

Abstract
Objective
To test the hypothesis that neutrophil adhesion to expanded polytetrafluoroethylene (ePTFE) and Dacron triggers cell death.
Summary Background Data
Vascular prosthetic infections are intransigent clinical dilemmas associated with excessive rates of death and complications. Impaired neutrophil function has been implicated in the infection of implanted cardiovascular devices. ePTFE and Dacron are potent neutrophil stimuli able to elicit activation responses such as reactive oxygen species production independent of exogenous/soluble agonists. Reactive oxygen species that are released into the medium when neutrophils are challenged by soluble agonists are known to cause self-destruction. The authors therefore sought to examine whether neutrophil adhesion to prosthetic graft materials decreases neutrophil viability by means of reactive oxygen species production.
Methods
Neutrophils were adhered to surfaces for up to 6 hours. Cell viability was monitored with propidium iodide staining and lactate dehydrogenase release.
Results
Within 6 hours of adhesion to ePTFE and Dacron, respectively, 59% ± 11% and 44% ± 5% (n = 7) of the neutrophils were stained by propidium iodide. Indistinguishable results were obtained with plasma-coated ePTFE and Dacron. In contrast, less than 2% of the neutrophils adherent to fibrinogen-, immunoglobin-, or fetal bovine serum-coated polystyrene surfaces for 6 hours were positive for propidium iodide. The increase in membrane permeability to propidium iodide was accompanied by a two- to threefold increase in lactate dehydrogenase release. Pretreatment of neutrophils with N-acetyl-L-cysteine, cytochalasin D, or cyclosporin A significantly reduced the number of propidium iodide-positive ePTFE and Dacron adherent neutrophils.
Conclusions
Neutrophil adhesion to ePTFE and Dacron triggers a rapid nonapoptotic cell death. The effect of ePTFE and Dacron on neutrophil viability appears to be caused by reactive oxygen species production. The premature death of graft-adherent neutrophils provides a novel explanation of the defect in neutrophil bacterial killing associated with vascular prosthetic grafts.
Vascular prosthetic infections are intransigent clinical complications associated with excessive rates of death and complications. It has been estimated that the number of vascular graft procedures performed in the United States per year exceeds 500,000, including peripheral vascular reconstruction and coronary bypass grafts. Because of the limitations of prosthetic materials in small vessel configurations, they are used in less than a third of these procedures. Infections occur in 2% to 12% of implanted vascular prostheses despite the use of systemic antibiotic prophylaxis. When these events unfold, they are associated with death rates of 30% to 50% and limb loss in approximately one third to one half of survivors. 1–4
The epidemiology of vascular prosthetic infections supports the hypothesis that implant infections occur because of contamination of the prosthesis with small numbers of bacteria that find the prosthetic surface favorable for survival. 5,6 The recognition of matrix proteins that coat implant surfaces by bacterial adhesion receptors 7; the complex three-dimensional structure of these surfaces, which may provide bacteria with niches in which to hide; and the fact that material-adherent bacteria become encapsulated in a secreted glycocalyx 8,9 all contribute to enhanced bacterial survival in the prosthetic graft milieu. An alternative explanation, not exclusive of the former, is that foreign bodies weaken the ability of host immune cells to kill bacteria. Using an implanted subcutaneous Teflon cage, Zimmerli et al 10 in 1984 observed that neutrophils adherent to the Teflon surface become impaired in their ability to kill bacteria, whereas nonadherent neutrophils recovered from the cage milieu displayed normal bacterial killing ability. Nonbiologic surfaces, including polystyrene, glass, expanded polytetrafluoroethylene (ePTFE), and Dacron are potent neutrophil stimuli able to elicit activation responses independent of exogenous/soluble agonists. 11–17 Neutrophils readily adhere to and spread on these surfaces. Additional intracellular signals triggered by these adhesive events result in degranulation and a massive production and secretion of reactive oxygen species into the surrounding medium. 11–13 Henson et al 18,19 coined the term “frustrated phagocytosis” to indicate that these activation responses may represent a failed attempt of neutrophils to phagocytose substrates too large to be engulfed.
Reactive oxygen species produced by neutrophils are central to their ability to kill bacteria. Patients with chronic granulomatous disease who fail to mount a respiratory burst are therefore subject to life-threatening bacterial infection. 20 Reactive oxygen species produced during phagocytosis are confined to phagosomes, whereas surface-adherent neutrophils appear to release their antibacterial substances, including reactive oxygen species, into the surrounding. 13,18 Henson and Johnston 19 contemplated whether reactive oxygen species that are released into the medium when neutrophils are challenged by stimulatory surfaces can cause damage to surrounding tissues. One can also speculate whether reactive oxygen species produced by neutrophils can cause self-destruction. Consistent with the latter, Tsan 21 showed that treatment of neutrophils with 12-myristate 13-acetate (phorbol ester) resulted in an increase in cell membrane permeability within a few hours. This correlated with hydrogen peroxide production by the activated neutrophils and was blocked by treatment of neutrophils with catalase, but not with superoxide dismutase. These data suggested that hydrogen peroxide produced by neutrophils could lead to self-killing. More recently, Takei et al 22 have shown that suspended neutrophils treated with phorbol ester die by a mechanism that they considered distinct from either apoptosis or necrosis. Nuclear changes such as swelling and fusion of nuclear lobes, followed by an increase in membrane permeability, were observed within minutes to hours of treatment. 22
The goal of this study was to test the hypothesis that neutrophil adhesion to prosthetic graft materials decreases neutrophil viability. We report that neutrophil adhesion to uncoated and plasma-coated ePTFE and Dacron results in a rapid cell death. In contrast, neutrophils adherent to polystyrene surfaces coated with fibrinogen, immunoglobin (IgG), or fetal bovine serum (FBS) for the same length of time remain viable. We also show that pretreatment of neutrophils with N-acetyl-L-cysteine (NAC), cytochalasin D, or cyclosporin A significantly improves neutrophil viability after adhesion to ePTFE and Dacron. NAC, a reactive oxygen intermediate scavenger, protects neutrophils from phorbol ester-induced cell death. 22 Cytochalasin D, an inhibitor of actin filament assembly, prevents the production of reactive oxygen intermediates by surface-adherent neutrophils. 23,24 Cyclosporin A is an inhibitor of calcineurin 25–29 and the mitochondrial megachannel. 30–32 Oxidative stress results in activation/opening of the mitochondrial megachannel. This in turn leads to a rapid membrane depolarization, uncoupling of oxidative phosphorylation, loss of metabolic intermediates, mitochondrial swelling, and ultimately cell death. 33,34 These data suggested that reactive oxygen intermediates produced by neutrophils adherent to ePTFE and Dacron affect cell viability. Further, within 3 hours of implantation into a subcutaneous pouch on the back of rats, numerous ePTFE- and Dacron-adherent neutrophils were dead. Taken together, these data demonstrate that prosthetic materials trigger a rapid cell death both in vitro and in vivo.
METHODS
Neutrophil Isolation
Blood was collected from healthy volunteer donors. Neutrophils were isolated using neutrophil isolation medium (Cardinal Associates, Inc., Santa Fe, NM) as previously described. 14 Neutrophils were resuspended in Hanks’ balanced salt solution (Gibco BRL Life Technologies Inc., Grand Island, NY) with Ca+2 and Mg+2. Neutrophil preparations were greater than 95% pure and 97% viable.
Neutrophil Adherence
Surfaces of 24-well polystyrene plates were coated for 18 hours at room temperature with IgG (50 μg/mL; Sigma, St. Louis, MO) or fibrinogen (100 μ g/mL; Sigma) in coating buffer (150 mmol/L NaCl, 20 mM NaPO4, pH 8.0). Control polystyrene surfaces were uncoated or coated with 10% FBS for 2 hours. ePTFE (donated by Impra Inc., Tempe, AZ) and Dacron (donated by Meadox Medicals Inc., Oakland, NJ) were gas-sterilized, autoclaved, and custom-fitted to line the bottom of 24-well plates. In some experiments, ePTFE, Dacron, and polystyrene surfaces were coated for 2 hours with platelet-poor plasma generated from the same blood source used to isolate the neutrophils for the respective experiment. To initiate adhesion, 2.5 × 106 neutrophils in 0.75 ml Hanks’ balanced salt solution were added to each well. Neutrophils were incubated in wells for 1, 3, and 6 hours.
Neutrophil Viability
To determine cell viability, neutrophils incubated in 24-well plates for 1, 3, and 6 hours were stained with a mix containing the green fluorescing nucleic acid dye SYTO 9 (5 μmol/L) and the membrane-impermeant red fluorescing nucleic acid dye propidium iodide (PI; 30 μmol/L). The fluorescent dyes were obtained from Molecular Probes (Eugene, OR). As a positive control for these experiments, neutrophils were treated with 100 ng/mL 12-myristate 13-acetate (phorbol ester; Sigma) for the indicated time. Once the dyes were applied to the wells, the ePTFE and Dacron surfaces were recovered, inverted, and placed into new wells containing Hanks’ balanced salt solution. ePTFE- and Dacron-adherent neutrophils were imaged using an inverted Zeiss Axiovert-10 fluorescence microscope (Zeiss Inc., Thornwood, NY). Neutrophils adherent to protein-coated polystyrene wells were visualized directly. Duplicate wells were set up for each data point. Three random microscopic fields were analyzed for each surface with more than 100 cells per field. Cells were counted using the NIH image program. The percentage of PI-positive (red; dead cells) relative to the total number of cells represented by PI-positive plus SYTO 9 (green cells) was calculated.
Where indicated, neutrophils were pretreated for 1 hour with NAC (1.25 mmol/L; Sigma), cytochalasin D (25 μmol/L; Aldreich, Milwaukee, WI), cyclosporin A (Novartis, East Hanover, NJ), and bisindolylmaleimide (BIS, 10 μmol/L; Calbiochem, La Jolla, CA). In other experiments, neutrophils were pretreated for 10 minutes with the PI 3-kinase inhibitor wortmannin (100 nmol/L; Biomol Inc., Plymouth Meeting, PA). Treated and untreated control neutrophils were adhered to surfaces for 6 hours.
Lactate Dehydrogenase Release Assay
Neutrophils were incubated in protein-coated polystyrene wells or in wells containing ePTFE and Dacron as described above. At the indicated time points, supernatants were recovered and analyzed for lactate dehydrogenase (LDH) release using the Cytox 96 nonradioactive cytotoxicity assay kit (Promega, Madison, WI). Samples were analyzed at 490 nm using a 96-well plate reader. Each data point was examined in triplicate in four independent experiments.
Neutrophil DNA Analysis
Neutrophils (2 × 106/mL) were rendered apoptotic by culturing them for 24 hours at 37°C in tissue culture plates in medium composed of DMEM, 1% penicillin and streptomycin, and 10% FBS. Freshly isolated suspended neutrophils and apoptotic neutrophils were spun at 5,000 g for 5 minutes. The resulting pellets were washed once with TBS buffer (137 mmol/L NaCl, 2.7 mmol/L KCl, 25 mmol/L Tris buffered saline, pH 7.4), and then lysed with 0.5 mL extraction buffer (10 mmol/L Tris, pH 8.0, 1 mmol/L EDTA, 0.5% SDS) containing proteinase K (0.5 mg/mL; Sigma). Neutrophils adherent to ePTFE and Dacron for 6 hours were similarly washed with TBS and then scraped and lysed in 0.5 mL extraction buffer containing proteinase K. Lysates were incubated for 18 hours at 50°C, and DNA was isolated by two successive phenol/chloroform extractions using gel lock tubes (5′3′, Inc. Boulder, CO). Samples were normalized for DNA content using the CyQuant method (Molecular probes), and equal DNA amounts were analyzed on a 1.6% agarose gel containing ethidium bromide. A 123-bp DNA ladder (Gibco) or a low DNA mass ladder was resolved on the same gel. The gel was photographed using ultraviolet transillumination.
Transmission and Scanning Electron Microscopy
For transmission electron microscopy, neutrophils were fixed in Trump McDowell fixative containing 3.7% formaldehyde and 10% glutaraldehyde in a phosphate buffer (pH 7.3) and postfixed in 1% OsO4. After dehydration, the samples were preinfiltrated with acetone, infiltrated with Spurr’s epoxy, and embedded. Thin samples were cut, mounted, and stained with methanolic uranyl acetate and Reynold’s lead citrate. Analysis was performed by Pathology International Group (Durham, NC).
For scanning electron microscopy, nonadherent cells were aspirated and the vascular graft materials were immediately fixed in 2% glutaraldehyde in 0.5 mol/L phosphate buffer at 5°C for 2 hours. The prosthetic materials were then rinsed in the phosphate buffer and postfixed in 1% aqueous osmium tetroxide at 5°C for 1 hour. After additional rinsing, the material was dehydrated in a Blazers QFD 020 critical point drier (Baltec Products Inc., Middlebury, CT). The samples were then sputter-coated with gold palladium using a Baltec SCD 004 sputter coater (Baltec Products Inc.). The ePTFE and Dacron with adherent neutrophils were then visualized using a Hitachi s450 scanning electron microscope (Hitachi Inc., Danbury, CT).
Implanted ePTFE and Dacron
Patches of ePTFE and Dacron (1 × 1 cm) were implanted in subcutaneous pouches on the back of rats following the protocol described by Shue et al. 35 The animal studies were approved by the Robert Wood Johnson Medical School Institutional Care and Use Committee. Briefly, the patches were implanted through a 1.5-cm incision along the back of rats, through which a subcutaneous pouch was created. After implantation, the pouches were closed by skin clips. To retrieve the patches, the animals were killed by exposure to CO2. The patches were retrieved through a second incision made approximately 1 cm caudal to the original incision. The patches were stained immediately with the vital dyes and examined by fluorescence microscopy.
Statistical Analysis
Data were analyzed with an unpaired t test using the StatView program (SAS Institute, Inc., San Francisco, CA). Significance was defined as P < .05. Data are expressed as mean ± SEM of at least three experiments.
RESULTS
We used a mix of two fluorescent dyes, PI and SYTO 9, to monitor changes in neutrophil cell membrane permeability after adhesion to ePTFE, Dacron, and protein-coated polystyrene surfaces. Viable cells are impermeable to PI, whereas cells with a damaged cell membrane are stained by PI and fluoresce red. SYTO 9 is a membrane-permeable green-fluorescing nucleic acid dye. When both dyes are present and the cell membrane is damaged, PI competes with SYTO 9 for nucleic acid binding sites. Dead cells therefore fluoresce red; live cells fluoresce green. Representative images of PI- and SYTO 9-stained ePTFE- and Dacron-adherent neutrophils are shown in Figure 1. The morphology of Dacron- and ePTFE-adherent neutrophils was evaluated by scanning electron microscopy. Neutrophils were fully spread on both Dacron and ePTFE.
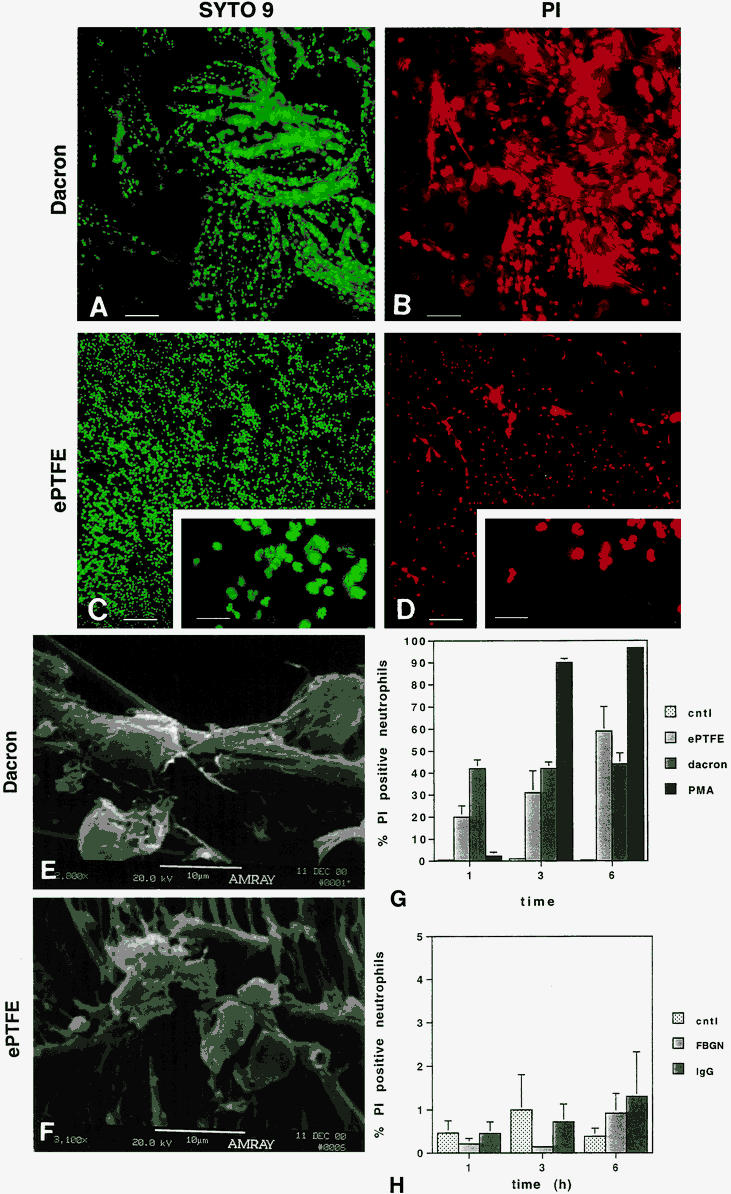
Figure 1. Neutrophils adherent to expanded polytetrafluoroethylene (ePTFE) and Dacron exhibit a time-dependent increase in cell death as determined by staining with propidium iodide (PI). Neutrophils were plated onto polystyrene plates prelined with Dacron (A, B) or ePTFE (C, D). At 1, 3, and 6 hours, neutrophils were stained with a mix of SYTO 9 and PI. Dead cells fluoresce red; live cells fluoresce green. Representative images of SYTO 9-stained (A, C) and PI-stained (B, D) Dacron- and ePTFE-adherent neutrophils at 6 hours are shown. Bar in A–D = 100 μm; bar in inserts in C and D = 30 μm. The morphology of Dacron- and ePTFE-adherent neutrophils was evaluated by scanning electron microscopy. Fully spread neutrophils were seen on both Dacron (E) and ePTFE (F). To quantify the percentage of PI-positive neutrophils relative to the total cell population (PI + SYTO 9 stained cells), neutrophils were plated onto wells precoated with fetal bovine serum (FBS; control), fibrinogen (FBGN), or immunoglobin (IgG) or prelined with ePTFE or Dacron (G, H). As a positive control, neutrophils plated onto a subset of control wells were treated with 12-myristate 13-acetate (phorbol ester; PMA) at the time of plating. At 1, 3, and 6 hours, neutrophils were stained with SYTO 9 and PI. Fluorescent cells in random microscopic fields were counted using the NIH image program, and the percentage of PI-positive cells relative to the total neutrophil population was determined. Data are expressed as mean ± SEM from seven independent experiments.
We sought to characterize the time course of change in neutrophil membrane permeability after adhesion to ePTFE and Dacron versus protein-coated polystyrene surfaces. For these studies, neutrophils were plated for 1, 3, and 6 hours onto wells prelined with ePTFE or Dacron or precoated with fibrinogen, IgG, or FBS. At each time point, the percentage of PI-positive (red-fluorescing) neutrophils was determined relative to the total neutrophil population (red- plus green-fluorescing cells). Neutrophils adherent to ePTFE and Dacron for 6 hours were, respectively, 59% ± 11% and 44% ± 5% PI positive (n = 7). In contrast, the percentage of neutrophils adherent to fibrinogen, IgG, or 10% FBS that were stained by PI did not exceed 1.5% at any time point.
Tsan 21 and Takei et al 22 have shown that phorbol ester-treated neutrophils die within 2 to 3 hours as a result of autotoxicity. We have similarly found that neutrophils become PI positive within less than 3 hours of exposure to phorbol ester. The appearance of phorbol ester-treated PI-positive neutrophils lagged behind that observed with ePTFE and Dacron surfaces. However, by the 3- and 6-hour time points, almost twice as many phorbol ester-treated neutrophils were PI positive compared with Dacron- and ePTFE-adherent neutrophils.
In vivo, implanted vascular devices are rapidly coated by plasma proteins. To examine the effect of plasma coating of ePTFE and Dacron on neutrophil viability, surfaces were precoated with platelet-poor plasma for 2 hours. As shown in Figure 2, neutrophils adherent to plasma-coated ePTFE and Dacron underwent a rapid time-dependent cell death. By 6 hours, there was no statistically significant difference between the percentage of PI-positive neutrophils adherent to uncoated versus plasma-coated ePTFE and Dacron. Taken together, these results indicate that neutrophil adhesion to uncoated or plasma-coated ePTFE and Dacron results in a rapid increase in cell membrane permeability, as monitored by PI staining.
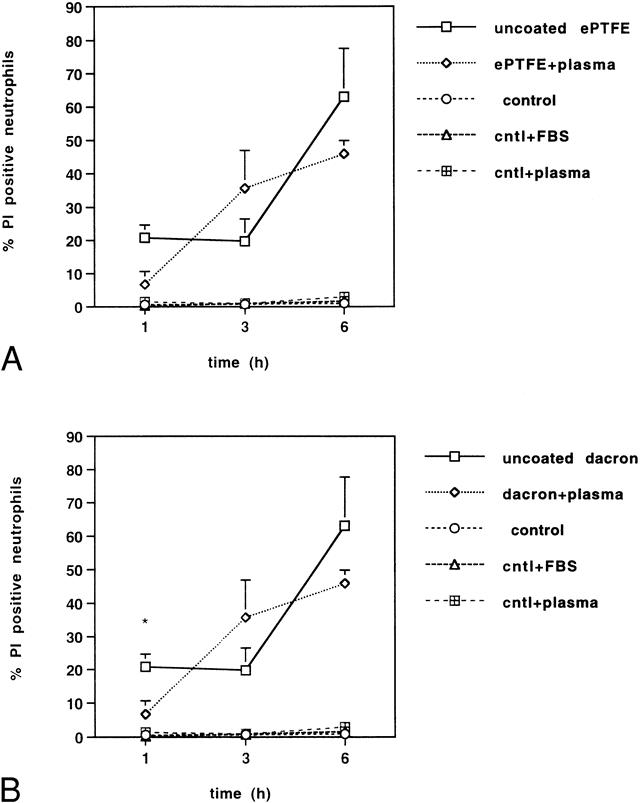
Figure 2. Neutrophils adherent to uncoated or plasma-coated (A) expanded polytetrafluoroethylene (ePTFE) and (B) Dacron exhibit a time-dependent increase in cell death. Neutrophils were plated onto polystyrene plates uncoated (control) or precoated with fetal bovine serum (control + FBS) or plasma (control + plasma). Parallel wells were prelined with ePTFE or Dacron. The ePTFE and Dacron surfaces were then untreated or plasma-coated. At 1, 3, and 6 hours, neutrophils were stained with SYTO 9 and propidium iodide (PI). Fluorescent cells in random microscopic fields were counted as described in the legend to Figure 1, and the percentage of PI-positive cells relative to the total neutrophil population was determined. Data are expressed as mean ± SEM from four independent experiments. *Significant difference (P < .001) between the percentage of PI-positive neutrophils adherent to untreated versus plasma-coated Dacron surfaces.
To assess membrane integrity by a method independent of vital dyes, we measured LDH release (Fig. 3). LDH is a stable cytosolic enzyme that is released when damage occurs to the cell membrane. 36 LDH levels reflect changes in membrane permeability of the total cell population, whereas vital dyes report changes in individual cells. Supernatants of neutrophils adherent to ePTFE, Dacron, and protein-coated polystyrene wells for 1, 3, and 6 hours were assayed for LDH release. LDH released by neutrophils incubated for 1 hour in wells precoated with 10% FBS was used as a point of reference for each experiment. Maximum LDH release was observed after phorbol ester treatment of neutrophils for 3 to 6 hours, with a 3.8- to 4.2-fold increase relative to control. Neutrophils adherent to ePTFE and Dacron for 6 hours released more LDH than control by factors of, respectively, 3.1 ± 0.4 and 2.1 ± 0.3. In contrast, there was no statistically significant difference between LDH released by fibrinogen-, IgG-, or FBS-adherent neutrophils compared with the control. Consistent with the results obtained with the fluorescent vital dyes, these data established that neutrophil adhesion to ePTFE and Dacron, but not to fibrinogen-, IgG- or FBS-coated polystyrene, triggers neutrophil activation responses that result in cell membrane damage.
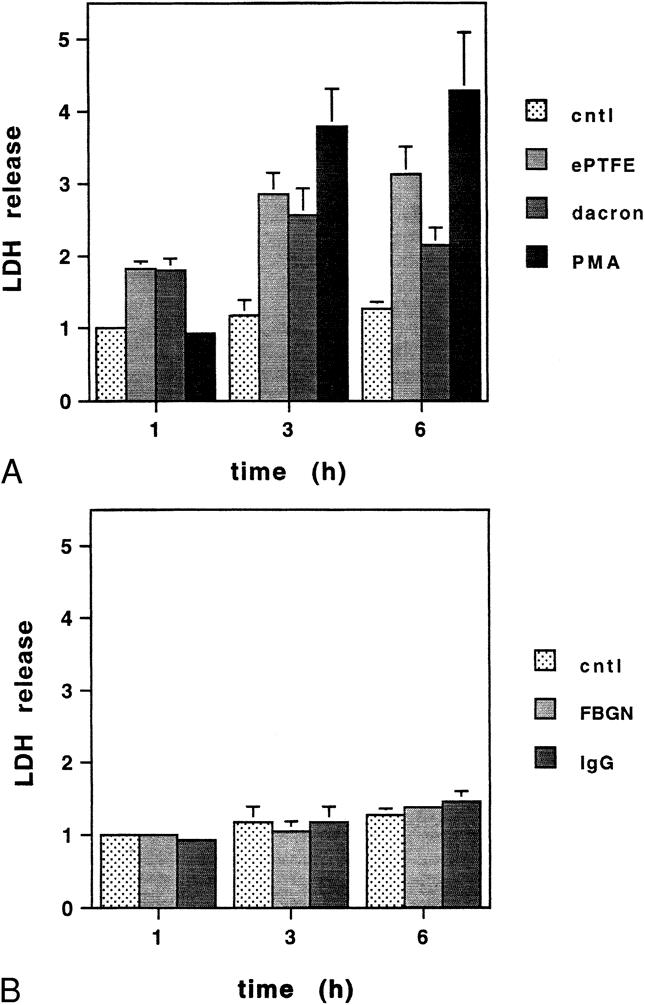
Figure 3. (A) Neutrophils adherent to expanded polytetrafluoroethylene (ePTFE) and Dacron exhibit a time-dependent increase in cell death as determined by lactate dehydrogenase (LDH) release. Neutrophils were plated onto plates precoated with fetal bovine serum (control), (B) fibrinogen (FBGN) or immunoglobin (IgG), or prelined with ePTFE and Dacron. As a positive control, neutrophils plated onto a subset of control wells were treated with phorbol ester (PMA) at the time of plating. At 1, 3, and 6 hours, supernatants were collected and analyzed for LDH release. The amount of LDH released by neutrophils plated onto control wells for 1 hour served as a point of reference for each experiment. Data are expressed as a ratio relative to the point of reference. Mean values ± SEM from four independent experiments are shown.
To identify time-dependent morphologic changes associated with increases in membrane permeability, neutrophils were adhered to ePTFE in the presence of PI and SYTO 9 and were observed at 15-minute intervals for 1 hour. The ePTFE-adherent neutrophils were viable at 15 minutes (Fig. 4). Changes in membrane permeability became visible within 45 minutes. Further, progressive increases in nuclear size and apparent chromatin decondensation were observed concomitant to the changes in membrane permeability. Fibrinogen- and IgG-adherent neutrophils observed in parallel did not undergo similar changes (not shown), nor did control suspended neutrophils. As expected, cultured neutrophils were apoptotic at 24 hours.
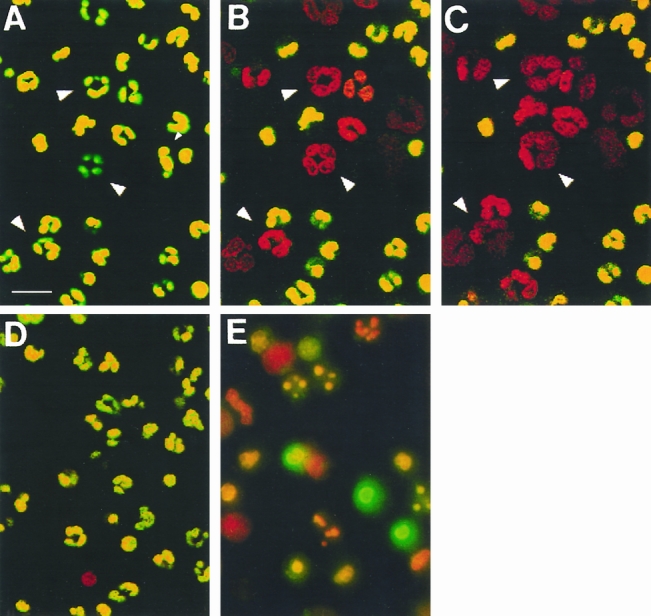
Figure 4. Neutrophil adhesion to expanded polytetrafluoroethylene (ePTFE) triggers time-dependent changes in membrane permeability and nuclear configuration. Neutrophils were adhered to ePTFE and observed at short intervals for 1 hour. SYTO 9 and propidium iodide (PI) were added to the neutrophils at t = 0, 30 minutes, and 45 minutes, and the neutrophils were observed within 15 minutes. (A–C) A time-lapse sequence at t = 15, 45, and 60 minutes. The cells rapidly become permeable to PI. Arrowheads mark three cells in which nuclear decondensation was observed. In contrast, neutrophils kept in suspension for 1 hour remained impermeable to PI (D), whereas many of the neutrophils cultured for 24 hours were apoptotic (E). Bar = 20 μm.
Ultrastructural changes were revealed through transmission electron micrographs of neutrophils adherent to ePTFE and Dacron (Fig. 5). In randomly sampled neutrophils, extensive changes were evident, including widespread cytoplasmic vacuolization, ill-defined organelles, and a reduced number of granules. Strikingly similar morphologic features were observed in phorbol ester-treated neutrophils. 37 Collectively, these findings provide additional support for an adhesion-induced cell damage defined by increased membrane permeability and ultrastructural changes in the nucleus and cytoplasm.
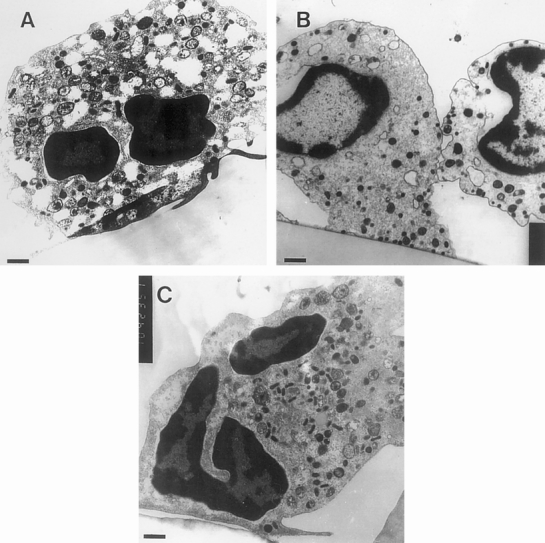
Figure 5. Ultrastructural cytoplasmic damage observed in neutrophils adherent to Dacron and expanded polytetrafluoroethylene (ePTFE). Neutrophils were adhered to ePTFE or Dacron for 1 hour and then fixed and processed for transmission electron microscopy. Representative images are shown of (A) ePTFE- and (B) Dacron-adherent neutrophils. Cytoplasmic vacuolization, poorly defined organelles, and a reduced number of granules were observed. For comparison, an example of a normal cell is shown in C. Bars (left corners) = 0.6 μm.
To define the mechanism by which the material-adherent neutrophils die, total DNA was isolated and analyzed by gel electrophoresis. Control DNA isolated from freshly purified neutrophils was compared with DNA isolated from neutrophils cultured for 24 hours or from neutrophils adherent to ePTFE and Dacron for 6 hours. DNA isolated from cultured apoptotic neutrophils revealed low-molecular-weight nucleosomal DNA ladders (Fig. 6). In contrast, DNA isolated from control neutrophils and neutrophils adherent to ePTFE and Dacron was of high molecular weight. These data indicate that ePTFE- and Dacron-induced cell death does not involve internucleosomal cleavage of DNA, consistent with a nonapoptotic cell death mechanism.
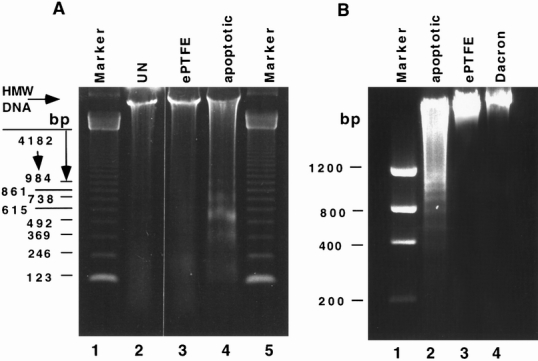
Figure 6. Lack of DNA fragmentation in DNA samples isolated from neutrophils adherent to Dacron and expanded polytetrafluoroethylene (ePTFE). DNA was isolated from untreated neutrophils (A, UN, lane 2), neutrophils adherent to ePTFE for 1 hour (A, ePTFE, lane 3), neutrophils cultured for 24 hours at 37°C to render them apoptotic (A and B, apoptotic, lanes 4 and 2), or neutrophils adherent to Dacron (B, lane 4) and ePTFE (B, lane 3) for 6 hours. After isolation, equal DNA amounts were resolved on agarose gels and visualized by ethidium bromide staining. A 123-bp DNA ladder was resolved in A, lane 1. A low-molecular-weight DNA ladder was resolved in B, lane 1. Fragmented DNA was seen only in the apoptotic groups.
To determine whether the production of reactive oxygen intermediates affects neutrophil viability on vascular prostheses, NAC, a potent superoxide scavenger, was used to treat neutrophils before adhesion to ePTFE and Dacron. Pretreatment of neutrophils with NAC reduced the number of ePTFE- and Dacron-adherent neutrophils that were PI positive by, respectively, 45% ± 5% (n = 6, P < .0001) and 26% ± 4% (n = 6, P < .0001) compared with untreated neutrophils (Fig. 7). These data suggest that the effect of ePTFE and Dacron on neutrophil viability could be linked to autotoxicity induced by reactive oxygen species.
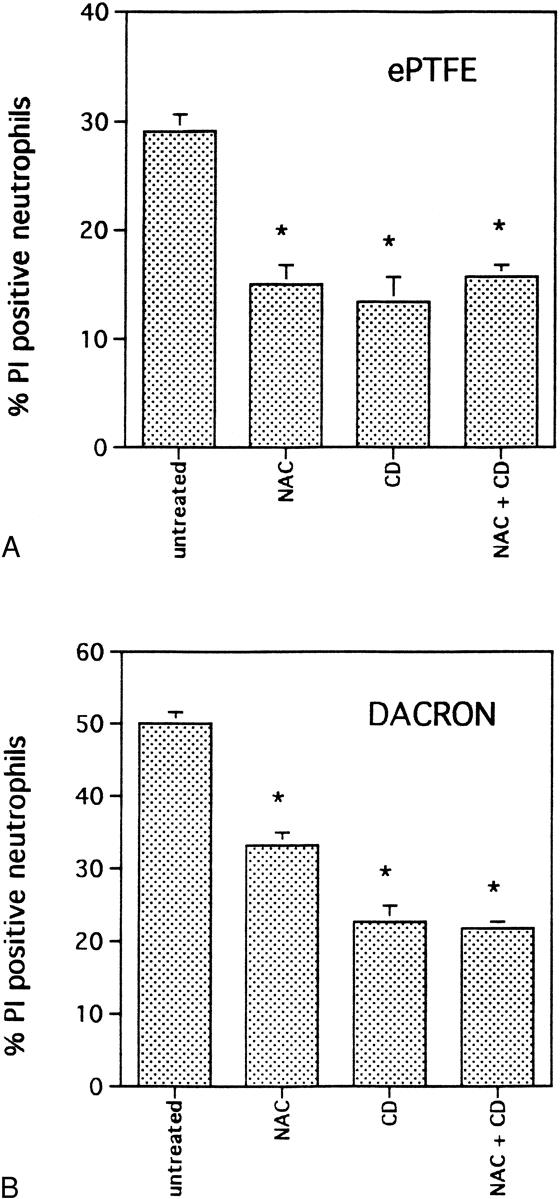
Figure 7. Pretreatment of neutrophils with N-acetyl-L-cysteine (NAC) and cytochalasin D (CD) ameliorated the effect of expanded polytetrafluoroethylene (ePTFE) and Dacron on neutrophil viability. Neutrophils were untreated or treated for 1 hour with NAC, CD, or a combination of the two before plating onto ePTFE (A) and Dacron (B) for 6 hours. Neutrophils were then stained with SYTO 9 and propidium iodide (PI). Fluorescent cells in random microscopic fields were counted as described in the legend to Figure 1, and the percentage of PI-positive cells relative to the total neutrophil population was determined. Data are expressed as mean ± SEM from four independent experiments. *Significant difference (P < .001) between untreated and treated neutrophil samples.
Demaurex et al 12 have reported that cytochalasin D, a potent inhibitor of actin filament assembly, prevented both cell spreading and superoxide production by adherent neutrophils. Consistent with the data obtained with NAC, we found that neutrophils pretreated with cytochalasin D adherent to ePTFE and Dacron exhibited, respectively, 53% ± 5% (n = 6, P < .0001) and 51% ± 5% (n = 6, P < .0001) fewer PI-positive cells compared with untreated neutrophils adherent to identical surfaces. The effects of NAC and cytochalasin D were not additive and were statistically indistinguishable from the results obtained with either NAC or cytochalasin D alone. The results obtained with cytochalasin D indicate that signaling events linked to the cytoskeleton organization affect the viability of ePTFE- and Dacron-adherent neutrophils. These data provide support for a model linking reactive oxygen species to neutrophil death after adhesion to ePTFE and Dacron.
Reactive oxygen species can induce mitochondrial damage and ultimately cell death by triggering the activation of the mitochondrial megachannel. Cyclosporin A is an inhibitor of calcineurin 25–29 and the mitochondrial megachannel. 30–32 Others 33 have reported that cyclosporin A protects cells from cell damage induced by oxidative stress. We therefore sought to examine the effect of cyclosporin A on ePTFE- and Dacron-adherent neutrophils (Fig. 8). For these studies, neutrophils were pretreated with several concentrations of cyclosporin A before plating onto ePTFE, Dacron, or protein-coated polystyrene. Neutrophils pretreated with 1 μmol/L cyclosporin A before plating onto Dacron exhibited 55% (n = 5, P < .001) fewer PI-positive neutrophils compared with untreated control samples. The effect of cyclosporin A was concentration-dependent: pretreatment with 0.5 μmol/L reduced the number of PI-positive neutrophils by 46% (n = 5, P < .001) relative to the untreated group, but pretreatment with 0.1 μmol/L had no effect on neutrophil viability. Neutrophils pretreated with 1.0 μmol/L or 0.5 μmol/L cyclosporin A failed to adhere to ePTFE. However, neutrophils pretreated with 0.1 μmol/L exhibited 44% (n = 7, P < .03) fewer PI-positive cells compared with control untreated samples. Cyclosporin A at a concentration of 1, 0.5, or 0.1 μmol/L did not affect neutrophil adhesion to fibrinogen- or IgG-coated polystyrene, nor did it affect neutrophil viability after adhesion to these surfaces (data not shown). These results suggest that a cyclosporin A-sensitive step that may involve activation of the mitochondrial megachannel and/or calcineurin contributes to neutrophil death after adhesion to ePTFE and Dacron.
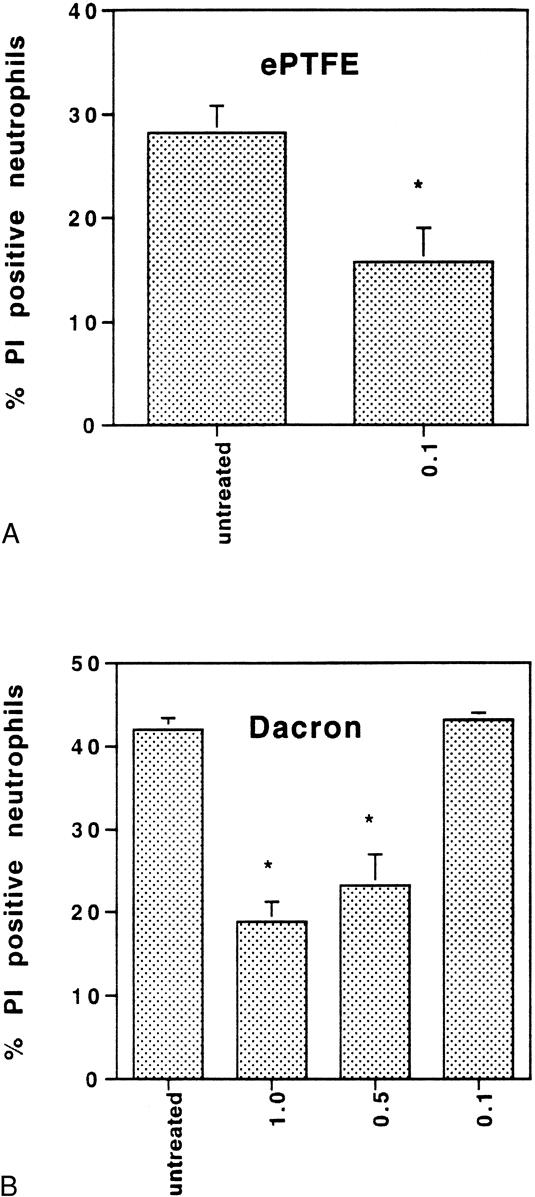
Figure 8. Pretreatment of neutrophils with cyclosporin A ameliorated the effect of expanded polytetrafluoroethylene (ePTFE) and Dacron on neutrophil viability. Neutrophils were untreated or treated for 1 hour with cyclosporin A at a concentration of 0.1, 0.5, or 1 μmol/L before plating onto ePTFE (A) and Dacron (B) for 6 hours. Neutrophils were then stained with SYTO 9 and propidium iodide (PI). Fluorescent cells in random microscopic fields were counted as described in the legend to Figure 1, and the percentage of PI-positive cells relative to the total neutrophil population was determined. Data are expressed as mean ± SEM from four independent experiments. *Significant difference (P < .001) between untreated and treated neutrophil samples.
Protein kinase C and PI 3-kinase regulate signaling pathways leading to the production of reactive oxygen species by agonist-stimulated neutrophils. 22,38 We therefore sought to examine whether inhibition of either one of these signaling components would affect the outcome of neutrophil adhesion to ePTFE and Dacron. For these studies, neutrophils were pretreated with BIS, 39 a protein kinase C inhibitor, and wortmannin, 40 a PI 3-kinase inhibitor. Pretreatment of neutrophils with BIS before exposure to phorbol ester reduced the number of PI-positive neutrophils from 73% to 23%. BIS, however, had no statistically significant effect on the number of PI-positive ePTFE- and Dacron-adherent neutrophils compared with untreated control neutrophils adherent to identical surfaces (data not shown). Wortmannin similarly did not significantly alter the outcome of neutrophil interaction with ePTFE and Dacron compared with untreated neutrophils adherent to identical surfaces (data not shown). Vlahos et al 38 reported that PI 3-kinase inhibitors failed to inhibit superoxide production by phorbol ester-stimulated neutrophils, but that the same inhibitors prevented formylated-methionyl-lencyl-phenylalanine–stimulated superoxide production. These data suggest that mechanisms leading to neutrophil death after adhesion to ePTFE and Dacron are independent of protein kinase C and PI 3-kinase.
We sought to examine whether the results obtained in vitro could be repeated in vivo. ePTFE and Dacron patches were implanted in a subcutaneous pouch on the back of rats. Neutrophils adherent to the implanted surfaces were detected within 3 hours. The neutrophil identity was confirmed based on its multilobular nuclear morphology (Fig. 9). A significant fraction of the ePTFE- and Dacron-adherent neutrophils were PI positive shortly after transmigration and adhesion to the implanted patches. These findings demonstrate that neutrophils adherent to ePTFE and Dacron undergo a rapid, nonapoptotic cell death both in vitro and in vivo.
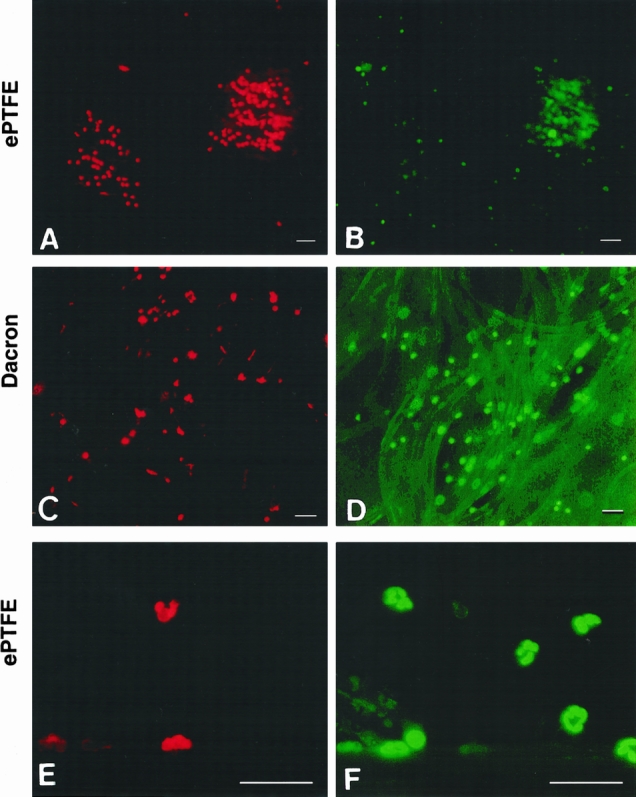
Figure 9. Neutrophils adherent to implanted expanded polytetrafluoroethylene (ePTFE) and Dacron exhibit a rapid cell death as determined by propidium iodide (PI) staining. Patches of ePTFE (A, B, E, F) and Dacron (C, D) were implanted into subcutaneous pouches on the back of rats. The patches were retrieved 3 hours after implantation and stained with PI (A, C, E) and SYTO 9 (B, D,F ) as described in the legend to Figure 1. Bar in A–D = 50 μm; bar in E and F = 120 μm.
DISCUSSION
Experiments performed 40 years ago showed that foreign bodies potentiate infection by opportunistic bacteria. 41 Subsequent studies focused on the interaction between the bacteria causing the infection (Staphylococcus aureus, Staphylococcus epidermidis, Pseudomonas aeruginosa) and the device surface. Receptors mediating bacterial adhesion to the surface were identified, and the ability of adherent bacteria to secrete an antibiotic-resistant glycocalyx was described. 42 More recently, impaired host defenses have been implicated in the etiology of graft infection, 10 raising the critical question of why the host immune system fails to kill contaminating bacteria.
This study demonstrates that neutrophil adhesion to two prosthetic graft materials, ePTFE and Dacron, impairs cell viability. This conclusion is based on four complementary analysis methods. First, we observed a time-dependent increase in the number of PI-positive neutrophils after adhesion to ePTFE or Dacron in vitro. Within 6 hours of adhesion to ePTFE and Dacron, 59% ± 11% and 44% ± 5%, respectively, of the neutrophils were PI positive. In contrast, neutrophils adherent to fibrinogen-, IgG-, or FBS-coated polystyrene surfaces were viable (PI negative). The results were the same in plasma-coated or uncoated ePTFE and Dacron. Second, we found that within 3 hours of implantation of ePTFE and Dacron patches into subcutaneous pouches on the back of rats, a significant fraction of the neutrophils adherent to these materials were PI positive and displayed nuclear disruption similar to that observed in vitro. Third, ePTFE- and Dacron-adherent neutrophils released two to three times more LDH than IgG-, fibrinogen-, or FBS-adherent neutrophils. Fourth, dramatic changes in the ultrastructure of ePTFE- and Dacron-adherent neutrophils were observed by transmission electron microscopy, including the appearance of numerous vacuoles and the loss of vesicles and granules. Strikingly similar morphologic features were previously observed in phorbol ester-stimulated neutrophils. 23 Taken together, these data demonstrate that prosthetic materials trigger a rapid, nonapoptotic cell death both in vitro and in vivo. Neutrophil death as a result of adhesion to prosthetic material may provide one mechanism to explain the inability of the neutrophil to kill bacteria that adhere to graft surfaces.
Neutrophils have a remarkable propensity to interact with nonphysiologic surfaces, including plastic, 11 nylon, 43 glass, 12 ePTFE, and Dacron. 14–16 How the neutrophils adhere to these surfaces is unknown. CD18-blocking antibodies failed to prevent neutrophil adhesion to glass, uncoated ePTFE and Dacron, as well as plasma-coated ePTFE and Dacron. 12,16,44 Fc receptor-blocking antibodies similarly failed to prevent neutrophil adhesion to uncoated or plasma-coated ePTFE. 44 Neutrophils adherent to nonphysiologic surfaces, 12,16 as well as neutrophils activated by soluble stimuli such as phorbol ester, produce reactive oxygen intermediates that include superoxide anions (O−2), hydrogen peroxide, and hydroxyl radicals (•OH). 21,22 Reactive oxygen intermediates and their derived products can produce extensive cell damage. Hydrogen peroxide, for example, is formed in large amounts by stimulated neutrophils. Although the reactivity of hydrogen peroxide is relatively low, its toxic effects are increased many orders of magnitude as a result of reactions with peroxidase (myeloperoxidase), halides, synergism with proteinases, and reactivity with ferrous iron to form hydroxyl radicals. 45,46 Hydrogen peroxide can cross the plasma membrane and travel by means of fluid to distant sites. 47 In addition, peroxidase/hydrogen peroxide/halide products can initiate oxidation of polyunsaturated fatty acids that can result in cell membrane leakage. 45,46 Further reduction of hydrogen peroxide results in the formation of hydroxyl radical, one of the most powerful oxidants known. Tsan 21 and Takei et al 22 demonstrated that hydrogen peroxide and other reactive oxygen species produced by phorbol ester-stimulated neutrophils lead to self-killing within several hours. The phorbol ester effect was reversed by treatment of the neutrophils with a variety of antioxidants, including NAC, thiourea, and catalase. We similarly found that pretreatment of neutrophils with NAC ameliorated the effect of ePTFE and Dacron on neutrophil viability, suggesting that reactive oxygen radicals produced as a result of neutrophil adhesion to these surfaces precipitated cell death.
Reactive oxygen species can induce activation/opening of a nonspecific, high-conductance channel (megachannel) in the mitochondrial inner membrane. 34,37 Opening of the megachannel increases the permeability of the inner membrane to small molecules and leads to a massive efflux of Ca+2 into the cytosol. This results in rapid membrane depolarization, uncoupling of oxidative phosphorylation, loss of metabolic intermediates, mitochondrial swelling, and ultimately cell death. 34 Cyclosporin A, an immunosuppressive cyclic oligopeptide, is a specific inhibitor/blocker of the mitochondrial megachannel. 31,32,48,49 Cyclosporin A protected hepatocytes from cytotoxicity brought about by hypoxia and ischemia/reperfusion injury. 37 Pretreatment of neutrophils with cyclosporin A at an optimal concentration reduced the number of neutrophils that died after adhesion to ePTFE and Dacron by approximately 50%. One possible explanation of these data is that reactive oxygen intermediates produced by ePTFE- and Dacron-adherent neutrophils activate the mitochondrial megachannel, leading to adherent neutrophil cell death.
Calcineurin, a calcium/calmodulin-dependent serine/threonine phosphatase, is a second potential target for cyclosporin A in neutrophils. 29 A dose-dependent inhibition of calcineurin was observed in neutrophils, with more than 90% inhibition at a concentration of 1 μg/mL cyclosporin A. 29 Calcineurins regulate the activation of transcription factors 25–28 and downstream events that affect cell motility and cytoskeleton organization. 50 Wang et al 51 recently showed that under certain conditions, calcineurin can dephosphorylate the proapoptotic Bcl-2 family member BAD to precipitate apoptosis. Inhibition of a calcineurin-dependent signaling event by cyclosporin A provides a second potential explanation for this compound’s effect on neutrophil viability. Considering the close correlation between oxidative stress-induced cell death and mitochondrial dysfunction, however, the former rather than the latter mechanism is more likely to explain the effect of cyclosporin A on neutrophil viability.
The effect of reactive oxygen intermediates on cells is apparently dependent on both cell type and concentration. Neutrophils, for example, are more sensitive to hydrogen peroxide than lymphocytic cells. 21 Other cell types may in fact benefit from hydrogen peroxide. Huot et al 52 have shown that exposure of vascular endothelial cells to hydrogen peroxide at a concentration of 250 μmol/L for 60 minutes triggered actin filament assembly and activation of MAP kinase pathways, whereas exposure of fibroblasts to a similar hydrogen peroxide concentration caused actin filament disassembly. Reactive oxygen intermediates are also necessary for Ras-transformed NIH 3T3 fibroblast cell cycle progression by means of a MAP kinase-dependent pathway. 53
During ingestion of microorganisms, reactive oxygen species are released into phagolysosomes. 18,19 This spatially regulated process serves to create a high local concentration of reactive oxygen intermediates while protecting the neutrophils from their toxic effect. In contrast, when interacting with particles or surfaces that are too large to be engulfed, the neutrophil’s phagolysosomes fuse with the plasma membrane, resulting in the release of granular constituents and reactive oxygen intermediates to the surroundings. 13,18 This aberrant activation response may explain at least in part the rapid death of surface-adherent neutrophils and phorbol ester-activated neutrophils that produce large amounts of reactive oxygen intermediates.
Human plasma and serum and FBS, as well as several plasma proteins (e.g., fibrinogen and fibronectin), suppress the neutrophil respiratory burst in response to polystyrene, whereas neutrophils adherent to uncoated or IgG-coated polystyrene surfaces produce similar amount of reactive oxygen intermediates. 11 Our data demonstrate that neutrophils adherent to fibrinogen-, IgG-, or FBS-coated polystyrene surfaces remain fully viable for at least 6 hours, whereas 30% to 50% of the neutrophils adherent to ePTFE and Dacron die within 3 to 6 hours. These data suggest that reactive oxygen intermediate production is most likely not the sole reason for neutrophil death after adhesion to ePTFE and Dacron. Catalase, glutathione redox enzymes, and superoxide dismutase protect neutrophils from the toxic effects of hydrogen peroxide and superoxide ions. 45 A major decrease in intracellular glutathione was observed in phorbol ester-stimulated neutrophils; treatment of neutrophils with NAC reversed the effect of phorbol ester on glutathione depletion and rescued the phorbol ester-treated neutrophils from death. 22,54,55 A similar mechanism may also explain why significantly fewer NAC-pretreated neutrophils died after adhesion to ePTFE and Dacron compared with untreated neutrophils. Neutrophil interaction with ePTFE and Dacron may be mediated by nonphysiologic adhesive mechanisms. The adhesive event may not only stimulate reactive oxygen intermediate production but may also interfere with the neutrophil’s ability to protect itself from reactive oxygen species-induced toxicity. Hanein et al 56 observed that epithelial cell adhesion to a calcium tetrahydrate crystal surface triggered a rapid apoptotic cell death. Epithelial cell adhesion to the crystal surface was integrin independent, which led these researchers to propose that cell death resulted from “excessive” nonintegrin-dependent interactions that prevented normal receptor–extracellular matrix contacts and the generation of signals necessary for cell survival. Adhesion to ePTFE and Dacron may similarly fail to generate signals necessary to protect neutrophils from the toxic effect of reactive oxygen intermediates through active downregulation or lack of activation of intrinsic antioxidative mechanisms. This hypothetical model will be tested in future studies.
The outcomes of neutrophil stimulation with phorbol ester or adhesion to ePTFE and Dacron are remarkably similar and consistent with a nonapoptotic cell death mechanism. Takei et al 22 observed nuclear changes such as swelling and fusion of nuclear lobes, followed by increased membrane permeability, within hours of neutrophil treatment with phorbol ester. Neutrophil adhesion to ePTFE and Dacron similarly led to changes in nuclear shape and size within minutes. These events unfolded in the absence of DNA fragmentation. Phorbol ester stimulates protein kinase C, leading to actin cytoskeleton organization and production of reactive oxygen intermediates. Neutrophil adhesion to ePTFE and Dacron is similarly linked to the cytoskeleton organization and production of reactive oxygen species. Thus, in both instances, cell death appears to be associated with cytoskeleton-dependent signaling events.
Majno and Joris 57 suggested that the old term “oncosis” referring to the swelling seen in nonapoptotic cell death should be used to describe the progression of cellular events leading to accidental, unprogrammed cell death. We propose the use of the term “synxenatosis” to indicate that cell death (death; tha nato) was precipitated by neutrophil attachment (syn desi) to a foreign (xe no) material. Analysis of the mechanism by which the adherent neutrophils die may allow the development of preventive therapeutic strategies, thus enhancing the host’s ability to kill bacteria.
Acknowledgment
The authors thank Ping Ji for expert technical assistance.
Footnotes
Correspondence: Beatrice Haimovich, PhD, Dept. of Surgery, Robert Wood Johnson Medical School, 675 Hoes Lane, Piscataway, NJ 08854.
Supported by a Focused Giving Award from Johnson & Johnson (RSG, BH). Dr. Haimovich is an Established Investigator of the American Heart Association.
Accepted for publication November 29, 1999.
References
- 1.Edwards MJ, Richardson JD, Klamer TW. Management of aortic prosthetic infections. Am J Surg 1988; 155:327–330. [DOI] [PubMed] [Google Scholar]
- 2.Young EJ, Sugarman B. Infections in prosthetic devices. Surg Clin North Am 1988; 68:167–180. [DOI] [PubMed] [Google Scholar]
- 3.Bandyk DF, Bergamini TM, Kinney EV, et al. In situ replacement of vascular prostheses infected by bacterial biofilms. J Vasc Surg 1991; 13:575–583. [PubMed] [Google Scholar]
- 4.Kikta MJ, Goodson SF, Bishara RA, et al. Mortality and limb loss with infected infrainguinal bypass grafts. J Vasc Surg 1987; 5:566–571. [DOI] [PubMed] [Google Scholar]
- 5.Sedwitz MM, Hye RJ, Stabile BE. The changing epidemiology of pseudoaneurysm. Arch Surg 1988; 123:473–476. [DOI] [PubMed] [Google Scholar]
- 6.Casey J, Flinn WR, Yao JST. Correlation of immune nutritional status with wound complications in patients undergoing vascular operations. Surgery 1983; 93:822–827. [PubMed] [Google Scholar]
- 7.Raja RH, Raucci G, Hook M. Peptide analogs to a fibronectin receptor inhibit attachment of Staphylococcus aureus to fibronectin-containing substrates. Infect Immun 1990; 58:2593–2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cramer EB. Cell biology of phagocyte migration from the bone marrow, out of the bloodstream, and across organ epithelia. In: Gallin JI, Goldstein IM, Snyderman R, eds. Inflammation: Basic Principles and Clinical Correlates. New York: Raven Press; 1992: 341–352.
- 9.Potera C. Forging a link between biofilms and disease. Science 1999; 283:1837,1839. [DOI] [PubMed] [Google Scholar]
- 10.Zimmerli W, Lew PD, Waldvogel FA. Pathogenesis of foreign body infection: evidence for a local granulocyte defect. J Clin Invest 1984; 73:1191–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nathan CF. Neutrophil activation on biological surfaces. J Clin Invest 1987; 80:1550–1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Demaurex N, Downey GP, Waddell TK, Grinstein S. Intracellular pH regulation during spreading of human neutrophils. J Cell Biol 1996; 133:1391–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu L, Elwing H, Karlsson A, et al. Surface-related triggering of the neutrophil respiratory burst. Characterization of the response induced by IgG adsorbed to hydrophilic and hydrophobic glass surfaces. Clin Exp Immunol 1997; 109:204–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Katz DA, Haimovich B, Greco RS. Neutrophil activation by expanded polytetrafluoroethylene is dependent on the induction of protein phosphorylation. Surgery 1994; 116:446–455. [PubMed] [Google Scholar]
- 15.Kaplan SS, Basford RE, Mora E, et al. Biomaterial-induced alterations of neutrophil superoxide production. J Biomed Materials Res 1992; 26:1039–1051. [DOI] [PubMed] [Google Scholar]
- 16.Kaplan SS, Basford RE, Jeong MH, Simmons RL. Mechanisms of biomaterial-induced superoxide release by neutrophils. J Biomed Materials Res 1994; 28:377–386. [DOI] [PubMed] [Google Scholar]
- 17.Kaplan SS, Basford RE, Jeong M, Simmons RL. Biomaterial–neutrophil interactions: dysregulation of oxidative functions of fresh neutrophils induced by prior neutrophil-biomaterial interaction. J Biomed Materials Res 1996; 30:67–75. [DOI] [PubMed] [Google Scholar]
- 18.Henson PM, Henson JE, Fittschen C, et al. Degranulation and secretion by phagocytic cells. In: Gallin JI, Goldstein IM, Snyderman R, eds. Inflammation: Basic Principles and Clinical Correlates. New York: Raven Press; 1992: 511–539.
- 19.Henson PM, Johnston RB Jr. Tissue injury in inflammation. J Clin Invest 1987; 79:669–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smith RM, Curnutte JT. Molecular basis of chronic granulomatous disease. Blood 1991; 77:673–686. [PubMed] [Google Scholar]
- 21.Tsan M-F. Phorbol myristate acetate induced neutrophil autotoxicity. J Cell Physiol 1980; 105:327–334. [DOI] [PubMed] [Google Scholar]
- 22.Takei H, Araki A, Watanabe H, et al. Rapid killing of human neutrophils by the potent activator phorbol 12-myristate 13-acetate (PMA) accompanied by changes different from typical apoptosis or necrosis. J Leuko Biol 1996; 59:229–240. [DOI] [PubMed] [Google Scholar]
- 23.Johnston RB Jr, Lehmeyer JE. Elaboration of toxic oxygen by-products by neutrophils in a model immune complex disease. J Clin Invest 1976; 57:836–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tsujimoto M, Yokota S, Vilcek J, Weissmann G. Tumor necrosis factor provokes superoxide anion generation from neutrophils. Biochem Biophys Res Commun 1986; 137:1094–1100. [DOI] [PubMed] [Google Scholar]
- 25.Liu J, Farmer JD Jr, Lane WS, et al. Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell 1991; 66:807–815. [DOI] [PubMed] [Google Scholar]
- 26.Fruman DA, Klee CB, Bierer BE, Burakoff SJ. Calcineurin phosphatase activity in T lymphocytes is inhibited by FK 506 and cyclosporin A. Proc Natl Acad Sci USA 1992; 89:3686–3690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.O’Keefe SJ, Tamura J, Kincaid RL, et al. FK-506- and CsA-sensitive activation of the interleukin-2 promoter by calcineurin. Nature 1992; 357:692–694. [DOI] [PubMed] [Google Scholar]
- 28.Clipstone NA, Crabtree GR. Identification of calcineurin as a key signaling enzyme in T-lymphocyte activation. Nature 1992; 357:695–697. [DOI] [PubMed] [Google Scholar]
- 29.Carballo M, Marquez G, Conde M, et al. Characterization of calcineurin in human neutrophils. Inhibitory effect of hydrogen peroxide on its enzyme activity and on NF-kappaB DNA binding. J Biol Chem 1999; 274:93–100. [DOI] [PubMed] [Google Scholar]
- 30.Fournier N, Ducet G, Crevat A. Action of cyclosporine on mitochondrial calcium fluxes. J Bioenerg Biomembr 1987; 19:297–303. [DOI] [PubMed] [Google Scholar]
- 31.Crompton M, Ellinger H, Costi A. Inhibition by cyclosporin A of a Ca+2-dependent pore in heart mitochondria activated by inorganic phosphate and oxidative stress. Biochem J 1988; 255:357–360. [PMC free article] [PubMed] [Google Scholar]
- 32.Szabo I, Zoratti M. The giant channel of the inner mitochondrial membrane is inhibited by cyclosporin A. Biochim Biophys Acta 1995; 1241:139–176.7640294 [Google Scholar]
- 33.Lemasters JJ, Nieminen A-L, Qian T, et al. The mitochondrial permeability transition in toxic, hypoxic and reperfusion injury. Mol Cell Biochem 1997; 174:159–165. [PubMed] [Google Scholar]
- 34.Hirsch T, Marzo SI, Marchetti P, et al. Mitochondrial permeability transition in apoptosis and necrosis. Cell Biol Toxicol 1998; 14:141–145. [DOI] [PubMed] [Google Scholar]
- 35.Shue WB, Worosilo SC, Donetz AP, et al. Prevention of vascular prosthetic infection with an antibiotic-bonded Dacron graft. J Vasc Surg 1988; 8:600–605. [PubMed] [Google Scholar]
- 36.Behl C, Davis JB, Lesley R, Schubert D. Hydrogen peroxide mediates amyloid-beta protein toxicity. Cell 1994; 77:817–827. [DOI] [PubMed] [Google Scholar]
- 37.Esaguy N, Aguas AP, Vilanova M, Silva MT. Activation of human neutrophils by phorbol ester decreases the cytoplasm compactness and the lactoferrin content of the granulocytes. J Leuko Biol 1991; 50:444–452. [DOI] [PubMed] [Google Scholar]
- 38.Vlahos CJ, Matter WF, Brown RF, et al. Investigation of neutrophil signal transduction using a specific inhibitor of phosphatidylinositol 3-kinase. J Immunol 1995; 154:2413–2422. [PubMed] [Google Scholar]
- 39.Toullec D, Pianetti P, Coste H, et al. The bisindolylmaleimide GF 109203X is a potent and selective inhibitor of protein kinase C. J Biol Chem 1991; 266:15771–15781. [PubMed] [Google Scholar]
- 40.Yano H, Nakanishi S, Kimura K, et al. Inhibition of histamine secretion by wortmannin through the blockade of phosphatidylinositol 3-kinase in RBL-2H3 cells. J Biol Chem 1993; 268:25846–25856. [PubMed] [Google Scholar]
- 41.Elek SD, Conen PE. The virulence of Staphylococcus pyogenes for man: a study of the problems of wound infection. Br J Exp Pathol 1957; 38:573–586. [PMC free article] [PubMed] [Google Scholar]
- 42.Katz DA, Greco RS. The pathobiology of infections associated with biomaterials. In: Klitzman B, McAllister L, eds. Problems in General Surgery. Philadelphia: JB Lippincott; 1994: 209–226.
- 43.Klock JC, Bainton DF. Degranulation and abnormal bactericidal function of granulocytes procured by reversible adhesion to nylon wool. Blood 1976; 48:149–161. [PubMed] [Google Scholar]
- 44.Katz DA, Haimovich B, Greco RS. FcγRII, FcγRIII, and CD18 receptors mediate in part neutrophil activation on a plasma coated expanded polytetrafluoroethylene surface. Surgery 1995; 118:154–161. [DOI] [PubMed] [Google Scholar]
- 45.Gutteridge JMC. Lipid peroxidation and antioxidants as biomarkers of tissue damage. Clin Chem 1995; 41:1819–1828. [PubMed] [Google Scholar]
- 46.Klebanoff SJ. Oxygen metabolites from phagocytes. In: Gallin JI, Goldstein IM, Snyderman R, eds. Inflammation: Basic Principles and Clinical Correlates. New York: Raven Press; 1992: 541–588.
- 47.Ohno Y, Gallin JI. Diffusion of extracellular hydrogen peroxide into intracellular compartment of human neutrophils. J Biol Chem 1985; 260:8438–8446. [PubMed] [Google Scholar]
- 48.Bernardi P, Broekemeier KM, Pfeiffer DR. Recent progress on regulation of the mitochondrial permeability transition pore; a cyclosporin-sensitive pore in the inner mitochondrial membrane. J Bioenerg Biomembr 1994; 26:509–517. [DOI] [PubMed] [Google Scholar]
- 49.Seaton TA, Cooper JM, Schapira AH. Cyclosporin inhibition of apoptosis induced by mitochondrial complex I toxins. Brain Res 1998; 809:12–17. [DOI] [PubMed] [Google Scholar]
- 50.Hojo M, Morimoto T, Maluccio M, et al. Cyclosporine induces cancer progression by a cell-autonomous mechanism. Nature 1999; 397:530–534. [DOI] [PubMed] [Google Scholar]
- 51.Wang H-G, Pathan N, Ethell IM, et al. Ca+2 induced apoptosis through calcineurin dephosphorylation of BAD. Science 1999; 284:339–343 [DOI] [PubMed] [Google Scholar]
- 52.Huot J, Houle F, Marceau F, Landry J. Oxidative stress-induced actin reorganization mediated by the p38 mitogen-activated protein kinase/heat shock protein 27 pathway in vascular endothelial cells. Circ Res 1997; 80:383–392. [DOI] [PubMed] [Google Scholar]
- 53.Irani K, Xia Y, Zweier JL, et al. Mitogenic signaling mediated by oxidants in ras-transformed fibroblasts. Science 1997; 275:1649–1652. [DOI] [PubMed] [Google Scholar]
- 54.Villagrasa V, Cortijo J, Marti-Cabrera M, et al. Inhibitory effects of N-acetylcysteine on superoxide anion generation in human polymorphonuclear leukocytes. J Pharm Pharmacol 1997; 49:525–529. [DOI] [PubMed] [Google Scholar]
- 55.Oddera S, Silvestri M, Sacco O, et al. N-acetylcysteine enhances in vitro the intracellular killing of Staphylococcus aureus by human alveolar macrophages and blood polymorphonuclear leukocytes and partially protects phagocytes from self-killing. J Lab Clin Med 1994; 124:293–301. [PubMed] [Google Scholar]
- 56.Hanein D, Yarden A, Sabanay H, et al. Cell adhesion to crystal surfaces. Cell Adhesion and Communication 1996; 4:341–354. [DOI] [PubMed] [Google Scholar]
- 57.Majno G, Joris I. Apoptosis, oncosis, and necrosis. An overview of cell death. Am J Pathol 1995; 146:3–15. [PMC free article] [PubMed] [Google Scholar]