

Abstract
Objective
To review the current knowledge on the genetic alterations involved in the development and progression of Barrett’s esophagus-associated neoplastic lesions.
Summary Background Data
Barrett’s esophagus (BE) is a premalignant condition in which the normal squamous epithelium of the esophagus is replaced by metaplastic columnar epithelium. BE predisposes patients to the development of esophageal adenocarcinoma. Endoscopic surveillance can detect esophageal adenocarcinomas when they are early and curable, but most of the adenocarcinomas are detected at an advanced stage. Despite advances in multimodal therapy, the prognosis for invasive esophageal adenocarcinoma is poor. A better understanding of the molecular evolution of the Barrett’s metaplasia to dysplasia to adenocarcinoma sequence may allow improved diagnosis, therapy, and prognosis.
Methods
The authors reviewed data from the published literature to address what is known about the molecular changes thought to be important in the pathogenesis of BE-associated neoplastic lesions.
Results
The progression of Barrett’s metaplasia to adenocarcinoma is associated with several changes in gene structure, gene expression, and protein structure. Some of the molecular alterations already showed promise as markers for early cancer detection or prognostication. Among these, alterations in the p53 and p16 genes and cell cycle abnormalities or aneuploidy appear to be the most important and well-characterized molecular changes. However, the exact sequence of events is not known, and probably multiple molecular pathways interact and are involved in the progression of BE to adenocarcinoma.
Conclusions
Further research into the molecular biology of BE-associated adenocarcinoma will enhance our understanding of the genetic events critical for the initiation and progression of Barrett’s adenocarcinoma, leading to more effective surveillance and treatment.
Since 1970, the incidence of adenocarcinomas of the esophagus has increased in many countries at a rate that exceeds that of any other malignancy. 1–5 It is now generally accepted that esophageal adenocarcinomas develop from a premalignant lesion of the esophagus, also referred to as Barrett’s esophagus (BE). BE is a metaplastic change of the normal squamous cell epithelium of the esophagus to a columnar type as a result of longstanding gastroesophageal reflux. Three subtypes of BE have been described, but the specialized intestinal type is the only subtype clearly associated with malignant transformation. 6,7 The risk of esophageal adenocarcinoma in patients with BE appears to be approximately 30- to 125-fold greater than that in the general population, with an estimated incidence of 1 in 180 patient-years. 8–11 Although high-grade dysplasia of BE is generally considered a precursor to invasive carcinoma, the assessment of novel biomarkers and better understanding of the pathophysiology of Barrett’s adenocarcinoma may help to identify patients at increased risk for malignant transformation. In addition, elucidating the genetic alterations leading to malignant transformation may someday lead to its prevention.
It is generally accepted that a multistep process of genetic and epigenetic alterations causes the transformation of a normal cell into a malignant tumor cell. These alterations render the cell independent of regulated proliferative and cell death pathways and infuse the cells with proliferative, invasive, and metastasizing capacities. As a consequence, a malignant tumor is generated composed of cells with an increased proliferative activity, a prolonged life span, and metastasizing capacity. At least 5 to 10 genetic alterations are necessary to generate the malignant phenotype, and most tumors are characterized by genomic instability, facilitating the accumulation of mutations. The genomic instability occurs in two different forms, one characterized by microsatellite instability (MSI) and the other by chromosomal instability. 12 The targets of the genomic instability are four classes of genes:
1. Protooncogenes: These are dominant genes that all act in signal transduction from extracellular stimuli to the nucleus or in regulation of gene expression, and they have a role in cell proliferation or inhibition of apoptosis. On activation of protooncogenes by mutation, amplification, translocation, and so forth, these genes turn into oncogenes with unregulated, constitutive activity. This results in excessive stimulation of cell proliferation or prevention of apoptosis, and both contribute to tumor formation.
2. Tumor suppressor genes: Tumor suppressor genes are normal cellular genes that primarily are involved in cell proliferation, apoptosis, cell adhesion, and gene expression regulation. These are recessive genes, which implies that both gene copies need to be inactivated to contribute to tumorigenesis. Functional inactivation of tumor suppressor genes can be caused by genetic as well as by epigenetic phenomena: mutation, deletion of part of the gene, and epigenetic silencing through promoter methylation.
3. Mismatch repair genes: Genetic instability can be caused by impairment in DNA repair. This deficiency is recognized in tumors by MSI or the replication error phenotype. The genes PMS1, PMS2, MLH1, MSH2, MSH6, and the recently discovered MBD4 (MED1) are all associated with MSI. 13,14 In tumors with underlying defects, contractions or expansions of short repeat sequences (microsatellites) can be found. 15 The mismatch repair deficiency leads to a genome-wide accumulation of mutations, also in protooncogenes and tumor suppressor genes, and contributes as such to tumorigenesis.
4. Mitotic checkpoint genes. An inactivating mutation in one copy of these genes has a dominant effect on the phenotype (dominant-negative). Eight human genes with a role in mitotic checkpoint control have been discovered. 16 Inactivation of mitotic checkpoint genes results in chromosomal instability and an abnormal chromosome number (aneuploidy). 16 Mutation analysis of the human mitotic checkpoint genes in aneuploid cancers revealed only a few alterations. It is therefore anticipated that genes yet to be discovered are responsible for most of the checkpoint defects found in aneuploid cancers.
Genomic instability at the nucleotide or chromosomal level ultimately leads to the activation of protooncogenes and inactivation of tumor suppressor genes. There are no protooncogenes or tumor suppressor genes that are activated or deleted in all cancers. Even comparable cancers from the same organ and cell type never share alterations in the same oncogenes and tumor suppressor genes completely. Although clear from a conceptual point of view, the relevant genetic alterations underlying comparable tumors, such as esophageal adenocarcinomas, show variation in the genes involved.
CELL PROLIFERATION AND APOPTOSIS
Dividing normal cell populations maintain the balance between cell proliferation and cell loss. This is important for maintaining a constant number of cells in a tissue. If there is increased proliferation, decreased apoptosis, or both, uncontrolled growth occurs, and this may result in tumor formation. 17
Cell Proliferation
To assess the amount and distribution of cell proliferation in paraffin-embedded tissues, monoclonal antibodies have been developed to detect cell cycle modulators. Several studies have used a monoclonal Ki-67 (MIB-1) and proliferating cell nuclear antigen (PCNA) antibody to study the proliferative properties in BE and adenocarcinomas. An increased number of proliferating cells and an expansion of the proliferative compartment were shown in BE and adenocarcinoma. 18–20 PCNA immunostaining was mainly seen in the basal cells of the neck/foveolar epithelial compartment of the glands in BE. However, in mucosa with high-grade dysplasia, the proliferative compartment extended upward into the superficial layers of the glands. 21–23 The Ki-67 staining pattern also correlated with the histologic findings in BE: the number and the localization of Ki-67-positive nuclei were significantly different between nondysplastic and low-grade and high-grade dysplastic BE and adenocarcinoma. 21,24–27
Apoptosis
Apoptosis, or programmed cell death, is one of the mechanisms responsible for cell loss. Apoptosis also provides a protective mechanism by removing senescent, DNA-damaged, or diseased cells that could either interfere with normal function or lead to neoplastic proliferation. Apoptosis itself can be detected by use of immunohistochemical detection of DNA fragmentation as markers for apoptosis. An increase has been found in the apoptotic rate with increasing histologic severity in intestinal metaplasia/dysplasia and carcinoma, 18 whereas others found few apoptotic cells in Barrett’s high-grade dysplasia and adenocarcinoma. 28,29
The Fas/APO-1 (CD95) gene encodes a transmembrane protein that is involved in apoptosis. Loss of its expression during carcinogenesis can result in interruption of the apoptotic pathway. Hughes et al 30 found that expression of Fas on the cell surface by esophageal adenocarcinomas was reduced or absent, whereas high levels of Fas mRNA were detected in these tumors. They also showed in an esophageal adenocarcinoma cancer cell line that wild-type Fas protein is retained in the cytoplasm. Apparently, retention of wild-type Fas protein in the cytoplasm may represent the mechanism by which malignant cells evade Fas-mediated apoptosis.
The bcl-2 protooncogene encodes a protein that blocks apoptosis. 31 Bcl-2 expression is increased in reflux esophagitis, nondysplastic BE, and low-grade dysplastic BE (70–100% of cases) but is low or virtually absent in high-grade dysplasia (0–25% of cases) and carcinomas (0–40% of cases). 21,26,29,32 Apparently, inhibition of apoptosis by overexpression of bcl-2 protein occurs early in the dysplasia-to-carcinoma sequence of BE. The resulting prolongation of cell survival may promote neoplastic progression. As malignancy appears, cells acquire other ways of avoiding apoptosis.
We can conclude that gradually increased and spatially distinguished cell proliferation is a well-established permanent alteration, whereas the role of apoptosis and bcl-2 seems less certain. The assessment of the ratio of proliferation to apoptosis may be more important than the isolated assessment of either, and this may be a useful and sensitive marker of neoplastic change in BE. 19
Telomerase
During normal somatic cell division, telomeres shorten. In contrast, immortalized and carcinoma cells show no loss of telomere length during cell division. Telomerase is a ribonucleoprotein complex that synthesizes telomeric DNA located at the chromosome ends, thereby maintaining telomere length. The increase in telomerase activity that accompanies most neoplastic and many preneoplastic conditions may permit the emergence of a population of immortalized cells, thereby facilitating the subsequent accumulation of genetic mutations. 33 By using in situ hybridization, Morales et al 34 detected only weak levels of telomerase RNA in cells of the basal layer of normal squamous esophageal epithelium, representing the population of stem cells. In contrast, moderate to strong telomerase RNA expression, similar to the zone of proliferation, was seen in Barrett’s metaplasia (70% of cases), in 90% of the low-grade dysplasias, and in 100% of the high-grade dysplasias and esophageal adenocarcinomas. Interestingly, cardiac and fundic-type Barrett’s mucosa, which is not associated with an increased risk of adenocarcinoma, showed no telomerase RNA.
DNA CONTENT AND CHROMOSOMAL ABNORMALITIES
DNA Content/Aneuploidy
A normal cell has a chromosome number of 2N, for which the term diploid is applied. Cells reproduce by duplicating their content (4N) and then dividing in two. This mammalian cell division cycle is divided into several distinct phases (Fig. 1). A cell with numeric aberrations is designated as aneuploid.
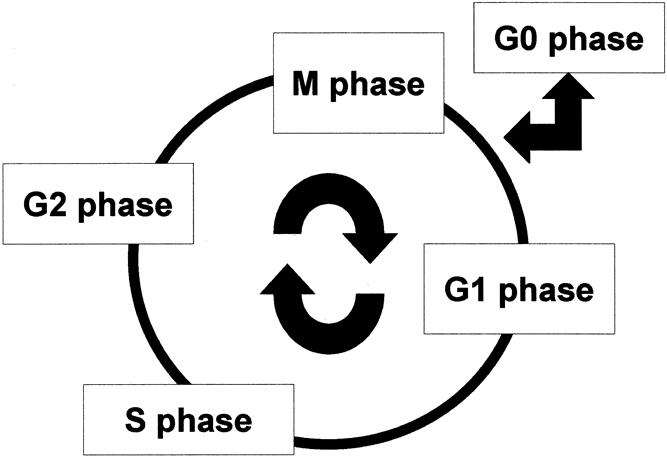
Figure 1. The cell cycle. Mitosis (M phase) is the process of nuclear division. Replication of DNA occurs in the S (synthesis) phase. The interval between M phase and S phase is called the G1 (gap) phase, and the interval between the end of the S phase and the beginning of the M phase is the G2 phase. Cells can exit the cell cycle and enter the G0 phase, which is the quiescent state.
When a suspension of single cells or nuclei is stained with quantitative fluorescent DNA dye, the amount of fluorescence is directly proportional to the amount of DNA in each cell. By using this technique, known as flow cytometry, it has been shown that the evolution from normal esophagus to premalignant Barrett’s metaplasia is frequently associated with abnormal DNA content (aneuploidy) and an increased G2/M fraction of the metaplastic cells. 35–37 Moreover, abnormal DNA content shows a correlation with the histologic diagnosis of dysplasia and carcinoma. 38,39 Flow cytometry also detects a subset of patients whose biopsy samples are histologically indefinite or negative for dysplasia and carcinoma but who have DNA content abnormalities similar to those otherwise seen only in dysplasia and carcinoma. 37,40 Therefore, this technique (in combination with histology) may be useful in screening patients with BE for early signs of malignant change. 41 Indeed, prospective studies have shown that both aneuploidy and dysplasia may be prognostic factors for malignant transformation in BE 36,41,42 : 70% of the patients with aneuploidy or increased G2/tetraploid fractions in biopsy specimens obtained during initial endoscopic evaluation developed high-grade dysplasia or cancer, whereas none of the patients without flow cytometric abnormalities on initial evaluation showed progression to invasive carcinoma or high-grade dysplasia. 36 Others have reported that histologic dysplasia and aneuploidy are often discordant. 43 Most discordance between several studies can be explained by methodologic differences.
Several studies have reported that Barrett’s adenocarcinomas with abnormal nuclear DNA content are associated with increased lymph node metastases, advanced disease, and poorer survival. 44–47 Others found no relationship between ploidy status and clinicopathologic parameters. 48,49
It has been suggested that chromosome instability, tetraploidization, and asymmetric chromosome segregation during cell division leading to aneuploidy are the result of deregulated cell cycle genes with multiple functions that normally exert active checks on the cell cycle processes, including apoptosis and chromosome stability (e.g., p53 and k-ras genes). 50 This is supported by the finding that patients with BE who show allelic loss of 17p (p53 gene locus) and p53 mutations in diploid cells develop increased 4N (G2/tetraploid) fractions and subsequently aneuploidy. 51,52 As discussed above, chromosomal instability is associated with defects in mitotic checkpoint genes. 16
Chromosomal Abnormalities
In BE and Barrett’s cancer, chromosomal alterations have also been described based on karyotyping and in situ hybridization studies. The most consistent numeric chromosomal abnormalities found in cytogenetic studies of dysplastic BE and adenocarcinoma is loss of the Y chromosome. 53–55 In esophageal adenocarcinoma, Y chromosome loss is found in 31% to 93% of the tumors. In one study, the frequency of Y chromosome loss in BE increased along with the grade of dysplasia. 27 Although BE and Barrett’s adenocarcinoma occur more commonly in men, no specific onco- or tumor suppressor genes have been assigned to the Y chromosome. Perhaps, as genetic instability increases during malignant transformation of BE, Y chromosome loss randomly occurs. Karyotyping revealed frequent structural rearrangements in esophageal adenocarcinomas in the 1p, 3q, 11p-13, and 22p regions. 53,56 Further, trisomies for chromosomes 5 and 7 and translocations involving chromosome 3 and 6 in BE have been described. 55 Other frequent numeric aberrations in esophageal adenocarcinomas are overrepresentation of chromosomes 6, 7, 8, 11, 12, 14, and 20 and loss of chromosomes 4, 17, 18, and 21. 53,54,56–58 In a subset of patients in whom premalignant lesions were examined, aneusomy of chromosomes 6, 7, 11, and 12 was found to be an early change, frequently present in both BE and dysplastic regions. It remains to be determined whether any of these abnormalities are predictive markers of progression to malignancy.
Microsatellite Instability
A form of genetic instability that has recently been identified is MSI. Microsatellites are mostly highly polymorph short, tandem repeat DNA sequences. They are abundantly and evenly distributed throughout the genome. MSI is caused by a failure of the DNA mismatch repair system to repair errors that occur during the replication of DNA and is characterized by the accumulation of single nucleotide mutations and alterations in length of the microsatellites. This widespread MSI is a characteristic feature of tumors from hereditary nonpolyposis coli cancer kindreds. 59
Meltzer et al 60 reported MSI at one or more chromosomal loci of the five dinucleotide microsatellite repeats tested in 2 of 28 (7%) patients with Barrett’s metaplasia and 8 of 36 (22%) esophageal adenocarcinomas. Among 25 flow cytometry-sorted adenocarcinomas, MSI occurred in 8 (32%). In four of these eight, the diploid component of the tumor showed MSI, suggesting that the MSI may develop as an early event in BE-associated neoplastic progression. However, confusion remains about how to define the MSI phenomenon—specifically, how many markers should be used and how many must display MSI before the tumor is defined as having MSI. According to more stringent MSI definition, Gleeson et al 61,62 found that MSI is infrequent in Barrett’s adenocarcinomas and adenocarcinomas of the gastric cardia. Several studies have confirmed the low prevalence of MSI (5–10%) in esophageal adenocarcinomas. 63–67 Interestingly, Wu et al 67 found a trend toward improved survival for patients with esophageal adenocarcinomas with MSI. It is not known which mismatch repair genes are responsible for the MSI observed in Barrett’s adenocarcinoma.
TUMOR SUPPRESSOR GENES
Microsatellite allelotyping or loss of heterozygosity analysis is a useful technique to define chromosomal regions of deletion in carcinomas. The loss of heterozygosity analysis uses the polymorphic microsatellite repeats, as mentioned above. These microsatellites are present on all chromosomes and differ in size in the population and usually between the paternal and maternal chromosome of an individual. Polymerase chain reaction amplification of the microsatellites, followed by size separation, can identify chromosomal arms or regions that are lost in the tumor compared with normal tissue from the same person. Frequent loss of one allele involving a chromosomal arm or locus suggests the presence, at or near that locus, of a tumor suppressor gene. Several groups have evaluated chromosomal regions for loss of heterozygosity in BE-associated neoplasia and adenocarcinomas of the gastroesophageal junction (Table 1). We will discuss the most common areas of chromosomal loss with their target genes.
Table 1. CHROMOSOMAL REGIONS THAT SHOW FREQUENT LOSS OF HETEROZYGOSITY

IFNA, interferon-alfa gene; NF1, neurofibromatosis gene; CSF3, colony stimulating factor 3; erbB-2, member of epidermal growth factor receptor family; ITB4, integrin-beta 4; MSH3, DNA mismatch repair gene; Rb, retinoblastoma; VHL, von Hippel Lindau gene; PPARγ, peroxidase proliferator-activated receptor-γ; MCC, mutated in colorectal cancer; APC, adenomatous polyposis coli; IRF-1, interferon regulatory factor 1; TOC, tylosis esophageal cancer gene; DCC, deleted in colorectal carcinoma; DPC4, deleted in pancreatic carcinoma, locus 4; TP53, p53 tumor suppressor gene; BRCA1, breast cancer gene; WT2, Wilms’ tumor suppressor gene; H19, gene involved in genomic imprinting; p57KIP2, a cyclin-dependent kinase inhibitor; HIC1, hypermethylated in cancer; OVCA1/2, ovarian cancer tumor suppressor genes 1 and 2; GH, growth hormone gene; Smad-2, signal transduction molecule involved in TGF-β signaling pathway.
Chromosome 3p
Fragile sites are genomic regions that predispose to structural chromosome aberrations such as translocations or deletions. It is hypothesized that genes at fragile sites are altered by chromosome rearrangements and thus contribute to neoplastic growth. 68 Chromosome band 3p14.3, encompassing the most inducible common fragile region, has been cloned and the fragile histidine triad (FHIT) gene characterized. In BE and associated adenocarcinomas, alterations of the FHIT transcripts (FHIT mRNA lacking one or more exons or homo- or hemizygous deletions of the gene) were observed in 86% and 93%, respectively. 69 Another study found low rates of alterations in the FHIT open reading frame in primary esophageal cancers, although lack of expression of FHIT transcripts was common in esophageal cancer cell lines. 70 However, aberrant FHIT transcripts have also been detected in normal, noncancerous tissues of the gastrointestinal tract, bringing into doubt the role of FHIT as a tumor suppressor. 71 Its apparent involvement might simply reflect its location in an unstable region of the genome, and FHIT might not be causally related to the tumorigenesis of the esophagus.
Other candidate tumor suppressor genes on 3p are the von Hippel Lindau (VHL) gene and the gene encoding the peroxidase proliferator activated receptor-gamma (PPARγ). The genes are mutated in VHL disease and colon carcinomas, respectively. 72,73 We screened adenocarcinomas of the esophagus and gastroesophageal junction for genetic alterations in the VHL and PPARγ gene but could not detect mutations in these genes. Because we and others detected a high percentage of loss of heterozygosity at the VHL locus, 66 other putative tumor suppressor genes at 3p could be involved in the carcinogenesis of esophageal adenocarcinomas.
Chromosome 5q
Loss of heterozygosity at the mutated in colorectal cancer (MCC) locus occurred in 63% of the patients with esophageal carcinoma. 74 So far, no reports have been published on MCC mutation analysis in esophageal adenocarcinomas. Observations from colorectal and gastric cancers suggest that despite the high frequency of loss of heterozygosity of the MCC gene, mutation of the retained MCC allele is uncommon. 75,76 This suggests that MCC does not function as a tumor suppressor gene in gastrointestinal malignancies.
The adenomatous polyposis coli (APC) gene is also a target of loss of heterozygosity on chromosome 5q21 to 22. Loss of heterozygosity of the APC locus on 5q has been found in esophageal adenocarcinomas and in the surrounding high-grade dysplastic BE. Further, the patterns of allelic loss of the APC gene were identical in all stages of neoplastic progression, suggesting the emergence of a clonal population of cells. 74 However, loss of heterozygosity has not been found in Barrett’s metaplasia or low-grade Barrett’s dysplasia. 77 Although APC mutations were found frequently in colorectal cancers, a low rate of APC mutations in esophageal cancers was detected, 77–79 although in most studies less than the entire coding sequence of the gene was screened for mutations. This raised the possibility that a gene distinct from APC may be the target of the frequent loss of heterozygosity on 5q. Deletion of the APC locus may simply be the result of large deletions on 5q and may not be important in esophageal carcinogenesis. 78 The interferon regulatory factor 1 (IRF-1) gene or other genes on 5q31.1 may be the true target of frequent deletions on 5q that may play an important role in the pathogenesis of most esophageal carcinomas. 78
Chromosome 9p
The p16 gene (MTS1, CDKN2A) encodes a 16-kD protein. It forms complexes with the cyclin-dependent kinases CDK4 and CDK6, inhibiting their ability to phosphorylate the retinoblastoma protein. Unphosphorylated retinoblastoma protein prevents the cell from entering the S phase of the cell cycle. Thus, inactivation of this gene may lead to uncontrolled cell growth. The p16 gene is located on chromosome 9p at 9p21, a locus at which frequent allelic loss occurs in esophageal adenocarcinomas. 77,80 Point mutations in exons 1 and 2 of the p16 gene are rare (approximately 5%) in esophageal adenocarcinomas, whereas p16 mutations were more frequent in squamous cell carcinomas. 70,81–83 Barrett et al 84 reported a higher prevalence (23%) of p16 gene mutations in adenocarcinomas with loss of heterozygosity of 9p21. However, in this study only aneuploid cell populations were investigated, which is not representative for esophageal carcinomas in general; this might explain the higher prevalence of p16 gene mutations. It is possible that p16 is inactivated by different mechanisms. Gonzalez et al 77 reported homozygous deletions of the p16 gene in 3 of 12 (25%) esophageal adenocarcinomas. However, these genetic changes were not present in patients with nondysplastic BE. Two studies showed that p16 promoter methylation (with or without p16 loss of heterozygosity) is a common mechanism of p16 inactivation during neoplastic progression in BE and is already present in nondysplastic premalignant BE. 85,86 p16 inactivation may indeed be a useful biomarker to stratify the risk of progression of Barrett’s metaplasia to esophageal cancer. 87
Another tumor suppressor gene on 9p, p15 (MTS2/CDKN2B), a close homolog of p16 and located 20 kb centromeric, is rarely altered in various types of human cancers, including esophageal adenocarcinomas. 81,84
Chromosome 13q
The protein coded for by the normal retinoblastoma gene is a critical regulatory molecule in the G1 phase of the cell cycle. Mutations in this gene result in uncontrollable cell proliferation and predispose to numerous human tumors. Loss of heterozygosity of 13q has been shown in esophageal adenocarcinomas 88 and was associated with an unfavorable survival rate. 89 There are no reports on mutation analysis of the retinoblastoma gene in esophageal adenocarcinomas, but loss of normal retinoblastoma protein expression was observed as the Barrett’s metaplasia progressed to dysplasia and carcinoma, indicating accumulation of unstainable aberrant protein. 18,90 Loss of heterozygosity of the retinoblastoma gene and retinoblastoma protein expression, as detected by immunohistochemistry, however, were not significantly correlated.
Chromosome 17p
Mutations and deletions of the p53 gene are the most common genetic lesions in human cancers. The p53 protein functions in a homotetrameric complex as a transcription factor that induces expression of genes that facilitate cell cycle arrest, DNA repair, and apoptosis (Fig. 2). One mutant p53 protein in the tetrameric p53 complex abolishes the function of the entire complex. Further, most mutant proteins have a much longer half-life than the wild-type protein. This implies that when a cell harbors one inactivating p53 mutation, the concentration of this protein will increase relative to the product of the wild-type allele, and the activity of the wild-type protein will be inhibited by complexing with the mutant protein (dominant-negative). The prolonged half-life of the mutant p53 protein and the concomitant increased cellular p53 concentration make visualization by immunohistochemistry possible. Studies on BE report a low percentage of metaplasia cases with p53 protein accumulation. 24,91–96 p53 accumulation increases in low-grade dysplasia to high-grade dysplasia from less than 10% to more than 70%. 93,94,96–98 From the results of numerous immunohistochemical studies, it is clear that in more than 50% of esophageal adenocarcinomas, pronounced p53 overexpression is present. 92,94–96,98–101 With molecular techniques to detect p53 gene alterations such as single-strand conformation polymorphism analysis, sequencing, and loss of heterozygosity analysis, occasional cases of metaplasia and low-grade dysplasia with p53 mutations have been found. 67,102,103 In high-grade dysplastic BE and invasive adenocarcinoma, the prevalence of p53 mutations exceeds 40%, and p53 locus loss of heterozygosity in these conditions is generally found at even higher rates. 51,67,87,99,100,102–104 In high-grade dysplasia and in esophageal adenocarcinoma, p53 mutations have been found even in diploid cell subpopulations from aneuploid tumors. 87,100 Further, allelic loss of chromosome 17p often occurs before the loss of 5q during neoplastic progression. 96,105 These findings suggest that p53 mutation is an early event in esophageal adenocarcinogenesis.
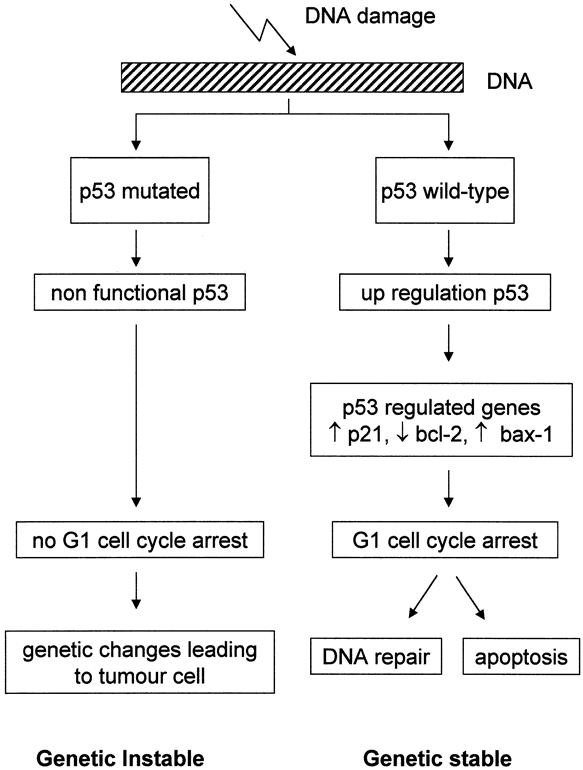
Figure 2. p53 and cell cycle regulation. With DNA damage there is upregulation of wild-type p53 protein. This leads to increased transcription of p53-regulated genes (e.g., p21, bax-1, bcl-2), which inhibit the cell cycle. This facilitates DNA repair, or the cell enters the apoptotic pathway. In this way p53 provides genomic stability. Mutations in the p53 gene render the p53 protein inactive, and the damaged DNA is transmitted.
In summary, there is overwhelming evidence that p53 gene alterations are early and frequent events in esophageal adenocarcinomas and that this gene is associated with malignant transformation of BE. Although the prognostic significance of p53 alterations has been suggested, p53 abnormality alone is not sufficient to predict progression to cancer or disease outcome. 106–108
Chromosome 18q
Allelotype analysis has shown that allelic loss of 18q is common in esophageal adenocarcinomas. However, mutational analysis of the deleted in pancreatic cancer gene (DPC4) revealed no mutations. 109 Therefore, DPC4 is unlikely to be the target gene on 18q21, and other candidates such as the deleted in colorectal cancer gene or as yet unidentified target genes may be involved.
PROTOONCOGENES
The protooncogenes encode a group of proteins involved in signal transduction (from the plasma membrane to the nucleus) or the regulation of gene expression. Mutated protooncogenes are called oncogenes because most mutations activate the proteins, resulting in overstimulation of growth (Fig. 3).
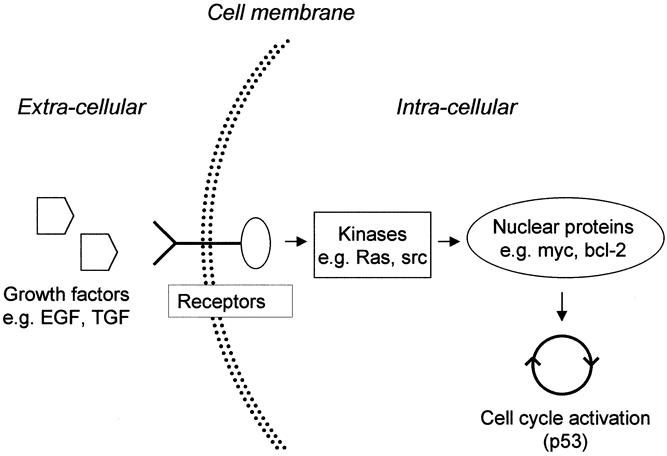
Figure 3. Signal transduction from the cell membrane to the nucleus and the proteins (only protooncogene products) involved. Activation (e.g., mutations) of the genes encoding growth factors, their receptors, or the signal transduction pathway genes (ras, src, myc, bcl-2) can lead to constitutive activation of the cell cycle. EGF, epidermal growth factor; TGF, transforming growth factor.
Growth Factors and Their Receptors
EGF, TGF-α, and EGFR
An important family of growth factors includes the ones that bind to the epidermal growth factor receptor (EGFR), including epidermal growth factor (EGF) and transforming growth factor alpha (TGF-α). EGF has a stimulatory effect on epithelial cell proliferation in the gastrointestinal tract and has been shown to be overexpressed in esophageal carcinomas. Although EGF is also expressed in BE, the expression of EGF does not discriminate between dysplastic and neoplastic epithelium. 110 Overexpression of EGFR in the esophagus correlated with the degree of mucosal dysplasia and the occurrence of adenocarcinoma, suggesting that high expression levels may reflect increased malignant transformation potential in BE. 25,110–112 Al-Kasspooles et al 113 found EGFR gene amplification in 30% of the esophageal adenocarcinomas and also in Barrett’s metaplasia, but no correlation with the level of EGFR expression by immunohistochemistry. TGF-α is structurally and functionally related to EGF. TGF-α expression is increased in metaplastic, dysplastic, and neoplastic tissue of the esophagus compared with normal mucosa. 110 The degree of abnormal expression becomes more marked as dysplasia increases and correlates with the proliferative indices in BE. 22,110,114 In summary, EGF/TGF-α and their receptor EGFR are important in the progression of normal esophageal squamous epithelium to metaplasia, dysplasia, and finally carcinoma and may be associated with autocrine growth regulation in normal gastrointestinal mucosa and neoplasia. 115
c-erbB2
The c-erbB2 protooncogene (HER2/neu; chromosome 17q21) encodes a transmembrane glycoprotein with intrinsic tyrosine kinase activity that is homologous to but distinct from EGFR. c-erbB2 protein overexpression or amplification of the c-erbB2 receptor gene occurs in approximately 10% to 70% of esophageal adenocarcinomas. 44,110,116–120 Overexpression of c-erbB2 was not demonstrated in dysplastic BE, suggesting it is a late event in the dysplasia-to-carcinoma sequence. 117 c-erbB2 overexpression in adenocarcinomas correlated significantly with tumor invasion, lymph node involvement, distant metastasis, and status of residual tumor after resection. 44,120
FGF
The fibroblast growth factors (FGFs) are potent mitogens that possess angiogenic properties and the ability to regulate growth and differentiation of various cell types. The expression of acidic and basic FGF (aFGF, bFGF) has also been studied in esophageal adenocarcinoma and Barrett’s metaplasia. FGF is generally sequentially accumulated in the progression from metaplasia to neoplasia. Esophageal adenocarcinomas and high-grade dysplastic BE showed enhanced expression of aFGF mRNA and protein (immunohistochemistry) but not of bFGF when compared with low-grade dysplasia and normal control epithelium. 121,122
TGF-β
In contrast to TGF-α, TGF-β is a potent inhibitor of cell proliferation, an inducer of differentiation in epithelial cells of the intestine in vitro, and a suppressor of genomic instability. 123 There is evidence that the TGF-β signaling pathway is involved in the initiation and progression of esophageal adenocarcinomas. First, TGF-β is expressed in nondysplastic BE as well as esophageal adenocarcinomas. 124,125 Second, inactivating mutations occur in MADR2, an important component of the signaling pathway for TGF-β, in colon cancers. 126 The chromosomal localization of the MADR2 gene is 18q21, which frequently shows loss of heterozygosity in esophageal adenocarcinomas. Finally, loss of expression of the functional receptor for TGF-β (TGF-β receptor type II) appears to be associated with BE and esophageal adenocarcinomas. 125,127,128 The exact role of TGF-β and its receptor in Barrett’s adenocarcinomas needs to be clarified.
ras Family
The ras families of protooncogenes (H, K, and N) encode specific proteins that appear to be essential components in normal cell division and differentiation. ras proteins act as signal-transducing molecules in the cytoplasm. ras has not been shown to be mutated in most studies on BE and esophageal adenocarcinomas. 129–131 However, increased H-ras expression in Barrett’s carcinoma and amplification of the K-ras gene in esophageal adenocarcinomas have been reported. 132–134 Two studies reported, for the first time, point mutations in K-ras in BE and in esophageal adenocarcinomas, but these were rare. 135,136 Activation of the ras protooncogenes seems to be of little importance in Barrett’s adenocarcinomas, contrary to what has been observed in other carcinomas of the gastrointestinal tract.
c-myc
The c-myc gene is located on chromosome 8q24 and encodes a nuclear protein thought to regulate the transcription of other genes important for cell growth. 137 Activation of the c-myc gene may contribute to tumor progression by preventing cells from entering the G0 resting phase. Studies suggest that c-myc is the target gene of the chromosome 8q high-level amplifications found in esophageal adenocarcinomas. 58,138–140 Using in situ hybridization, Abdelatif et al 134 found enhanced c-myc expression in dysplastic BE and adenocarcinomas but not in nondysplastic Barrett’s mucosa. In contrast, c-myc could not be detected immunohistochemically in esophageal adenocarcinomas or BE. 132 It is unclear whether amplification or mutation of c-myc plays a significant role in the malignant progression of BE, but it appears to have limited prognostic value in human esophageal carcinomas. 141
src
The cellular oncogene c-src and its viral homolog v-src encode 60-kD, cytoplasmic, membrane-associated, protein-tyrosine kinases. A close correlation exists between elevated specific kinase activity and cell transformation. src may deregulate cell adhesion by anchorage-dependent growth control, thereby maintaining cells in the proliferative state. 142 src activity was found to be three to fourfold greater in BE and sixfold greater in esophageal adenocarcinomas than in control tissues. 143 Moreover, Jankowski et al 132 found that 20% of the cases of esophageal adenocarcinomas and BE expressed src. These data suggest a role for src in the malignant transformation of BE and warrant further investigation.
Prostaglandins
Cyclooxygenase (COX) catalyzes the rate-limiting step in prostaglandin synthesis. There are two different isoforms of COX: COX-1 and COX-2. COX-1 is constitutively expressed and is involved, for example, in cytoprotection of the gastric mucosa. In contrast, COX-2 is normally absent in most tissues but can be induced by proinflammatory or mitogenic stimuli. COX-2 is involved in many processes fundamental for tumor development: apoptosis, cell adhesion, invasion and metastasis, and angiogenesis. 144 Chronic esophagitis is associated with the excessive mucosal production of prostaglandin E2, and bile acids stimulate COX-2 expression in esophageal cells in vitro. 145,146 Further, COX-2 is expressed (determined by immunohistochemistry or polymerase chain reaction or Western blotting) in 70% to 80% of esophageal adenocarcinomas and also incorresponding Barrett’s metaplasia. 147,148 Inhibition of COX-2 in esophageal cancer cell lines induced apoptotic cell death and reduced the proliferative activity and synthesis of prostaglandin E2. 147,148 Among regular aspirin users, a 40% to 50% or even greater reduction in esophageal cancer risk was found. 149,150 These data indicate that the chemopreventive potential of nonsteroidal antiinflammatories in esophageal adenocarcinomas, by repressing the induction of COX-2 enzymes in esophagitis and Barrett’s metaplasia, deserves further attention.
GENES INVOLVED IN CONTROLLING THE CELL CYCLE
The progression of cells through the cell cycle is governed by genes encoding proteins transmitting positive (e.g., activated cyclins and cyclin-dependent kinases [CDKs]) and negative (e.g., inhibitors of CDK) signals (Fig. 4). Cyclins form a family of proteins that complex with CDKs. Phosphorylation of the retinoblastoma protein by cyclin D1-CDK4/6 is correlated with the transition across the G1 checkpoint. Cyclin D1-CDK4/6 activity is regulated by phosphorylation events and by CDK inhibitors, which bind to the cyclin–CDK complex and inhibit its activity. This cyclin–CDK inhibition impairs retinoblastoma protein phosphorylation and thereby prevents the cell from entering the cell cycle (S phase). The CDK inhibitors p15 and p16 have been already discussed. Another group of CDK inhibitors is the Cip/Kip family and includes p21, p27, and p57.
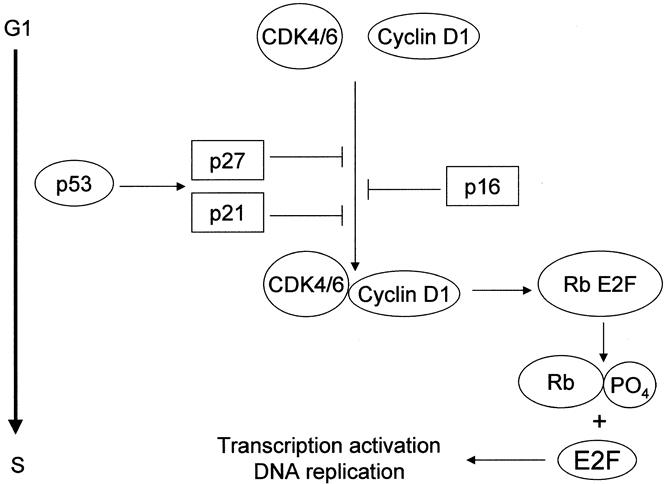
Figure 4. Genes involved in cell cycle progression and inhibition. Cell cycle progression from G1 into S phase requires activation of the cyclin-dependent kinases (CDK4/6) in association with cyclin D1. This active complex phosphorylates the retinoblastoma protein (Rb). Phosphorylated Rb releases Rb-bound transcription factors (E2F family). Free E2Fs transactivate genes that are essential for entry into the S phase and DNA replication. At the G1 checkpoint, there are also negative regulatory signals controlling the cell cycle, namely inhibitors (p16, p21, and p27) of activated cyclin–CDK complexes.
Cyclin D1
Cyclin D1 abnormalities, either gene amplification or overexpression, lead to constitutive activation of the cyclin D1-CDK4/6 pathway. Increased nuclear expression of cyclin D1 is observed in 22% to 64% of esophageal adenocarcinomas and is already present in Barrett’s metaplasia. 89,151–153 The increased expression of cyclin D1 is especially common in intestinal-type lesions and in tumors with early T stage. 151,154 Amplification of cyclin D1 gene was observed in 16% to 26% of esophageal adenocarcinomas. 154,155 However, cyclin D1 immunoreactivity was not always associated with gene amplification. 89,156 Therefore, an additional regulatory mechanism of protein expression probably exists.
p27Kip-1
The p27Kip1 (p27) gene is located on chromosome 12p13. Overexpression of p27 induces a block during G1 in the cell cycle. Singh et al 157 found p27 protein expression and p27 mRNA to be increased in intensity and distributed throughout the glands of high-grade dysplastic BE, indicating transcriptional upregulation of p27. In contrast, low p27 protein expression but elevated levels of p27 mRNA were found in 83% of esophageal adenocarcinomas, possibly because of posttranscriptional regulation of the gene. In addition to nuclear staining, cytoplasmic staining of p27 was noted in 48% and 26% of cases of dysplasia and carcinoma, respectively. 157 Loss of nuclear or cytoplasmic staining for p27 correlated with higher histologic grade, depth of invasion, presence of lymph node metastasis, and shorter survival. 157 These findings suggest that the cell cycle inhibitor p27 may be overexpressed to counteract proliferative stimuli in BE-associated dysplasia. Loss of p27 or altered subcellular localization as the process becomes invasive suggests an important role for this CDK inhibitor in preventing the progression of BE to adenocarcinoma. 124
p21WAF1/CIP1
The G1-S phase of the cell cycle can also be downregulated by inhibition of CDKs by p21WAF1/CIP1 (p21). Nuclear expression of p21 is upregulated by the wild-type p53 tumor suppressor but not mutated p53. 158 p21 expression was elevated in Barrett’s tissue classified as indefinite for dysplasia, low-grade dysplasia, high-grade dysplasia, and Barrett’s adenocarcinoma, but not in BE negative for dysplasia. 159 No relationship between p21 and p53 staining in esophageal adenocarcinomas was found, indicating that there are also p53-independent pathways for the upregulation of p21. 91,159 The elevated nuclear p21 expression in BE and adenocarcinoma probably does not represent mutated protein. 160 p21 expression was significantly associated with prognosis: patients with p21-positive tumors showed a better prognosis than patients with p21-negative tumors. 153
CELL–CELL ADHESION
It has long been known that cell–cell adhesion is generally reduced in human cancers. Reduced cell–cell adhesiveness removes contact inhibition of proliferation, thus allowing escape from growth control signals. Moreover, invasion and metastases, which are life-threatening properties of malignant tumors, are considered to be later but critical carcinogenic steps.
E-Cadherin-Catenin Complex
The E-cadherin-catenin complex is the prime mediator of calcium-dependent cell–cell adhesion in normal epithelial cells. In nonmalignant epithelia, E-cadherin and the catenins show a membranous localization at intercellular borders. In Barrett’s adenocarcinomas, reduced membranous expression of E-cadherin as well as the catenins is observed in 60% to 80% of tumors. 161–164 Moreover, reduced expression of E-cadherin and α- and β-catenin correlated significantly with unfavorable tumor stage, tumor grade, lymph node metastases, and survival. 164 Reduced expression has also been shown to be associated with greater degrees of dysplasia in BE. 162,165 This suggests that the E-cadherin-catenin complex may be useful as a marker for neoplastic progression from Barrett’s metaplasia to adenocarcinoma and metastases. No mutations could be detected in esophageal adenocarcinomas, despite frequent loss of heterozygosity of the E-cadherin locus at 16q22. 166
Besides establishing cell–cell adhesion, β-catenin was shown recently to function in cell signaling. 167,168 Under normal conditions, β-catenin is bound to the cytoplasmic tail of E-cadherin. Free, unbound β-catenin in the cytoplasm is kept low by rapid degradation of unbound β-catenin. To be degraded, β-catenin is phosphorylated by a protein complex, of which the APC protein is one of the members. Inactivation of APC leads to an increase in cellular free β-catenin that enters the nucleus of the cell, directly binds to transcription factors, and activates gene expression. These target genes are involved in promoting cellular proliferation and migration, such as the c-myc oncogene and the cell cycle regulator cyclin D1. Besides inactivation of APC, mutations in phosphorylation sites of the β-catenin gene can also lead to stabilization of the protein. In esophageal adenocarcinomas, increased cytoplasmic and nuclear localization of β-catenin has been observed, 162 implying involvement of APC inactivation or β-catenin mutations with subsequent activation of the signal transduction pathway. However, inactivation of APC is rare, and no mutations in β-catenin could be detected in esophageal adenocarcinomas. 79,169 This implies that other proteins that function in this pathway should be involved.
Serine Protease System
The serine protease system has been shown to play an important role in the invasive potential of a variety of tumors by breaking down the extracellular matrix. Urokinase plasminogen activator is a serine protease. High levels of urokinase plasminogen activator were found in esophageal adenocarcinomas 170,171 and correlated with pTNM category, tumor stage, lymphatic invasion, and survival. 172 Therefore, urokinase plasminogen activator antigen content could identify esophageal adenocarcinoma patients who will develop early tumor recurrences, thus providing a more accurate estimation of prognosis.
CD44 Protein Family
CD44 is a family of glycoproteins involved in cell–cell adhesion and cell–matrix interactions. As a result of alternative splicing of 10 exons (v1–10), more than 20 isoforms have been described. CD44 standard (CD44s) and its abnormal transcripts (CD44v) have been detected in esophageal adenocarcinoma. 173–175 Increased CD44s expression was seen in 50% to 66% of esophageal adenocarcinomas (Dr. K.K. Krishnadath, personal communication, November 1997). 173,175 In BE, CD44s but not CD44v6 expression increases along with dysplasia and the proliferation rate, and increased CD44v6 was seen in an early stage of malignant transformation (Dr. K.K. Krishnadath, personal communication, November 1997). 175 A significant correlation between CD44s and v6 and v10 expression and clinicopathologic characteristics has been reported (Dr. K.K. Krishnadath, personal communication, November 1997), 173,175 but further studies on a larger patient cohort are required to validate the usefulness of CD44s and isoforms in clinical decision making.
Cathepsin B
The cysteine protease cathepsin B (CTSB) gene, which maps to 8p22, codes for a lysosomal enzyme that has been shown to be overexpressed or to exhibit altered localization in cancers. 176 Overexpression or altered localization of CTSB is thought to result in degradation of the basement membrane, facilitating tumor invasion and metastasis. Hughes et al 177 found an amplicon at chromosome 8p22 to 23 resulting in CTSB gene amplification and overexpression. Moreover, abundant extracellular expression of CTSB protein was found in 29 of 40 (73%) esophageal adenocarcinoma specimens. 177 These data support an important role for CTSB gene amplification and CTSB protein overexpression in esophageal adenocarcinomas.
SUMMARY AND CONCLUSIONS
There is a need for improved understanding of the molecular biology of BE and adenocarcinoma. Despite ongoing efforts to characterize the molecular changes in BE, its pathogenesis remains poorly understood. A wide variety of genetic events and mechanisms appear to play a role in the development and progression of BE-associated neoplastic lesions (Fig. 5), but there is still no uniform molecular pathway of progression. In fact, a surprising degree of clonal heterogeneity in premalignant BE has been found, consistent with a complex pattern of evolution of neoplastic cell lineages rather than a simple linear pathway of progression.

Figure 5. Genetic alterations involved in the progression of Barrett’s metaplasia toward Barrett’s adenocarcinoma. Alterations that occur in the early stages are usually also present in the advanced histologic stages. In each histologic category, the alterations are placed randomly; there is no hierarchical order.
Meaningful clinical intervention in patients with BE is still predicated on accurate histologic descriptions, but this information can be supplemented by biomarkers of cell proliferation and abnormalities in protooncogenes and tumor suppressor genes. Many of these markers do not have this potential: dysfunction of ras families of protooncogenes and cell cycle genes such as c-myc and cyclin D1 are not predictive of malignant transformation of BE. However, development of Barrett’s adenocarcinoma is associated with losses on chromosomes; for example, losses of 4q, 5q, 16q, and 18q are frequently observed. Results of these studies are promising but need further attention. The same can be said about cell–cell adhesion molecules and growth factors and their receptors (e.g., EGFR, c-erbB2, src, and the prostaglandins). Abnormalities involving the p16 and p53 tumor suppressor genes and aneuploidy or increased 4N populations are among the most common somatic genetic lesions in the progression from BE to esophageal adenocarcinoma. These biomarkers are potential candidates for objective molecular markers that can be used in combination with histologic staging to stratify a patient’s risk of progressing to esophageal adenocarcinoma.
In the next several years, the subsequent genetic events critical for the initiation and progression of Barrett’s adenocarcinoma will be characterized and may be clinically useful as biomarkers for early cancer detection or prognostication. With the disturbing increase in the incidence of Barrett’s adenocarcinomas, further research into this area is vital.
Footnotes
Correspondence: Prof. dr. H.W. Tilanus, Department of Surgery, University Hospital Dijkzigt, Dr Molewaterplein 40, 3015 GD Rotterdam, The Netherlands.
E-mail: tilanus@hlkd.azr.nl
Accepted for publication November 13, 2000.
References
- 1.Devesa SS, Blot WJ, Fraumeni JF Jr. Changing patterns in the incidence of esophageal and gastric carcinoma in the United States. Cancer 1998; 83: 2049–2053. [PubMed] [Google Scholar]
- 2.Armstrong RW, Borman B. Trends in incidence rates of adenocarcinoma of the oesophagus and gastric cardia in New Zealand, 1978–1992. Int J Epidemiol 1996; 25: 941–947. [DOI] [PubMed] [Google Scholar]
- 3.Hansson LE, Sparen P, Nyren O. Increasing incidence of both major histological types of esophageal carcinomas among men in Sweden. Int J Cancer 1993; 54: 402–407. [DOI] [PubMed] [Google Scholar]
- 4.Hansen S, Wiig JN, Giercksky KE, et al. Esophageal and gastric carcinoma in Norway 1958–1992: incidence time trend variability according to morphological subtypes and organ subsites. Int J Cancer 1997; 71: 340–344. [DOI] [PubMed] [Google Scholar]
- 5.Dolan K, Sutton R, Walker SJ, et al. New classification of oesophageal and gastric carcinomas derived from changing patterns in epidemiology. Br J Cancer 1999; 80: 834–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Paull A, Trier JS, Dalton MD, et al. The histologic spectrum of Barrett’s esophagus. N Engl J Med 1976; 295: 476–480. [DOI] [PubMed] [Google Scholar]
- 7.Thompson JJ, Zinsser KR, Enterline HT. Barrett’s metaplasia and adenocarcinoma of the esophagus and gastroesophageal junction. Hum Pathol 1983; 14: 42–61. [DOI] [PubMed] [Google Scholar]
- 8.Cameron AJ, Ott BJ, Payne WS. The incidence of adenocarcinoma in columnar-lined (Barrett’s) esophagus. N Engl J Med 1985; 313: 857–859. [DOI] [PubMed] [Google Scholar]
- 9.Hameeteman W, Tytgat GN, Houthoff HJ, et al. Barrett’s esophagus: development of dysplasia and adenocarcinoma. Gastroenterology 1989; 96: 1249–1256. [DOI] [PubMed] [Google Scholar]
- 10.Menke-Pluymers MB, Hop WC, Dees J, et al. Risk factors for the development of an adenocarcinoma in columnar-lined (Barrett) esophagus. The Rotterdam Esophageal Tumor Study Group. Cancer 1993; 72: 1155–1158. [DOI] [PubMed] [Google Scholar]
- 11.Kim R, Weissfeld JL, Reynolds JC, Kuller LH. Etiology of Barrett’s metaplasia and esophageal adenocarcinoma. Cancer Epidemiol Biomarkers Prev 1997; 6: 369–377. [PubMed] [Google Scholar]
- 12.Lengauer C, Kinzler KW, Vogelstein B. Genetic instabilities in human cancers. Nature 1998; 396: 643–649. [DOI] [PubMed] [Google Scholar]
- 13.Riccio A, Aaltonen LA, Godwin AK, et al. The DNA repair gene MBD4 (MED1) is mutated in human carcinomas with microsatellite instability [letter]. Nat Genet 1999; 23: 266–268. [DOI] [PubMed] [Google Scholar]
- 14.Lynch HT, de La Chapelle A. Genetic susceptibility to non-polyposis colorectal cancer. J Med Genet 1999; 36: 801–818. [PMC free article] [PubMed] [Google Scholar]
- 15.Modrich P, Lahue R. Mismatch repair in replication fidelity, genetic recombination, and cancer biology. Annu Rev Biochem 1996; 65: 101–133. [DOI] [PubMed] [Google Scholar]
- 16.Cahill DP, Lengauer C, Yu J, et al. Mutations of mitotic checkpoint genes in human cancers. Nature 1998; 392: 300–303. [DOI] [PubMed] [Google Scholar]
- 17.Sinicrope FA, Roddey G, McDonnell TJ, et al. Increased apoptosis accompanies neoplastic development in the human colorectum. Clin Cancer Res 1996; 2: 1999–2006. [PubMed] [Google Scholar]
- 18.Soslow RA, Remotti H, Baergen RN, et al. Suppression of apoptosis does not foster neoplastic growth in Barrett’s esophagus. Mod Pathol 1999; 12: 239–250. [PubMed] [Google Scholar]
- 19.Whittles CE, Biddlestone LR, Burton A, et al. Apoptotic and proliferative activity in the neoplastic progression of Barrett’s oesophagus: a comparative study. J Pathol 1999; 187: 535–540. [DOI] [PubMed] [Google Scholar]
- 20.Gillen P, McDermott M, Grehan D, et al. Proliferating cell nuclear antigen in the assessment of Barrett’s mucosa. Br J Surg 1994; 81: 1766–1768. [DOI] [PubMed] [Google Scholar]
- 21.Lauwers GY, Kandemir O, Kubilis PS, et al. Cellular kinetics in Barrett’s epithelium carcinogenic sequence: roles of apoptosis, bcl-2 protein, and cellular proliferation. Mod Pathol 1997; 10: 1201–1208. [PubMed] [Google Scholar]
- 22.Jankowski J, McMenemin R, Yu C, et al. Proliferating cell nuclear antigen in oesophageal diseases; correlation with transforming growth factor alpha expression. Gut 1992; 33: 587–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hong MK, Laskin WB, Herman BE, et al. Expansion of the Ki-67 proliferative compartment correlates with degree of dysplasia in Barrett’s esophagus. Cancer 1995; 75: 423–429. [DOI] [PubMed] [Google Scholar]
- 24.Polkowski W, van Lanschot JJ, Ten Kate FJ, et al. The value of p53 and Ki67 as markers for tumour progression in the Barrett’s dysplasia-carcinoma sequence. Surg Oncol 1995; 4: 163–171. [DOI] [PubMed] [Google Scholar]
- 25.Yacoub L, Goldman H, Odze RD. Transforming growth factor-alpha, epidermal growth factor receptor, and MiB-1 expression in Barrett’s-associated neoplasia: correlation with prognosis. Mod Pathol 1997; 10: 105–112. [PubMed] [Google Scholar]
- 26.Rioux-Leclercq N, Turlin B, Sutherland F, et al. Analysis of Ki-67, p53 and Bcl-2 expression in the dysplasia-carcinoma sequence of Barrett’s esophagus. Oncol Rep 1999; 6: 877–882. [DOI] [PubMed] [Google Scholar]
- 27.Krishnadath KK, Tilanus HW, van Blankenstein M, et al. Accumulation of genetic abnormalities during neoplastic progression in Barrett’s esophagus. Cancer Res 1995; 55: 1971–1976. [PubMed] [Google Scholar]
- 28.Wetscher GJ, Schwelberger H, Unger A, et al. Reflux-induced apoptosis of the esophageal mucosa is inhibited in Barrett’s esophagus. Am J Surg 1998; 176: 569–573. [DOI] [PubMed] [Google Scholar]
- 29.Katada N, Hinder RA, Smyrk TC, et al. Apoptosis is inhibited early in the dysplasia-carcinoma sequence of Barrett esophagus. Arch Surg 1997; 132: 728–733. [DOI] [PubMed] [Google Scholar]
- 30.Hughes SJ, Nambu Y, Soldes OS, et al. Fas/APO-1 (CD95) is not translocated to the cell membrane in esophageal adenocarcinoma. Cancer Res 1997; 57: 5571–5578. [PubMed] [Google Scholar]
- 31.Reed JC. Bcl-2 and the regulation of programmed cell death. J Cell Biol 1994; 124: 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goldblum JR, Rice TW. bcl-2 protein expression in the Barrett’s metaplasia-dysplasia-carcinoma sequence. Mod Pathol 1995; 8: 866–869. [PubMed] [Google Scholar]
- 33.Oshimura M, Barrett JC. Multiple pathways to cellular senescence: role of telomerase repressors. Eur J Cancer 1997; 33: 710–715. [DOI] [PubMed] [Google Scholar]
- 34.Morales CP, Lee EL, Shay JW. In situ hybridization for the detection of telomerase RNA in the progression from Barrett’s esophagus to esophageal adenocarcinoma. Cancer 1998; 83: 652–659. [PubMed] [Google Scholar]
- 35.Levine DS, Reid BJ, Haggitt RC, et al. Correlation of ultrastructural aberrations with dysplasia and flow cytometric abnormalities in Barrett’s epithelium. Gastroenterology 1989; 96: 355–367. [DOI] [PubMed] [Google Scholar]
- 36.Reid BJ, Blount PL, Rubin CE, et al. Flow-cytometric and histological progression to malignancy in Barrett’s esophagus: prospective endoscopic surveillance of a cohort. Gastroenterology 1992; 102: 1212–1219. [PubMed] [Google Scholar]
- 37.Reid BJ, Haggitt RC, Rubin CE, et al. Barrett’s esophagus. Correlation between flow cytometry and histology in detection of patients at risk for adenocarcinoma. Gastroenterology 1987; 93: 1–11. [PubMed] [Google Scholar]
- 38.Gimenez A, Minguela A, Parrilla P, et al. Flow cytometric DNA analysis and p53 protein expression show a good correlation with histologic findings in patients with Barrett’s esophagus. Cancer 1998; 83: 641–651. [DOI] [PubMed] [Google Scholar]
- 39.Montgomery EA, Hartmann DP, Carr NJ, et al. Barrett esophagus with dysplasia. Flow cytometric DNA analysis of routine, paraffin-embedded mucosal biopsies. Am J Clin Pathol 1996; 106: 298–304. [DOI] [PubMed] [Google Scholar]
- 40.Haggitt RC, Reid BJ, Rabinovitch PS, et al. Barrett’s esophagus. Correlation between mucin histochemistry, flow cytometry, and histologic diagnosis for predicting increased cancer risk. Am J Pathol 1988; 131: 53–61. [PMC free article] [PubMed] [Google Scholar]
- 41.Teodori L, Gohde W, Persiani M, et al. DNA/protein flow cytometry as a predictive marker of malignancy in dysplasia-free Barrett’s esophagus: thirteen-year follow-up study on a cohort of patients. Cytometry 1998; 34: 257–263. [DOI] [PubMed] [Google Scholar]
- 42.James PD, Atkinson M. Value of DNA image cytometry in the prediction of malignant change in Barrett’s oesophagus. Gut 1989; 30: 899–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fennerty MB, Sampliner RE, Way D, et al. Discordance between flow cytometric abnormalities and dysplasia in Barrett’s esophagus. Gastroenterology 1989; 97: 815–820. [DOI] [PubMed] [Google Scholar]
- 44.Nakamura T, Nekarda H, Hoelscher AH, et al. Prognostic value of DNA ploidy and c-erbB-2 oncoprotein overexpression in adenocarcinoma of Barrett’s esophagus [published erratum appears in Cancer 1994 Oct 15;74: 2396]. Cancer 1994; 73: 1785–1794. [DOI] [PubMed] [Google Scholar]
- 45.Schneeberger AL, Finley RJ, Troster M, et al. The prognostic significance of tumor ploidy and pathology in adenocarcinoma of the esophagogastric junction. Cancer 1990; 65: 1206–1210. [DOI] [PubMed] [Google Scholar]
- 46.Finley RJ, Inculet RI. The results of esophagogastrectomy without thoracotomy for adenocarcinoma of the esophagogastric junction. Ann Surg 1989; 210: 535–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bottger T, Dutkowski P, Kirkpatrick CJ, et al. Prognostic significance of tumor ploidy and histomorphological parameters in adenocarcinoma of Barrett’s esophagus. Dig Surg 1999; 16: 180–185. [DOI] [PubMed] [Google Scholar]
- 48.Gleeson CM, Sloan JM, McManus DT, et al. Comparison of p53 and DNA content abnormalities in adenocarcinoma of the oesophagus and gastric cardia. Br J Cancer 1998; 77: 277–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sarbia M, Molsberger G, Willers R, et al. The prognostic significance of DNA ploidy in adenocarcinomas of the esophagogastric junction. J Cancer Res Clin Oncol 1996; 122: 186–188. [DOI] [PubMed] [Google Scholar]
- 50.Giaretti W. Aneuploidy mechanisms in human colorectal preneoplastic lesions and Barrett’s esophagus. Is there a role for K-ras and p53 mutations? Anal Cell Pathol 1997; 15: 99–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Blount PL, Galipeau PC, Sanchez CA, et al. 17p allelic losses in diploid cells of patients with Barrett’s esophagus who develop aneuploidy. Cancer Res 1994; 54: 2292–2295. [PubMed] [Google Scholar]
- 52.Galipeau PC, Cowan DS, Sanchez CA, et al. 17p (p53) allelic losses, 4N (G2/tetraploid) populations, and progression to aneuploidy in Barrett’s esophagus. Proc Natl Acad Sci USA 1996; 93: 7081–7084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rodriguez E, Mathew S, Reuter V, et al. Cytogenetic analysis of 124 prospectively ascertained male germ cell tumors. Cancer Res 1992; 52: 2285–2291. [PubMed] [Google Scholar]
- 54.Raskind WH, Norwood T, Levine DS, et al. Persistent clonal areas and clonal expansion in Barrett’s esophagus. Cancer Res 1992; 52: 2946–2950. [PubMed] [Google Scholar]
- 55.Garewal HS, Sampliner R, Liu Y, et al. Chromosomal rearrangements in Barrett’s esophagus. A premalignant lesion of esophageal adenocarcinoma. Cancer Genet Cytogenet 1989; 42: 281–286. [DOI] [PubMed] [Google Scholar]
- 56.Menke-Pluymers MB, van Drunen E, Vissers KJ, et al. Cytogenetic analysis of Barrett’s mucosa and adenocarcinoma of the distal esophagus and cardia. Cancer Genet Cytogenet 1996; 90: 109–117. [DOI] [PubMed] [Google Scholar]
- 57.Krishnadath KK, Tilanus HW, Alers JC, et al. Detection of genetic changes in Barrett’s adenocarcinoma and Barrett’s esophagus by DNA in situ hybridization and immunohistochemistry. Cytometry 1994; 15: 176–184. [DOI] [PubMed] [Google Scholar]
- 58.Persons DL, Croughan WS, Borelli KA, et al. Interphase cytogenetics of esophageal adenocarcinoma and precursor lesions. Cancer Genet Cytogenet 1998; 106: 11–17. [DOI] [PubMed] [Google Scholar]
- 59.Aaltonen LA, Peltomaki P, Mecklin JP, et al. Replication errors in benign and malignant tumors from hereditary nonpolyposis colorectal cancer patients. Cancer Res 1994; 54: 1645–1648. [PubMed] [Google Scholar]
- 60.Meltzer SJ, Yin J, Manin B, et al. Microsatellite instability occurs frequently and in both diploid and aneuploid cell populations of Barrett’s-associated esophageal adenocarcinomas. Cancer Res 1994; 54: 3379–3382. [PubMed] [Google Scholar]
- 61.Gleeson CM, Sloan JM, McGuigan JA, et al. Ubiquitous somatic alterations at microsatellite alleles occur infrequently in Barrett’s-associated esophageal adenocarcinoma. Cancer Res 1996; 56: 259–263. [PubMed] [Google Scholar]
- 62.Gleeson CM, Sloan JM, McGuigan JA, et al. Widespread microsatellite instability occurs infrequently in adenocarcinoma of the gastric cardia. Oncogene 1996; 12: 1653–1662. [PubMed] [Google Scholar]
- 63.Keller G, Rotter M, Vogelsang H, et al. Microsatellite instability in adenocarcinomas of the upper gastrointestinal tract. Relation to clinicopathological data and family history. Am J Pathol 1995; 147: 593–600. [PMC free article] [PubMed] [Google Scholar]
- 64.Muzeau F, Flejou JF, Belghiti J, et al. Infrequent microsatellite instability in oesophageal cancers. Br J Cancer 1997; 75: 1336–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Barrett MT, Galipeau PC, Sanchez CA, et al. Determination of the frequency of loss of heterozygosity in esophageal adenocarcinoma by cell sorting, whole genome amplification and microsatellite polymorphisms. Oncogene 1996; 12: 1873–1878. [PubMed] [Google Scholar]
- 66.Dolan K, Garde J, Gosney J, et al. Allelotype analysis of oesophageal adenocarcinoma: loss of heterozygosity occurs at multiple sites. Br J Cancer 1998; 78: 950–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wu TT, Watanabe T, Heitmiller R, et al. Genetic alterations in Barrett esophagus and adenocarcinomas of the esophagus and esophagogastric junction region. Am J Pathol 1998; 153: 287–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Huebner K, Garrison PN, Barnes LD, et al. The role of the FHIT/FRA3B locus in cancer. Annu Rev Genet 1998; 32: 7–31. [DOI] [PubMed] [Google Scholar]
- 69.Michael D, Beer DG, Wilke CW, et al. Frequent deletions of FHIT and FRA3B in Barrett’s metaplasia and esophageal adenocarcinomas. Oncogene 1997; 15: 1653–1659. [DOI] [PubMed] [Google Scholar]
- 70.Zou TT, Lei J, Shi YQ, et al. FHIT gene alterations in esophageal cancer and ulcerative colitis (UC). Oncogene 1997; 15: 101–105. [DOI] [PubMed] [Google Scholar]
- 71.Chen YJ, Chen PH, Lee MD, et al. Aberrant FHIT transcripts in cancerous and corresponding non-cancerous lesions of the digestive tract. Int J Cancer 1997; 72: 955–958. [DOI] [PubMed] [Google Scholar]
- 72.Maher ER, Kaelin WG, Jr. von Hippel-Lindau disease. Medicine (Baltimore) 1997; 76: 381–391. [DOI] [PubMed] [Google Scholar]
- 73.Vamecq J, Latruffe N. Medical significance of peroxisome proliferator-activated receptors. Lancet 1999; 354: 141–148. [DOI] [PubMed] [Google Scholar]
- 74.Zhuang Z, Vortmeyer AO, Mark EJ, et al. Barrett’s esophagus: metaplastic cells with loss of heterozygosity at the APC gene locus are clonal precursors to invasive adenocarcinoma. Cancer Res 1996; 56: 1961–1964. [PubMed] [Google Scholar]
- 75.Sud R, Talbot IC, Delhanty JD. Infrequent alterations of the APC and MCC genes in gastric cancers from British patients. Br J Cancer 1996; 74: 1104–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Curtis LJ, Bubb VJ, Gledhill S, et al. Loss of heterozygosity of MCC is not associated with mutation of the retained allele in sporadic colorectal cancer. Hum Mol Genet 1994; 3: 443–446. [DOI] [PubMed] [Google Scholar]
- 77.Gonzalez MV, Artimez ML, Rodrigo L, et al. Mutation analysis of the p53, APC, and p16 genes in the Barrett’s oesophagus, dysplasia, and adenocarcinoma. J Clin Pathol 1997; 50: 212–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ogasawara S, Tamura G, Maesawa C, et al. Common deleted region on the long arm of chromosome 5 in esophageal carcinoma. Gastroenterology 1996; 110: 52–57. [DOI] [PubMed] [Google Scholar]
- 79.Powell SM, Papadopoulos N, Kinzler KW, et al. APC gene mutations in the mutation cluster region are rare in esophageal cancers. Gastroenterology 1994; 107: 1759–1763. [DOI] [PubMed] [Google Scholar]
- 80.Tarmin L, Yin J, Zhou X, et al. Frequent loss of heterozygosity on chromosome 9 in adenocarcinoma and squamous cell carcinoma of the esophagus. Cancer Res 1994; 54: 6094–6096. [PubMed] [Google Scholar]
- 81.Esteve A, Martel-Planche G, Sylla BS, et al. Low frequency of p16/CDKN2 gene mutations in esophageal carcinomas. Int J Cancer 1996; 66: 301–304. [DOI] [PubMed] [Google Scholar]
- 82.Muzeau F, Flejou JF, Thomas G, Hamelin R. Loss of heterozygosity on chromosome 9 and p16 (MTS1, CDKN2) gene mutations in esophageal cancers. Int J Cancer 1997; 72: 27–30. [DOI] [PubMed] [Google Scholar]
- 83.Suzuki H, Zhou X, Yin J, et al. Intragenic mutations of CDKN2B and CDKN2A in primary human esophageal cancers. Hum Mol Genet 1995; 4: 1883–1887. [DOI] [PubMed] [Google Scholar]
- 84.Barrett MT, Sanchez CA, Galipeau PC, et al. Allelic loss of 9p21 and mutation of the CDKN2/p16 gene develop as early lesions during neoplastic progression in Barrett’s esophagus. Oncogene 1996; 13: 1867–1873. [PubMed] [Google Scholar]
- 85.Klump B, Hsieh CJ, Holzmann K, et al. Hypermethylation of the CDKN2/p16 promoter during neoplastic progression in Barrett’s esophagus. Gastroenterology 1998; 115: 1381–1386. [DOI] [PubMed] [Google Scholar]
- 86.Wong DJ, Barrett MT, Stoger R, et al. p16INK4a promoter is hypermethylated at a high frequency in esophageal adenocarcinomas. Cancer Res 1997; 57: 2619–2622. [PubMed] [Google Scholar]
- 87.Galipeau PC, Prevo LJ, Sanchez CA, et al. Clonal expansion and loss of heterozygosity at chromosomes 9p and 17p in premalignant esophageal (Barrett’s) tissue. J Natl Cancer Inst 1999; 91: 2087–2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Boynton RF, Huang Y, Blount PL, et al. Frequent loss of heterozygosity at the retinoblastoma locus in human esophageal cancers. Cancer Res 1991; 51: 5766–5769. [PubMed] [Google Scholar]
- 89.Roncalli M, Bosari S, Marchetti A, et al. Cell cycle-related gene abnormalities and product expression in esophageal carcinoma. Lab Invest 1998; 78: 1049–1057. [PubMed] [Google Scholar]
- 90.Coppola D, Schreiber RH, Mora L, et al. Significance of Fas and retinoblastoma protein expression during the progression of Barrett’s metaplasia to adenocarcinoma. Ann Surg Oncol 1999; 6: 298–304. [DOI] [PubMed] [Google Scholar]
- 91.Moskaluk CA, Heitmiller R, Zahurak M, et al. p53 and p21(WAF1/CIP1/SDI1) gene products in Barrett esophagus and adenocarcinoma of the esophagus and esophagogastric junction. Hum Pathol 1996; 27: 1211–1220. [DOI] [PubMed] [Google Scholar]
- 92.Krishnadath KK, van Blankenstein M, Tilanus HW. Prognostic value of p53 in Barrett’s oesophagus. Eur J Gastroenterol Hepatol 1995; 7: 81–84. [PubMed] [Google Scholar]
- 93.Krishnadath KK, Tilanus HW, van Blankenstein M, et al. Accumulation of p53 protein in normal, dysplastic, and neoplastic Barrett’s oesophagus. J Pathol 1995; 175: 175–180. [DOI] [PubMed] [Google Scholar]
- 94.Rice TW, Goldblum JR, Falk GW, et al. p53 immunoreactivity in Barrett’s metaplasia, dysplasia, and carcinoma. J Thorac Cardiovasc Surg 1994; 108: 1132–1137. [PubMed] [Google Scholar]
- 95.Symmans PJ, Linehan JM, Brito MJ, et al. p53 expression in Barrett’s oesophagus, dysplasia, and adenocarcinoma using antibody DO-7. J Pathol 1994; 173: 221–226. [DOI] [PubMed] [Google Scholar]
- 96.Ramel S, Reid BJ, Sanchez CA, et al. Evaluation of p53 protein expression in Barrett’s esophagus by two- parameter flow cytometry. Gastroenterology 1992; 102: 1220–1228. [PubMed] [Google Scholar]
- 97.Jones DR, Davidson AG, Summers CL, et al. Potential application of p53 as an intermediate biomarker in Barrett’s esophagus. Ann Thorac Surg 1994; 57: 598–603. [DOI] [PubMed] [Google Scholar]
- 98.Younes M, Lebovitz RM, Lechago LV, et al. p53 protein accumulation in Barrett’s metaplasia, dysplasia, and carcinoma: a follow-up study. Gastroenterology 1993; 105: 1637–1642. [DOI] [PubMed] [Google Scholar]
- 99.Hamelin R, Flejou JF, Muzeau F, et al. TP53 gene mutations and p53 protein immunoreactivity in malignant and premalignant Barrett’s esophagus. Gastroenterology 1994; 107: 1012–1018. [DOI] [PubMed] [Google Scholar]
- 100.Neshat K, Sanchez CA, Galipeau PC, et al. p53 mutations in Barrett’s adenocarcinoma and high-grade dysplasia. Gastroenterology 1994; 106: 1589–1595. [DOI] [PubMed] [Google Scholar]
- 101.Flejou JF, Paraf F, Potet F, et al. p53 protein expression in Barrett’s adenocarcinoma: a frequent event with no prognostic significance. Histopathology 1994; 24: 487–489. [DOI] [PubMed] [Google Scholar]
- 102.Campomenosi P, Conio M, Bogliolo M, et al. p53 is frequently mutated in Barrett’s metaplasia of the intestinal type. Cancer Epidemiol Biomarkers Prev 1996; 5: 559–565. [PubMed] [Google Scholar]
- 103.Casson AG, Manolopoulos B, Troster M, et al. Clinical implications of p53 gene mutation in the progression of Barrett’s epithelium to invasive esophageal cancer. Am J Surg 1994; 167: 52–57. [DOI] [PubMed] [Google Scholar]
- 104.Hardwick RH, Shepherd NA, Moorghen M, et al. Adenocarcinoma arising in Barrett’s oesophagus: evidence for the participation of p53 dysfunction in the dysplasia/carcinoma sequence. Gut 1994; 35: 764–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Blount PL, Meltzer SJ, Yin J, et al. Clonal ordering of 17p and 5q allelic losses in Barrett dysplasia and adenocarcinoma. Proc Natl Acad Sci USA 1993; 90: 3221–3225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Coggi G, Bosari S, Roncalli M, et al. p53 protein accumulation and p53 gene mutation in esophageal carcinoma. A molecular and immunohistochemical study with clinicopathologic correlations. Cancer 1997; 79: 425–432. [DOI] [PubMed] [Google Scholar]
- 107.Kimura H, Konishi K, Maeda K, et al. Flow cytometric analysis and immunohistochemical staining for the p53 protein and proliferating cell nuclear antigen in submucosal carcinoma of the esophagus. Hepato-Gastroenterology 1999; 46: 285–289. [PubMed] [Google Scholar]
- 108.Kubba AK, Poole NA, Watson A. Role of p53 assessment in management of Barrett’s esophagus. Dig Dis Sci 1999; 44: 659–667. [DOI] [PubMed] [Google Scholar]
- 109.Barrett MT, Schutte M, Kern SE, et al. Allelic loss and mutational analysis of the DPC4 gene in esophageal adenocarcinoma. Cancer Res 1996; 56: 4351–4353. [PubMed] [Google Scholar]
- 110.Jankowski J, Hopwood D, Wormsley KG. Expression of epidermal growth factor, transforming growth factor alpha and their receptor in gastro-oesophageal diseases. Dig Dis 1993; 11: 1–11. [DOI] [PubMed] [Google Scholar]
- 111.Jankowski J, Coghill G, Tregaskis B, et al. Epidermal growth factor in the oesophagus. Gut 1992; 33: 1448–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Jankowski J, Murphy S, Coghill G, et al. Epidermal growth factor receptors in the oesophagus. Gut 1992; 33: 439–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.al-Kasspooles M, Moore JH, Orringer MB, et al. Amplification and over-expression of the EGFR and erbB-2 genes in human esophageal adenocarcinomas. Int J Cancer 1993; 54: 213–219. [DOI] [PubMed] [Google Scholar]
- 114.Jankowski J, Hopwood D, Wormsley KG. Flow-cytometric analysis of growth-regulatory peptides and their receptors in Barrett’s oesophagus and oesophageal adenocarcinoma. Scand J Gastroenterol 1992; 27: 147–154. [DOI] [PubMed] [Google Scholar]
- 115.Tahara E. Growth factors and oncogenes in human gastrointestinal carcinomas. J Cancer Res Clin Oncol 1990; 116: 121–131. [DOI] [PubMed] [Google Scholar]
- 116.Hardwick RH, Barham CP, Ozua P, et al. Immunohistochemical detection of p53 and c-erbB-2 in oesophageal carcinoma; no correlation with prognosis. Eur J Surg Oncol 1997; 23: 30–35. [DOI] [PubMed] [Google Scholar]
- 117.Hardwick RH, Shepherd NA, Moorghen M, et al. c-erbB-2 overexpression in the dysplasia/carcinoma sequence of Barrett’s oesophagus. J Clin Pathol 1995; 48: 129–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Duhaylongsod FG, Gottfried MR, Iglehart JD, et al. The significance of c-erb B-2 and p53 immunoreactivity in patients with adenocarcinoma of the esophagus. Ann Surg 1995; 221: 677–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Flejou JF, Paraf F, Muzeau F, et al. Expression of c-erbB-2 oncogene product in Barrett’s adenocarcinoma: pathological and prognostic correlations. J Clin Pathol 1994; 47: 23–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Polkowski W, van Sandick JW, Offerhaus GJ, et al. Prognostic value of Lauren classification and c-erbB-2 oncogene overexpression in adenocarcinoma of the esophagus and gastroesophageal junction. Ann Surg Oncol 1999; 6: 290–297. [DOI] [PubMed] [Google Scholar]
- 121.Soslow RA, Ying L, Altorki NK. Expression of acidic fibroblast growth factor in Barrett’s esophagus and associated esophageal adenocarcinoma. J Thorac Cardiovasc Surg 1997; 114: 838–843. [DOI] [PubMed] [Google Scholar]
- 122.Soslow RA, Nabeya Y, Ying L, et al. Acidic fibroblast growth factor is progressively increased in the development of oesophageal glandular dysplasia and adenocarcinoma. Histopathology 1999; 35: 31–37. [DOI] [PubMed] [Google Scholar]
- 123.Glick AB, Weinberg WC, Wu IH, et al. Transforming growth factor beta 1 suppresses genomic instability independent of a G1 arrest, p53, and Rb [published erratum appears in Cancer Res 1997 May 15;57: 2079]. Cancer Res 1996; 56: 3645–3650. [PubMed] [Google Scholar]
- 124.Ellis FH Jr, Loda M. Role of surveillance endoscopy, biopsy and biomarkers in early detection of Barrett’s adenocarcinoma. Dis Esophagus 1997; 10: 165–171. [DOI] [PubMed] [Google Scholar]
- 125.Triadafilopoulo G, Kumble S. Transforming growth factor-b (TGFb) expression is enhanced in gastroesophageal reflux disease, Barrett’s esophagus, and esophageal adenocarcinoma [abstract]. Gastroenterology 1996; 110: 1126. [Google Scholar]
- 126.Eppert K, Scherer SW, Ozcelik H, et al. MADR2 maps to 18q21 and encodes a TGF-beta-regulated MAD-related protein that is functionally mutated in colorectal carcinoma. Cell 1996; 86: 543–552. [DOI] [PubMed] [Google Scholar]
- 127.Souza RF, Garrigue-Antar L, Lei J, et al. Alterations of transforming growth factor-beta 1 receptor type II occur in ulcerative colitis-associated carcinomas, sporadic colorectal neoplasms, and esophageal carcinomas, but not in gastric neoplasms. Hum Cell 1996; 9: 229–236. [PubMed] [Google Scholar]
- 128.Garrigue-Antar L, Souza RF, Vellucci VF, et al. Loss of transforming growth factor-beta type II receptor gene expression in primary human esophageal cancer. Lab Invest 1996; 75: 263–272. [PubMed] [Google Scholar]
- 129.Jiang W, Kahn SM, Guillem JG, et al. Rapid detection of ras oncogenes in human tumors: applications to colon, esophageal, and gastric cancer. Oncogene 1989; 4: 923–928. [PubMed] [Google Scholar]
- 130.Meltzer SJ, Zhou D, Weinstein WM. Tissue-specific expression of c-Ha-ras in premalignant gastrointestinal mucosae. Exp Mol Pathol 1989; 51: 264–274. [DOI] [PubMed] [Google Scholar]
- 131.Meltzer SJ, Mane SM, Wood PK, et al. Activation of c-Ki-ras in human gastrointestinal dysplasias determined by direct sequencing of polymerase chain reaction products. Cancer Res 1990; 50: 3627–3630. [PubMed] [Google Scholar]
- 132.Jankowski J, Coghill G, Hopwood D, et al. Oncogenes and onco-suppressor gene in adenocarcinoma of the oesophagus. Gut 1992; 33: 1033–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Sorsdahl K, Casson AG, Troster M, et al. p53 and ras gene expression in human esophageal cancer and Barrett’s epithelium: a prospective study. Cancer Detect Prev 1994; 18: 179–185. [PubMed] [Google Scholar]
- 134.Abdelatif OM, Chandler FW, Mills LR, et al. Differential expression of c-myc and H-ras oncogenes in Barrett’s epithelium. A study using colorimetric in situ hybridization. Arch Pathol Lab Med 1991; 115: 880–885. [PubMed] [Google Scholar]
- 135.Trautmann B, Wittekind C, Strobel D, et al. K-ras point mutations are rare events in premalignant forms of Barrett’s oesophagus. Eur J Gastroenterol Hepatol 1996; 8: 799–804. [PubMed] [Google Scholar]
- 136.Casson AG, Wilson SM, McCart JA, et al. ras mutation and expression of the ras-regulated genes osteopontin and cathepsin L in human esophageal cancer. Int J Cancer 1997; 72: 739–745. [DOI] [PubMed] [Google Scholar]
- 137.Dang CV, Resar LM, Emison E, et al. Function of the c-Myc oncogenic transcription factor. Exp Cell Res 1999; 253: 63–77. [DOI] [PubMed] [Google Scholar]
- 138.Moskaluk CA, Hu J, Perlman EJ. Comparative genomic hybridization of esophageal and gastroesophageal adenocarcinomas shows consensus areas of DNA gain and loss. Genes Chromosomes Cancer 1998; 22: 305–311. [PubMed] [Google Scholar]
- 139.van Dekken H, Geelen E, Dinjens WN, et al. Comparative genomic hybridization of cancer of the gastroesophageal junction: deletion of 14Q31–32.1 discriminates between esophageal (Barrett’s) and gastric cardia adenocarcinomas. Cancer Res 1999; 59: 748–752. [PubMed] [Google Scholar]
- 140.Lu SH, Hsieh LL, Luo FC, et al. Amplification of the EGF receptor and c-myc genes in human esophageal cancers. Int J Cancer 1988; 42: 502–505. [DOI] [PubMed] [Google Scholar]
- 141.Miyazaki S, Sasno H, Shiga K, et al. Analysis of c-myc oncogene in human esophageal carcinoma: immunohistochemistry, in situ hybridization and northern and Southern blot studies. Anticancer Res 1992; 12: 1747–1755. [PubMed] [Google Scholar]
- 142.Takekura N, Yasui W, Yoshida K, et al. pp60c-src protein kinase activity in human gastric carcinomas. Int J Cancer 1990; 45: 847–851. [DOI] [PubMed] [Google Scholar]
- 143.Kumble S, Omary MB, Cartwright CA, Triadafilopoulos G. Src activation in malignant and premalignant epithelia of Barrett’s esophagus. Gastroenterology 1997; 112: 348–356. [DOI] [PubMed] [Google Scholar]
- 144.Tsujii M, Kawano S, Tsuji S, et al. Cyclooxygenase regulates angiogenesis induced by colon cancer cells [published erratum appears in Cell 1998 Jul 24;94: 271]. Cell 1998; 93: 705–716. [DOI] [PubMed] [Google Scholar]
- 145.Morgan GP, Williams JG. Inflammatory mediators in the oesophagus. Gut 1994; 35: 297–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Zhang F, Subbaramaiah K, Altorki N, et al. Dihydroxy bile acids activate the transcription of cyclooxygenase-2. J Biol Chem 1998; 273: 2424–2428. [DOI] [PubMed] [Google Scholar]
- 147.Zimmermann KC, Sarbia M, Weber AA, et al. Cyclooxygenase-2 expression in human esophageal carcinoma. Cancer Res 1999; 59: 198–204. [PubMed] [Google Scholar]
- 148.Wilson KT, Fu S, Ramanujam KS, et al. Increased expression of inducible nitric oxide synthase and cyclooxygenase-2 in Barrett’s esophagus and associated adenocarcinomas. Cancer Res 1998; 58: 2929–2934. [PubMed] [Google Scholar]
- 149.Funkhouser EM, Sharp GB. Aspirin and reduced risk of esophageal carcinoma. Cancer 1995; 76: 1116–1119. [DOI] [PubMed] [Google Scholar]
- 150.Thun MJ, Namboodiri MM, Calle EE, et al. Aspirin use and risk of fatal cancer. Cancer Res 1993; 53: 1322–1327. [PubMed] [Google Scholar]
- 151.Arber N, Gammon MD, Hibshoosh H, et al. Overexpression of cyclin D1 occurs in both squamous carcinomas and adenocarcinomas of the esophagus and in adenocarcinomas of the stomach. Hum Pathol 1999; 30: 1087–1092. [DOI] [PubMed] [Google Scholar]
- 152.Arber N, Lightdale C, Rotterdam H, et al. Increased expression of the cyclin D1 gene in Barrett’s esophagus. Cancer Epidemiol Biomarkers Prev 1996; 5: 457–459. [PubMed] [Google Scholar]
- 153.Kuwahara M, Hirai T, Yoshida K, et al. p53, p21(Waf1/Cip1) and cyclin D1 protein expression and prognosis in esophageal cancer. Dis Esophagus 1999; 12: 116–119. [DOI] [PubMed] [Google Scholar]
- 154.Morgan RJ, Newcomb PV, Hardwick RH, et al. Amplification of cyclin D1 and MDM-2 in oesophageal carcinoma. Eur J Surg Oncol 1999; 25: 364–367. [DOI] [PubMed] [Google Scholar]
- 155.Shimada Y, Imamura M, Shibagaki I, et al. Genetic alterations in patients with esophageal cancer with short- and long-term survival rates after curative esophagectomy. Ann Surg 1997; 226: 162–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Adelaide J, Monges G, Derderian C, et al. Oesophageal cancer and amplification of the human cyclin D gene CCND1/PRAD1. Br J Cancer 1995; 71: 64–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Singh SP, Lipman J, Goldman H, et al. Loss or altered subcellular localization of p27 in Barrett’s associated adenocarcinoma. Cancer Res 1998; 58: 1730–1735. [PubMed] [Google Scholar]
- 158.el-Deiry WS, Tokino T, Velculescu VE, et al. WAF1, a potential mediator of p53 tumor suppression. Cell 1993; 75: 817–825. [DOI] [PubMed] [Google Scholar]
- 159.Hanas JS, Lerner MR, Lightfoot SA, et al. Expression of the cyclin-dependent kinase inhibitor p21(WAF1/CIP1) and p53 tumor suppressor in dysplastic progression and adenocarcinoma in Barrett esophagus. Cancer 1999; 86: 756–763. [DOI] [PubMed] [Google Scholar]
- 160.Shiohara M, el-Deiry WS, Wada M, et al. Absence of WAF1 mutations in a variety of human malignancies. Blood 1994; 84: 3781–3784. [PubMed] [Google Scholar]
- 161.Jian WG, Darnton SJ, Jenner K, et al. Expression of E-cadherin in oesophageal carcinomas from the UK and China: disparities in prognostic significance. J Clin Pathol 1997; 50: 640–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Bailey T, Biddlestone L, Shepherd N, et al. Altered cadherin and catenin complexes in the Barrett’s esophagus dysplasia-adenocarcinoma sequence: correlation with disease progression and dedifferentiation. Am J Pathol 1998; 152: 135–144. [PMC free article] [PubMed] [Google Scholar]
- 163.Jankowski JA, Newham PM, Kandemir O, et al. Differential expression of E-cadherin in normal, metaplastic and dysplastic oesophageal mucosa: a putative biomarker. Int J Oncol 1994; 441–448. [DOI] [PubMed] [Google Scholar]
- 164.Krishnadath KK, Tilanus HW, van Blankenstein M, et al. Reduced expression of the cadherin-catenin complex in oesophageal adenocarcinoma correlates with poor prognosis. J Pathol 1997; 182: 331–338. [DOI] [PubMed] [Google Scholar]
- 165.Washington K, Chiappori A, Hamilton K, et al. Expression of beta-catenin, alpha-catenin, and E-cadherin in Barrett’s esophagus and esophageal adenocarcinomas. Mod Pathol 1998; 11: 805–813. [PubMed] [Google Scholar]
- 166.Wijnhoven BP, de Both NJ, van Dekken H, et al. E-cadherin gene mutations are rare in adenocarcinomas of the oesophagus. Br J Cancer 1999; 80: 1652–1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Pennisi E. How a growth control path takes a wrong turn to cancer [news; comment] [published erratum appears in Science 1998 Sep 18;281: 1809]. Science 1998; 281: 1438–1441. [DOI] [PubMed] [Google Scholar]
- 168.Wijnhoven BP, Pignatelli M. E-cadherin-catenin: more than a “sticky” molecular complex. Lancet 1999; 354: 356–357. [DOI] [PubMed] [Google Scholar]
- 169.Wijnhoven BPL, Nollet F, de Both NJ, et al. Genetic alterations involving exon 3 of the beta-catenin gene do not play a role in adenocarcinomas of the oesophagus. Int J Cancer 2000; 86: 533–537. [DOI] [PubMed] [Google Scholar]
- 170.Sier CF, Verspaget HW, Griffioen G, et al. Plasminogen activators in normal tissue and carcinomas of the human oesophagus and stomach. Gut 1993; 34: 80–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Hewin DF, Savage PB, Alderson D, Vipond MN. Plasminogen activators in oesophageal carcinoma. Br J Surg 1996; 83: 1152–1155. [DOI] [PubMed] [Google Scholar]
- 172.Nekarda H, Schlegel P, Schmitt M, et al. Strong prognostic impact of tumor-associated urokinase-type plasminogen activator in completely resected adenocarcinoma of the esophagus. Clin Cancer Res 1998; 4: 1755–63. [PubMed] [Google Scholar]
- 173.Bottger TC, Youssef V, Dutkowski P, et al. Expression of CD44 variant proteins in adenocarcinoma of Barrett’s esophagus and its relation to prognosis. Cancer 1998; 83: 1074–1080. [DOI] [PubMed] [Google Scholar]
- 174.Castella E, Ariza A, Fernandez-Vasalo A, et al. Expression of CD44H and CD44v3 in normal oesophagus, Barrett mucosa and oesophageal carcinoma. J Clin Pathol 1996; 49: 489–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Lagorce-Pages C, Paraf F, Dubois S, et al. Expression of CD44 in premalignant and malignant Barrett’s oesophagus. Histopathology 1998; 32: 7–14. [DOI] [PubMed] [Google Scholar]
- 176.Keppler D, Sameni M, Moin K, et al. Tumor progression and angiogenesis: cathepsin B and C. Biochem Cell Biol 1996; 74: 799–810. [DOI] [PubMed] [Google Scholar]
- 177.Hughes SJ, Glover TW, Zhu XX, et al. A novel amplicon at 8p22–23 results in overexpression of cathepsin B in esophageal adenocarcinoma. Proc Natl Acad Sci USA 1998; 95: 12410–12415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Gleeson CM, Sloan JM, McGuigan JA, et al. Barrett’s oesophagus: microsatellite analysis provides evidence to support the proposed metaplasia-dysplasia-carcinoma sequence. Genes Chromosomes Cancer 1998; 21: 49–60. [PubMed] [Google Scholar]
- 179.Gleeson CM, Sloan JM, McGuigan JA, et al. Allelotype analysis of adenocarcinoma of the gastric cardia. Br J Cancer 1997; 76: 1455–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Rumpel CA, Powell SM, Moskaluk CA. Mapping of genetic deletions on the long arm of chromosome 4 in human esophageal adenocarcinomas. Am J Pathol 1999; 154: 1329–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Hammoud ZT, Kaleem Z, Cooper JD, et al. Allelotype analysis of esophageal adenocarcinomas: evidence for the involvement of sequences on the long arm of chromosome 4. Cancer Res 1996; 56: 4499–4502. [PubMed] [Google Scholar]
- 182.Boynton RF, Blount PL, Yin J, et al. Loss of heterozygosity involving the APC and MCC genetic loci occurs in the majority of human esophageal cancers. Proc Natl Acad Sci USA 1992; 89: 3385–3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Peralta RC, Casson AG, Wang RN, et al. Distinct regions of frequent loss of heterozygosity of chromosome 5p and 5q in human esophageal cancer. Int J Cancer 1998; 78: 600–605. [DOI] [PubMed] [Google Scholar]
- 184.Morgan RJ, Newcomb PV, Bailey M, et al. Loss of heterozygosity at microsatellite marker sites for tumour suppressor genes in oesophageal adenocarcinoma. Eur J Surg Oncol 1998; 24: 34–37. [DOI] [PubMed] [Google Scholar]
- 185.Huang Y, Boynton RF, Blount PL, et al. Loss of heterozygosity involves multiple tumor suppressor genes in human esophageal cancers. Cancer Res 1992; 52: 6525–6530. [PubMed] [Google Scholar]
- 186.Dunn J, Garde J, Dolan K, et al. Multiple target sites of allelic imbalance on chromosome 17 in Barrett’s oesophageal cancer. Oncogene 1999; 18: 987–993. [DOI] [PubMed] [Google Scholar]
- 187.Blount PL, Ramel S, Raskind WH, et al. 17p allelic deletions and p53 protein overexpression in Barrett’s adenocarcinoma. Cancer Res 1991; 51: 5482–5486. [PubMed] [Google Scholar]
- 188.Swift A, Risk JM, Kingsnorth AN, et al. Frequent loss of heterozygosity on chromosome 17 at 17q11.2-q12 in Barrett’s adenocarcinoma. Br J Cancer 1995; 71: 995–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Petty EM, Kalikin LM, Orringer MB, et al. Distal chromosome 17q loss in Barrett’s esophageal and gastric cardia adenocarcinomas: implications for tumorigenesis. Mol Carcinog 1998; 22: 222–228. [DOI] [PubMed] [Google Scholar]