherent leukocytes. Am J Physiol 1990; 258:G158–G163. [DOI] [PubMed] [Google Scholar]
- 18.Jaeschke H, Schini VB, Farhood A. Role of nitric oxide in the oxidant stress during ischemia/reperfusion injury of the liver. Life Sci 1992; 50:1797–1804. [DOI] [PubMed] [Google Scholar]
- 19.Horie Y, Wolf R, Granger DN. Role of nitric oxide in gut ischemia-reperfusion–induced hepatic microvascular dysfunction. Am J Physiol 1997; 273:G1007–G1013. [DOI] [PubMed] [Google Scholar]
- 20.Mayumi T, Chan CK, Clemens MG, Bulkley GB. Zonal heterogeneity of hepatic injury following shock/resuscitation: relationship of xanthine oxidase activity to localization of neutrophil accumulation and central lobular necrosis. Shock 1996; 5:324–332. [PubMed] [Google Scholar]
- 21.Wang P, Singh G, Rana MW, et al. Preheparinization improves organ function after hemorrhage and resuscitation. Am J Physiol 1990; 259:R645–R650. [DOI] [PubMed] [Google Scholar]
- 22.Bulkley GB, Amano H, Hamilton SR, Zuidema GD. Microvascular thrombosis preventing reperfusion: a factor limiting intestinal recovery from acute ischemic injury [abstract]. Microvasc Res 1982;245. [Google Scholar]
- 23.Tan S, Yokoyama Y, Dickens E, et al. Xanthine oxidase activity in the circulation of rats following hemorrhagic shock. Free Radic Biol Med 1993; 15:407–414. [DOI] [PubMed] [Google Scholar]
- 24.Whigham H, Weil MH. A model for the study of hemorrhagic shock in the rat: development of the method. J Appl Physiol 1966; 21:1860–1863. [DOI] [PubMed] [Google Scholar]
- 25.Wigger CJ. Physiology of Shock. New York: The Commonwealth Fund; 1950.
- 26.Grode GA, Anderson SJ, Grotta HM, Falb RD. Nonthrombogenic materials via a simple coating process. Transactions of the American Society for Artificial Internal Organs 1998; 15:1–6. [PubMed] [Google Scholar]
- 27.Belinsky SA, Popp JA, Kauffman FC, Thurman RG. Trypan blue uptake as a new method to investigate hepatotoxicity in periportal and pericentral regions of the liver lobule: studies with allyl alcohol in the perfused liver. J Pharmacol Exp Ther 1984; 230:755–760. [PubMed] [Google Scholar]
- 28.Tietze F. Enzymic method for quantitative determination of nanogram amounts of total and oxidized glutathione: applications to mammalian blood and other tissues. Anal Biochem 1969; 27:502–522. [DOI] [PubMed] [Google Scholar]
- 29.Griffith OW. Determination of glutathione and glutathione disulfide using glutathione reductase and 2-vinylpyridine. Anal Biochem 1980; 106:207–212. [DOI] [PubMed] [Google Scholar]
- 30.Mullane KM, Kraemer R, Smith B. Myeloperoxidase activity as a quantitative assessment of neutrophil infiltration into ischemic myocardium. J Pharmacol Methods 1985; 14:157–167. [DOI] [PubMed] [Google Scholar]
- 31.Bradley PP, Priebat DA, Christensen RD, Rothstein G. Measurement of cutaneous inflammation: estimation of neutrophil content with an enzyme marker. J Invest Dermatol 1982; 78:206–209. [DOI] [PubMed] [Google Scholar]
- 32.Moloney WC, McPherson K, Fliegelman L. Esterase activity in leukocytes demonstrated by the use of naphthol AS-D chloroacetate substrate. J Histochem Cytochem 1960; 8:200–207. [DOI] [PubMed] [Google Scholar]
- 33.Katayama I, Mochino T. Naphthol AS-D chloroacetate esterase reaction. Systemic modifications for optimization to smears and sections. Acta Pathol Jpn 1983; 33:381–393. [PubMed] [Google Scholar]
- 34.Amador E, Franey RJ, Massod MF. Serum glutamic-oxaloacetic transaminase activity: diagnostic accuracy of the revised spectrophotometric and the dinitrophenyl-hydrazine methods. Clin Chem 1966; 12:475–481. [PubMed] [Google Scholar]
- 35.Babson AL, Arndt EG, Sharkey LJ. A revised colorimetric glutamic-oxalacetic transaminase assay. Clin Chim Acta 1969; 26:419–423. [DOI] [PubMed] [Google Scholar]
- 36.Yamauchi M, Potter JJ, Mezey E. Lobular distribution of alcohol dehydrogenase in the rat liver. Hepatology 1988; 8:243–247. [DOI] [PubMed] [Google Scholar]
- 37.Kato S, Ishii H, Aiso S, et al. Histochemical and immunohistochemical evidence for hepatic zone 3 distribution of alcohol dehydrogenase in rats. Hepatology 1990; 12:66–69. [DOI] [PubMed] [Google Scholar]
- 38.Vickers S, Schiller HJ, Hildreth JEK, Bulkley GB. Immunoaffinity localization of the enzyme xanthine oxidase on the outside surface of the endothelial cell plasma membrane. Surgery 1998; 124:551–560. [PubMed] [Google Scholar]
- 39.Jaeschke H. The therapeutic potential of glutathione in hepatic ischemia-reperfusion injury. Transplantation 1993; 56:256–257. [DOI] [PubMed] [Google Scholar]
- 40.Okuda M, Lee H-C, Chance B, Kumar C. Glutathione and ischemia-reperfusion injury in the perfused rat liver. Free Radic Biol Med 1992; 12:271–279. [DOI] [PubMed] [Google Scholar]
- 41.Moorhouse PC, Grootveld M, Halliwell B, et al. Allopurinol and oxypurinol are hydroxyl radical scavengers. FEBS Lett 1987; 213:23–28. [DOI] [PubMed] [Google Scholar]
- 42.Zimmerman BJ, Parks DA, Grisham MB, Granger DN. Allopurinol does not enhance antioxidant properties of extracellular fluid. Am J Physiol 1988; 255:H202–H206. [DOI] [PubMed] [Google Scholar]
- 43.Klein AS, Joh JW, Rangan U, et al. Allopurinol: discrimination of antioxidant from enzyme inhibitory activities. Free Radic Biol Med 1996; 21:713–717. [DOI] [PubMed] [Google Scholar]
- 44.Toledo-Pereyra LH, Simmons RL, Najarian JS. Effect of allopurinol on the preservation of ischemic kidneys perfused with plasma or plasma substitutes. Ann Surg 1974; 180:780–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kusumoto K, Morimoto T, Minor T, et al. Allopurinol effects in rat liver transplantation on recovery of energy metabolism and free radical-induced damage. Eur Surg Res 1995; 27:285–291. [DOI] [PubMed] [Google Scholar]
- 46.Karwinski W, Farstad M, Ulvik R, Soreide O. Sixty-minute normothermic liver ischemia in rats. Evidence that allopurinol improves liver cell energy metabolism during reperfusion but that timing of drug administration is important. Transplantation 1991; 52:231–234. [PubMed] [Google Scholar]
- 47.Sun X, Hsueh W. Platelet-activating factor produces shock, in vivo complement activation, and tissue injury in mice. J Immunol 1991; 147:509–514. [PubMed] [Google Scholar]
- 48.Zhang C, Hsueh W, Caplan MS, Kelly A. Platelet activating factor-induced shock and intestinal necrosis in the rat: role of endogenous platelet-activating factor and effect of saline infusion. Crit Care Med 1991; 19:1067–1072. [DOI] [PubMed] [Google Scholar]
- 49.Hsueh W, Gonzalez-Crussi F, Arroyave JL. Platelet-activating factor: an endogenous mediator for bowel necrosis in endotoxemia. FASEB J 1987; 1:403–405. [DOI] [PubMed] [Google Scholar]
- 50.Bautista AP, Spitzer JJ. Platelet activating factor stimulates and primes the liver, Kupffer cells and neutrophils to release superoxide anion. Free Rad Res Comm 1992; 17:195–209. [DOI] [PubMed] [Google Scholar]
- 51.Komatsu Y, Shiratori Y, Hikiba Y, et al. Role of platelet-activating factor in pathogenesis of galactosamine-lipopolysaccharide-induced liver injury. Dig Dis Sci 1996; 41:1030–1037. [DOI] [PubMed] [Google Scholar]
- 52.Walcher F, Marzi I, Fisher R, et al. Platelet-activating factor is involved in the regulation of pathological leukocyte adhesion after liver transplantation. J Surg Res 1996; 61:244–249. [DOI] [PubMed] [Google Scholar]
- 53.Kubes P, Suzuki M, Granger DN. Modulation of PAF-induced leukocyte adherence and increased microvascular permeability. Am J Physiol 1990; 259:G859–G864. [DOI] [PubMed] [Google Scholar]
- 54.Nilsson UA, Schoenberg MH, Aneman A, et al. Free radicals and pathogenesis during ischemia and reperfusion of the cat small intestine. Gastroenterology 1994; 106:629–636. [DOI] [PubMed] [Google Scholar]
- 55.Hernandez LA, Granger DN. Role of antioxidants in organ preservation and transplantation. Crit Care Med 1988; 16:543–549. [DOI] [PubMed] [Google Scholar]
- 56.Korthuis RJ, Anderson DC, Granger DN. Role of neutrophil-endothelial cell adhesion in inflammatory disorders. J Crit Care 1998; 9:47–71. [DOI] [PubMed] [Google Scholar]
- 57.Romson JL, Hook BG, Kunkel SL, et al. Reduction of the extent of ischemic myocardial injury by neutrophil depletion in the dog. Circulation 1983; 67:1016–1023. [DOI] [PubMed] [Google Scholar]
- 58.Terada LS, Arnold PD. Xanthine oxidase does not mediate the antiproliferative effects of interferon-gamma in human umbilical vein endothelial cells. J Interferon Res 1998; 13:419–422. [DOI] [PubMed] [Google Scholar]
- 59.Inauen W, Granger DN, Meininger CJ, Schelling ME, Granger HJ, Kvietys PR. An in vitro model of ischemia/reperfusion-induced microvascular injury. Am J Physiol 1990; 259:G134–G139. [DOI] [PubMed] [Google Scholar]
- 60.Inauen W, Granger DN, Meininger CJ, Schelling ME, Granger HJ, Kvietys PR. Anoxia-reoxygenation-induced, neutrophil-mediated endothelial cell injury: role of elastase. Am J Physiol 1990; 259:H925–H931. [DOI] [PubMed] [Google Scholar]
- 61.Roebuck KA, Rahman A, Lakshminarayanan V, et al. H2O2 and tumor necrosis factor-α activate intercellular adhesion molecule 1 (ICAM-1) gene transcription through distinct cis-regulatory elements within the ICAM-1 promoter. J Biol Chem 1995; 270:18966–18974. [DOI] [PubMed] [Google Scholar]
- 62.Morita Y, Clemens MG, Miller LS, et al. Reactive oxidants mediate TNF-α-induced leukocyte adhesion to rat mesenteric venular endothelium. Am J Physiol 1995; 269:H1833–H1842. [DOI] [PubMed] [Google Scholar]
- 63.Arai T, Kelly SA, Brengman ML, et al. Ambient but not incremental oxidant generation effects intercellular adhesion molecule-1 (ICAM-1) induction by TNF-α in endothelium. Biochem J 1998; 331:853–861. [DOI] [PMC free article] [PubMed] [Google Scholar]