

Abstract
Objective
To investigate whether transforming growth factor beta (TGFβ) signaling is disrupted in human pancreatic cancer cells, and to study the role of TGFβ receptors and Smad genes.
Summary Background Data
TGFβ is a known inhibitor of pancreatic growth. Disruption of the TGFβ signaling pathway may play a role in pancreatic cancer development.
Methods
The effect of TGFβ on the BxPC-3, MiaPaCa-2, and PANC-1 pancreatic cancer cell lines was evaluated by [3H]thymidine incorporation and a TGFβ-responsive reporter assay. Expression of TGFβ receptors and Smads 2 and 3 was assessed by cross-linking assays and reverse transcriptase–polymerase chain reaction (RT-PCR). The ability to restore TGFβ responsiveness was evaluated by transfection of TGFβ signaling components.
Results
TGFβ produced little inhibition of DNA synthesis and did not activate a TGFβ-responsive reporter in pancreatic cancer cell lines. 125TGFβ cross-linking and RT-PCR confirmed the presence of TGFβ receptors and Smad2 and Smad3 transcripts. Transfection of TGFβ receptors or Smads 2 and 3 did not restore responsiveness. However, transfection of Smad4 into the BxPC-3 pancreatic cancer cell line restored TGFβ responsiveness.
Conclusions
Pancreatic cancer cells show loss of TGFβ responsiveness. Smads 2 and 3 and TGFβ receptors are not defective in the cell lines studied. Transfection of Smad4 into one of the cell lines restored TGFβ responsiveness, suggesting an important role for Smad4 in pancreatic cancer. It is likely that other, as yet unidentified genes are important in TGFβ resistance in pancreatic cancer cells.
Carcinoma of the pancreas is the fifth leading cause of cancer death in the United States, with a dismal 5-year survival rate of less than 5%. The poor prognosis may be related to several factors. The diagnosis of pancreatic cancer is often delayed, with 80% to 90% of patients already having metastatic disease at the time of initial presentation, precluding the possibility of surgical resection. In addition, adjuvant chemotherapy and irradiation have not had a significant impact on improving disease survival. 1 Better understanding of pancreatic cancer cell biology is needed to provide insight into the treatment of this disease.
The transforming growth factor beta (TGFβ) signaling pathway may be important in the development of pancreatic cancer. TGFβ is an ubiquitous growth factor that plays a key role in regulation of growth and differentiation in a wide variety of cell types, including the pancreas. TGFβ exerts its biologic effects by interacting with two types of transmembrane receptors (type I and II) with protein serine/threonine kinase activity. 2 The type II receptor is involved in initial ligand binding; once ligand is bound, type II receptors bind to type I receptors, forming a complex. Type I receptors are then phosphorylated by the kinase domain of type II receptors, resulting in propagation of downstream signals.
Recently, a novel family of proteins, the Smad family, was discovered as intracellular molecules required for signal propagation by the TGFβ family. The term “Smad” is derived from a combination of the gene names from Caenorhabditis elegans, sma, and Drosophila, Mad. Activated, phosphorylated TGFβ type I receptors transiently associate with and phosphorylate Smad2, Smad3, or both. Phosphorylation of Smad2, Smad3, or both allows complex formation with Smad4 (also known as deleted in pancreatic carcinoma 4, or DPC4), and movement into the nucleus. In the nucleus, Smad complexes can act as transcriptional activators. TGFβ is thus able to induce the expression of growth inhibitory proteins that regulate cell cycle progression, such as the cyclin-dependent kinase (CDK) inhibitors p15INK4B, p21WAF1, and p27KIP1. 3–5 In addition, TGFβ has been shown to inhibit cell growth by causing tyrosine phosphorylation and inactivation of CDK4 and CDK6 by repression of the CDK tyrosine phosphatase Cdc25A. 6
TGFβ is a potent inhibitor of pancreatic acinar and duct cell proliferation in vitro. 7,8 Genetic mutations in a component or components of the TGFβ signaling pathway may lead to loss of TGFβ responsiveness and may contribute to tumor development. Initial studies investigating genetic mutations in TGFβ receptors have suggested this possibility. For example, in approximately 90% of colon cancers that exhibit microsatellite instability, and in several TGFβ-resistant human gastric cancers, mutations causing inactivation of TGFβ receptor II have been identified. 9,10 Alterations in the type I receptor have also been found in prostate, colon, and gastric cancer cells that are insensitive to TGFβ. 11 Similar to the receptors, intracellular components of the TGFβ signaling pathway may possess a tumor suppressor function. Recently, DPC4 (Smad4) was identified as a tumor suppressor gene that is commonly inactivated in pancreatic cancers. 12 Inactivating mutations in another member of this gene family (Smad2) have been found in colorectal carcinomas. 13
Many human pancreatic cancer cell lines have been shown to be refractory to the growth inhibitory effects of TGFβ. 14,15 Resistance to the growth inhibitory effects of TGFβ may be explained by either inactivating mutations in the TGFβ receptors or defects in components of the signaling pathway downstream of the TGFβ receptors, such as the Smads. Studies that determine mechanisms of escape from TGFβ regulation may provide insight into the molecular genetics of pancreatic cancer.
In the current study, we examined the effects of TGFβ on normal pancreatic acinar cells and three well-characterized human pancreatic cancer cell lines: BxPC-3, MiaPaCa-2, and PANC-1. We sought to determine the mechanism or mechanisms by which human pancreatic cancer cells lose responsiveness to TGFβ. We found that the pancreatic cancer cell lines investigated had defective responses to TGFβ, as measured by [3H]thymidine incorporation and TGFβ-sensitive reporter assays. TGFβ type I and II receptors and Smads 2 and 3 were not involved in the loss of TGFβ responsiveness. Introduction of a functional Smad4 into a Smad4-defective pancreatic cell line restored TGFβ-mediated responses.
METHODS
Materials
The following were purchased: type 1 TGFβ from R & D Systems (Minneapolis, MN); disuccinimidyl suberate from Pierce Inc. (Rockford, IL); 125TGFβ (1,228 Ci/mmol) and [3H]thymidine (25 Ci/mmol) from Amersham (Arlington Heights, IL); basic fibroblast growth factor from Collaborative Research (Waltham, MA); and cholecystokinin from Research Plus (Bayonne, NJ); all other chemicals were from Sigma Chemical (St. Louis, MO).
DNA Constructs
Smad2 in CS2 and Smad4 in pCMV5 were provided by J. Massague (Sloan-Kettering); Smad3 in pcDNA3 was provided by W. Vale (Salk Institute); type I TGFβ receptor in pSV7d was provided by K. Miyazono (Ludwig Institute); and type II TGFβ receptor in pcDNA1 was provided by X. F. Wang (Duke University). 3TP-Lux was as described. 16
Cell Lines
The pancreatic cancer cell lines BxPC-3, MiaPaCa-2, and PANC-1 and the colon cancer cell line HCT 116 were obtained from the American Type Culture Collection (Rockville, MD). Mv1Lu mink lung epithelial cells and their derivative L17 cells, which lack a functional TGFβ type 1 receptor, were kindly provided by J. Massague (Sloan-Kettering).
Preparation of Acini
Pancreatic acini were isolated from Wistar rats as previously described. 17 In brief, pancreatic tissue was digested with collagenase, mechanically dispersed, and passed through a 150-μm mesh nylon cloth. Acini were then purified by centrifugation at 50 g for 3 minutes in a solution containing 4% bovine serum albumin and were resuspended in enhanced Waymouth media. Cells were seeded onto 24-well culture plates coated with air-dried rat-tail collagen. Hormones and growth factors were added for indicated times.
Measurement of DNA Synthesis
MiaPaCa-2, PANC-1, and BxPC-3 were seeded onto 12-well tissue culture plates at a density of 5 × 104 cells/well. Twenty-four hours after plating, cells were exposed to TGFβ1 (1–100 pmol/L) for 24 hours. For acinar cell studies, cells were exposed to TGFβ (100 pmol/L) in either the absence of growth factors (control) or in the presence of cholecystokinin (10 nmol/L) or basic fibroblast growth factor (1 nmol/L) for 24 hours. [3H]thymidine assays were performed as described. 18
Luciferase Assays
Five times 104 MiaPaCa-2, PANC-1, and BxPC-3 cells (human pancreatic cancer cell lines) and L17 cells (mink lung epithelial cells that lack functional type 1 TGFβ receptors) were plated and transfected with 1 μg each p3TP-Lux and pRSVβgal using lipofectamine. In one group of L17 cells, 1 μg TGFβ receptor type 1 or blank vector was included in the transfection (positive control). After transfection, cells recovered for 24 hours, and in ligand-treated groups, 100 pmol/L TGFβ was added to induce transcription of 3TP-Lux. The next day, cells were harvested and luciferase activity was measured and normalized to β-galactosidase activity as described. 16 Transfection experiments with Smads and TGFβ receptors type I and II were performed in a similar fashion, as described above.
Reverse Transcriptase–Polymerase Chain Reaction (RT-PCR)
mRNA was isolated from cell lines using the Micro-FastTrack Kit (Invitrogen, Carlsbad, CA). First-strand cDNA synthesis using 1 μg mRNA was performed using random hexamers and Superscript II (Gibco BRL Life Technology, Gaithersburg, MD). The Smad2 primers were 5′-TGT TAA CCG AAA TGC CAC GG-3′ (sense) and 5-TCT TAT GGT GCA CAT TCT AG-3′ (antisense), amplifying nucleotides 953 to 1245 (293 bp) as described. 13 Smad3 primers were 5′- CCA GCC ATG TCG TCC ATC C-3′ (sense) and 5′-TTT TCC CCA AGC CTG CCC TC-3′ (antisense), amplifying the entire coding sequence as described. 19 Each PCR reaction (25 μL) included 5 μL of the cDNA synthesis product, 100 ng of each primer, 1.0 units of Taq polymerase (Boehringer Mannheim, Indianapolis, IN), and 2 mmol/L dNTPs (Boehringer Mannheim). For Smad2 reactions, an initial denaturing period of 5 minutes at 94°C was followed by 30 cycles of denaturing for 1 minute at 94°C and annealing/extension for 1 minute at 55°C and 1 minute at 72°C. For Smad3 reactions, an initial denaturing period of 5 minutes at 94°C was followed by 30 cycles of denaturing for 1 minute at 94°C and annealing/extension for 1 minute at 58°C and 1 minute at 72°C. As a control for cDNA synthesis, RT-PCR was also performed using primers specific for glyceraldehyde-3-phosphate dehydrogenase (GAPDH). 20 PCR products were loaded onto 1% agarose gels and visualized with ethidium bromide. Specificity was confirmed by Southern blotting with Smad2 and Smad3 cDNAS.
Receptor Cross-Linking
BxPC-3, MiaPaCa-2, and PANC-1 grown in 35-mm wells were incubated for 3 hours at 4°C with 200 pmol/L human [125I]TGFβ and then cross-linked to its receptors using 0.4 mmol/L disuccinimidyl suberate as described. 9 Where noted, cells were competed with 50-fold molar excess of unlabeled TGFβ to control for nonspecific binding. Mv1Lu cells served as a control. Cells were then solubilized in 300 μL 1% Triton-X 100. Equal amounts of cell lysate protein underwent electrophoresis in a 5% to 12% gradient SDS-polyacrylamide gel and were exposed for autoradiography.
RESULTS
To determine the effect of TGFβ on normal pancreatic cell growth, rat pancreatic acinar cells were treated with 100 pmol/L TGFβ alone, or with cholecystokinin (10 nmol/L) and basic fibroblast growth factor (10 pmol/L). Cells were treated for 24 hours, followed by a [3H]thymidine incorporation assay to assess DNA synthesis. In rat acinar cells, TGFβ inhibited basal [3H]thymidine incorporation and completely blocked increases induced by cholecystokinin and basic fibroblast growth factor (Fig. 1).
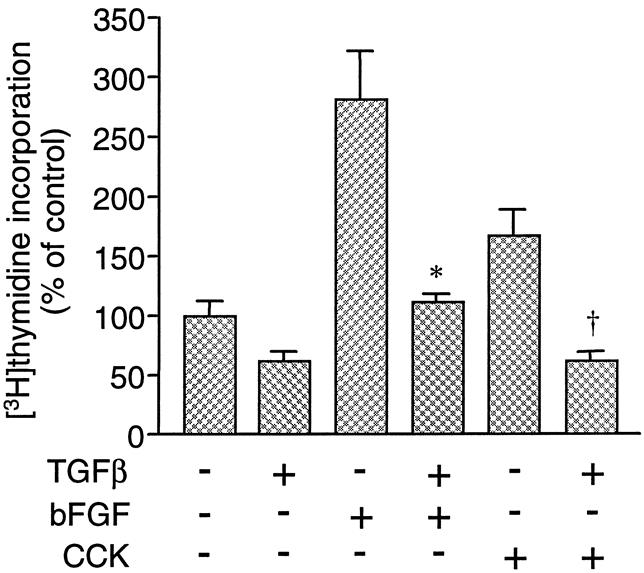
Figure 1. Effects of transforming growth factor beta (TGFβ) on [3H]thymidine incorporation in pancreatic acinar cells in vitro. Cells were cultured for 24 hours in the presence or absence of 100 pmol/L TGFβ, and in the presence of either no growth factor (control), 10 pmol/L basic fibroblast growth factor, or 1 nmol/L cholecystokinin. Values are expressed as percentage of control [3H]thymidine incorporation. Results are means ± standard error for three experiments. * and + represent P < .05 vs. basic fibroblast growth factor and cholecystokinin, respectively.
The effect of TGFβ on [3H]thymidine incorporation in BxPC-3, MiaPaCa-2, and PANC-1 cells was examined. At a maximal dose (100 pmol/L), TGFβ produced no or only partial inhibition of serum-induced DNA synthesis (Fig. 2), whereas this dose was sufficient to suppress DNA synthesis completely in normal pancreatic cells in vitro. MiaPaCa-2 cells showed no inhibition of DNA synthesis, whereas at maximal doses of TGFβ, PANC-1 and BxPC-3 cells had a limited reduction in [3H]thymidine incorporation, inhibited by 13% and 41%, respectively.
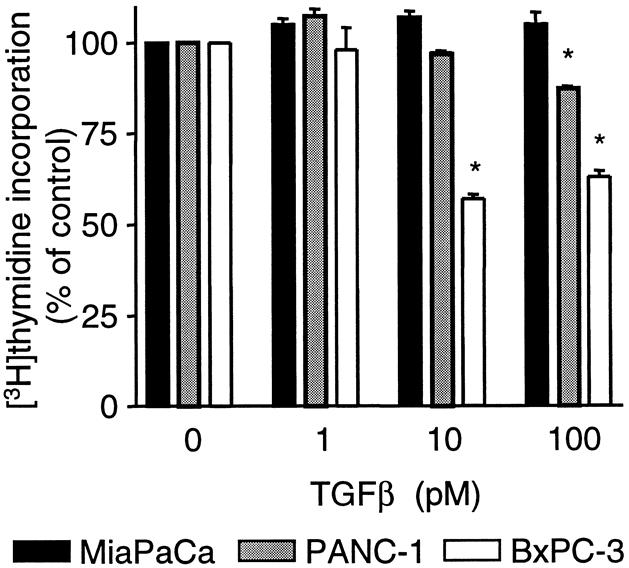
Figure 2. Dose response for transforming growth factor beta (TGFβ) inhibition of serum-induced [3H]thymidine incorporation in the three human pancreatic cancer cell lines MiaPaCa-2, PANC-1, and BxPC-3. Cells were incubated in the absence or presence of TGFβ (1–100 pmol/L) for 24 hours. Values are expressed as percentage of control [3H]thymidine incorporation and are the means ± standard error for four experiments. *P < .05 vs. control.
Diminished TGFβ responsiveness in the pancreatic cancer cell lines was further substantiated by transfection of pancreatic cancer cell lines with 3TP-Lux, a luciferase reporter construct under the control of a TGFβ signal-responsive promoter. The TGFβ-responsive 3TP-Lux construct contains three repeats of a 12-O-tetradecanoylphorbol 13-acetate responsive element plus the plasminogen activator inhibitor promoter linked to a luciferase reporter gene. 21 TGFβ did not induce luciferase expression in 3TP-Lux-transfected MiaPaCa-2 and PANC-1 cells and had a minimal effect on 3TP-Lux-transfected BxPC-3 cells (Fig. 3). L17 cells, mink lung epithelial cells lacking a functional TGFβ type 1 receptor, were used as a control. TGFβ had no effect on luciferase activity in 3TP-Lux-transfected L17 cells, but transfection of TGFβ type 1 receptors into L17 cells restored TGFβ’s ability to induce luciferase.
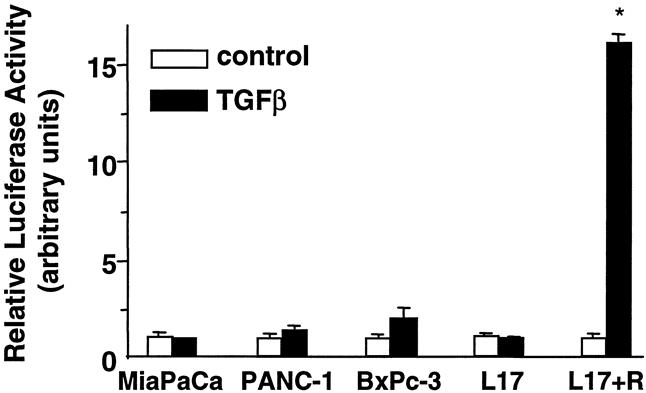
Figure 3. Effect of transforming growth factor beta (TGFβ) on 3TP-Lux expression. MiaPaCa-2, PANC-1, BxPC-3, and L17 cells were transiently transfected with 3TP-Lux and treated with 100 pmol/L TGFβ for 24 hours. Luciferase activity was measured and normalized with β-gal activity. White bars, negative TGFβ; black bars, positive TGFβ. In one condition, L17 cells were transfected with the type I TGFβ receptor ALK5 to restore the TGFβ signaling pathway. The data are normalized with the level of expression in the absence of ligand set to one. Values were determined in triplicate and represent the means ± standard error for three or four experiments. *P < .05 vs. control (no TGFβ).
Resistance to TGFβ has been shown to be due to lack of receptor expression in several cell types, including breast and colon cancer cells. 9,22 To determine whether the three pancreatic cancer cell lines investigated have defective TGFβ responsiveness because of altered expression of TGFβ receptors, we performed cross-linking experiments with 125I-labeled TGFβ in normal mink lung epithelial cells, Mv1Lu, which have a robust TGFβ growth inhibitory response, and in the pancreatic cancer cell lines. All three of the pancreatic cancer cell lines express TGFβ receptors types I and II, corresponding to the molecular weights of 68 and 85 Kd, respectively (Fig. 4). A third TGFβ receptor, shown recently to enhance responsiveness to TGFβ23 and to regulate autocrine TGFβ activity, 24 is expressed in all the cell lines (280 Kd). Addition of excess, unlabeled TGFβ competes out the bands, demonstrating specificity (see Fig. 4, lane 2).
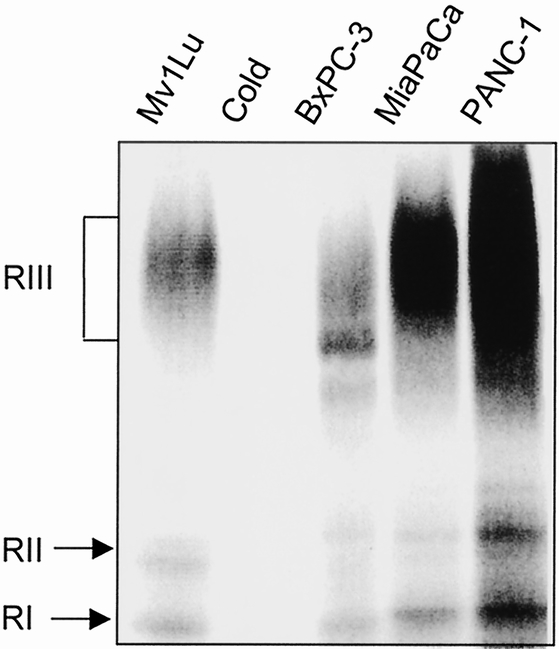
Figure 4. Analysis of cell surface receptors in Mv1Lu mink lung epithelial cells and the human pancreatic cancer cell lines MiaPaCa-2, PANC-1, and BxPC-3. Monolayers of cells were affinity-labeled with 200 pmol/L 125I-transforming growth factor beta (TGFβ) and disuccinimidyl suberate in the absence (lanes 1, 3, 4, 5) and presence (lane 2) of 50-fold molar excess unlabeled TGFβ. The 125I-TGFβ affinity-labeled receptors were visualized by 5–12% gradient SDS-polyacrylamide gel electrophoresis under reducing conditions and autoradiography. The bracket indicates the location of the type III TGFβ receptor, and the arrows note the locations of the type I and II receptors. The results are representative of three independent experiments.
Recent reports have identified TGFβ receptor-inactivating mutations in cell lines defective in TGFβ signaling while retaining ligand binding ability, as determined by cross-linking of receptor-bound [125I]TGFβ. 25 To ascertain whether this type of mutation was present in TGFβ receptors in the pancreatic cancer cell lines used in this study, cells were transfected with wild-type TGFβ receptor type I or type II and 3TP-Lux, and luciferase assays were performed. Ligand-dependent signaling was not restored in any of the pancreatic cancer cell lines when wild-type I TGFβ receptor was introduced, whereas transfection of type I receptor into TGFβ type I receptor-defective L17 cells restored sensitivity to TGFβ, as measured by activation of the 3TP-Lux reporter (Fig. 5A). Similarly, transfection of TGFβ type II receptor into the TGFβ type II-defective HCT 116 cell line restored a strong TGFβ-induced response but did not produce a significant response in the pancreatic cancer cell lines (see Fig. 5B). These results demonstrate that loss of TGFβ signaling in the pancreatic cancer cell lines is not due to the presence of defective TGFβ type I or II receptors.

Figure 5. Effect of transfection of transforming growth factor beta (TGFβ) type I and II receptors on TGFβ-dependent 3TP-Lux expression. (A) MiaPaCa-2, PANC-1, and L17 cells were transiently transfected with 3TP-Lux and either blank vector or TGFβ type I receptor in the absence or presence of TGFβ (100 pmol/L). (B) MiaPaCa-2, PANC-1, and HCT 116 cells were transiently transfected with 3TP-Lux and either blank vector or TGFβ type II receptor in the absence or presence of TGFβ (100 pmol/L). Luciferase activity was measured and normalized with β-gal activity. White bars, negative TGFβ; black bars, positive TGFβ. Data are normalized with the expression in the absence of ligand and in the absence of TGFβ type I receptor set to one. Values were determined in triplicate and represent the means ± standard error for three experiments. *P < .05 vs. control (no TGFβ).
Smad2, Smad3, and Smad4 are important intracellular components in the TGFβ signaling pathway. Previous studies investigating Smad4 mutations in human pancreatic cancer cell lines have demonstrated a homologous deletion of the Smad4 gene in BxPC-3 cells 12; MiaPaCa-2 and PANC-1 cell lines have no mutations in the Smad4 gene. 26 The presence of Smad2 and Smad3 in these cell lines has not previously been reported. We performed RT-PCR to analyze the expression of Smad2 and Smad3. RT-PCR showed the presence of Smad2 and Smad3 transcripts in all three cell lines (Fig. 6), suggesting that loss of Smad2 or Smad3 is unlikely to play a role in TGFβ resistance.
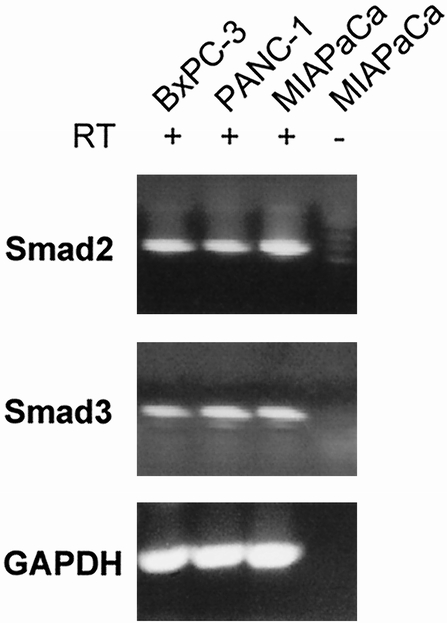
Figure 6. Reverse transcriptase–polymerase chain reaction (RT-PCR) of Smad2 and Smad3 in pancreatic cancer cell lines. MiaPaCa-2, PANC-1, and BxPC-3 cells were assayed for Smad2 and Smad3 mRNA by RT-PCR and Southern blotting. Each RNA sample was subjected to first-strand complementary DNA synthesis in the presence (+) or absence (-) of RT, followed by PCR. In separate experiments, a single band corresponding to the expected size of the PCR product (293 bp) of Smad2 (A) and a single band corresponding to the expected size of the PCR product (1,324 bp) of Smad3 (B), respectively, was detected in all three pancreatic cancer cell lines and only in reactions that included RT. GAPDH was used as a control.
To determine whether Smad4 can restore the TGFβ signaling pathway in pancreatic cancer cells, we assessed the ability of TGFβ to activate the TGFβ-inducible reporter 3TP-Lux in the absence and presence of wild-type Smad4. Mv1Lu cells demonstrated a robust response in 3TP-Lux activity in response to TGFβ (Fig. 7A). Transfection of Smad4 into Mv1Lu cells did not further augment the response to TGFβ. MiaPaCa-2 cells, which are not responsive to TGFβ but have a normal Smad4 gene, demonstrated little activation of 3TP-Lux in response to TGFβ in either the absence or presence of Smad4. TGFβ displayed little ability to activate 3TP-Lux in BxPC-3 cells, but transfection of BxPC-3 cells with Smad4 was able to restore an effective response. BxPC-3 cells have previously been shown to possess a homologous deletion of the Smad4 gene. 12 Transfection of Smad2 or Smad3 did not restore TGFβ signaling in MiaPaCa-2 or PANC-1 cells, ruling out the possibility that point mutations in Smad2 or Smad3 account for loss of TGFβ responsiveness (see Fig. 7B). These results suggest the importance of Smad4 as a key molecule in the TGFβ signaling pathway in some human pancreatic cancer cells.

Figure 7. (A) Effect of Smad4 on transforming growth factor beta (TGFβ)-dependent 3TP-Lux expression. Mv1Lu, MiaPaCa-2, and BxPC-3 cells were transiently transfected with 3TP-Lux and either blank vector or Smad4 in the absence or presence of TGFβ (100 pmol/L). (B) Effect of Smad2 and Smad3 on TGFβ-dependent 3TP-Lux expression. Mv1Lu, MiaPaCa-2, and PANC-1 cells were transiently transfected with 3TP-Lux and either blank vector or Smad2 or Smad3 in the absence or presence of TGFβ (100 pmol/L). Luciferase activity was measured and normalized with β-gal activity. White bars, negative TGFβ; black bars, positive TGFβ. Data are normalized with the expression in the absence of ligand and in the absence of Smad4 set to one. Values were determined in triplicate and represent the means ± standard error for two or three experiments. *P < .05 vs. control (no TGFβ).
DISCUSSION
TGFβ is a potent inhibitor of pancreatic cell proliferation. An important step in tumorigenesis in the pancreas may involve the loss of sensitivity to negative growth regulators, such as TGFβ. In fact, most pancreatic cancer cell lines are refractory to the growth inhibitory effects of TGFβ. A better understanding of the molecular events associated with loss of TGFβ responsiveness in pancreatic tumors may provide insight into the mechanisms of tumor development.
We examined the effects of TGFβ on three pancreatic cancer cell lines and compared the results with those obtained in normal pancreatic cells. These three cell lines were chosen because they have been widely studied and are fairly well characterized. To investigate the effect of TGFβ, we initially performed DNA synthesis studies on pancreatic acinar cells to confirm TGFβ’s effect on growth on normal pancreatic cells in vitro. TGFβ inhibited basal levels of [3H]thymidine incorporation and completely blocked increases induced by cholecystokinin and basic fibroblast growth factor. These results are consistent with those previously reported in acinar cells. 7 In contrast, the ability of TGFβ to inhibit serum-induced DNA synthesis was diminished or absent in the three pancreatic cell lines. These results are consistent with previous work by Beauchamp et al, 14 who noted no effect of TGFβ on soft agar growth of MiaPaCa-2 and PANC-1 cell lines. Interestingly, the authors did observe a twofold induction of TGFβ mRNA in PANC-1 cells. This suggests that in PANC-1 cells, some but not all TGFβ responses are lost. In two other studies, 15,27 TGFβ had no effect on growth of MiaPaCa-2 cells but did have a modest inhibitory effect on PANC-1 cells, with a 25% growth inhibition after 72 hours. No inhibitory effect was evident at 48 hours, consistent with our observation of no effect at 24 hours. Studies on the growth inhibitory effects of TGFβ on BxPC-3 cells have not previously been published.
Recent studies have indicated that alterations in TGFβ receptor expression or function may be important in several types of cancer. For example, TGFβ receptor type II inactivation induces escape from TGFβ-mediated growth inhibition in several cell types. 9 Moreover, gene transfer of TGFβ receptor type II suppresses the in vivo tumorigenicity of receptor-negative breast and colon cancer cell lines. 28,29 Examination of primary human pancreatic cancers suggests that abnormalities in TGFβ type II receptors are uncommon. In a study of 17 human pancreatic cancer samples, Friess et al 30 demonstrated normal or enhanced expression of type II TGFβ receptors in human pancreatic cancers samples using Northern blotting and in situ hybridization. There are conflicting data regarding the receptor status of several pancreatic cancer cell lines, including two of the pancreatic cancer cell lines used in this study. Freeman et al, 31 using RNase protection assays, reported a lack of TGFβ type II receptor mRNA in MiaPaCa-2 cells, with normal expression in PANC-1 cells. In a separate study, type I receptor mRNA was present in MiaPaCa-2 cells and PANC-1 cells, but the type II receptor transcript was absent in MiaPaCa-2 cells. In contrast, Beauchamp et al 14 determined that both MiaPaCa-2 and PANC-1 cell lines have specific, saturable TGFβ binding sites with similar binding affinities. Scatchard analysis revealed a similar number of TGFβ receptors for both cell lines (2.1 and 9.9 × 103/cell, respectively). They reported that binding sites were present with a Kd and receptor numbers comparable to those reported for TGFβ-sensitive cells. Therefore, lack of high-affinity binding sites did not appear to explain the lack of TGFβ responsiveness in these cells. In the current study, we demonstrated the presence of TGFβ type I and II receptors in all three pancreatic cancer cell lines using the technique of affinity cross-linking. This technique demonstrates specific TGFβ binding.
Taken together, these results suggest that disruption of the TGFβ signaling pathway in these pancreatic cancer cell lines is not due to a lack of TGFβ receptors. However, TGFβ receptors capable of binding agonist but deficient in signaling have been reported. 25 To evaluate this possibility, reconstitution experiments were performed by transfecting the pancreatic cancer cell lines with wild-type TGFβ type I and II receptors and comparing the results with two cell lines known to have defective TGFβ receptors, L17 (defective type I) and HCT 116 (defective type II). The pancreatic cancer cell lines showed no return of TGFβ responsiveness, whereas transfection of wild-type receptors restored full responsiveness in L17 and HCT 116 cell lines. This demonstrates that the lack of TGFβ responsiveness in the pancreatic cancer cell lines is not due to a lack of functional TGFβ receptors or decreased expression of receptors.
Recently, Smads have been identified in a variety of species as important signaling components in the TGFβ pathway. Pancreatic cancers and pancreatic cancer cell lines often contain mutations in Smad4, which may be important in loss of responsiveness to growth inhibition by TGFβ. In a study by Hahn et al, 12 25 of 84 pancreatic cancers had a homologous deletion in Smad4 (DPC4), and 6 of 27 pancreatic cancers had inactivating somatic mutations, consistent with the fact that Smad4 is a tumor suppressor gene. Although Smad4 inactivation is prevalent in pancreatic carcinoma, it is distinctly uncommon in other tumor types, such as colon, breast, and lung. 26 The tumor suppressor function of Smad proteins has been further supported by the findings of mutations of Smad2 in colon and lung cancers. 13,19
Mutational analysis has previously demonstrated a homologous deletion of Smad4 in BxPC-3 cells; both MiaPaCa-2 and PANC-1 cells possess wild-type Smad4. 12,26 To determine whether a lack of Smad2 or Smad3 might account for TGFβ resistance in any of our pancreatic cancer cell lines, RT-PCR was performed. We demonstrated the presence of Smad2 and Smad3 transcripts in all three cell lines; therefore, it is unlikely that Smad2 or Smad3 plays a role in TGFβ resistance in these cells. This was confirmed by transfection of wild-type Smad2 and Smad3 into pancreatic cancer cell lines: TGFβ responsiveness was not restored. These results are supported by the findings of Riggins et al, 19 who performed mutational analysis of Smads on 167 cancer cell lines, either passaged in vitro or as xenografts in nude mice, including 12 cancers of the pancreas. No mutations in Smad2 or Smad3 were identified.
To demonstrate the role of Smad4 in TGFβ signaling, BxPC-3 (known to lack Smad4), MiaPaCa-2, and Mv1Lu cells were transfected with Smad4 or empty vector. TGFβ responsiveness was determined using the 3TP-Lux reporter assay. Transfection with Smad4 restored TGFβ responsiveness in BxPC-3 cells but had no effect on MiaPaCa-2 cells. These results show that loss of TGFβ responsiveness, at least in some pancreatic cancers, correlates with loss of Smad4 expression. This has also been observed in the TGFβ-resistant breast tumor cell line MDA-MB-468, from which Smad4 has been deleted. 32 Transfection of Smad4 in this cell line restored both growth inhibition and induction of 3TP-Lux. The key role of Smad4 in TGFβ signaling is further supported by a recent study in which the Smad4 gene was deleted through homologous recombination in human colorectal cancer cells. 33 The deletion rendered cells unresponsive to TGFβ, as measured by a TGFβ-responsive reporter assay and a BrdUrd incorporation assay. Interestingly, TGFβ had a partial growth inhibitory effect, as measured by [3H]thymidine incorporation, in BxPC-3 cells but little effect on 3TP-Lux activity without Smad4. This uncoupling of effects on cell growth and transcriptional responses suggests that TGFβ may be able to mediate some of its growth inhibitory effects through signaling pathways other than Smad4 in BxPC-3 pancreatic cancer cells.
In summary, the results from this study demonstrate that the TGFβ signaling pathway is important in growth inhibition of normal pancreatic acinar cells but is defective in human pancreatic cell lines. Like the cell lines used in our study, most pancreatic cancer cell lines do not respond to TGFβ. However, defined genetic defects, such as Smad4 mutations or abnormal TGFβ receptors, have been identified in relatively few of these cell lines. 33,34 This demonstrates an incomplete understanding of the basis of TGFβ responsiveness in most cancer cells. In our study of three pancreatic cancer cell lines, one of the three lines has a known defect in Smad4, and in this cell line, transfection with wild-type Smad4 restored TGFβ responsiveness using a 3TP-Lux reporter assay. This supports the general concept that Smad4 is a tumor suppressor gene that is important for TGFβ signaling. However, the lack of responsiveness to TGFβ in MiaPaCa-2 and PANC-1 cells remains unclear. It is likely that other, as yet unidentified genes important in the TGFβ pathway may be found to play a role in cancer development.
Note Added in Proof
After submission of this manuscript, a study by Le Dai et al corroborated our findings that TGFβ has growth-inhibiting effects on BxPC-3 cells. 35
Acknowledgments
The authors thank L. Mathews for critically reading the manuscript, J. Massague for providing the Smad2 and Smad4 constructs and L17 cells, W. Vale for providing the Smad3 construct, K. Miyazono for providing the TGFβ type I receptor construct, and X. F. Wang for providing the TGFβ type II receptor construct.
Footnotes
Correspondence: Diane M. Simeone, MD, Dept. of Surgery, TC 2922D, Box 0331, University of Michigan Medical Center, 1500 E. Medical Center Dr., Ann Arbor, MI 48109.
Supported in part by University of Michigan Gastrointestinal Peptide Research Center Grant 5 P30 DK34933 (D.S.) and American College of Surgeons Faculty Research Fellowship (D.S.).
E-mail: simeone@umich.edu
Accepted for publication December 21, 1999.
References
- 1.Yeo CJ. Pancreatic cancer: 1998 update. J Am Coll Surg 1998; 187:429–442. [PubMed] [Google Scholar]
- 2.Massague J, Attisano L, Wrana JL. The TGFβ family and its composite receptors. Trends Cell Biol 1994; 4:172–178. [DOI] [PubMed] [Google Scholar]
- 3.Hannon GJ, Beach D. P15INK4B is a potential effector of TGF-β-induced cell cycle arrest. Nature 1994; 371:257–261. [DOI] [PubMed] [Google Scholar]
- 4.Datto MB, Yu Y, Wang XF. Functional analysis of the transforming growth factor β responsive elements in the WAF1/Cip1/p21 promoter. J Biol Chem 1995; 270:28623–28628. [DOI] [PubMed] [Google Scholar]
- 5.Reynisdottir I, Polyak K, Iavarone A, Massague J. Kip/Cip and Ink4 CDK inhibitors cooperate to induce cell cycle arrest in response to TGF-β. Genes Dev 1995; 9:1831–1845. [DOI] [PubMed] [Google Scholar]
- 6.Iavarone A, Massague J. Repression of the CDK activator Cdc25A and cell-cycle arrest by cytokine TGF-β in cells lacking the CDK inhibitor p15. Nature 1997; 387:417–426. [DOI] [PubMed] [Google Scholar]
- 7.Logsdon CD, Keyes L, Beauchamp RD. Tranforming growth factor-β (TGF-β1) inhibits pancreatic acinar cell growth. Am J Physiol 1992; 262:G364–368. [DOI] [PubMed] [Google Scholar]
- 8.Bisgaard Hc, Thorgeirsson SS. Evidence for a common cell of origin for primitive epithelial cells isolated from rat liver and pancreas. J Cell Physiol 1991; 147:333–343. [DOI] [PubMed] [Google Scholar]
- 9.Markowitz S, Wang J, Myeroff L, et al. Inactivation of the type II TGF-β receptor in colon cancer cells with microsatellite instability. Science 1995; 268:1336–1338. [DOI] [PubMed] [Google Scholar]
- 10.Park K, Kim SJ, Bang YJ, et al. Genetic changes in the transforming growth factor β (TGFβ) type II receptor gene in human gastric cancer cells: correlation with sensitivity to growth inhibition by TGFβ. Proc Natl Acad Sci U S A 1994; 91:8772–8776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hoodless PA, Wrana JL. Mechanism and function of signaling by the TGFβ superfamily. Curr Topics Microbiol Immunol 1998; 228:235–272. [DOI] [PubMed] [Google Scholar]
- 12.Hahn SA, Schutte M, Hoque S, et al. DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science 1996; 271:350–353. [DOI] [PubMed] [Google Scholar]
- 13.Eppert K, Scherer SW, Ozcelik H, et al. MADR2 maps to 18q21 and encodes a TGFβ-regulated MAD-related protein that is functionally mutated in colorectal cancer. Cell 1996; 86:543–552. [DOI] [PubMed] [Google Scholar]
- 14.Beauchamp RD, Lyons RM, Yang EY, et al. Expression of and response to growth regulatory peptides by two human pancreatic carcinoma cell lines. Pancreas 1990; 5:369–380. [DOI] [PubMed] [Google Scholar]
- 15.Baldwin RL, Friess H, Yokoyama M, et al. Attenuated ALK5 receptor expression in human pancreatic cancer: correlation with resistance to growth inhibition. Int J Cancer 1996; 67:283–288. [DOI] [PubMed] [Google Scholar]
- 16.Willis SA, Zimmerman CM, Li L, Mathews LS. Formation and activation by phosphorylation of activin receptor complexes. Mol Endocrinol 1996; 10:367–379. [DOI] [PubMed] [Google Scholar]
- 17.Williams JA, Korc M, Dormer RL. Action of secretagogues on a new preparation of functionally intact, isolated pancreatic acini. Am J Physiol 1978; 253:E517–E524. [DOI] [PubMed] [Google Scholar]
- 18.Logsdon CD, Williams JA. Pancreatic acinar cells in monolayer culture: direct trophic effects of cerulein in vitro. Am J Physiol 1986; 250:G440–G447. [DOI] [PubMed] [Google Scholar]
- 19.Riggins GJ, Kinzler KW, Vogelstein B, Thiagalingam S. Frequency of Smad gene mutations in human cancers. Cancer Res 1997; 57:2578–2580. [PubMed] [Google Scholar]
- 20.Mata M, Jin CF, Fink DJ. Axotomy increases CNTF receptor mRNA in rat spinal cord. Brain Res 1993; 610:162–165. [DOI] [PubMed] [Google Scholar]
- 21.Wrana JL, Attisano L, Carcamo J, et al. TGF beta signals through a heteromeric protein kinase receptor complex. Cell 1992; 71:1003–1014. [DOI] [PubMed] [Google Scholar]
- 22.Kalkhoven E, Roelen BA, de Winter JP, et al. Resistance to transforming growth factor b and activin due to reduced receptor expression in human breast tumor cell lines. Cell Growth Differ 1995; 6:1151–1161. [PubMed] [Google Scholar]
- 23.Lopez-Casillas F, Wrana JL, Massague J. Betaglycan presents ligand to the TGF beta signaling receptor. Cell 1993; 73:1435–1444. [DOI] [PubMed] [Google Scholar]
- 24.Sun L, Chen C. Expression of transforming growth factor β type III receptor suppresses tumorigenicity of human breast cancer MDA-MB-231 cells. J Biol Chem 1997; 272:25367–25372. [DOI] [PubMed] [Google Scholar]
- 25.Weis-Garcia F, Massague J. Complementation between kinase-defective and activation-defective TGF-β receptors reveals a novel form of receptor cooperativity essential for signaling. EMBO J 1996; 15:276–289. [PMC free article] [PubMed] [Google Scholar]
- 26.Schutte M, Hruban RH, Hedrick L, et al. DPC4 gene in various tumor types. Cancer Res 1996; 56:2527–2530. [PubMed] [Google Scholar]
- 27.Grau AM, Zhang L, Wang W, et al. Induction of p21WAF1 expression and growth inhibition by transforming growth factor β involve the tumor suppressor gene DPC4 in human pancreatic adenocarcinoma cells. Cancer Res 1997; 57:3929–3934. [PubMed] [Google Scholar]
- 28.Wang J, Sun L, Myeroff L, et al. Demonstration that mutation of the type II transforming growth factor beta receptor inactivates its tumor suppressor activity in replication error-positive colon carcinoma cells. J Biol Chem 1995; 270:22044–22049. [DOI] [PubMed] [Google Scholar]
- 29.Sun L, Wu G, Willson JKV, et al. Expression of transforming growth factor b type II receptor leads to reduced malignancy in human breast cancer MCF-7 cells. J Biol Chem 1994; 269:26449–26455. [PubMed] [Google Scholar]
- 30.Friess H, Yamanaka Y, Buchler M, et al. Enhanced expression of the type II transforming growth factor β receptor in human pancreatic cancer cells without alteration of type III receptor expression. Cancer Res 1993; 53:2704–2707. [PubMed] [Google Scholar]
- 31.Freeman J, Mattingly CA, Strodel WE. Increased tumorigenicity in the human pancreatic cell line MIA PaCa-2 is associated with an aberrant regulation of an IGF-1 autocrine loop and lack of expression of the TGF-beta type II receptor. J Cell Physiol 1995; 165:155–163. [DOI] [PubMed] [Google Scholar]
- 32.DeWinter JP, Roelen B, Dijke P, et al. DPC4 (SMAD4) mediates transforming growth factor-β1 (TGFβ1) induced growth inhibition and transcriptional response in breast tumour cells. Oncogene 1997; 14:1891–1899. [DOI] [PubMed] [Google Scholar]
- 33.Zhou S, Buckhaults P, Zawel L, et al. Targeted deletion of Smad4 shows it is required for transforming growth factor beta and activin signaling in colorectal cancer cells. Proc Natl Acad Sci U S A 1998; 95:2412–2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rodeck U, Nishiyama T, Mauviel A. Independent regulation of growth and SMAD-mediated transcription by transforming growth factor β in human melanoma cells. Cancer Res 1999; 59:547–550. [PubMed] [Google Scholar]
- 35.Le Dai J, Schutte M, Bansai RK, et al. Transforming growth factor-β responsiveness in DPC4/SMAD4-null cells. Mol Carcinog 1999; 26:37–43. [DOI] [PubMed] [Google Scholar]