

Abstract
Objective
To investigate the expression of interferon regulatory factors 1 and 2 (IRF-1 and IRF-2) in human breast cancer.
Summary Background Data
Interferon regulatory factors 1 and 2 are transcription factors in the interferon gamma signal transduction pathway. IRF-1 acts as the effector arm of the interferon gamma response; IRF-2 binds to the same DNA consensus sequence and opposes IRF-1 activity. Previous work in the authors’ laboratory has shown the tumor suppressor activity of IRF-1 expression and the oncogenic effect of IRF-2 in human and murine tumor models, including human breast cancer cell lines. The authors’ hypothesis is that this pathway is involved in human tumor development, and alterations in the expression of IRF-1 and IRF-2 may occur in breast cancer tissue compared with normal breast tissue, and between more and less differentiated breast cancers.
Methods
Formalin-fixed paraffin-embedded human archival tissue specimens were obtained from 33 patients with pure ductal carcinoma in situ (DCIS) and 49 women with invasive ductal cancer. Adjacent areas of normal breast tissue were assayed in 31 women. These specimens were stained with polyclonal IRF-1 and IRF-2 antibodies using an avidin–biotin–peroxidase complex technique after epitope retrieval.
Results
Most normal breast tissue showed expression of IRF-1 and no expression of IRF-2 by immunohistochemistry. High-grade DCIS or node-positive invasive ductal cancers were less likely to express the tumor suppressor IRF-1 than normal tissue. More strikingly, high-grade DCIS and invasive ductal cancers were much more likely to express the oncogenic IRF-2 protein than was normal tissue.
Conclusions
Expression of IRF-1 and IRF-2 is altered in human breast cancer compared with normal adjacent tissue. The loss of IRF-1 expression is consistent with tumor suppressor loss and the development of IRF-2 expression with oncogenic activation. These data support the hypothesis that this pathway is involved in human breast oncogenesis, which warrants further investigation regarding prognostic and therapeutic implications.
Neoplasia occurs as a result of cellular changes that perturb normal balances of cell growth and cell death. As the pathways regulating growth control have become understood, alterations in these pathways have been identified that characterize breast cancer and can help to select therapies for patients. Examples of measurements that have been used to describe breast cancer growth include DNA ploidy, S-phase fraction, p53 status, her2/neu expression, and estrogen receptor expression. In this study, we investigated two additional factors using immunohistochemical techniques that may allow clinical assessment of the intactness of interferon gamma (IFN-γ)-based immunity in individual host–tumor relationships.
Interferon gamma is a cytokine, made by T cells and natural killer cells, that has a variety of effects on different cells. Among its actions are antitumor effects, although these have been difficult to translate into clinical use. The effects of IFN-γ on tumor cells can be separated into direct effects on the tumor cells themselves and indirect effects that increase tumor immune recognition and immune reactivity of tumors. The fact that IFN-γ has clear antitumor effects requiring tumor cell IFN-γ signal transduction has been shown in a variety of models, including the observation of increased tumor growth rate in tumors engineered to express decreased numbers of functional IFN-γ receptors. 1,2
Interferon regulatory factors 1 and 2 (IRF-1 and IRF-2) are nuclear transcription factors that respond to IFN-γ (Fig. 1). IRF-1 acts as the effector arm of the IFN-γ response in tumor cells; IRF 2 binds to the same DNA consensus sequence and opposes IRF-1 activity. IRF-1 is a transcriptional activator of gene expression, whereas IRF-2 appears to downregulate these processes. IFN-γ has been found to be a powerful inducer of IRF-1, 3–5 and IRF-1 appears to be a necessary mediator of many IFN-γ effects on cells. These include inducible nitric oxide synthase (iNOS) expression by macrophages, 6,7 guanylate binding protein expression, 8 major histocompatibility complex class I expression, 9–11 major histocompatibility complex class II expression, 12–14 and IFN-α/β expression. 11,15,16 In contrast, IRF-2 is induced later than IRF-1 and appears to play a role in the feedback inhibition of IFN-γ effects mediated by IRF-1. 17 Its effects, therefore, are oncogenic. For example, NIH3T3 cells with overexpressed IRF-2 became transformed and were more tumorigenic in nude mice, implicating IRF-2 as a potential oncoprotein. 18 Further, IRF-1 overexpression in the IRF-2 transformed cells reversed this phenotype, implicating IRF-1 as a tumor suppressor. 18

Figure 1. Effects of interferon gamma (IFN-γ) on cells. The signal is transduced through the IFN-γ receptor, which consists of two distinct proteins (α and β subunits). This activates the Jak/Stat-1 pathway (Jak, Justin A kinase; STAT1, S ignal T ransducer and A ctivator of T ranscription-1), and the phosphorylated STAT1 translocates to the nucleus, where it induces the transcription of primary IFN-γ response genes. Interferon regulatory factor 1 (IRF-1) is a primary response gene; interferon regulatory factor 2 (IRF-2) induction is later and is influenced by other, poorly understood factors, including IRF-1. IRF-1 and IRF-2 bind to the same nuclear binding site on other secondary genes; IRF-1 is a transcription activator, and IRF-2 blocks the IRF-1 action. Actions distal to the IRF activity include MHC class I expression, inducible nitric oxide synthase (iNOS) expression, intercellular adhesion molecule-1 (ICAM-1) expression, and cell growth effects. There are conflicting data regarding control of expression of MHC class II transactivator (CIITA). TAP-1, transporter associated with antigen processing-1.
The control of IRF-1 and IRF-2 expression is not understood. In response to IFN-γ, some tumor cell lines express solely IRF-1, whereas others express these transcription factors in nearly equal amounts. We previously showed that IRF-1 and IRF-2 expression patterns in response to IFN-γ support the idea that they have opposing roles in IFN-γ growth inhibition. 19 We observed that IRF-1 overexpression in a nonimmunogenic murine tumor cell line suppressed the malignant phenotype in vitro and enhanced immunogenicity in syngeneic mice. 14 Conversely, IRF-2 overexpression in an immunogenic murine tumor created a more aggressive, faster-growing tumor that was not affected by exogenous or endogenous IFN-γ. 17 Thus, IRF-1 expression correlates with inhibition of growth and IRF-2 opposes inhibition. We have also performed in vitro studies on human breast cancer cell lines that parallel the murine in vitro data. To date, the only study of IRF-1 and IRF-2 in human solid tumors is our study of IRF-1 and IRF-2 expression in archival melanoma samples. 20 However, there have been no studies before this one that assess the expression of IRF-1 and IRF-2 in human breast cancer tissue.
Our hypothesis in this study was that some breast cancers escape the growth-control mechanisms normally induced by endogenous IFN-γ because of alterations in the signaling pathway (see Fig. 1). If the alteration occurs proximal to IRF-1 and IRF-2 induction, then the expression of IRF-1 and IRF-2 may be perturbed and could be measured by immunohistochemistry.
METHODS
Staining and Slide Review
Archival specimens from 1995 were obtained from the Barnes-Jewish Hospital pathology files. The formalin-fixed, paraffin-embedded archival tissue specimens were from 33 patients with ductal carcinoma in situ (DCIS) and 49 patients with invasive ductal breast cancer. These were then stained with polyclonal IRF-1 (1:800) and IRF-2 (1:200) antibodies (Santa Cruz Biotechnology, Santa Cruz, CA) using an avidin–biotin–peroxidase complex technique after epitope retrieval (0.01 mol/L citrate buffer [pH 6.0] for 8 minutes in a commercial microwave oven). The slides were reviewed and characterized by one pathologist (L.B.), who was unaware of the details regarding each patient’s characteristics, such as estrogen receptor status or nodal status. Normal breast tissue adjacent to the DCIS was characterized in 31 patients.
Clinical Review
Review of further pathology data, including grade as scored by the original clinical pathologist, estrogen receptor status, and node status, was performed under a protocol approved by the Siteman Cancer Center Protocol Review and Monitoring Committee and the Human Studies Committee. Of the 49 invasive tumors, 6 patients had no Page grade assigned, 9 patients had no estrogen receptor status determined, and 8 patients had no axillary lymph node staging.
Statistics
Statistical analysis used two-tailed Fisher exact tests, with significance defined as P ≤ .05.
RESULTS
Normal breast epithelium showed diffuse cytoplasmic staining for IRF-1 and no IRF-2 staining in the cells lining the ducts (Fig. 2). Of the 31 patients whose normal breast epithelium was stained and assessed, 30 had diffuse IRF-1 staining and only 2 had IRF-2 staining. This result is consistent with the concept that IRF-1 is a tumor suppressor protein involved in the control of cell growth. In the neoplastic tissue, staining, when present, was nearly always diffuse both in the cytoplasm and nucleus for both IRF-1 and IRF-2. In a few specimens, the staining was limited to 10% to 25% of cells, but still including both the cytoplasm and nucleus. Because the number with focal staining was small (three patients), they were analyzed as positive rather than as a separate result.
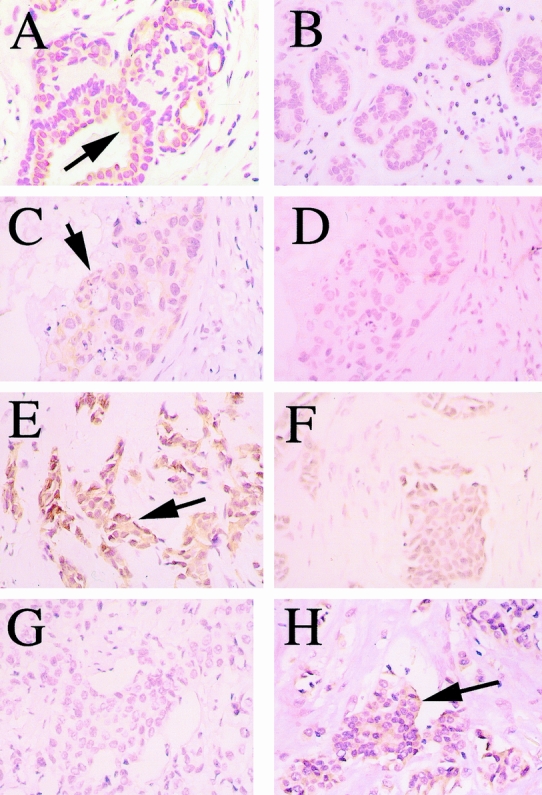
Figure 2. Immunohistochemical staining of human breast specimens; the positive staining is represented by the yellow-brown peroxidase stain in contrast to the hematoxylin and eosin counterstain. Examples of positive staining are indicated by dark arrows. (A, B) Normal breast tissue stained with antibodies to interferon regulatory factor 1 (IRF-1) (+) and interferon regulatory factor 2 (IRF-2) (-), respectively. (C, D) Ductal carcinoma in situ stained with antibodies to IRF-1 (+) and IRF-2 (-), respectively. (E, F) Node-negative invasive ductal breast cancer showing positive staining for IRF-1 in A and negative staining for IRF-2 in B. (G, H) Node-positive invasive ductal breast cancer showing negative staining for IRF-1 in A and positive staining for IRF-2 in B.
Of the 33 patients with DCIS, only 23 had staining for IRF-1, which was significantly different from the staining of normal breast epithelium (Table 1). This result is consistent with the loss of IRF-1 tumor suppressor expression in at least some breast tumors as being part of the neoplastic process. Even more interesting, when subdivided by nuclear grade, nearly all the tumors of low nuclear grade expressed IRF-1 (10/11), but a substantial proportion of the tumors of high nuclear grade (9/22) lacked IRF-1 expression. Thus, the loss of IRF-1 tumor suppressor expression appeared to segregate with the nuclear grade.
Table 1. IRF-1 AND IRF-2 IMMUNOHISTOCHEMICAL EXPRESSION IN DUCTAL CARCINOMA IN SITU
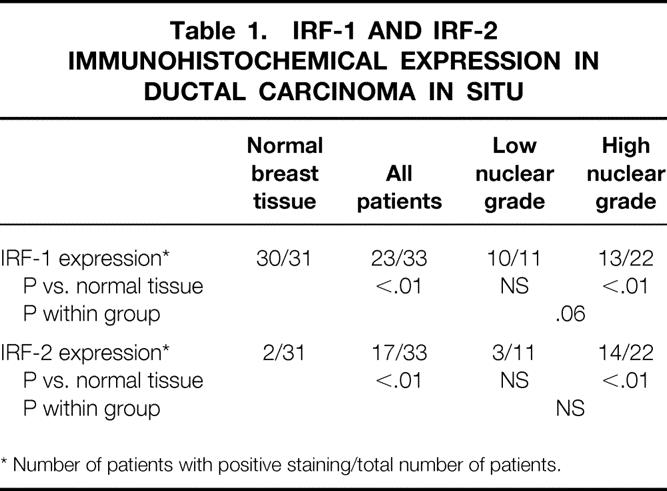
* Number of patients with positive staining/total number of patients.
IRF-2 expression followed a similar but inverse pattern in DCIS specimens. About half (17/33) of the DCIS specimens had staining for IRF-2, markedly different from the normal tissue. Again, the gain of IRF-2 expression was mainly segregated to the tumors of high nuclear grade (14/22) compared with 3 of the 11 low-grade tumors. Thus, the gain of oncogenic IRF-2 expression tended to segregate with nuclear grade, as did the loss of IRF-1 expression. There was no consistent correlation of IRF-1 and IRF-2 staining with one another.
Most invasive breast cancers retained expression of IRF-1 in the tumor tissue (42/49), which was similar to normal breast tissue. Evaluation of IRF-1 expression in relation to other tumor characteristics, including Page grade and estrogen receptor status, showed no correlations. Interestingly, however, six of the seven patients with IRF-1-negative invasive breast cancers had lymph node metastasis, leading to a significant difference in frequency of IRF-1 expression between node-positive tumors and normal breast tissue, and patients with node-positive and node-negative tumors (Table 2). The absence of tumor suppressive IRF-1 expression correlated with the presence of lymph node metastases.
Table 2. IRF-1 AND IRF-2 IMMUNOHISTOCHEMICAL EXPRESSION IN INVASIVE BREAST CANCER

* Number of patients with positive staining/total number of patients.
IRF-2 expression by immunohistochemistry in the invasive breast cancers was similar to the staining pattern in DCIS. Of the 49 patients, 28 had IRF-2 staining. This was markedly different from the staining in normal breast tissue. However, the presence of IRF-2 staining did not segregate with differences in the Page grade or estrogen receptor status, or the presence of lymph node metastases.
DISCUSSION
Our hypothesis was that some breast cancers escape the growth-control mechanisms of endogenous IFN-γ-based immunity by altering the pathway shown in Figure 1. If the interruption occurs proximal to IRF-1 and IRF-2 induction, then these changes may be reflected in the expression of IRF-1 and IRF-2 as measured by immunohistochemistry. Our results support the hypothesis that alterations occur in the IFN-γ/Jak/Stat/IRF pathway that contribute to the development and progression of some breast neoplasias. There were substantial differences in the frequency of IRF-1 and IRF-2 expression between normal tissue and neoplastic tissue. These differences were consistent with the proposed roles of IRF-1 as a tumor suppressor protein and IRF-2 as an oncoprotein. As further evidence, there were differences in the IRF expression between low-grade and high-grade DCIS, and node-negative and node-positive invasive cancers.
The hypothesis advanced here grew from laboratory evidence that IFN-γ has antitumor effects both in vitro and in vivo. At least a part of that antitumor effect is based on direct effects of IFN-γ on the tumor cell, mediated through the IFN-γ receptor. 1,2 Our ongoing work evaluating the signal transduction pathway of IFN-γ in tumor cells led us to hypothesize that evaluation of IRF expression in human tumors might reflect the intactness of this growth-control mechanism in that patient–tumor system.
Proof of this hypothesis emphasizes the relevance to human cancers of this set of endogenous immune effects, which has been previously confirmed in murine models. Notably, interruption or suppression of this pathway at any point between release of IFN-γ by cells of the immune system (T cells and natural killer cells) and the final intratumoral cell growth effects could allow neoplastic cells to “escape” this growth-control mechanism. For example, lack of endogenous IFN-γ at the tumor site or inactivation of the components of the IFN-γ receptor or of the Jak or STAT proteins could affect the relative expression of IRF-1 or IRF-2. This could lead to a selective advantage for those mutated cells. The indirect evidence presented in this article is useful in supporting the idea that the IFN-γ signal transduction pathway is a target site for changes leading to more aggressive growth.
We investigated IRF-1 and IRF-2 expression in melanoma in a prior study. 20 In that article, we reported that IRF-1 expression was most common in early forms of melanoma (thin lesions, lower AJCC stage) and less common in more advanced lesions. IRF-2 staining did not follow any clear pattern in that small study. One advantage of this study over the melanoma study was the opportunity to study IRF expression in adjacent normal tissue. The comparison of IRF expression in the normal tissue with the expression in neoplastic tissue provides important insight into the changes that have taken place in the tumor.
In conclusion, IRF-1 and IRF-2 are expressed in breast epithelium and neoplasms, and the differing frequencies of expression support a role for this pathway in the growth control of neoplasia. For neoplasms that do not have changes in IRF-1 or IRF-2 expression, we would hypothesize that alterations in the IFN-γ signaling pathway either do not occur or occur distal to IRF production. This is a small study and does not purport to show any clinical role for the assessment of IRF-1 and IRF-2. Future studies should include examinations of larger archival specimen collections. This might include comparisons of matched pairs of affected breast and nodal tissues to confirm our findings and to allow correlation of IRF-1 and IRF-2 expression with the subsequent patient course so that the prognostic significance of these findings can be evaluated. In addition, the expression of IRF-1 and IRF-2 in other human tumors should be investigated.
Acknowledgments
The authors thank Judith Connett, PhD, and Steven Hunt, MD, for their careful reading and constructive comments regarding the manuscript.
Discussion
DR. KIRBY I. BLAND (Birmingham, Alabama): I congratulate Dr. Doherty and his colleagues from Washington University in presenting this splendid research effort to elucidate previously undefined transcriptional pathways in human mammary carcinoma. I also would like to thank them for forwarding this manuscript in advance.
The authors have previously investigated interferon regulatory factors 1 and 2 expression in melanoma studies. And, as mentioned as a correlate of this study, they have reported that IRF-1 expression was most commonly observed in early variants of melanoma; that is, those with early or thin lesions, low AJCC stage, and whereas the IRF-2 had no correlate in this small population.
To my knowledge, Jerry, this is the first study to characterize activity of nuclear transcription factors IRF-1 and IRF-2 for human mammary cancer. This study suggests that IRF-1 acts as the effector arm of the interferon gamma response in tumor cells, while IRF-2 binds to the same DNA sequence to oppose this IRF activity. Thus, IRF-1 is a transcriptional activator of gene expression, while IRF-2 appears to be a downregulator of this process.
I feel that the authors have been forthright in their analysis in suggesting that there is a lot that is currently undefined in IRF-1 and IRF-2 regulation. I have some questions concerning the authors’ hypothesis that certain breast cancer cell lines escape growth-control mechanisms of endogenous interferon gamma-based immunity by interrupting the pathway that they have shown you.
First, normal breast epithelium has diffuse cytoplasmic staining for IRF-1, with lack of IRF-2 staining. Is this low frequency of IRF-2 staining consistent with the concept that IRF-1 acts as a tumor suppressor protein involved in the molecular control of cell growth and subsequent carcinogenesis? Secondly, it is evident from the analysis of DCIS and invasive lesions that there is a correlate of increasing expression of IRF-1 in low-nuclear-grade tumors, while the converse is true, with a reduction of IRF-1 in these high-grade tumors. The loss of IRF-1 tumor suppressor expression appears to segregate with high-nuclear-grade tumors. However, in reviewing the tables included in the manuscript, it is unclear why ER negative in 10 of those 12 as well as lymph node-positive invasive tumors possess such a high expression of IRF-1. Please comment: Does this have to do with the methodologies used in immunohistochemistry analysis, or do you think that there is variable expression of these nuclear transcription factors? What is your explanation for the reason that the presence of IRF-2 staining did not segregate with differences in the Page nuclear grade, ER status, or the presence of lymph node metastasis?
Finally, the evidence presented today suggests that IFN-γ/Jak/STAT/IRF signal transduction pathways are target sites for mutations that lead to more aggressive phenotypic neoplastic growth. As IRF-1 and IRF-2 are expressed in normal breast epithelium and neoplasms, the differing frequencies of these expressions in your study support a role for this pathway to influence the growth and control of metastasis. It would be helpful to the audience if you would indicate what studies you consider important for us to retrieve archival specimens that allow correlations with the clinical course of these patients with breast neoplasms. Additionally, what other molecular markers should be correlated with these nuclear transcription factors in order that we may more precisely define their role in breast carcinogenesis and prognosis?
I enjoyed this very important paper and I thank the audience for the opportunity to discuss it.
DR. MICHAEL J. EDWARDS (Louisville, Kentucky): I want to say congratulations to Dr. Doherty and his group for what I believe to be a very important line of investigation, since his group is the first to demonstrate the expression of IRF-1 and 2 in breast epithelium and in breast neoplasms. It turns out, with the passage of time, immunotherapy has had a checkered past. It’s one associated with much hope and promise, but early positive results have almost uniformly been followed by initial enthusiasm which led to disappointment. All the hype and hope has been followed by disappointment, many times associated with terrible toxicity as well. I think we now have to get back to the concept of studying the basic science of immunotherapy, since it appears that there is no magic bullet. I congratulate Dr. Doherty for an early career dedication to the whole field; it’s the hard work that needs to be done now. We realize that it took nearly 30 years for us to find out where 5-FU played a role in reducing the risk of death from colon cancer by a third. I think this is exciting, basic scientific work that importantly began first in murine tumor cell lines, was expanded to murine preclinical models, then to human cancer cell lines and then to the clinical setting. It has the potential to help us elucidate mechanisms of carcinogenesis. It could directly impact growth rates and the basic biological nature of cancer. I am encouraged by this.
My questions, however, relate to some of the potential limitations of the data. Jerry, of the 31 samples of normal tissue, there were two patients that expressed IRF-2, and that was somewhat unexpected. There was one patient that did not express IRF-1, raising the issue of a specificity problem on the order of 10% or so. What are your explanations for this expression? Could it be related to the predisposition of that particular breast endothelium to the development of other tumors? Specifically, was there any evidence of a genetic predisposition in those patients? The findings in the breast proper that you reported today are a little bit different than what you previously reported in melanoma. In melanoma, the findings were not nearly so clear as in the breast paper. I would have expected that this would have applied more to melanoma, or perhaps a tumorlike renal cell carcinoma, both of which have a greater tendency to spontaneously regress and hence are thought to be more immunosensitive. So I would ask you to respond to those two questions, but I think that this is a very significant line of work. It reflects significant dedication beginning at the most basic of scientific research, and it is wonderful to see it conclude today finding a great potential in the clinical arena. Thank you.
DR. MARCIA MOORE (Charlottesville, Virginia): Thank you for the opportunity for a guest to discuss a paper. I just very much enjoyed Dr. Doherty’s presentation and have a simple clinical question for him. I am interested in whether the loss of IRF-1 expression precedes lymph node metastasis. Therefore, would it complement or replace sentinel node surgery? Also, what is the cost of the determination, and how would that cost compare to sentinel node surgery? Thank you.
VICE PRESIDENT LAWRENCE: Are there any other comments or questions? If not, Dr. Doherty, we’d like to have you come forward, and I am hoping you will comment on any data you have on patient outcome.
DR. GERARD M. DOHERTY (St. Louis, Missouri): I will start with your question then, Dr. Lawrence. We don’t have any information about patient outcomes in these patients, and that is somewhat deliberate. This is a small group of patients, and you hate to take a small group of patients and slice things too thinly and maybe imply things that aren’t true. I think, as you point out, that is going to be the critical question. The next study, I believe, needs to be a larger study on a well-characterized group of patients in terms of their molecular markers as well as good information about their outcomes. This is really a preliminary observation of this issue.
Just to address things in order, Dr. Bland asked, I think, if this was the first study regarding breast cancer, and it is, to my knowledge. Nobody else has looked at either normal breast epithelium or breast carcinomas for these factors.
Both Dr. Bland and Dr. Edwards asked about the IRF-1 and IRF-2 staining in the normal tissue. I would say that one weakness of this study is that our normal tissue was adjacent normal tissue in a patient with an abnormality. I would like to have a normal control that was, for example, people who had benign breast biopsies and didn’t have some other neoplastic pathology that may be altering their immune response to breast epithelium. I think that is an important thing to do in the next study.
On the other hand, the findings that we had are completely consistent with the roles of IRF-1 and IRF-2 as tumor suppressor and oncogenic proponent, respectively. The limitations that Mike referred to in terms of the staining of the normal tissue: 2 out of the 31 were IRF-2 positive and 1 out of the 31 was IRF-1 negative. Things weren’t entirely consistent. I don’t know if that is a problem with staining methodology, or with specimens, or the patient’s genetic predispositions, as you suggest. Again, that may be a good reason to have completely normal patient specimens for the next assessment. Those are the data, and I can’t change them.
Dr. Bland’s next comments were about why there weren’t differences between the ER positive and ER negative and the Page grade things that we assessed in the paper that I didn’t talk about too much in the presentation today. I don’t know why there aren’t differences there, but we have seen in other cancers that there are multiple pathways that may be altered in the development of a cancer, but not all of them are altered in every cancer. It is not that a cancer is undifferentiated and has loss of function of all pathways; they seem to sort of pick and choose the ones that get knocked out. There did not seem to be an association between alterations in the estrogen receptor expression pathway and this interferon gamma pathway.
In terms of the next studies, I have already alluded to our desire to find a larger number of patients who have got a long period of follow-up and are well characterized in their original pathology. I think that is the next study. Currently, I would not propose that there is any clinical utility to this. I would hope that we are describing something that is important and that may be useful at some time in the future.
In terms of preserving specimens, we did this on paraffin-embedded, routinely processed archival specimens, which was a part of our design. We wanted to have something that was fairly simple.
Mike’s last question was about the differences between this and the melanoma study. I think the most important difference between the breast cancer study and the melanoma study is that we didn’t have a good normal tissue to assess for the melanoma. It is difficult to go back and say this is the normal melanocyte and here is the staining pattern. So we didn’t have the benefit of knowing what normal tissue staining looks like, and I think that is an important benefit that we have had in describing the biology here.
Finally, Dr. Moore’s question was about the node-positive patients and their IRF-1 expression and whether this might replace sentinel node biopsy as a way of determining node positivity. I certainly would not propose that based on seven patients with IRF-1 staining, but it is an intriguing result. This requires a lot more study and confirmation in larger numbers of patients. It does not have to replace sentinel node biopsy to be an important prognostic factor, or something that might allow us to choose an effective therapy designed for this pathway. We have known for a long time that HER-2 was a prognostic factor in breast cancer, but it wasn’t very important until we could treat the patients with HERceptin to try and address that particular pathway alteration.
In terms of the cost, this is quite inexpensive. This is just immunohistochemical stain.
Thank you very much.
Footnotes
Presented at the 112th Annual Meeting of the Southern Surgical Association, December 4–6, 2000, Palm Beach, Florida.
Supported by National Cancer Institute grants R29-CA 73846-01A1 and 5T32 CA-09621 and a research award from the Breast Health Center of Barnes-Jewish Hospital.
Correspondence: Gerard Doherty, MD, Washington University School of Medicine, 660 S. Euclid Ave., Box 8109, St. Louis, MO 63110.
E-mail: dohertyg@msnotes.wustl.edu
Accepted for publication December 2000.
References
- 1.Dighe AS, Richards E, Old LJ, et al. Enhanced in vivo growth and resistance to rejection of tumor cells expressing dominant negative IFN-gamma receptors. Immunity 1994; 1: 447–456. [DOI] [PubMed] [Google Scholar]
- 2.Doherty GM, Tsung K, McCluskey B, et al. Endogenous interferon gamma acts directly on tumor cells in vivo to suppress growth. J Surg Res 1996; 64: 68–74. [DOI] [PubMed] [Google Scholar]
- 3.Geller DA, Nguyen D, Shapiro RA, et al. Cytokine induction of interferon regulatory factor-1 in hepatocytes. Surgery 1993; 114: 235–242. [PubMed] [Google Scholar]
- 4.Pine R. Constitutive expression of an ISGF2/IRF1 transgene leads to interferon-independent activation of interferon-inducible genes and resistance to virus infection. J Virol 1992; 66: 4470–4478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Coccia EM, Marziali G, Stellacci E, et al. Cells resistant to interferon-beta respond to interferon-gamma via the stat1-IRF-1 pathway. Virology 1995; 211: 11–122. [DOI] [PubMed] [Google Scholar]
- 6.Kamijo R, Harada H, Matsuyama T, et al. Requirement for transcription factor IRF-1 in NO synthase induction in macrophages. Science 1994; 263: 1612–1615. [DOI] [PubMed] [Google Scholar]
- 7.Martin E, Nathan C, Xie Q-W. Role of interferon regulatory factor 1 in induction of nitric oxide synthase. J Exp Med 1994; 180: 977–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Briken V, Ruffner H, Schultz U, et al. Interferon regulatory factor 1 is required for mouse Gbp gene activation by gamma interferon. Mol Cell Biol 1995; 15: 975–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chang C-H, Hammer J, Loh JE, et al. The activation of major histocompatibility complex class I genes by interferon regulatory factor-1 (IRF-1). Immunogenetics 1992; 35: 378–384. [DOI] [PubMed] [Google Scholar]
- 10.Lim SP, Hui KM. Characterization of a novel IRF-1-deficient mutant cell line. Immunogenetics 1994; 39: 168–177. [DOI] [PubMed] [Google Scholar]
- 11.Reis LFL, Harada H, Wolchok JD, et al. Critical role of a common transcription factor, IRF-1, in the regulation of IFN-beta and IFN-inducible genes. EMBO J 1992; 11: 185–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hobart M, Ramassar V, Goes N, et al. IFN regulatory factor-1 plays a central role in the regulation of the expression of class I and II MHC genes in vivo. J Immunol 1997; 158: 4260–4269. [PubMed] [Google Scholar]
- 13.Hobart M, Ramassar V, Goes N, et al. The induction of class I and II major histocompatibility complex by allogeneic stimulation is dependent on the transcription factor interferon regulatory factor 1 (IRF-1). Transplantation 1996; 62: 1895–1901. [DOI] [PubMed] [Google Scholar]
- 14.Yim JH, Wu SJ, Casey MJ, et al. Interferon regulatory factor-1 gene transfer into an aggressive, nonimmunogenic sarcoma suppresses the malignant phenotype and enhances immunogenicity in syngeneic mice. J Immunol 1997; 158: 1284–1292. [PubMed] [Google Scholar]
- 15.Harada H, Willison K, Sakakibara J, et al. Absence of the type 1 IFN system in EC cells: transcriptional activator (IRF-1) and repressor (IRF-2) genes are developmentally regulated. Cell 1990; 63: 303–312. [DOI] [PubMed] [Google Scholar]
- 16.Fujita T, Kimura Y, Miyamoto M, et al. Induction of endogenous IFN-alpha and IFN-beta genes by a regulatory transcription factor, IRF-1. Nature 1989; 337: 270–272. [DOI] [PubMed] [Google Scholar]
- 17.Yim JH, Wu SJ, Lowney JK, et al. Enhancing in vivo tumorigenicity of B16 melanoma by overexpressing interferon regulatory factor-2: resistance to endogenous interferon-γ. J Interferon Cytokine Res 1999; 19: 723–729. [DOI] [PubMed] [Google Scholar]
- 18.Harada H, Kitagawa M, Tanaka N, et al. Anti-oncogenic and oncogenic potentials of interferon regulatory factors-1 and -2. Science 1993; 259: 971–974. [DOI] [PubMed] [Google Scholar]
- 19.Doherty GM, McCluskey B, Tsung K, et al. Correlation of interferon regulatory factors 1 and 2 (IRF-1 and -2): expression and murine tumor growth in the presence of interferon-gamma. Surg Forum 1995; 46: 544–546. [Google Scholar]
- 20.Lowney JK, Boucher LD, Swanson PE, et al. Interferon regulatory factor 1 and 2 expression in human melanoma specimens. Ann Surg Oncol 1999; 6: 604–608. [DOI] [PubMed] [Google Scholar]