

Abstract
Objective
To determine whether blocking the cell surface expression of intracellular adhesion molecules (ICAM-1) in established severe acute pancreatitis (AP) would ameliorate pulmonary injury.
Summary Background Data
Lung injury in AP is in part mediated by infiltrating leukocytes, which are directed to lung tissue by ICAM-l. The authors’ laboratory has previously demonstrated that AP results in overproduction of inflammatory cytokines, upregulation of pulmonary ICAM-1 expression, and a concomitant infiltration of neutrophils, which results in lung injury.
Methods
Young female mice were fed a choline-deficient/ethionine-supplemented diet to induce AP and were treated with a blocking dose of monoclonal antibody specific to the ICAM-1 receptor. Antibody treatment was administered at 72, 96, and 120 hours after beginning the diet, and all animals were killed at 144 hours. The degree of pancreatitis was evaluated by serum biochemical and tumor necrosis factor α levels as well as histology. The dual radiolabeled monoclonal antibody method was used to quantitate ICAM-1 cell surface expression in pulmonary tissue. Lung injury was assessed histologically and by determining lung microvascular permeability by measuring accumulated 125I-radiolabeled albumin. Pulmonary neutrophil sequestration was determined by the myeloperoxidase assay.
Results
All mice developed severe AP, and pancreatic injury was equally severe in both treated and untreated groups. Pulmonary ICAM-1 expression was significantly upregulated in animals with AP compared with controls. Treatment with a blocking dose of anti-ICAM-1 antibody after the induction of AP resulted in inhibited ICAM-1 cell surface expression to near control levels. Compared to untreated animals with AP, mice treated with anti-ICAM-1 mice had significantly reduced histologic lung injury and neutrophil sequestration, and a decreased microvascular permeability by more than twofold.
Conclusions
These results demonstrate for the first time that treatment targeting the cell surface expression of ICAM-1 after the induction of AP ameliorates pulmonary injury, even in the face of severe pancreatic disease.
The high rates of death and complications associated with severe acute pancreatitis (AP) stem from an overwhelming inflammatory response that can cause injury to distant organs such as lung tissue. 1,2 Patients with severe AP develop pulmonary dysfunction, which clinically resembles the acute respiratory distress syndrome of sepsis. 3 Despite advances in the diagnosis and treatment of inflammatory pancreatic disease, supportive care remains the only treatment for patients with pulmonary complications. 4,5
The role of adhesion molecules in the development of AP-associated organ injury has been verified in several experimental models using mice that are genetically deficient of selectin or intracellular adhesion molecules (ICAM-1), as well as in models where these molecules are blocked with specific monoclonal antibodies (mAbs). For example, Werner et al 6 highlighted the potential for adhesion molecule inhibitors in pancreatitis-associated lung injury. Frossard et al 7 showed that inhibiting neutrophil action reduces the severity of the lung injury that occurs in mice with AP. They also showed that AP is less severe when induced in mice deficient in ICAM-1 compared with their normal counterparts. Together, these results implicate both the neutrophil and the ICAM-1 receptor as significant mediators of local and distant injury in AP. 7–9
Starting with the discovery of an overproduction of inflammatory cytokines in AP, our laboratory has demonstrated that systemic manifestations of AP can be ameliorated by cytokine blockade given before 10,11 or immediately after the onset of experimental AP. However, because of our interest in devising clinically relevant therapies, our focus has shifted to more downstream events in AP. Despite their utility in experimental models, therapies such as antiproteases, somatostatin, and antiplatelet activating factor administered late in the disease course have little effect on clinical outcome. 12 Similarly, anticytokine treatment given later in the disease course appears to be of little benefit. 13 One of the major effects by which cytokine upregulation mediates distant injury is by the adhesion molecule overexpression on endothelial cells. Specifically, adhesion molecules mediate leukocyte activation and can instigate lung injury. 7,14 We reasoned that intervening in the events downstream to cytokine activation might afford effective tools for clinical disease management.
In the current study, we used a novel approach to demonstrate the significance of adhesion molecule blockade in ameliorating the lung manifestations associated with AP. Specifically, we evaluated lung injury in mice treated with anti-ICAM-1 mAb several days after the onset of AP. So far, other investigators have reported that blocking ICAM-1 expression before the onset of AP diminishes the severity of the disease, 6 thus confirming the role of ICAM-1-mediated organ injury. However, the clinical relevance of anti-ICAM-1 treatment after the onset of pancreatitis remained questionable until now. This work is the first demonstration of a delayed treatment during AP that ameliorates pancreatitis-associated pulmonary injury without affecting the inflammatory process seen in the pancreas.
METHODS
Animals and Diet
Young female Swiss-Webster mice were purchased from Harlan Laboratory (Madison, WI). A choline-deficient/ethionine-supplemented (CDE) diet (Harlan Teklad, Madison, WI) in powder form was fed to the fasted animals ad libitum. Care of the animals was in accordance with NIH standards published in the “Guide for Care and Use of Laboratory Animals” (NIH 85–23, 1985). The CDE diet used in these experiments was modeled after that described by Lombardi et al. 15,16
Antibodies
Purified endotoxin-free mAb against mouse CD54 (ICAM-1) clone 3E2B was purchased from Endogen (Woburn, MA) and used for all blocking experiments and for determining ICAM-1 expression in lung tissue. A blocking dose of 2 mg/kg was given intraperitoneally. The nonbinding (isotype) antibody clone A19 to 3 (Pharmingen, San Diego, CA) was used in conjunction with the binding antibody to determine ICAM-1 expression quantitatively. As previously described, we used the iodogen method 17 to radiolabel the binding and nonbinding mAbs with 125I and 131I, respectively (DuPont NEN, Boston, MA). The albumin was radiolabeled with 125I (DuPont NEN).
Experimental Protocol and Measurements
In experiment 1, the dual radiolabeled mAb technique was used in mice (n = 15) to determine the quantitative expression of pulmonary ICAM-1 at 144 hours after induction of AP with the CDE diet. Experiment 2 was designed to determine whether a blocking dose of anti-ICAM-1 mAb would ameliorate AP-associated lung injury. Animal groups in experiment 2 were normal controls (n = 10), mice with AP (n = 29), and AP mice treated with intraperitoneal injections of anti-ICAM-1 mAb at 72, 96, and 120 hours after initiating the CDE diet (n = 14) (Fig. 1). To prove that the reduced pulmonary injury observed in AP-treated animals was not caused by reduced pancreatic injury associated with anti-ICAM-1 antibody treatment, biochemical markers (amylase and glucose), serum tumor necrosis factor α (TNFα), and pancreatic morphologic evaluation was performed at 144 hours in both treated and untreated groups, which ensured that pancreatitis was equally severe. Similarly, at this time point (144 hours), lung tissue of the animals was evaluated for myeloperoxidase activity, microvascular permeability, and histology, as detailed below. In addition, to prove that severe pancreatic injury occurred before anti-ICAM-1 antibody treatment, a separate group of animals (n = 5) was killed at 72 hours and sections of pancreatic tissue stained with hematoxylin and eosin were examined for morphologic changes associated with AP.
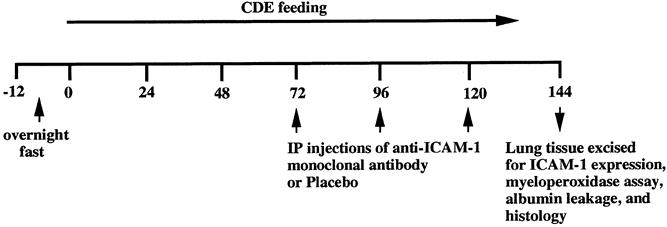
Figure 1. Experimental design of ICAM-1 blocking study. The line represents the time course of the experiment in hours. CDE diet feeding to mice started at 0 hours, and the anti-ICAM-1 blocking antibody was administered intraperitoneally at 72 hours after feeding and given at 96 and 120 hours as well. Lung ICAM-1 measurement and injury assessment were performed at 144 hours.
The procedure for determining ICAM-1 expression has been previously described. 18 For measuring ICAM-1 expression, 10 μg 125I-radiolabeled ICAM-1 and 40 μg cold ICAM-1 were used with 400,000 to 600,000 counts per minute of the nonbinding mAb labeled with 131I. The method used to calculate the expression of ICAM-1 has also been described previously. 19 Calculation of expression in terms of μg mAb per gram dry tissue can be obtained. The equation used to calculate the ICAM-1 and expression was as follows: ICAM-1 expression (μg mAb per gram tissue) = (125I cpm/g/125I cpm injected) minus (131I cpm/g/131I cpm injected) × total injected binding mAb (μg)/100.
Thirty minutes before the mice were killed, anesthesia was administered and the jugular vein and carotid artery were cannulated. One milligram of albumin 125I was administered intravenously and allowed to circulate for 30 minutes, after which time the animal was exsanguinated. Lung tissue was removed and weighed before being placed in a gamma counter, along with the blood sample. The activity of lung albumin 125I to blood 125I was calculated and reported as mean value ± standard error. This measurement was used as a marker of microvascular permeability.
Myeloperoxidase extraction was performed by methods previously described. 20 Frozen tissue was thawed, weighed, and placed in 20 mmol/L potassium phosphate buffer (pH 7.4), which was then homogenized and centrifuged. The pellet was resuspended and sonicated in 50 mmol/L phosphate buffer containing 0.5% hexadecylmethylammonium bromide (HETAB) purchased from Sigma (St. Louis, MO). Using a mixture of water, HETAB, tetramethylbenzidine (TMB), sodium acetate buffer, and H2O2, we assayed the supernatants of the samples for myeloperoxidase activity. Activity units (AU) per gram tissue were calculated.
When the animals were killed, pancreatic tissue was removed and placed in a formalin fixative until paraffin embedding was performed. After this step, 4-μm paraffin sections were cut and stained with hematoxylin and eosin. Histologic sections of the pancreas were evaluated by a pathologist who was unaware of the assignment of the animals to demonstrate the presence or absence of pancreatic injury.
For histologic evaluation of lung tissue, the trachea of the animals were cannulated with an 18-gauge angiocatheter and a 5% formalin solution was flushed into the bronchial system under slow, constant pressure to ensure homogeneous lung distention. Once fully inflated, lung tissue was excised and placed into a formalin solution until paraffin embedding and staining with hematoxylin and eosin were performed. At this time, 4-μm sections were cut and stained and subsequently examined by a pathologist who was unaware of the assignment of the animals. Lung sections were examined for the magnitude of alveolar collapse, intravascular congestion, and alveolar hemorrhage. Five regions from each specimen were examined, and an injury score of 0 to 3 (0, normal; 1, mild; 2, moderate; 3, severe) was assigned to each of the three parameters and then added to obtain a total score of 0 to 9.
Amylase was measured by a quantitative enzymatic assay (Sigma, No. 575-UV) and results are expressed as International Units per liter (U/L). Serum glucose levels were measured using the Accu-check 111 kit (Boehringer Mannheim Biochemica, Indianapolis, IN) and reported in mg/dL. Serum samples were measured for TNFα by a standard enzyme-linked immunoassay method, 21 and levels are reported as pg/mL ± standard error.
Analysis
Results are expressed as mean ± standard error of the mean. Data for response variables were analyzed using the Student unpaired t test when two groups were compared. Analysis of variance and the Fisher protected least significant difference post hoc tests were used to determine the significance between more than two groups. Statistical significance was set at P < .05. Significance of nonparametric data including histologic analysis was evaluated by the Mann-Whitney test. Significance was set at P < .05.
RESULTS
Normal mice had no histopathologic changes in the pancreas (Fig. 2). The animals fed the CDE diet and killed at 72 hours had pancreatic morphologic changes associated with severe pancreatitis (results not shown). Both AP-untreated and AP-treated groups that were fed the CDE diet for 144 hours developed similar morphologic changes in the pancreas associated with AP, which included acinar cell vacuolization, cellular necrosis, pyknosis, and intralobular hemorrhage. Similarly, there were no significant differences in the serum amylase, glucose, and TNFα levels obtained from both groups (AP-treated and AP-untreated) (Table 1).
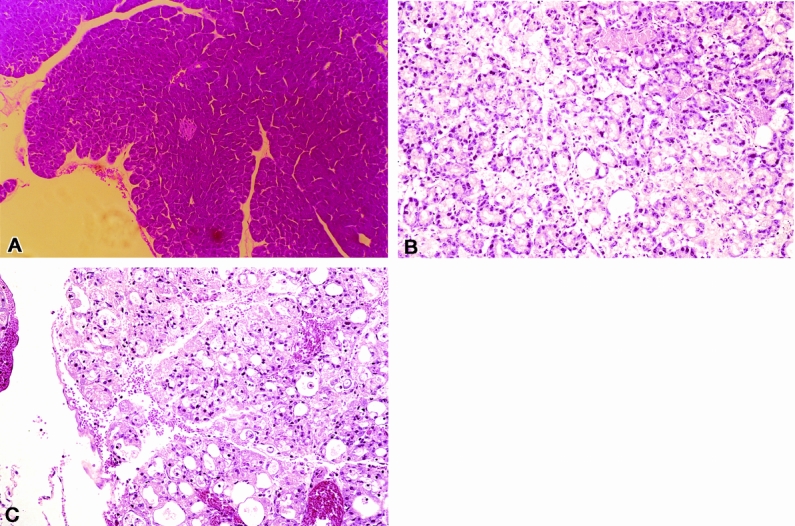
Figure 2. Morphologic determination of CDE diet-induced pancreatitis. (A) Normal pancreatic tissue from a mouse fed a normal diet. (B, C) Histopathologic changes in pancreatic tissue from untreated mice and mice treated with anti-ICAM-1 that had been fed the CDE diet for 144 hours. This was performed to ensure that both groups developed equally severe disease. Both groups had severe pancreatic injury, including acinar cell death, vacuolization, and inflammatory cell infiltration throughout the gland.
Table 1. AMYLASE, GLUCOSE, AND TNFα in PANCREATITIS TREATED AND UNTREATED ANIMALS
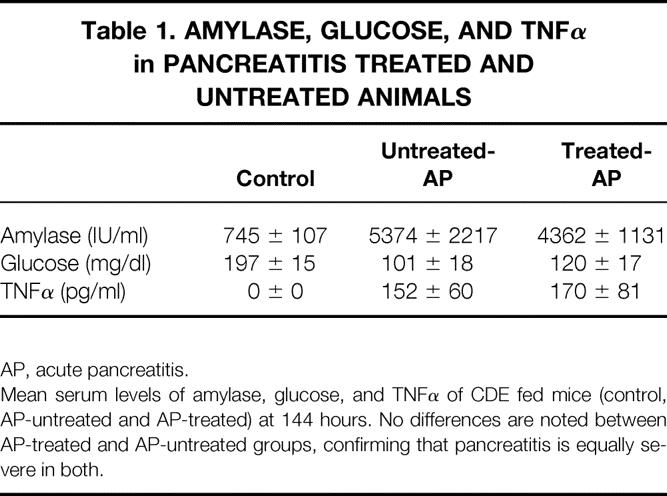
AP, acute pancreatitis.
Mean serum levels of amylase, glucose, and TNFα of CDE fed mice (control, AP-untreated and AP-treated) at 144 hours. No differences are noted between AP-treated and AP-untreated groups, confirming that pancreatitis is equally severe in both.
Cell surface pulmonary ICAM-1 expression was upregulated in AP-untreated mice compared with normal control animals at 144 hours (122.2 ± 30.4 vs. 17.3 ± 7.3 μg mAb/g;P < .01) (Fig. 3). By contrast, the levels of ICAM-1 expression in the lung tissue of anti-ICAM-1-treated animals with AP were not significantly different from those found in the lung tissue of control mice without AP (33.2 ± 18.2 vs. 17.3 ± 7.3 μg mAb/g;P = .68).
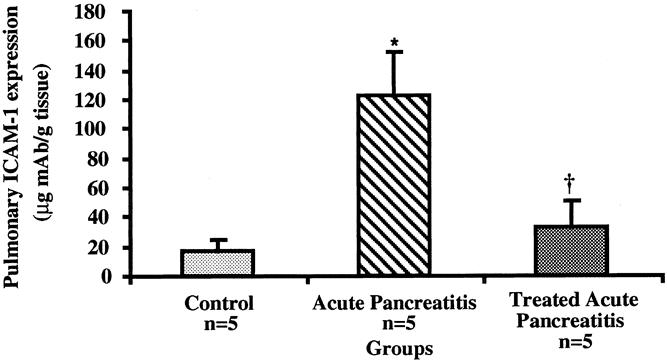
Figure 3. Determination of pulmonary ICAM-1 expression in CDE-induced pancreatitis at 144 hours. Mice with acute pancreatitis had elevated levels of endothelial cell ICAM-1 expression vs. control normal animals as measured by the dual radiolabeled monoclonal antibody technique (*;P < .01). Similarly, mice treated with ICAM-1 antibody had blocked cell surface ICAM-1 receptors compared with mice with acute pancreatitis who did not receive treatment (†;P < .02). All values are given as mean values ± standard error of the mean.
Measurement of neutrophil accumulation by the myeloperoxidase assay revealed that AP-untreated mice had an increase of myeloperoxidase activity almost fourfold greater than normal control animals (23.8 ± 2.7 vs. 6.4 ± 1.5 AU/g tissue;P < .002) (Fig. 4). Anti-ICAM-1-treated animals with AP, however, showed markedly lower levels of neutrophil accumulation in pulmonary tissue at 144 hours compared with untreated mice with AP at the same time point (13.9 ± 1.3 vs. 23.8 ± 2.7 AU/g tissue;P < .01). Further, there was no significant difference in myeloperoxidase activity between anti-ICAM-1-treated animals with established AP and normal control mice (P = .19).
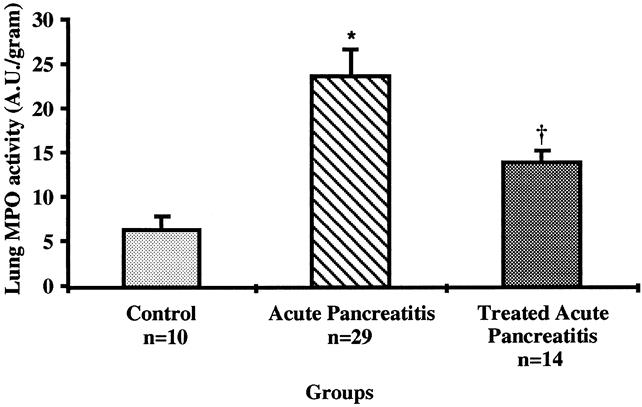
Figure 4. Determination of neutrophil sequestration in lung tissue by the myeloperoxidase (MPO) enzyme assay at 144 hours. MPO activity was significantly elevated in mice with acute pancreatitis vs. control normal mice (*;P < .002) and was lower in animals treated with anti-ICAM-1 compared with those who did not receive therapy (†;P < .01). All values are reported as mean values ± standard error of the mean.
Untreated CDE-fed mice had a significant increase in microvascular permeability compared with normal controls, as measured by extravasated radiolabeled albumin into pulmonary tissue (1.66 ± 0.42 vs. 0.22 ± 0.03 lung albumin 125I/blood albumin 125I;P < .007) (Fig. 5). In contrast, ICAM-1-treated mice with AP had significantly less pulmonary albumin accumulation compared with AP-untreated mice (0.58 ± 0.16 vs. 1.66 ± 0.42 activity lung 125I/blood 125I;P < .04). Again, no difference in microvascular permeability between treated mice and normal control mice was seen (P = .49).
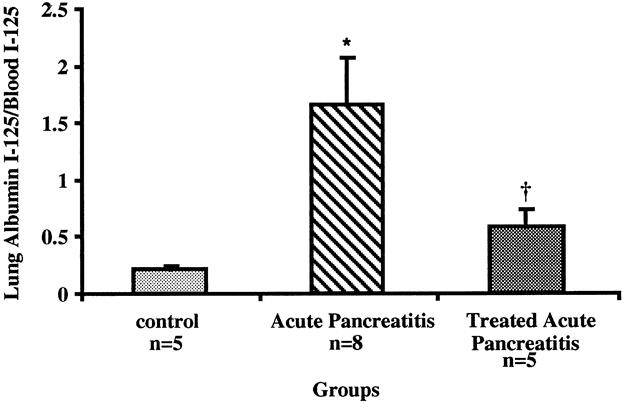
Figure 5. Determination of microvascular permeability in lung tissue at 144 hours. Animals with acute pancreatitis had more pulmonary microvascular permeability than normal controls (*;P < .007). Animals treated with anti-ICAM-1 had significantly less albumin leakage than untreated animals (†;P < .04).
Compared with normal control mice, which were shown to have normal lung morphology, animals in the AP-untreated group had changes associated with severe lung injury, including alveolar collapse, intraalveolar hemorrhage, and intravascular congestion (P < .003) (Fig. 6). However, the AP-treated group had fewer morphologic changes associated with lung injury than the AP-untreated group (P < .03). Normal mice who were fed standard laboratory chow had normal lung morphology; AP-untreated animals had severe lung injury compared with AP-treated animals (Fig. 7).
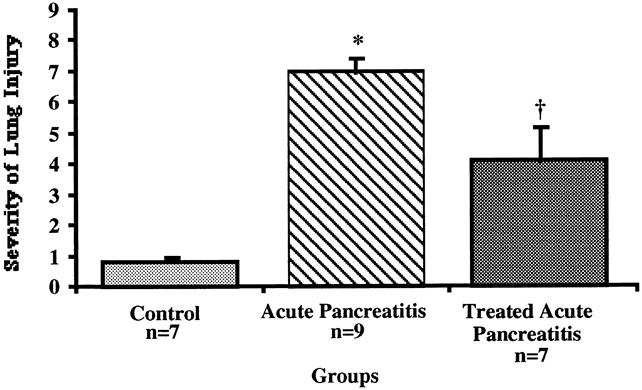
Figure 6. Severity of lung injury in untreated vs. treated animals. Significant morphologic changes occurred in the untreated group vs. the normal animal group (*;P < .003) and the group treated with ICAM-1 (†;P < .03).
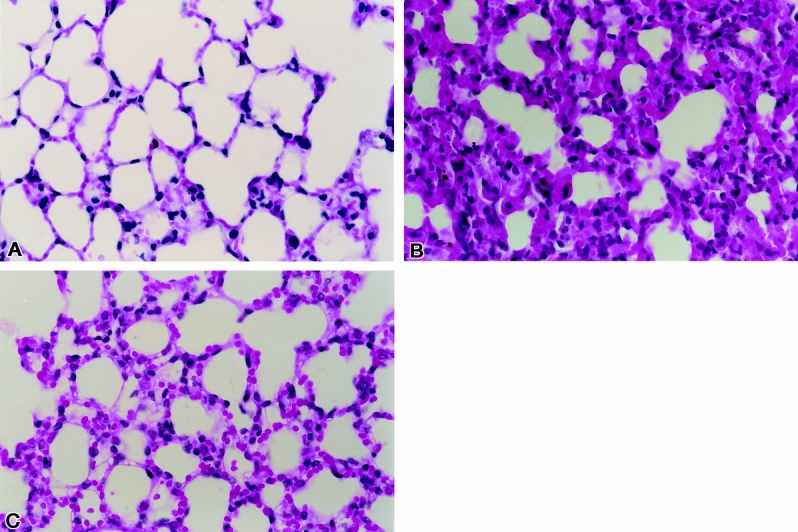
Figure 7. Pulmonary injury after diet-induced acute pancreatitis. (A) Section of lung tissue from normal mouse. (B) Section of lung tissue from a mouse with diet-induced acute pancreatitis at 144 hours (hematoxylin and eosin stain). (C) Representative lung tissue section from a mouse with diet-induced acute pancreatitis at 144 hours treated with anti-ICAM-1 antibody at 72, 96, and 120 hours.
DISCUSSION
Acute pancreatitis results in an overwhelming inflammatory response characterized by overproduction of cytokines such as TNFα, interleukin 1β, and interleukin 6, which are believed to mediate organ injury through the overexpression of adhesion molecules on endothelial cells. 22 These inflammatory cytokines elicit the migration of activated leukocytes in stimulated tissues. 23,24 Many studies have demonstrated the importance of these cytokines in AP by monitoring their expression during the disease. 12 Several groups, including our own, have demonstrated that anticytokine treatment administered before or immediately after the onset of AP ameliorated disease progression. 25 However, because of the narrow therapeutic window of time in which anticytokine treatment in humans can be given to ameliorate disease, 12,13 this mode of therapy might be difficult. Failure of late anticytokine intervention is presumably due to the fact that downstream inflammatory events secondary to cytokine stimulation are already in progress and are not affected by treatment administered late in the course of the disease. This situation has led us to examine the effects of intervention in these downstream events, with special emphasis on adhesion molecule upregulation and leukocyte migration. Until now, there have been no reports of an effective antiadhesion molecule intervention to treat AP after the onset of severe pancreatic injury.
Our goal was to initiate therapy with anti-ICAM-1 antibody after AP induction but before severe pulmonary injury occurred. Thus, the first time point for anti-ICAM-1 antibody therapy (72 hours) was chosen to coincide with established severe pancreatitis and the first appearance of pulmonary neutrophil infiltration, as determined in a previous study. 26 Treatment with blocking antibody was continued through the end of the experimental period. The dual mAb assay demonstrated that anti-ICAM-1 antibodies blocked ICAM-1 receptors on pulmonary endothelia. This in turn reduced neutrophil infiltration, as demonstrated by myeloperoxidase reduction, and subsequently caused amelioration of morphologic evidence of lung injury. Similarly, pulmonary endothelial cell damage, as measured by albumin leakage, was decreased in lung tissue after anti-ICAM-1 antibody treatment.
Recently, we examined the kinetics of upregulation of ICAM-1 expression in the pulmonary endothelia of animals with diet-induced AP. Pulmonary ICAM-1 expression was found to occur after 48 hours of ingestion of the CDE diet and correlated with the presence of pancreatic injury and increased levels of TNFα. 26 The upregulation of pulmonary ICAM-1 preceded a subsequent increase in neutrophil sequestration and progressive lung injury, which occurred at 72 hours after starting the CDE diet, thus strongly supporting the role of ICAM-1 expression as an early inciting event for lung injury in AP. 26 Other investigators have supported this finding by demonstrating that before AP induction, treatments directed against ICAM-1 upregulation cause inhibition of leukocyte migration into distant tissues, thus ameliorating organ injury occurring with AP. 7,27,28 Based on these findings, we used our data regarding the kinetics of ICAM-1 expression and the appearance of leukocyte infiltration to design a novel current therapy schedule.
Mice fed the CDE diet develop histologic changes in the pancreas consistent with AP. These changes include acinar swelling, cellular vacuolization, intralobular hemorrhage, cellular pyknosis, and necrosis, which are consistent with previous reports. 15 When the CDE diet is fed to young (3–4-week-old) female mice, the death rate approaches 100% by 96 hours. Manipulation of the feeding period and the age of the mice has reportedly decreased the death rate, but pancreatic injury is still present in these animals. 16,29 In our study, we used 6-week-old female mice, which decreased the death rate while maintaining severe pancreatic injury by 48 to 72 hours. This model was chosen because AP-associated pulmonary injury is a major component of the natural progression in this model. 26 This point is particularly important because it allowed us to study the effects of treatment on severe lung injury in a nonlethal, noninvasive form of AP.
We demonstrated that neutrophils are sequestrated in lung tissue in animals with AP. Myeloperoxidase is an enzyme that is extracted from neutrophils and has been used in many experimental studies to demonstrate the extent of cellular tissue infiltration. 20,30,31 In AP, leukocyte migration into lung tissue results in the appearance of reactive oxygen species and injury to vascular endothelial cells with resultant extravasation of proteins and fluid into the interstitial space. 32 The occurrence of pulmonary vascular endothelial injury was measured directly by the extent of albumin leakage (microvascular permeability) into the lung interstitium. These changes are related to the upregulation of ICAM-1 expression because albumin leakage is reduced after the administration of anti-ICAM-1 mAb.
Lung tissue from the AP-untreated animals showed changes that were severe: alveolar collapse, intraalveolar hemorrhage, and vascular congestion. This finding of lung injury occurs in the CDE model of experimental acute pancreatitis. 30,31 The severity of these findings in the lung and their reversal by anti-ICAM-1 antibody treatment suggest that changes that occur in distant organs, such as the lung, may be amenable to intervention after the onset of disease, even during a severe inflammatory event. An argument could be made that anti-ICAM-1 antibody treatment reduced pancreatic inflammation and resulted in a blunted systemic inflammatory response; hence, less pulmonary injury would occur. However, because there was no significant difference in biochemical and cytokine markers, and pancreatic morphologic findings were the same between the two groups (AP-treated and AP-untreated), this possibility is less likely and therefore strengthens our conclusions.
Important to the evaluation of the results of this study is that despite demonstrating dramatic reduction in various markers of lung injury, we could not eliminate neutrophil sequestration, albumin leakage, or morphologic changes in treated animals. These results suggests that other factors are involved in the progression of lung injury associated with AP, such as other adhesion molecules such as the P and E selectins, 21 other inflammatory molecules such as cytokines, and chemokines. 33–35 Similarly, it is difficult to conclude that neutrophils are the only cell mediators of pulmonary dysfunction when in fact other leukocytes, such as lymphocytes and macrophages, could be involved in the progression of injury. 36 More experiments will be needed to answer this question.
In addition, we did not design this study to demonstrate that anti-ICAM-1 therapy reduces the overall death rate from AP. This is partly a limitation of the CDE model that we used, because it was specifically designed to accentuate lung injury while reducing the death rate from the disease. Clearly, demonstrating a survival advantage as a result of the decreased parameters of lung injury would be important in the context of the clinical applicability of these findings, and this will be addressed in future studies.
In conclusion, our results show for the first time that treatment with anti-ICAM-1 antibodies in established AP, before the onset and progression of lung injury, is associated with reduced inflammatory cell infiltration in the lungs and amelioration of associated inflammatory changes. We believe this information may affect the management of patients with AP.
Footnotes
Supported in part by NIH grant # (5RO1GM53439)kk.
Correspondence: A. Osama Gaber, MD, Department of Surgery, Division of Transplantation, University of Tennessee, Memphis, 956 Court Ave., Suite A202, Memphis, TN 38163. E-mail: agaber@utmem.edu
Accepted for publication July 5, 2000.
References
- 1.Ranson JH, Turner JW, Roses DF, et al. Respiratory complications in acute pancreatitis. Ann Surg 1974; 179: 557–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Renner IG, Savage WTD, Pantoja JL, et al. Death due to acute pancreatitis. A retrospective analysis of 405 autopsy cases. Dig Dis Sci 1985; 30: 1005–1018. [DOI] [PubMed] [Google Scholar]
- 3.Guice KS, Oldham KT, Johnson KJ, et al. Pancreatitis-induced acute lung injury. An ARDS model. Ann Surg 1988; 208: 71–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fernandez-del Castillo C, Rattner DW, Warshaw AL. Acute pancreatitis. Lancet 1993; 342 (8869): 475–479. [DOI] [PubMed] [Google Scholar]
- 5.Geokas MC, Baltaxe HA, Banks PA, et al. Acute pancreatitis. Ann Intern Med 1985; 103: 86–100. [DOI] [PubMed] [Google Scholar]
- 6.Werner J, Z’Graggen K, Fernandez-del Castillo C, et al. Specific therapy for local and systemic complications of acute pancreatitis with monoclonal antibodies against ICAM-1. Ann Surg 1999; 229: 834–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Frossard JL, Saluja A, Bhagat L, et al. The role of intercellular adhesion molecule 1 and neutrophils in acute pancreatitis and pancreatitis-associated lung injury. Gastroenterology 1999; 116: 694–701. [DOI] [PubMed] [Google Scholar]
- 8.Bhatia M, Saluja AK, Hofbauer B, et al. The effects of neutrophil depletion on a completely noninvasive model of acute pancreatitis-associated lung injury. Int J Pancreatol 1998; 24: 77–83. [DOI] [PubMed] [Google Scholar]
- 9.Inoue S, Nakao A, Kishimoto W, et al. Anti-neutrophil antibody attenuates the severity of acute lung injury in rats with experimental acute pancreatitis. Arch Surg 1995; 130: 93–98. [DOI] [PubMed] [Google Scholar]
- 10.Hughes CB, Gaber LW, Mohey el-Din AB, et al. Inhibition of TNF alpha improves survival in an experimental model of acute pancreatitis. Am Surg 1996; 62: 8–13. [PubMed] [Google Scholar]
- 11.Norman J, Franz M, Messina J, et al. Interleukin-1 receptor antagonist decreases severity of experimental acute pancreatitis. Surgery 1995; 117: 648–655. [DOI] [PubMed] [Google Scholar]
- 12.Norman JG. New approaches to acute pancreatitis: role of inflammatory mediators. Digestion 1999; 60 (suppl S1): 57–60. [DOI] [PubMed] [Google Scholar]
- 13.Norman J. The role of cytokines in the pathogenesis of acute pancreatitis. Am J Surg 1998; 175: 76–83. [DOI] [PubMed] [Google Scholar]
- 14.Kusske AM, Rongione AJ, Reber HA. Cytokines and acute pancreatitis [editorial; comment]. Gastroenterology 1996; 110: 639–642. [DOI] [PubMed] [Google Scholar]
- 15.Lombardi B, Estes LW, Longnecker DS. Acute hemorrhagic pancreatitis (massive necrosis) with fat necrosis induced in mice by DL-ethionine fed with a choline-deficient diet. Am J Pathol 1975; 79: 465–480. [PMC free article] [PubMed] [Google Scholar]
- 16.Lombardi B, Rao NK. Acute hemorrhagic pancreatic necrosis in mice. Influence of the age and sex of the animals and of dietary ethionine, choline, methionine, and adenine sulfate. Am J Pathol 1975; 81: 87–99. [PMC free article] [PubMed] [Google Scholar]
- 17.Fraker PJ, Speck JC Jr. Protein and cell membrane iodinations with a sparingly soluble chloroamide, 1,3,4,6-tetrachloro-3a,6a-diphrenylglycoluril. Biochem Biophys Res Commun 1978; 80: 849–857. [DOI] [PubMed] [Google Scholar]
- 18.Eppihimer MJ, Wolitzky B, Anderson DC, et al. Heterogeneity of expression of E- and P-selectins in vivo. Circ Res 1996; 79: 560–569. [DOI] [PubMed] [Google Scholar]
- 19.Henninger DD, Panes J, Eppihimer M, et al. Cytokine-induced VCAM-1 and ICAM-1 expression in different organs of the mouse. J Immunol 1997; 158: 1825–1832. [PubMed] [Google Scholar]
- 20.Bradley PP, Priebat DA, Christensen RD, et al. Measurement of cutaneous inflammation: estimation of neutrophil content with an enzyme marker. J Invest Dermatol 1982; 78: 206–209. [DOI] [PubMed] [Google Scholar]
- 21.Lundberg AH, Granger DN, Russell J, et al. Quantitative measurement of P- and E-selectin adhesion molecules in acute pancreatitis: correlation with distant organ injury. Ann Surg 2000; 231: 213–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Masamune A, Shimosegawa T, Kimura K, et al. Specific induction of adhesion molecules in human vascular endothelial cells by rat experimental pancreatitis-associated ascitic fluids. Pancreas 1999; 18: 141–150. [DOI] [PubMed] [Google Scholar]
- 23.Granger DN, Kubes P. The microcirculation and inflammation: modulation of leukocyte-endothelial cell adhesion. J Leukoc Biol 1994; 55: 662–675. [PubMed] [Google Scholar]
- 24.Springer TA. Traffic signals on endothelium for lymphocyte recirculation and leukocyte emigration. Annu Rev Physiol 1995; 57: 827–872. [DOI] [PubMed] [Google Scholar]
- 25.Hughes CB, Grewal HP, Gaber LW, et al. Anti-TNF-alpha therapy improves survival and ameliorates the pathophysiologic sequelae in acute pancreatitis in the rat. Am J Surg 1996; 171: 274–280. [DOI] [PubMed] [Google Scholar]
- 26.Lundberg AH, Granger N, Russell J, et al. Temporal correlation of tumor necrosis factor-alpha release, upregulation of pulmonary ICAM-1 and VCAM-1, neutrophil sequestration, and lung injury in diet-induced pancreatitis. J Gastrointest Surg 2000; 4: 248–257. [DOI] [PubMed] [Google Scholar]
- 27.Doerschuk CM, Quinlan WM, Doyle NA, et al. The role of P-selectin and ICAM-1 in acute lung injury as determined using blocking antibodies and mutant mice. J Immunol 1996; 157: 4609–4614. [PubMed] [Google Scholar]
- 28.Inoue S, Nakao A, Kishimoto W, et al. LFA-1 (CD11a/CD18) and ICAM-1 (CD54) antibodies attenuate superoxide anion release from polymorphonuclear leukocytes in rats with experimental acute pancreatitis. Pancreas 1996; 12: 183–188. [DOI] [PubMed] [Google Scholar]
- 29.Niederau C, Luthen R, Niederau MC, et al. Acute experimental hemorrhagic-necrotizing pancreatitis induced by feeding a choline-deficient, ethionine-supplemented diet. Methodology and standards. Eur Surg Res 1992; 24 (suppl 1): 40–54. [DOI] [PubMed] [Google Scholar]
- 30.Todd KE, Lewis MP, Gloor B, et al. An ETa/ETb endothelin antagonist ameliorates systemic inflammation in a murine model of acute hemorrhagic pancreatitis. Surgery 1997; 122: 443–450. [DOI] [PubMed] [Google Scholar]
- 31.Gloor B, Todd KE, Lane JS, et al. Mechanism of increased lung injury after acute pancreatitis in IL-10 knockout mice. J Surg Res 1998; 80: 110–114. [DOI] [PubMed] [Google Scholar]
- 32.Guice KS, Oldham KT, Caty MG, et al. Neutrophil-dependent, oxygen-radical mediated lung injury associated with acute pancreatitis. Ann Surg 1989; 210: 740–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gerard C, Frossard JL, Bhatia M, et al. Targeted disruption of the beta-chemokine receptor CCR1 protects against pancreatitis-associated lung injury. J Clin Invest 1997; 100: 2022–2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ward PA. Role of complement, chemokines, and regulatory cytokines in acute lung injury. Ann NY Acad Sci 1996; 796: 104–112. [DOI] [PubMed] [Google Scholar]
- 35.Luster AD. Chemokines–chemotactic cytokines that mediate inflammation. N Engl J Med 1998; 338: 436–445. [DOI] [PubMed] [Google Scholar]
- 36.Closa D, Sabater L, Fernandez-Cruz L, et al. Activation of alveolar macrophages in lung injury associated with experimental acute pancreatitis is mediated by the liver. Ann Surg 1999; 229: 230–236. [DOI] [PMC free article] [PubMed] [Google Scholar]