

Abstract
Objective
To review present knowledge of intracellular mechanisms and molecular regulation of muscle cachexia.
Summary Background Data
Muscle cachexia, mainly reflecting degradation of myofibrillar proteins, is an important clinical feature in patients with severe injury, sepsis, and cancer. The catabolic response in skeletal muscle may result in muscle wasting and weakness, delaying or preventing ambulation and rehabilitation in these patients and increasing the risk for pulmonary complications.
Results
Muscle cachexia, induced by severe injury, sepsis, and cancer, is associated with increased gene expression and activity of the calcium/calpain- and ubiquitin/proteasome-proteolytic pathways. Calcium/calpain-regulated release of myofilaments from the sarcomere is an early, and perhaps rate-limiting, component of the catabolic response in muscle. Released myofilaments are ubiquitinated in the N-end rule pathway, regulated by the ubiquitin-conjugating enzyme E214k and the ubiquitin ligase E3α, and degraded by the 26S proteasome.
Conclusions
An understanding of the mechanisms regulating muscle protein breakdown is important for the development of therapeutic strategies aimed at treating or preventing muscle cachexia in patients with severe injury, sepsis, cancer, and perhaps other catabolic conditions as well.
Muscle cachexia is a common metabolic response to several disease states seen in surgical patients, including sepsis, severe injury, renal failure, and cancer. Although not identical, the intracellular mechanisms and molecular regulation of muscle atrophy are similar in many of these conditions. The catabolic response in skeletal muscle is important from a clinical standpoint for several reasons. During the early phase of muscle cachexia, the release of amino acids from muscle tissue may be beneficial to the organism because it provides the liver, intestinal mucosa, and probably other tissues as well with amino acids for gluconeogenesis and acute-phase protein synthesis and provides an important energy source for enterocytes and cells in the immune system. 1–3 Prolonged and severe muscle cachexia, however, has significant deleterious consequences and results in muscle wasting and weakness, delaying or preventing ambulation and rehabilitation in catabolic patients. When the respiratory muscles are involved, prolonged ventilatory support may be needed and pulmonary complications, including pneumonia from aspiration, may develop. 4 In patients with cancer, weight loss and muscle cachexia may be the first manifestations of the disease. In addition, there is evidence that the response to chemotherapy is impaired in patients with cachexia. 5 It has been estimated that almost one third of deaths in patients with cancer are related to muscle catabolism and weakness, particularly if the respiratory muscles are involved. 6 An understanding of the mechanisms and molecular regulation of muscle cachexia, therefore, has important implications for the care of surgical patients.
Although reduced protein synthesis and inhibited uptake of amino acids contribute to the catabolic response in skeletal muscle, increased protein breakdown, in particular breakdown of the myofibrillar proteins actin and myosin, is the most important mechanism of muscle cachexia. 7,8 Intracellular protein degradation is regulated by multiple proteolytic pathways, including lysosomal, energy-ubiquitin-dependent, and calcium-calpain-dependent mechanisms. 9 A relatively large number of studies have been published in recent years providing evidence that the ubiquitin-proteasome pathway plays an important role in the development of muscle cachexia in sepsis, 10–12 severe injury, including burn injury, 13,14 renal failure, 15 and cancer. 16,17
It is the purpose of this review to provide current knowledge of the mechanisms involved in the regulation of muscle cachexia. In particular, recent evidence suggesting that calcium-calpain-mediated release of myofilaments from the sarcomere may be an initial and perhaps rate-limiting step in muscle cachexia will be discussed. First, however, recent observations, including those in human muscle, regarding the ubiquitin-proteasome pathway and different enzymes and mechanisms involved in the ubiquitination of muscle proteins are reviewed.
PROTEIN UBIQUITINATION AND DEUBIQUITINATION
The ubiquitin-proteasome pathway and its role in the regulation of muscle proteolysis were described in recent review articles from this and other laboratories 9,18–20 and will therefore be discussed only briefly here. Proteins degraded by this mechanism are first conjugated to multiple molecules of ubiquitin, which is a 76-amino acid, 8.5-kDa residue, highly conserved and present in all eukaryotic cells. Ubiquitinated proteins are recognized and degraded by the large proteolytic 26S proteasome. The catalytic core of the 26S proteasome is the 20S proteasome, a barrel-shaped particle consisting of four stacked rings with seven subunits in each ring. 21,22 The 19S capping protein, attached to the end of the 20S proteasome, recognizes, binds, and unfolds ubiquitinated proteins that are subsequently funneled through the central channel of the 20S proteasome and hydrolyzed. A more detailed description of the 26S proteasome and its various components can be found elsewhere. 23
Ubiquitination of proteins is regulated by at least three different enzymes: ubiquitin activating enzyme, E1; ubiquitin conjugating enzyme, E2; and ubiquitin ligase, E3. The structure of the ubiquitin system is hierarchical in that a single E1 carries out activation of ubiquitin for all modifications, whereas substrate and tissue specificity are accounted for by different E2s and E3s. Individual E2s work in concert with specific E3s, which in turn have substrate specificity. A simplified scheme of the ubiquitin-proteasome pathway 24 and the different steps involved in the ubiquitination of proteins are depicted in Figure 1. Note that energy (ATP) is needed at multiple steps during the process.
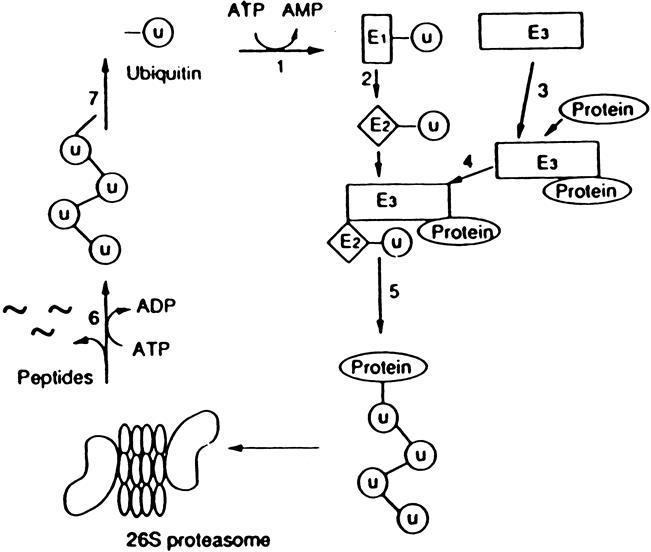
Figure 1. Simplified scheme of the ubiquitin-proteasome proteolytic pathway. In this pathway, ubiquitinated proteins are recognized and degraded by the 26S proteasome. The steps involved in the breakdown of proteins by this mechanism include 1) activation of ubiquitin by the ubiquitin activating enzyme E1; 2) transfer of ubqiutin to the ubiquitin conjugating enzyme E2; 3) interaction between the substrate protein and the ubiquitin ligase E3; 4) interaction between E2 and E3 resulting in 5) multiubiquitination of the substrate protein; 6) degradation of the ubiquitinated protein by the 26S proteasome; and 7) deubiquitination resulting in the release and reuse of ubiquitin in the pathway. Energy is required for at least two steps in the pathway: activation of ubiquitin by E1 (step 1) and the proteolytic activity in the 26S proteasome (step 6). (From Hershko A. Lessons from the discovery of the ubiquitin system. Trends Biochem Sci 1996; 21:445–449, with permission.)
Recent studies suggest that the 14-kDa ubiquitin conjugating enzyme E214k and the ubiquitin ligase E3α are particularly important for the degradation of muscle proteins. 25–27 In studies from this laboratory, the gene expression of E214k28 and E3α29 was upregulated in septic muscle, supporting the concept that ubiquitination of muscle proteins is increased during sepsis. Additional evidence for an increased rate of ubiquitination of proteins in cachectic muscle was found in other experiments. 30
As illustrated in Figure 1, ubiquitin is released from the substrate protein after its degradation, and because ubiquitin is a stable protein, it can be recycled in the proteolytic pathway. The deubiquitination process is highly regulated by several deubiquitinating enzymes that actually make up the largest known family in the ubiquitin system. 31 Recent studies suggest that the deubiquitination may influence the breakdown rate of ubiquitinated proteins. 31 This process, therefore, offers an additional point of regulation of the ubiquitin-proteasome pathway. The influence of sepsis, injury, cancer, and other catabolic conditions on deubiquitination of muscle proteins is not known but is an important area for future studies.
THE N-END RULE PATHWAY
In addition to the activity of enzymes involved in the ubiquitination and deubiquitination of proteins, certain features of a protein can make it susceptible to degradation. Such features are called degradation signals, or degrons. Varshavsky 32 reported that there was a correlation between specific features of the N-terminal of a protein and the half-life of the protein, the so-called N-end rule. Subsequent studies provided evidence that certain N-terminal basic amino acids (Arg, Lys, His) and bulky hydrophobic amino acids (Phe, Leu, Trp, Tyr, Ile) made the proteins susceptible to ubiquitination and degradation by the proteasome. Ubiquitination of proteins in the N-end rule pathway is regulated by the ubiquitin-conjugating enzyme E214k and the ubiquitin ligase E3α (Fig. 2).
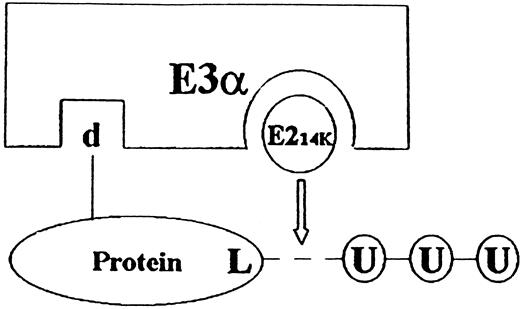
Figure 2. Simplified scheme of the interaction between protein substrate, E214k, and E3α in the N-end rule pathway. Proteins with a destabilizing N-end (d) are ligated to ubiquitin ligase E3α. Specific interaction between E3α and the ubiquitin conjugating enzyme E214k (ubiquitin carrier protein) allows for the transfer and conjugation of ubiquitin to a lysine residue (L) of the substrate protein. This is followed by the conjugation of additional ubiquitin molecules, resulting in a multiubiquitinated protein that is subsequently recognized and degraded by the 26S proteasome.
Because most proteins do not have a destabilizing N-terminal under normal conditions, the role of the N-end rule pathway was initially questioned. 33 However, more recent studies using specific E3α blockers in cell-free systems suggest that a large portion of muscle proteins can be ubiquitinated in the N-end rule pathway and that proteins in cachectic muscles are degraded by this mechanism. 30,34 In other studies, increased gene expression of E214k and E3α provided further evidence for the involvement of the N-end rule pathway in muscle cachexia. 28,29
In addition to the interaction between E214k and E3α, other “pairs” of enzymes regulating the ubiquitination of proteins have been described. For example, recent studies characterized a novel 18-kDa ubiquitin conjugating enzyme, E2-F1, that acts in concert with the ubiquitin ligase E3L in the ubiquitination of several non–N-end rule protein substrates. 35,36 Those substrates include actin, troponin T, and MyoD, suggesting that E2-F1 and E3L may be involved in the degradation of muscle proteins. The role of these enzymes in the development of muscle cachexia is not known.
THE 20S PROTEASOME
The 20S proteasome is the catalytic core of the 26S proteolytic complex and is central for the breakdown of ubiquitinated proteins. 21,22 The 20S proteasome is an abundant particle, making up approximately 1% of cell proteins. Multiple proteolytic activities have been ascribed to the 20S proteasome, including chymotrypsinlike, trypsinlike, and peptidyl-glutamyl peptidase activities. Interestingly, recent studies suggest that the proteasome active sites regulate each other. 37 The initial and rate-limiting step in the digestion of a polypeptide when it enters the proteasome is the cleavage by a chyomotrypsinlike site. This generates fragments that are cleaved further by the other active sites, including the peptidyl-glutamyl peptidase activity (caspase activity). The chymotrypsinlike activity stimulates the downstream caspase activity, whereas caspase activity inhibits chymotrypsinlike activity. These observations are consistent with a cyclical “bite–chew” mechanism for protein breakdown. 37 The chymotrypsinlike activity initially cleaves (“bites”) the polypeptide, thereby stimulating the caspaselike activity. This activation increases further cleavage (“chewing”) of the fragments while the chymotrypsinlike activity is temporarily inhibited. When further caspase activity is not possible, the chymotrypsinlike site is reactivated and the cycle is repeated.
In experiments in our laboratory, the activity of isolated 20S proteasomes was determined by measuring the activity against the fluorogenic substrates succinyl-leucine-leucine-valine-tyrosine-7-amido-4-methylcoumarin (LLVY) and N-carbenzoxy-leucine-leucine-glutamate-7-amido-4-methylcoumarin (LLE). These substrates are preferentially hydrolyzed by the chymotrypsinlike and peptidyl-glutamyl peptidase activities of the 20S proteasome, respectively. Sepsis in rats resulted in an approximately 50% increase in proteasome activity against LLVY and a doubling of the activity against LLE in skeletal muscle 38 (Fig. 3). The involvement of the 20S proteasome in the catabolic response in skeletal muscle was further supported by increased gene expression of several 20S proteasome α and β subunits (Fig. 4) and by inhibition of sepsis-induced muscle protein breakdown by proteasome blockers. 39 Evidence for increased expression of the 20S proteasome in skeletal muscle has been reported in other catabolic conditions as well, including cancer, 17 burn injury, 40 and metabolic acidosis. 41
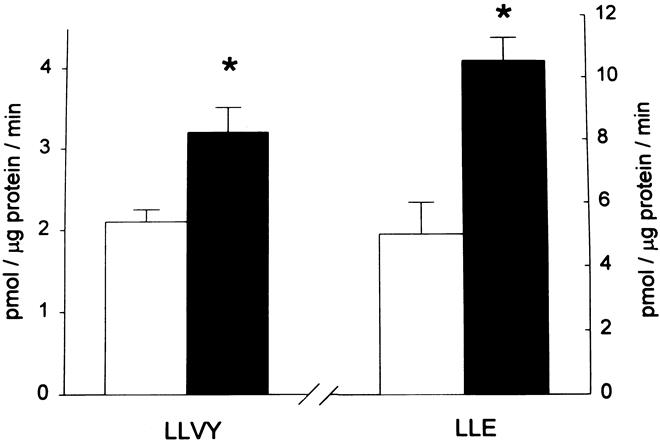
Figure 3. Sepsis results in increased activity of the 20S proteasome in skeletal muscle. 20S proteasomes were isolated from muscles of sham-operated (open bars) or septic rats (filled bars), and the chymotrypsinlike and peptidyl-glutamyl peptidase activities were determined by using the fluorogenic substrates LLVY and LLE, respectively. * P < .05. (Hobler SC, Williams AB, Fischer D, et al. Activity and expression of the 20S proteasome are increased in skeletal muscle during sepsis. Am J Physiol 1999; 277:R434–R440, with permission.)
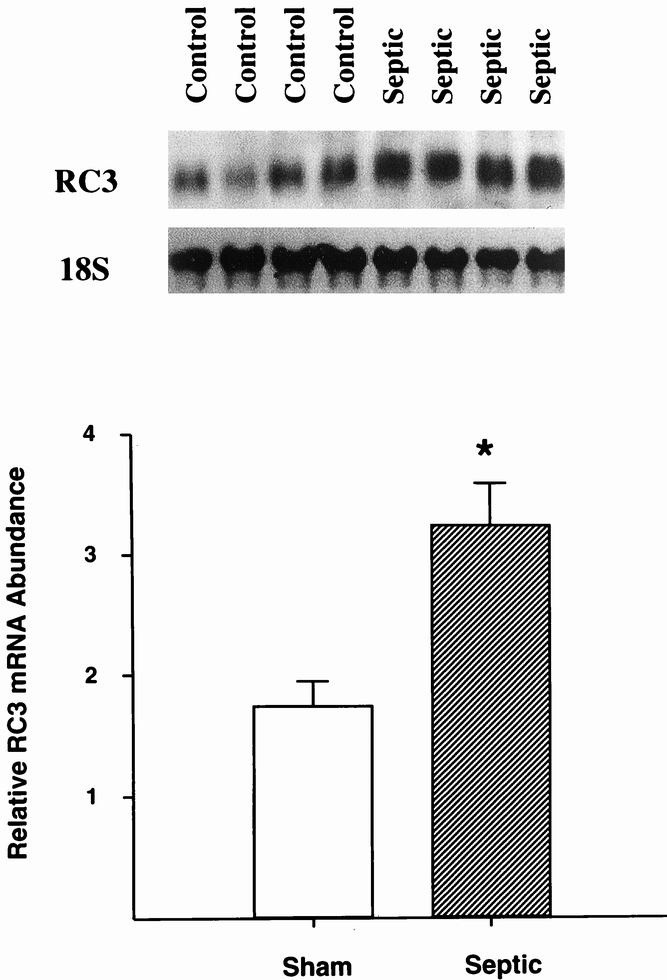
Figure 4. Sepsis results in increased gene expression of the 20S α subunit RC3 in skeletal muscle. Northern blots from four sham-operated control rats and four septic rats (sepsis induced by cecal ligation and puncture) are shown in the upper panel, and quantitation of the blots is shown in the lower panel. * P < .05. (Data from Hobler SC, Williams AB, Fischer D, et al. Activity and expression of the 20S proteasome are increased in skeletal muscle during sepsis. Am J Physiol 1999; 277:R434–R440, with permission.)
PROTEASOME INHIBITORS
Pharmacologic inhibitors of the proteasome have been available only since 1994. Different types of proteasome inhibitors and their use in cell biology research were reviewed recently. 42 Four classes of proteasome inhibitors have been described: peptide aldehydes, lactacystin/β-lactone, vinyl sulfones, and dipeptide boronic acid analogs. The peptide aldehydes are reversible inhibitors of the proteasome that block primarily the chymotrypsinlike activity of the proteasome. Other inhibitors, including lactacystin/β-lactone and vinyl sulfones, block the proteasome irreversibly.
Most studies published so far and in which the effects of proteasome inhibitors have been examined have been performed in vitro in cell-free systems, cultured cells, or incubated muscles. In experiments in our 39,43 and other 44 laboratories, treatment of incubated cachectic rat skeletal muscles with aldehydes (e.g., LLnL) or lactacystin (and its active product β-lactone) inhibited protein degradation (Fig. 5), providing further evidence that muscle cachexia at least in part reflects proteasome-dependent proteolysis. Only a few studies have been reported in which a proteasome blocker was administered in vivo to an intact animal, and in none of those studies was the effect of the proteasome inhibitor on muscle cachexia examined. 45,46 In recent experiments in our laboratory, treatment of septic rats in vivo with the proteasome inhibitor N-benzyloxycarbonyl-Ile-Glu-(O-t-butyl)-Ala-leucinal (PSI) reduced the sepsis-induced muscle proteolysis and inhibited the chymotrypsinlike proteasome activity. 47 These results are important because they suggest that it may be possible to prevent or treat the catabolic response in skeletal muscle during sepsis (and perhaps other catabolic conditions as well) with a proteasome blocker. An interesting observation reported recently was that treatment of mice with the proteasome inhibitor PS-341 (a boronic acid analog) reduced or even abolished experimental tumors. 46
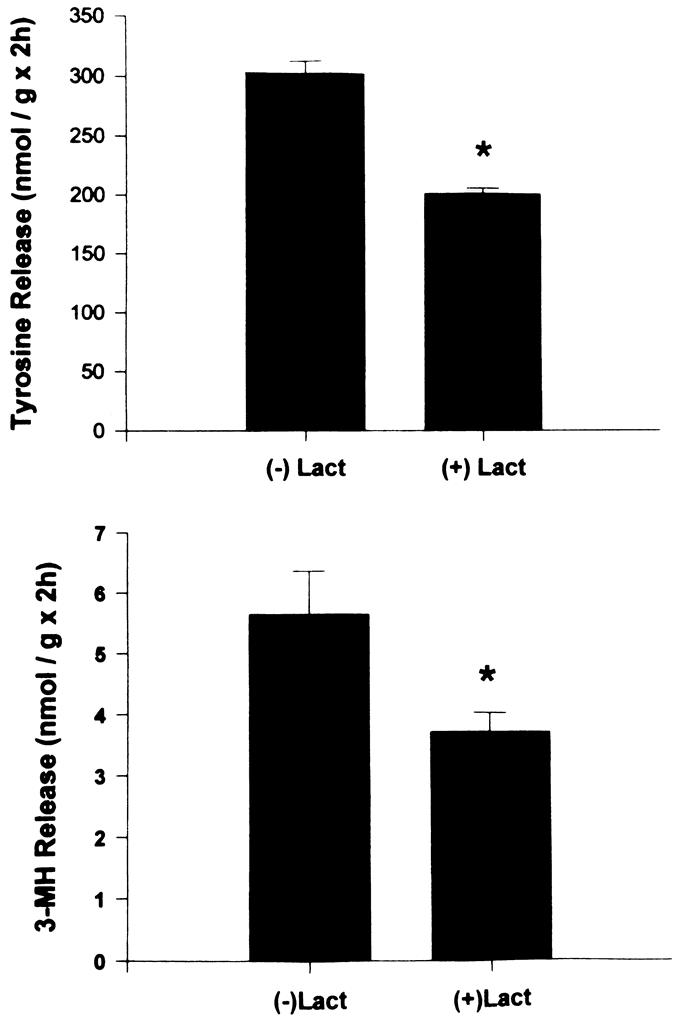
Figure 5. Total (upper panel) and myofibrillar (lower panel) protein breakdown in incubated muscles from septic rats is inhibited by the specific proteasome blocker lactacystin (100 μM). * P < .05. (Hobler SC, Tiao G, Fischer JE, et al. The sepsis-induced increase in muscle proteolysis is blocked by specific proteasome inhibitors. Am J Physiol 1998; 274:R30–R37, with permission.)
THE UBIQUITIN-PROTEASOME PATHWAY IN HUMAN SKELETAL MUSCLE
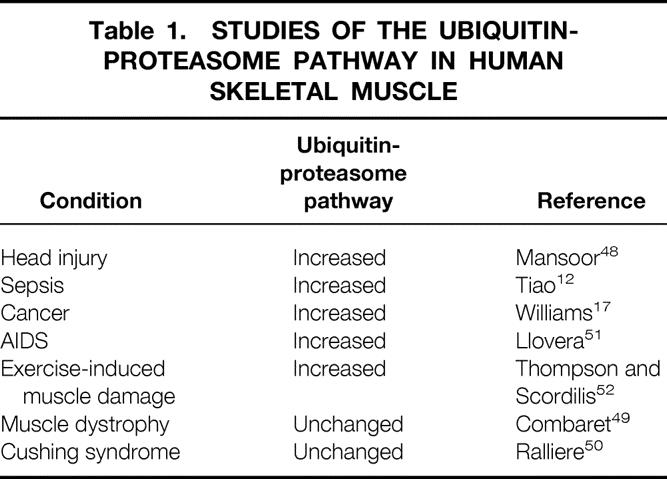
Most previous studies on the role of the ubiquitin-proteasome pathway in muscle cachexia were performed in experimental animals. Several recent studies have been published suggesting that muscle cachexia is associated with upregulated expression of the ubiquitin-proteasome pathway in patients as well. For example, in patients with severe head injury, the catabolic response in skeletal muscle was associated with increased gene expression of the ubiquitin-proteasome pathway. 48 In studies in our laboratory, similar results were observed in patients with sepsis 12 and cancer. 17 Other reports in which the ubiquitin-proteasome pathway was examined in human skeletal muscle are summarized in Table 1. 49–52 In most but not all conditions, the results were similar in muscles from patients and experimental animals. Thus, ubiquitin-proteasome-dependent proteolysis probably plays an important role in the development of muscle cachexia in different groups of catabolic patients, including patients with severe injury, sepsis, and cancer.
Table 1. STUDIES OF THE UBIQUITIN-PROTEASOME PATHWAY IN HUMAN SKELETAL MUSCLE
CALCIUM/CALPAIN-DEPENDENT RELEASE OF MYOFILAMENTS
Since the publication of a previous review in this journal that focused on the role of the ubiquitin-proteasome pathway in muscle cachexia, 18 our understanding of other important aspects of the proteolytic process has improved substantially. Because it is mainly the myofibrillar proteins actin and myosin that are degraded in cachectic muscle, 7,8 an apparently conflicting observation reported previously is that the proteasome does not degrade intact myofibrils. For example, incubation of ovine skeletal muscle with isolated proteasomes did not result in changes indicative of myofibrillar breakdown. 53 In other studies, isolated muscle proteasomes degraded free actin and myosin but not intact myofibrils. 54 These results suggest that actin and myosin need to dissociate from the myofibrils before they can be ubiquitinated (possibly in the N-end rule pathway) and degraded by the 26S proteasome.
To understand this hypothesis, we will briefly review the ultrastructure of skeletal muscle here. The sarcomere, extending from one Z-band to the next, is the rudimentary “contractile unit” of striated muscles (Fig. 6). The sarcomeres are approximately 2 to 3 μm long and 1 to 2 μm in diameter and are linked end to end to form long, thin strands known as myofibrils. Many parallel myofibrils together make up a muscle fiber. Current concepts of the architecture and function of the muscle sarcomere were reviewed recently. 55
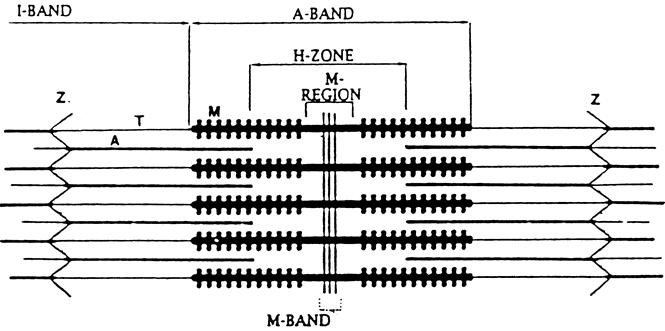
Figure 6. Simplified scheme of the muscle sarcomere. The sarcomere extends from one Z-disk to the next. Myosin (M) is anchored to the Z-disk by titin (T) and actin (A) by nebulin. Z, Z-band.
The Z-disks serve to anchor the myofilaments and to link thin myofilaments (actin) from one sarcomere to the next along the myofibril. Myosin is anchored to the Z-disk by the large protein titin. It is easy to understand, therefore, that disintegration or disruption of the Z-disk may result in the release of actin and myosin. Indeed, in recent experiments in our laboratory, morphologic evidence was found for disintegration of the Z-disk in cachectic muscle from septic rats (Fig. 7). 56 This effect of sepsis was associated with the release of myofilaments from the myofibrils, determined by measuring the fraction of “easily releasable myofilaments.” In the same study, calcium concentrations and the gene expression of μ- and m-calpain and the muscle-specific calpain p94 were increased in septic muscle (Fig. 8). Treatment of septic rats with dantrolene (a substance that inhibits the release of calcium from intracellular stores) prevented the sepsis-induced release of myofilaments. These observations are consistent with a model of muscle cachexia in which an early, calcium-calpain-dependent release of myofilaments from the myofibrils is followed by ubiquitination of the myofilaments (possibly in the N-end rule pathway) and subsequent degradation by the proteasome (Fig. 9). This model would also explain why both calcium antagonists and proteasome inhibitors may block protein breakdown in cachectic muscle. Even though dantrolene inhibits the release of calcium from intracellular stores and reduces cytoplasmic calcium levels and the release of myofilaments from the sarcomere, it may also exert anticatabolic effects through other mechanisms as well. For example, treatment of septic mice with dantrolene reduced plasma levels of interleukin-1 and tumor necrosis factor, 57 substances that are at least in part responsible for sepsis-induced muscle cachexia.

Figure 7. Electron microscopy of muscles from sham-operated (A) and septic (B, C) rats. Note disintegration or complete loss (arrow in C) of Z-bands in muscles from septic rats. Swollen mitochondria (*) were frequently seen in septic muscles. (From Williams AB, DeCourten-Myers GM, Fischer JE, et al. Sepsis stimulates release of myofilaments in skeletal muscle by a calcium-dependent mechanism. FASEB J 1999; 13:1435–1443, with permission.)
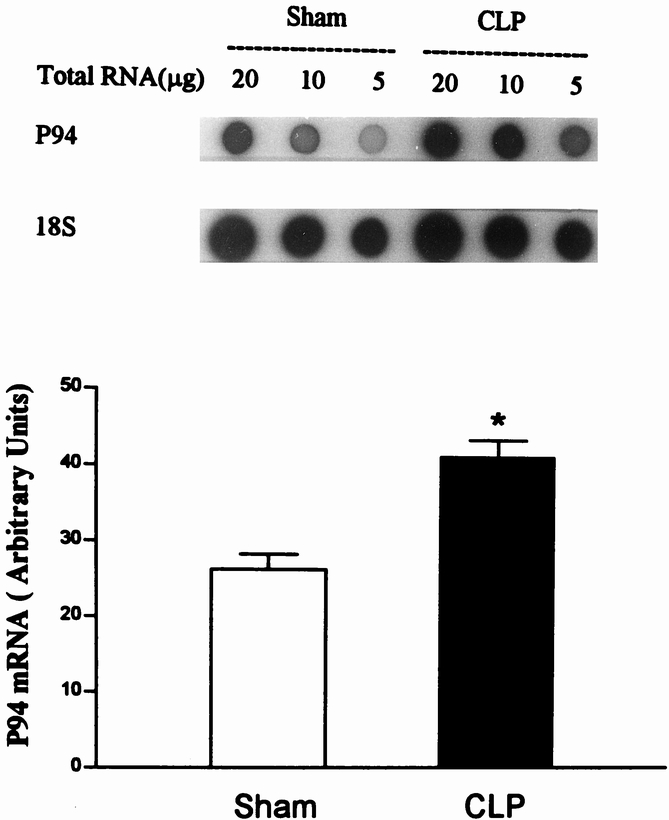
Figure 8. Sepsis results in increased gene expression of the muscle-specific calpain p94. Dot blots of RNA extracted from control and septic muscles are shown in the upper panel and quantitation of the blots is shown in the lower panel. (From Williams AB, DeCourten-Myers GM, Fischer JE, et al. Sepsis stimulates release of myofilaments in skeletal muscle by a calcium-dependent mechanism. FASEB J 1999; 13:1435–1443, with permission.)
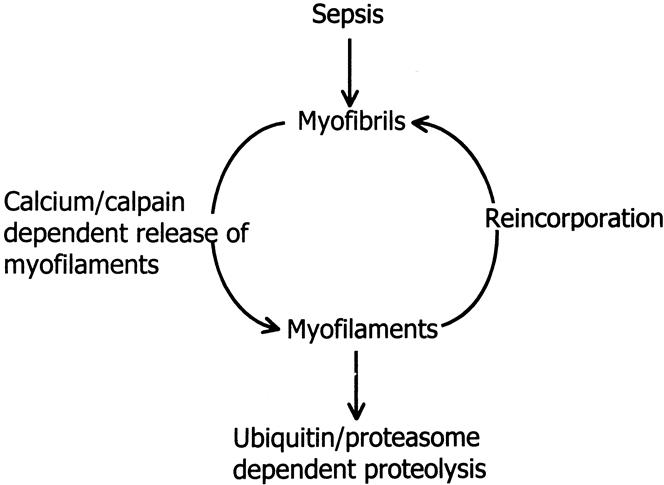
Figure 9. Model of sepsis-induced muscle cachexia. In this model, sepsis results in calcium/calpain-dependent release of myofilaments from the sarcomere. The myofilaments are ubiquitinated in the N-end rule pathway and degraded by the 26S proteasome or reincorporated into the myofibrils. This model offers two levels of possible therapeutic intervention: inhibition of the calcium/calpain-dependent release of myofilaments (e.g., with dantrolene) or inhibition of the ubiquitin-proteasome pathway (e.g.,with proteasome inhibitor).
The model proposed here suggests that increased expression and activity of the ubiquitin-proteasome pathway may be the response to an increased amount of available substrates (released and perhaps modified myofilaments) rather than the cause of muscle cachexia. Interestingly, in early studies, the role of the ubiquitin-proteasome pathway was believed to be breakdown of abnormal and potentially harmful proteins. 58 It may therefore be argued that inhibition of the proteasome in cachectic muscle may cause more harm than good. However, myofilaments released from the sarcomere can be reincorporated into the myofibril 59 (see Fig. 9), and it is possible that this process is enhanced if the proteasome-dependent breakdown of the myofilaments is inhibited. More studies need to be performed to define the relation between calpain-dependent release of myofilaments and ubiquitin-proteasome-dependent protein degradation and their roles in the development of muscle cachexia. Such studies will be extremely important from a clinical standpoint because they may help develop treatments targeting different levels of the proteolytic process in patients with muscle breakdown.
TRANSCRIPTION FACTORS AND SIGNALING PATHWAYS
Because the catabolic response in skeletal muscle is associated with altered expression of different genes, most notably the genes for ubiquitin, 10,11 several of the proteasome subunits, 12 enzymes involved in the ubiquitination of proteins, 28,29 and calpains, 56 it is likely that different transcription factors play an important role in the development of muscle cachexia. Little is known about the influence of catabolic signals on transcription factors in muscle tissue. In a previous study, treatment of cultured muscle cells with tumor necrosis factor-α resulted in activation of the “inflammatory” transcription factor NF-κB. 60 In recent experiments in our laboratory, the expression and activity of the cytosolic glucocorticoid receptor (which acts as a transcription factor after hormone binding) were increased in cachectic muscle from septic rats. 61 In other studies, we have found evidence that DNA binding activity of NF-κB, AP-1, and C/EBP (NF-IL6) may be upregulated in septic muscle. 62
Mediators that have been implicated in the development of muscle cachexia include tumor necrosis factor-α, 63 interleukin-1, 64 glucocorticoids, 11 and a cachectic factor isolated from patients with cancer. 65 Among these factors, glucocorticoids are probably the most important signal substance, and the catabolic effects of some of the other agents, in particular tumor necrosis factor-α, are mediated by glucocorticoids. 66 It is not known which signaling pathways or transcription factors are activated by the various cachectic mediators. It is possible that multiple transcription factors are activated in catabolic muscle and that an interaction between them is important for the metabolic response. Evidence for cross-talk between transcription factors and between transcription factors and other proteins exists in other cell types 67 and may be important in muscle cells as well. Indeed, recent observations suggest that an interaction between the activated glucocorticoid receptor and AP-1 may regulate myofibrillar protein breakdown in muscle cells. 68
It is likely that more knowledge regarding transcription factors and signaling pathways in cachectic muscle will be generated in the near future, and such knowledge will enhance our ability to prevent or treat muscle cachexia.
CONCLUSIONS
Muscle cachexia is a significant clinical characteristic of patients with severe injury, sepsis, and cancer. The catabolic response in skeletal muscle, mainly reflecting the breakdown of myofibrillar proteins, can give rise to muscle wasting and fatigue that may delay or complicate the recovery in patients with injury and sepsis and contribute to the rate of death and complications in patients with cancer. An understanding of the mechanisms regulating muscle breakdown in these conditions, therefore, has important clinical implications. Studies performed during the past decade have provided insight into the cellular and molecular mechanisms leading to muscle cachexia. Protein degradation in cachectic muscle is regulated by multiple mechanisms and is associated with increased gene expression for several proteolytic pathways, including the calcium-dependent calpain system and the ubiquitin-proteasome pathway. Recent studies in experimental animals suggest that it is possible to reduce the catabolic response in skeletal muscle by interfering with the specific molecular mechanisms underlying increased muscle proteolysis. Such observations are important for the development of therapeutic strategies aimed at reducing the catabolic response in patients with muscle cachexia.
Footnotes
Supported in part by NIH grant DK37908 and by grants from the Shriners of North America, Tampa, Florida.
Correspondence: Per-Olof Hasselgren, MD, Department of Surgery, University of Cincinnati, 231 Bethesda Ave., ML 0558, Cincinnati, OH 45267-0558. E-mail: hasselp@uc.edu
Accepted for publication July 18, 2000.
References
- 1.Rosenblatt S, Clowes GHA, George BC,et al. Exchange of amino acids by muscle and liver in sepsis. Comparative studies in vivo and in vitro. Arch Surg 1983; 118: 167–175. [DOI] [PubMed] [Google Scholar]
- 2.Windmueller HG, Spaeth AE. Respiratory fuels and nitrogen metabolism in vivo in small intestine of fed rats. J Biol Chem 1980; 255: 107–112. [PubMed] [Google Scholar]
- 3.Newsholme EA, Parry-Billings M. Properties of glutamine release from muscle and its importance for the immune system. J Parenteral Enteral Nutr 1990; 14: 63S–67S. [DOI] [PubMed] [Google Scholar]
- 4.Reid WD, MacGowan NA. Respiratory muscle injury in animal models and humans. Mol Cell Biochem 1998; 179: 63–80. [DOI] [PubMed] [Google Scholar]
- 5.Andreyev HJN, Norman AR, Oates J, Cunningham D. Why do patients with weight loss have a worse outcome when undergoing chemotherapy for gastrointestinal malignancies? Eur J Cancer 1998; 34: 503–509. [DOI] [PubMed] [Google Scholar]
- 6.Warren S. The immediate cause of death in cancer. Am J Med Sci 1932; 184: 610–613. [Google Scholar]
- 7.Hasselgren PO, James JH, Benson DW,et al. Total and myofibrillar protein breakdown in different types of rat skeletal muscle: effects of sepsis and regulation by insulin. Metabolism 1989; 38: 634–640. [DOI] [PubMed] [Google Scholar]
- 8.Long CL, Birkhahn RH, Geiger JW,et al. Urinary excretion of 3-methylhistidine: an assessment of muscle protein catabolism in adult normal subjects and during malnutrition, sepsis and skeletal trauma. Metabolism 1981; 30: 765–776. [DOI] [PubMed] [Google Scholar]
- 9.Attaix D, Taillandier D. The critical role of the ubiquitin-proteasome pathway in muscle wasting in comparison to lysosomal and Ca2+-dependent systems. Adv Mol Cell Biol 1998; 27: 235–266. [Google Scholar]
- 10.Tiao G, Fagan JM, Samuels N,et al. Sepsis stimulates nonlysosomal, energy-dependent proteolysis and increases ubiquitin mRNA levels in rat skeletal muscle. J Clin Invest 1994; 94: 2255–2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tiao G, Fagan JM, Roegner V,et al. Energy-ubiquitin-dependent muscle proteolysis during sepsis in rats is regulated by glucocorticoids. J Clin Invest 1996; 97: 339–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tiao G, Hobler S, Wang JJ,et al. Sepsis is associated with increased mRNAs of the ubiquitin-proteasome proteolytic pathway in human skeletal muscle. J Clin Invest 1997; 99: 163–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fang CH, Tiao G, James JH,et al. Burn injury stimulates multiple proteolytic pathways in skeletal muscle including the ubiquitin-energy-dependent pathway. J Am Coll Surg 1995; 180: 161–170. [PubMed] [Google Scholar]
- 14.Fang CH, Li BG, Tiao G,et al. The molecular regulation of protein breakdown following burn injury is different in fast- and slow-twitch skeletal muscle. Int J Mol Med 1998; 1: 163–169. [DOI] [PubMed] [Google Scholar]
- 15.Mitch WE, Medina R, Grieber S,et al. Metabolic acidosis stimulates muscle protein degradation by activating the adenosine triphosphate-dependent pathway involving ubiquitin and proteasomes. J Clin Invest 1994; 93: 2127–2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baracos VE, DeVivo C, Hoyle DHR, Goldberg AL. Activation of the ATP-ubiquitin-proteasome pathway in skeletal muscle of cachectic rats bearing a hepatoma. Am J Physiol 1995; 268: E996–E1006. [DOI] [PubMed] [Google Scholar]
- 17.Williams A, Sun X, Fischer JE, Hasselgren PO. The expression of genes in the ubiquitin-proteasome proteolytic pathway is increased in skeletal muscle from patients with cancer. Surgery 1999; 126: 744–750. [PubMed] [Google Scholar]
- 18.Hasselgren PO, Fischer JE. The ubiquitin-proteasome pathway. Review of a novel intracellular mechanism of muscle protein breakdown during sepsis and other catabolic conditions. Ann Surg 1997; 225: 307–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hasselgren PO. Role of the ubiquitin-proteasome pathway in sepsis-induced muscle catabolism. Mol Biol Rep 1999; 26: 71–76. [DOI] [PubMed] [Google Scholar]
- 20.Hasselgren PO. Pathways of muscle protein breakdown in injury and sepsis. Curr Opin Clin Nutr Metab Care 1999; 2: 155–160. [DOI] [PubMed] [Google Scholar]
- 21.Tanaka K. Molecular biology of proteasomes. Mol Biol Rep 1995; 21: 21–26. [DOI] [PubMed] [Google Scholar]
- 22.Weissman JS, Sigler PB, Horwich AL. From the cradle to the grave: ring complexes in the life of a protein. Science 1995; 268: 523–524. [DOI] [PubMed] [Google Scholar]
- 23.Rechsteiner M. The 26S proteasome. In: Peters JM, Harris JR, Finley D,eds. Ubiquitin and the biology of the cell. New York: Plenum Press; 1998: 147–189.
- 24.Hershko A. Lessons from the discovery of the ubiquitin system. Trends Biochem Sci 1996; 21: 445–449. [DOI] [PubMed] [Google Scholar]
- 25.Wing SS, Banville D. 14 kDa ubiquitin-conjugating enzyme: structure of the rat gene and regulation upon fasting and by insulin. Am J Physiol 1994; 267: E39–E48. [DOI] [PubMed] [Google Scholar]
- 26.Wing SS, Bedard N. Insulin-like growth factor I stimulates degradation of an mRNA transcript encoding the 14 kDa ubiquitin-conjugating enzyme. Biochem J 1996; 319: 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kwon YT, Reiss Y, Fried VA,et al. The mouse and human genes encoding the recognition component of the N-end rule pathway. Proc Natl Acad Sci USA 1998; 95: 7898–7903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hobler S, Wang JJ, Williams AB,et al. Sepsis is associated with increased ubiquitin conjugating enzyme E214k mRNA in skeletal muscle. Am J Physiol 1999; 276: R476–473. [DOI] [PubMed] [Google Scholar]
- 29.Fischer D, Sun X, Gang G, et al. The gene expression of ubiquitin ligase E3α is upregulated in skeletal muscle during sepsis in rats. Potential role of glucocorticoids. Biochem Biophys Res Commun 2000; 267:504–508. [DOI] [PubMed]
- 30.Solomon V, Baracos V, Sarraf P, Goldberg AL. Rates of ubiquitin conjugation increase when muscles atrophy, largely through activation of the N-end rule pathway. Proc Natl Acad Sci USA 1998; 95: 12602–12607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wilkinson KD, Hochstrasser M. The deubiquitinating enzymes. In: Peters JM, Harris JR, Finley D,eds. Ubiquitin and the biology of the cell. New York: Plenum Press; 1998: 99–125.
- 32.Varshavsky A. The N-end rule pathway of protein degradation. Genes to Cells 1997; 2: 13–28. [DOI] [PubMed] [Google Scholar]
- 33.Ciechanover A. The ubiquitin-proteasome proteolytic pathway. Cell 1994; 79: 13–21. [DOI] [PubMed] [Google Scholar]
- 34.Solomon V, Lecker SH, Goldberg AL. The N-end rule pathway catalyzes a major fraction of the protein degradation in skeletal muscle. J Biol Chem 1998; 273: 25216–25222. [DOI] [PubMed] [Google Scholar]
- 35.Blumenfeld N, Gonen H, Mayer A,et al. Purification and characterization of a novel species of ubiquitin-carrier protein, E2, that is involved in degradation of non-“N-end rule” protein substrates. J Biol Chem 1994; 269: 9574–9581. [PubMed] [Google Scholar]
- 36.Gonen H, Stancovski I, Shkedy D,et al. Isolation, characterization, and partial purification of a novel ubiquitin-protein ligase, E3. Targeting of protein substrates via multiple and distinct recognition signals and conjugating enzymes. J Biol Chem 1996; 271: 302–310. [DOI] [PubMed] [Google Scholar]
- 37.Kisselev F, Akopian TN, Castillo V, Goldberg AL. Proteasome active sites allosterically regulate each other, suggesting a cyclical bite-chew mechanism for protein breakdown. Mol Cell 1999; 4: 395–402. [DOI] [PubMed] [Google Scholar]
- 38.Hobler SC, Williams AB, Fischer D,et al. Activity and expression of the 20S proteasome are increased in skeletal muscle during sepsis. Am J Physiol 1999; 277: R434–R440. [DOI] [PubMed] [Google Scholar]
- 39.Hobler SC, Tiao G, Fischer JE,et al. The sepsis-induced increase in muscle proteolysis is blocked by specific proteasome inhibitors. Am J Physiol 1998; 274: R30–R37. [DOI] [PubMed] [Google Scholar]
- 40.Fang CH, Li BG, Fischer DR,et al. Burn injury upregulate the activity and gene expression of the 20S proteasome in rat skeletal muscle. Clin Sci 2000; 99: 181–187. [PubMed] [Google Scholar]
- 41.Price SR, England BK, Bailey JL,et al. Acidosis and glucocorticoids concomitantly increase ubiquitin and proteasome subunit mRNAs in rat muscle. Am J Physiol 1994; 267: C955–C960. [DOI] [PubMed] [Google Scholar]
- 42.Lee DH, Goldberg AL. Proteasome inhibitors: valuable new tools for cell biologists. Trends Cell Biol 1998; 8: 397–403. [DOI] [PubMed] [Google Scholar]
- 43.Fang CH, Wang JJ, Hobler S,et al. Proteasome blockers inhibit protein breakdown in skeletal muscle after burn injury in rats. Clin Sci 1998; 95: 225–233. [PubMed] [Google Scholar]
- 44.Tawa NE, Odessey R, Goldberg AL. Inhibitors of the proteasome reduce the accelerated proteolysis in atrophying rat skeletal muscles. J Clin Invest 1997; 100: 197-203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Campbell B, Adams J, Shin YK, Lefer AM. Cardioprotective effects of a novel proteasome inhibitor following ischemia and reperfusion in the isolated perfused rat heart. J Mol Cell Cardiol 1999; 31: 467–476. [DOI] [PubMed] [Google Scholar]
- 46.Adams J, Palombella VJ, Sansville EA,et al. Proteasome inhibitors: a novel class of potent and effective antitumor agents. Cancer Res 1999; 59: 2615–2622. [PubMed] [Google Scholar]
- 47.Fischer DR, Gang G, Pritts T, Hasselgren PO. Sepsis-induced muscle proteolysis is prevented by a proteasome inhibitor in vivo. Biochem Biophys Res Commun 2000; 270: 215–221. [DOI] [PubMed] [Google Scholar]
- 48.Mansoor O, Beaufrere B, Boirie Y,et al. Increased mRNA levels for components of the lysosomal, calcium-activated and ATP-ubiquitin-dependent proteolytic pathways in skeletal muscle from head trauma patients. Proc Natl Acad Sci USA 1996; 93: 2714–2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Combaret L, Taillandier D, Voisin L,et al. No alteration in gene expression of components of the ubiquitin-proteasome proteolytic pathway in dystrophin-deficient muscle. FEBS Lett 1996; 393: 292–296. [DOI] [PubMed] [Google Scholar]
- 50.Ralliere C, Tauveron I, Taillandier D,et al. Glucocorticoids do not regulate the expression of proteolytic genes in skeletal muscle from Cushing syndrome patients. J Clin Endocrinol Metab 1997; 82: 3161–3164. [DOI] [PubMed] [Google Scholar]
- 51.Llovera M, Garcia-Martinez C, Agell N,et al. Ubiquitin and proteasome gene expression is increased in skeletal muscle of slim AIDS patients. Int J Mol Med 1998; 2: 69–73. [PubMed] [Google Scholar]
- 52.Thompson HS, Scordilis SP. Ubiquitin changes in human biceps muscles following exercise-induced damage. Biochem Biophys Res Commun 1994; 204: 1193–1198. [DOI] [PubMed] [Google Scholar]
- 53.Koohmaraie M. Ovine skeletal muscle multicatalytic proteinase complex (proteasome): purification, characterization and comparison of its effects on myofibrils with μ-calpains. J Anim Sci 1992; 70: 3697–3708. [DOI] [PubMed] [Google Scholar]
- 54.Solomon V, Goldberg AL. Importance of the ATP-ubiquitin-proteasome pathway in the degradation of soluble and myofibrillar proteins in rabbit muscle extracts. J Biol Chem 1996; 271: 26690–26697. [DOI] [PubMed] [Google Scholar]
- 55.Squire JM. Architecture and function in the muscle sarcomere. Curr Opin Struct Biol 1997; 7: 247–257. [DOI] [PubMed] [Google Scholar]
- 56.Williams AB, DeCourten-Myers GM, Fischer JE,et al. Sepsis stimulates release of myofilaments in skeletal muscle by a calcium-dependent mechanism. FASEB J 1999; 13: 1435–1443. [DOI] [PubMed] [Google Scholar]
- 57.Hotchkiss RS, Osborne DF, Lappas GD, Karl IE. Calcium antagonists decrease plasma and tissue concentrations of tumor necrosis factor-α, interleukin-1B, and interleukin-1α in a mouse model of endotoxin. Shock 1995; 3: 337–342. [PubMed] [Google Scholar]
- 58.Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem 1998; 67: 425–479. [DOI] [PubMed] [Google Scholar]
- 59.Van der Westhuyzen DR, Matsumoto K, Etlinger JD. Easily releasable myofilaments from skeletal and cardiac muscles maintained in vitro. Role in myofibrillar assembly and turnover. J Biol Chem 1981; 256: 11791–11797. [PubMed] [Google Scholar]
- 60.Sen CK, Khanna S, Resznick AZ,et al. Glutathione regulation of tumor necrosis factor α-induced NF-κB activation in skeletal muscle-derived L6 cells. Biochem Biophys Res Commun 1997; 237: 645–649. [DOI] [PubMed] [Google Scholar]
- 61.Fischer DR, Sun X, Pritts TA, Hasselgren PO. The amount of the glucocorticoid receptor (GR) and its hormone binding activity are increased in skeletal muscle during sepsis. Surg Forum 1999; 50: 232–234. [Google Scholar]
- 62.Gang G, Penner CG, Fischer DR, Hasselgren PO. Sepsis upregulates the DNA binding activity of the inflammatory transcription factors NF-κB, C/EBP and AP-1 in rat skeletal muscle. Surg Forum 2000; 51: 214–216. [Google Scholar]
- 63.Zamir O, Hasselgren PO, Kunkel SL,et al. Evidence that tumor necrosis factor participates in the regulation of muscle proteolysis during sepsis. Arch Surg 1992; 127: 170–174. [DOI] [PubMed] [Google Scholar]
- 64.Zamir O, O’Brien W, Thompson R,et al. Reduced muscle protein breakdown in septic rats following treatment with interleukin-1 receptor antagonist. Int J Biochem 1994; 26: 943–950. [DOI] [PubMed] [Google Scholar]
- 65.Lorite MJ, Cariuk P, Tisdale MJ. Induction of muscle protein degradation by a tumour factor. Br J Cancer 1997; 76: 1035–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zamir O, Hasselgren PO, Higashiguchi T,et al. Tumor necrosis factor (TNF) and interleukin-1 (IL-1) induce muscle proteolysis through different mechanisms. Med Inflam 1992; 1: 247–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Vig E, Barrett TJ, Vedeckis WV. Coordinate regulation of glucocorticoid receptor and c-jun mRNA levles: evidence for cross-talk between two signaling pathways at the transcriptional level. Mol Endocrinol 1994; 8: 1336–1346. [DOI] [PubMed] [Google Scholar]
- 68.Thompson JG, Palmer RM. Signaling pathways regulating protein turnover in skeletal muscle. Cell Signal 1998; 10: 1–11. [DOI] [PubMed] [Google Scholar]