

Abstract
Objective
To investigate whether a high-fat/high-protein diet (HFPD) acts as a promoter of the natural course of cancer growth in the 7,12-dimethylbenzanthracene (DMBA)-induced ductal pancreatic cancer model in rats.
Summary Background Data
DMBA implantation to the rat pancreas induces ductal adenocarcinoma. Information regarding the effects of diet and the presence of K-ras mutation in this model is not available.
Methods
Rats were randomly assigned to regular rat chow or a diet with a 30% content in fat and protein (HFPD). The presentation of cancer, the histologic spectrum of neoplasia at 1 and 9 months, and the prevalence of cancer in relation to diet were assessed. Histologic specimens comprising normal ducts, hyperplasia, dysplasia/carcinoma in situ, or carcinoma were designated by a pathologist and microdissected. Genomic DNA was extracted, and K-ras and H-ras gene mutations were determined by a mutant-enriched polymerase chain reaction assay and direct sequencing.
Results
Rats fed HFPD increased their weight significantly compared with controls. DMBA induced characteristic stages of neoplasia at the implant site but not elsewhere. Macroscopic cancers of the pancreatic head presented regularly with common bile duct and gastric outlet obstruction. The prevalence of K-ras mutations was proportional to the degree of epithelial abnormality. K-ras mutations were significantly more frequent in cancer than in normal and hyperplastic ducts. H-ras mutations were not found. At 1 month in the HFPD-fed rats, the prevalence of cancer (16%) and dysplasia (16%) was not significantly different from the prevalence of cancer (29%) and dysplasia (8%) in the chow-fed rats. At 9 months the prevalence of cancer in the HFPD-fed rats increased to 29%, whereas that in the chow-fed rats decreased to 17%. The combined prevalence of cancer and dysplasia at 9 months in the HFPD-fed rats (34%) significantly exceeded that in the chow-fed rats.
Conclusions
DMBA induces characteristic stages of neoplasia in the evolution of ductal pancreatic cancer in rats. K-ras mutations occur progressively in the ladder of oncogenesis, as in human pancreatic neoplasms. The addition of a diet with a high fat and protein content acts as a promoter of carcinogenesis, possibly by interfering with repair mechanisms and natural regression of early lesions.
Our understanding of the development of early stages of pancreatic cancer and of the molecular mechanisms in pancreatic carcinogenesis is incomplete. The analysis of precursor lesions of pancreatic cancer in humans is limited by its recognition only at advanced tumor stages in more than 95% of patients. Mucinous cystic neoplasms and intraductal papillary mucinous tumors of the pancreas, which may serve as surrogate models of pancreatic carcinogenesis, display an evolutionary spectrum of neoplasia, including stages of dysplasia and cancer, and analysis of these tumors indicates that certain oncogenic molecular changes are associated with those stages. 1,2 Animal models of pancreatic ductal adenocarcinoma also can provide an opportunity to investigate evolutionary stages of neoplasia and the sequence of genetic changes involved in their development. These models have the additional advantage of allowing the study of promoters of carcinogenesis either by targeting specific genes or by varying environmental and nutritional factors.
DMBA (7,12-dimethylbenzanthracene) is known to induce pancreatic cancer of ductal phenotype in rats, 3 but its ductal origin was debated until recently. 4 DMBA has been shown to induce cancer through pathways that consistently involve K-ras gene mutations, as in human pancreatic cancer. It seems particularly suitable for recreating a controllable approximation of the human counterpart.
A diet high in fat and protein content has been identified as a potential risk factor for pancreatic adenocarcinoma. 5 We therefore tested the hypothesis that a high-fat/high-protein diet (HFPD) acts as a promoter of carcinogenesis by examining the histologic response of the pancreas to DMBA and the appearance of characteristic ras mutations in rats fed either a normal diet or the fat diet.
METHODS
Experimental Design
Previous observations 5 and pilot studies suggested that the prevalence of cancer in the normal-diet group would be 25% and would increase to at least 40% in the HFPD group. Considering a likely attrition of 35% to 40%, we designed the study to include 155 rats in each arm to achieve significance at an α-level of 0.05 with 80% power (two-tailed chi-square test). Death within 4 days of surgery was defined as perioperative. The death rate was analyzed for 3-month intervals. Based on prior experience with this model, the end point of the study was set at 9 months after carcinogen implantation. An additional 50 rats randomly assigned to the two different diets were killed at 1 month after carcinogen implantation.
Animal care was provided in accordance with the procedures outlined in the Guide for Care and Use of Laboratory Animals (NIH publication 85–23, 1985). Male Sprague-Dawley rats (Charles River Laboratories, Cambridge, MA) weighing 100 to 125 g were housed in cages and randomly allocated to two different diets 2 weeks before carcinogen implantation. The rats were allowed free access to food and water and were exposed to 12-hour light/dark sleep cycles. After DMBA implantation, the rats were inspected daily for complications and weighed monthly. Sick animals and rats with a loss of more than 20% of body weight in a 1-month period were killed. All animals underwent autopsy to determine the cause of death and weight loss, to assess tumor extent and spread, and to excise the pancreas for histopathology and molecular analysis. The subcommittee on animal research of the Massachusetts General Hospital approved the design of the study.
Diets
The two diets fed to the rats were similar with respect to vitamin and mineral profile but differed considerably in fat and protein content and composition. The normal diet involved a standard rodent formula with a protein content of 23.4% and a fat content of 4.5% (Laboratory Rodent Diet, PMI Feeds, Inc., St. Louis, MO). The HFPD (Rodent Diet Modified, AIN-93G, Bio-Serve) contained 30% protein and 30% fat.
Tumor Induction
DMBA was implanted directly into the pancreas of rats according to a previously established protocol. 3 In brief, surgical anesthesia was induced with vaporized ether and maintained by an intramuscular injection of pentobarbital (20 mg/kg) and ketamine (40 mg/kg). The rats underwent a midline laparotomy with exposure of the pancreatic head segment. The parenchyma was incised parallel to the course of the common duct, and a pocket was developed in the pancreatic parenchyma at this site. DMBA crystals (5 mg) were implanted and secured in place with a 6-0 prolene pursestring suture.
Pathologic Examination
At autopsy the abdomen was explored with particular attention to pancreatic changes, enlargement of peripancreatic lymph nodes, and metastasis to the liver and the peritoneal cavity. The pleural cavity and lungs were inspected for possible tumor spread. The pancreaticoduodenal segment, including peripancreatic lymph nodes, was excised for histologic examination. Macroscopic nodules were separated and examined histologically. The carcinogen implant site, marked by a nonabsorbable prolene stitch, was separated from the rest of the pancreatic head for examination for microscopic neoplastic foci.
All pancreatic nodules and adjacent pancreas were fixed in formalin and embedded in paraffin, and multiple 5-μm sections were prepared and stained with hematoxylin and eosin for routine histology. All slides were reviewed by a single pathologist (F.G.-C.) and assessed for the presence of pancreatic dysplasia, neoplasia, and inflammation. Changes at the implant site and of the main pancreatic duct were separately recorded.
Hyperplasia was defined as an increase in the number of epithelial cells lining the duct, with crowding and subsequent micropapillary growth. Atypia was defined by nuclear or cytoplasmic abnormalities, including nuclear enlargement (often with nucleolus) and nuclear crowding without hyperchromatism. Dysplasia represented changes reminiscent of adenoma, with nuclear crowding, hyperchromatism, pseudostratification, and often mitotic activity. Carcinoma in situ was diagnosed when architectural changes such as cribriforming epithelial growth were seen in conjunction with nuclear changes of malignancy, such as large nuclei with nucleoli. Designation as invasive adenocarcinoma required the presence of infiltrating irregular neoplastic glands within a desmoplastic stroma.
Histologic analysis 1 month after carcinogen implantation consistently showed a marked inflammatory reaction at the site of carcinogen implantation. Tubular complexes were consistently found in rats within 1 month of DMBA implantation; they have been shown as the precursor formations of neoplasia in this model. 4 The (chemical) pancreatitis made it difficult to distinguish reactive cellular atypia from dysplasia. For this reason, histologic lesions with pronounced inflammatory tissue reaction, although having criteria consistent with high-grade dysplasia, were rated as indefinite for dysplasia.
Gene Analysis
Histologic specimens from 41 rats comprising normal ducts (n = 10), hyperplasia (n = 14), dysplasia/carcinoma in situ (n = 6), and carcinoma (n = 11) were designated by a pathologist (F.G.-C.). Consecutive unstained 10-μm sections were microdissected under an operating microscope at 30× to 45× magnification. The designated epithelium was carefully harvested with exclusion of associated fibrous reactive tissue. Disposable fine-tipped scalpels were used for microdissection to prevent contamination. After microdissection, the slides containing the residual tissues were stained with hematoxylin and eosin to confirm the accuracy of the collection.
Microdissected samples were deparaffinized in xylene and gradually rehydrated. Tissues were then digested with proteinase K (200 mg/mL) for 16 to 18 hours at 50°C. Genomic DNA was extracted sequentially with phenol, phenol:chloroform (1:1), and chloroform. Subsequently, DNA was precipitated with 100% ethanol and washed with 70% ethanol. After centrifugation, each sample was allowed to dry for at least 1 hour at room temperature and then resuspended in 25 μL Tris-EDTA buffer. DNA concentration and purity were measured by optical density.
K-ras and H-ras codon 12 gene mutations were detected by a species-adapted mutant-enriched nested polymerase chain reaction (PCR), as described previously. 1,2 In brief, an initial PCR using mismatched 5' primers was performed, yielding a 174- and 164-base pair product corresponding to exon 1 of the K-ras and H-ras gene, respectively. These mismatched primers were designated to introduce a BstN1 restriction site in wild-type codon 12 PCR products. The products were then digested with BstN1, thereby enriching the mutated DNA sequences. A second PCR amplification followed, using the same mismatched 5' primer and a 3' primer upstream of the one previously used. The second PCR reaction yielded a 152- and 120- base pair product. PCR products were confirmed on a 2% low-melt agarose gel. The following primers were used: K-ras assay: sense primer, 5'-ACTGAGTATAAACTTGTGGTAGTTGGACCT-3'; antisense primer 1, 5'-CTTTTTCAAACAAAGGATGACT-3'; antisense primer 2, 5'-GCCACCCTTTACAAATTGTAC-3'; H-ras assay: sense primer 1, 5'-GACAGAATACAAGCTTGTGGTGGGCCCT-3'; antisense primer 1, 5'-GACTCTAACCCATGACCACT-3'; sense primer 2, 5'-CAAGCTTGTGGTGGTGGGCCCT-3'; antisense primer 2, 5'-GGCAGGTAGTCAGAGCTCAC-3'.
The individual bands on the agarose gel were cut using sterile disposable scalpels and transferred to tubes. PCR products were then extracted and diluted to a concentration of 3 ng/μL and 10 μL per reaction. Products obtained were sequenced by the dye terminator method using an ABI 373a automatic sequencer, as previously described. 1,2
All PCR reactions were 50 μL in volume and included 5 μL DNA (10–50 μg/mL), 1× PCR buffer A, 200 μmol/L each deoxynucleotide triphosphate, 1 μmol/L concentration of each primer, and 0.5 U Taq polymerase. Each PCR run included a sample with no template DNA to control for carryover contamination. Samples were subjected to 30 cycles of amplification in a DNA thermal cycler at the following parameters: 45 seconds at 94°C, 45 seconds at 55°C, 1 minute at 72°C. After amplification, 20% of PCR reaction mixture was digested with BstN1. Each BstN1 digestion totaled 50 μL in volume, containing 10 μL PCR product and 50 U BstN1 in 1× NE buffer 2 supplemented with 100 μg/mL bovine serum albumin. Incubation was at 60°C for 3 hours. PCR fragments and BstN1 digestion products were electrophoresed on ethidium bromide-stained 2% low-melt agarose gels.
Statistical Analysis
Differences between groups were analyzed by two-tailed chi-square tests with Yates correction and two-tailed Fisher exact tests when one group contained five or fewer observations. Differences in weight were calculated by Student t test. Significance was defined at P < .05.
RESULTS
Death Rate
The death rate of the rats in the 1-month short-term study was 4% (1/25) in the chow group and 16% (4/25) in the HFPD group by 30 days. Among the 310 rats in the long-term study, the overall death rate was identical in both groups: chow 39% (61/155) and HFPD 41% (63/155). The perioperative death rate was 5% (7/155 in the chow group) and 3% (5/155 in the HFPD group). The times and causes of deaths were not significantly different (two-tailed chi-square test) in the two groups. The death rate was 20% (31/155) in the chow group versus 17% (26/155) in the HFPD group at 3 months, 8% (12/155) in the chow group versus 12% (19/155) in the HFPD group between 4 and 6 months, and equal in both groups at 12% (18/155) between 7 and 9 months.
The main cause of death through month 3 was necrotizing pancreatitis (67%). In the second three-month period, pneumonia was the principal cause of death (9/31 [29%]) and common bile duct obstruction with purulent cholangitis and mesenteric ischemia were each found in 10%. During the last 3 months, animals were most often killed for cancer progression. Tumor-related complications included weight loss from gastric outlet obstruction in 25% (9/36) and the presence of extrapancreatic tumors in 19% (7/36). None of these tumor-related complications occurred during the first 6 months.
Body Weight
At all time points, body weight was significantly greater in the HFPD group (Fig. 1;P < .01).
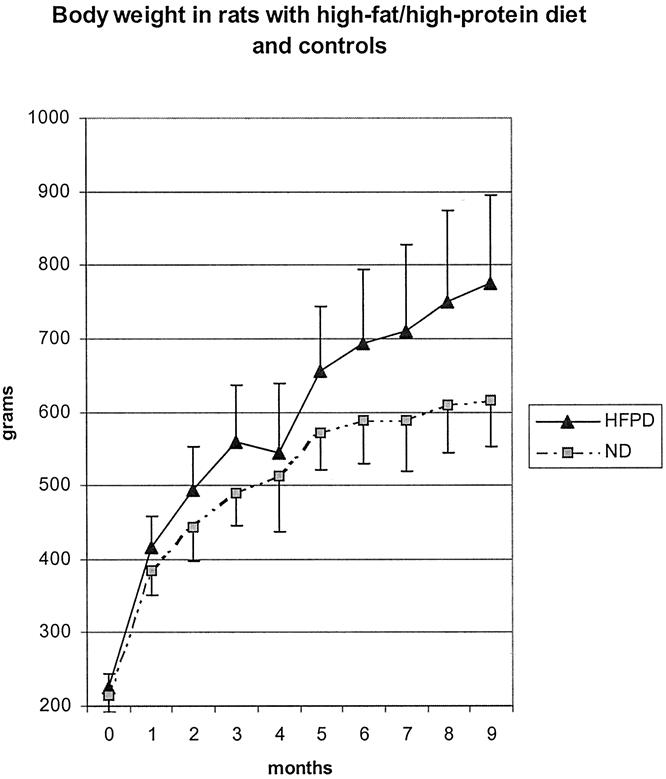
Figure 1. Weight gain of rats fed a high-fat/high-protein diet (HFPD) or a diet with normal fat and protein content (ND).
Macroscopic Findings
In animals killed at 1 month, the pancreas consistently showed a fibrous nodule 2 to 5 mm in size at the location of the DMBA implant. None of these nodules showed histologic characteristics of cancer. Local inflammatory reactions frequently resulted in adhesions to adjacent organs, predominantly to the colon. In the long-term group, macroscopic nodules of the pancreatic head were detected in 20% (62/310), 17% (26/155) in the chow group and 23% (36/155) in the HFPD group (P = .31). Although most were proven histologically to be adenocarcinomas, five (3%) were inflammatory pseudotumors in both groups, 2 to 30 mm in size. There were also two cases of sarcoma of the head of the pancreas and one case of a carcinosarcoma of the head and body of the pancreas.
The pancreatic adenocarcinomas measured 2 to 38 mm (mean diameter 19.8 mm) in the chow group and 2 to 58 mm (mean 15.0;P = .22) in the HFPD group. Obstruction of the common bile duct was observed in 51% (25/49) and gastric outlet obstruction was found in 31% (15/49) of rats with macroscopic pancreatic adenocarcinoma. In 14% (7/49) of rats, the tumors invaded into adjacent abdominal organs, including the duodenum (n = 5), colon (n = 1), and liver (n = 1). Macroscopic metastatic disease was present in 14% of rats (7/49) with macroscopic tumors: three animals had lymph node metastasis and four had peritoneal carcinomatosis. Metastases were observed in 5% (1/19) in the chow group and 20% (6/30;P = .15) in the HFPD group. No other metastases were detected.
Histologic Analysis
Microscopic analysis of the pancreas 1 month after carcinogen implantation revealed characteristic changes of central necrosis surrounded by a zone of tubular complexes and normal peripheral pancreatic tissue (Fig. 2). The degree of inflammatory reaction at the site of the implant, in particular in the area of tubular complexes, was pronounced, whether reactive cellular atypia, dysplasia, or cancer was found. Carcinoma in situ or microinvasive adenocarcinoma was found in 29% (chow) and 16% (HFPD) of the rats and indefinite-for-dysplasia lesions in 8% (chow) and 16% (HFPD) (Fig. 3). A 16% (chow) and 8% (HFPD) prevalence of hyperplasia was found in the main pancreatic duct, but dysplasia and adenocarcinoma were detected only in the area of tubular complexes. The prevalence of these histologic abnormalities in the two groups was not significantly different (P > .4).

Figure 2. Preneoplastic tubular complexes 1 month after DMBA implantation. HFPD, high-fat/high-protein diet; ND, normal diet.

Figure 3. Distribution of induced histologic changes at 1 month after implantation of DMBA in the pancreas. ND, normal diet; HFPD, high-fat/high-protein diet.
Histologic analysis of the 310 rats in the long-term study showed various stages of pancreatic neoplasia and ductal adenocarcinoma (Fig. 4) in 23% (71/310) overall, 17% in the chow-fed rats and 33% in the HFPD group. The prevalence of adenocarcinoma was greater in HFPD rats at all time points, and the difference was significant at 9 months (P = .02;Fig. 5). Compared with the 1-month animals, the prevalence of cancer increased in the HFPD group from 16% (4/21) to 33% (45/155;P = .02); that in chow-fed rats decreased from 29% (7/24) to 17% (26/155;P = .02). Considering the indefinite-for-dysplasia lesions as precursors of adenocarcinoma, the prevalence of the two categories was combined. As a consequence, the chow-fed group had a significantly increased prevalence of neoplasia at 1 month (37%; 9/24) compared with 17% at 9 months (P = .017), but the prevalence of neoplasia in the HFPD group was not different (33% vs. 29%;P = .39) (Fig. 6). The combined prevalence of cancer and dysplasia at 9 months in the HFPD group (34%) significantly exceeded that in the chow group (21%;P < .02).
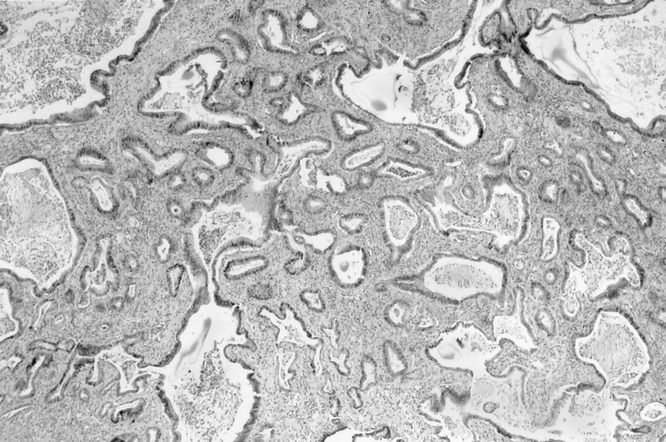
Figure 4. Invasive pancreatic ductal adenocarcinoma 9 months after DMBA implantation.

Figure 5. Occurrence of pancreatic ductal adenocarcinoma during the 9-month experimental period. The prevalence of cancer was higher at all intervals in the high-fat/high-protein diet group (HFPD) than in the normal diet group (ND).

Figure 6. Prevalence of adenocarcinoma and precursor lesions in the normal diet (ND) group and the high-fat/high-protein diet (HFPD) group at 1 month versus 9 months.
Although the carcinogen implant was placed adjacent to the pancreatic duct, dysplastic lesions in the main duct were rare (1% each for low- and high-grade dysplasia). No cancer was found in the main pancreatic duct or in its side branches. Hyperplasia of the main pancreatic duct occurred in 26% of animals (79/310) and was associated with pancreatic cancer in 15% (12/79).
Most of the pancreatic cancers were moderately differentiated (grade 2) mucinous ductal adenocarcinomas, but 10% (7/71) of the cancers showed an intestinal differentiation (grade 1) that displayed a characteristic increase in the number of neuroendocrine cells, as determined by silver stains. Microscopic adenocarcinoma was detected in 27% (7/26) of all cancers in the chow group and 33% (15/45) of all cancers in the HFPD group; the difference was not significant.
Gene Analysis
H-ras mutations were not found in any of the 39 samples analyzed (2 samples were technically unsuccessful). The prevalence of K-ras mutations was proportional to the degree of epithelial abnormality. Normal ducts unrelated to neoplastic lesions did not display K-ras mutations (0/10). The prevalence of K-ras mutations was 21% in ductal hyperplasia (3/14), 50% in dysplastic or carcinoma in situ samples (3/6), and 91% in invasive cancer (10/11). K-ras mutations were significantly more frequent in cancer than in normal and hyperplastic ducts (P < .001). The mutations were always detected in codon 12 and consisted of GGT to GAT and AGT gene mutations.
DISCUSSION
Although the progenitor cell of human pancreatic adenocarcinomas is still controversial, experimental evidence indicates that early pancreatic cancer of ductal phenotype arises in small pancreatic ducts or ductules. 4 The transformation of ductules to tubular complexes represents the earliest stage in the evolution of DMBA-induced pancreatic cancer and can be consistently found as early as 14 days after carcinogen placement. 6 Surrounded by a marked inflammatory reaction, dysplastic changes evolve in tubular complexes within the first month after carcinogen implantation, and early dysplasia and cancer develop through these distinctive stages in 30% to 40% of animals. The model displays positive ductal, negative acinar, and negative islet cell markers in tubular complexes and pancreatic cancer. 4
DMBA induces cancer by covalent binding to DNA, forming DNA adducts that can be measured by 32P postlabeling techniques. 7 DNA adduct formation of carcinogens can lead to mutations in oncogenes and tumor suppressor genes. 8 Carcinogen DNA adducts have been found in human and rat organs, and increased levels of aromatic and lipid peroxidation-related DNA adducts in pancreas adjacent to cancers in human tissues are consistent with a pathogenetic role of DNA adducts in pancreatic cancer. 9
Interactions of carcinogens with DNA can be altered by dietary and environmental factors. A Western-style diet has been shown to induce hyperproliferation of interlobular ducts and intralobular ducts but not of centroacinar cells in rodents, and may sensitize the pancreas to the effects of carcinogens. 10,11 A high-fat diet also acts as a promoter in a model of pancreatic cancer that seems to arise in islets. 12 A promoting effect of a high-fat/high-protein diet on tumor development, as shown in this study, is also suspected in human pancreatic cancer. Various promoting and protective genetic, dietary, and environmental factors have an impact on the multistep process of carcinogenesis, but experimental and clinical data support the significant influence of diet in pancreatic cancer. 5
A high-fat diet and obesity, both increasingly prevalent in Western societies, have been suspected risk factors for human pancreatic cancer and other cancers. 5,13 A constant positive energy balance leading to obesity and specific lipid effects on tumor promotion are thought to be pathogenetically important. 14,15 Whereas the specific mechanisms involved are not known, a negative effect of a high-fat diet on gene repair mechanisms or enhanced DNA repair by constant energy restriction has previously been proposed. 16 The dietary influence on the integrity of the p53 tumor suppressor gene 17 and on the efficiency of repairing DNA fragments of various genes, including ras genes, supports this hypothesis. 18 High dietary fat ingestion may also promote tumorigenesis by increasing COX-2 and ras-p21 expression and membrane localization of ras-p21, which is essential for ras function. The influence of dietary factors on ras pathways may be particularly important in a model with a high prevalence of activating K-ras mutations. 19–21
The hypothesis that dietary factors can enhance pathologic ras pathways and concurrently inhibit repair mechanisms by interfering with tumor suppressor genes is supported by our results. The HFPD acted as a promoter of carcinogenesis in the DMBA rat model of pancreatic cancer. At 9 months the prevalence of pancreatic adenocarcinoma (29%) was significantly increased compared with normal-diet controls (17%). The cancer prevalence in HFPD rats also increased from 16% at 1 month to 29% at 9 months, whereas that in control rats decreased to 17%, indicating that regression of neoplastic lesions may occur as a normal process in a subgroup of animals fed a normal diet. If the dysplastic precursor lesions are included in the analysis, the cancer-promoting effect of the HFPD and the apparent regression in normal diet conditions is retained.
In summary, this carcinogen-induced rodent model of ductal adenocarcinoma provides further support for the epidemiologic observation that an HFPD promotes the development of pancreatic cancer, perhaps by interfering with natural reparative defense mechanisms that would otherwise abort the oncogenic progression.
Acknowledgments
The authors thank Ms. S. Kupa for her technical assistance.
Discussion
DR. MARSHALL M. URIST (Birmingham, Alabama): In 1945, Albert Tannenbaum published a paper in Cancer Research entitled ‘The Dependence of Tumor Formation on the Composition of the Calorie-Restricted Diet As Well As on the Degree of Restriction.‘ He examined both total caloric and relative composition influences on the development of chemically-induced and spontaneous mammary and skin tumors in mice. He concluded that elevated fat content in the diet increased the incidence of tumors, even in the face of the inhibitory effect of calorie restriction. This hallmark paper described two independent factors, total calorie consumption and high fat diet.
This is the same fact that Dr. Warshaw and colleagues have described in a rat model of pancreatic cancer induced by implanting DMBA directly in into the pancreas. The difference between study designs lies in the total amount of calories consumed by the two groups of control animals. Dr. Tannenbaum showed that the effect can be due to increased consumption of total calories alone.
The primary question for the authors, therefore, is: how does the total calorie intake compare between your two groups? Could the effect you observed be due to higher total calorie intake rather than the specific content of the high fat/high protein Westernized rat diet?
You also described that some animals become ill for a period of time after the pellet implantation. Did this lead to a change in the calorie consumption between the two groups?
Second, you described very nicely the transitory nature of the tumors seen in the control group. Why do early proliferative lesions not lead to cancer even in the control group? How long is the DMBA released into the local tissues?
The authors are to be congratulated on developing an interesting model of pancreatic cancer and using this to investigate factors that are known to be associated with the development of this disease.
I would like to thank the Association for the opportunity to comment on the paper and the authors for the opportunity to review their manuscript.
DR. ALEX S. ROSEMURGY, II (Tampa, Florida): It is a privilege to have the opportunity to discuss this. In summary, this paper shows that pancreatic adenocarcinomas did occur with a normal diet after implantation of a strong carcinogen, but the rats were able to prevent clinical progression of the cancer in the absence of a high-fat/high-protein diet. Several questions come to mind.
What is the deleterious factor in the high-fat/high protein diet? It seems that it is not the high protein, since the protein concentrations in the two diets were quite similar; whereas, the fat content in the diets was quite dissimilar. Is diet different than obesity, as was just touched upon? It would be interesting to know if the rats were exercised to weight control, if there would be a difference in the progression of the cancer, since it is not the appearance of the cancer that seems to be different.
Can you speculate about analogous dietary factors that we might be exposed to that would make a difference in our clinical population, as pancreatic cancer seems to be increasing in this ever increasing obese population of ours?
How does the high-fat diet and the factors in that diet exert its effect? Obviously, it seems to have its effect through some process through genetic repair, but that is not necessarily directly apparent.
Lastly, how can we use utilize it in our treatment to protect from or treat cancers that are induced by dietary factors?
Thank you very much.
DR. COURTNEY M. TOWNSEND, JR. (Galveston, Texas): In Pour’s study using the Syrian golden hamster also treated with carcinogen (but by systemic administration), the changes in that model, particularly with the RAS mutations, were identical to changes in human cancers. In addition, there was a high incidence of perineural invasion in the Syrian hamster model just like there is in humans. Now you have developed a different animal model, a different species, and it seems like the changes are identical to human and to another animal model. It’s a really neat model and ought to provide significant information. I have two questions.
One, are the changes, particularly the precancerous changes, limited to the area of local application of the carcinogen, or are there changes throughout the pancreas?
Second, have you looked for or found any perineural invasion in the animals that have developed tumors and died from tumors?
Thank you.
DR. ANDREW L. WARSHAW (Boston, Massachusetts): I want to first thank Dr. Townsend for asking those questions, because up until that moment this was going to be a unique experience for me in not being able to answer a single question asked in the course of the discussion. So you can anticipate many of my comments.
Dr. Urist, in effect, questioned whether it could be the caloric difference alone rather than the fat, and the answer is yes, our experimental model does not distinguish between those two. There clearly was an increased total calorie intake as manifested by weight gain in those rats, so I can’t answer that question, Dr. Urist.
Could it have been some effect of other illness, whether it was the pneumonia or pancreatitis? I don’t think so because those rats in general died, and they were autopsied, and their presence or absence of cancer was included in the cumulative figures, and that did not affect the overall curve.
Your third question was why don’t the tumors progress in the normal diet, a similar question to what Dr. Rosemurgy asked. The answer can only be speculated on the basis of previous work of others cited on COX-2 and on DNA error repair and a variety of other speculative possibilities.
Dr. Rosemurgy asked what is the toxin in this, whether it be the calorie, the fat, or something else. I don’t know, and we have an observation without an explanation.
What potential human dietary factors might we address so that none of us get pancreatic cancer? I don’t know. I wish I did. Many years ago we proved it wasn’t coffee, thank goodness.
What is the mechanism of repair? I have tried to address that in my limited fashion. What dietary modifications might be implied? Again, I don’t know.
Dr. Townsend, the hamster, the Syrian Golden hamster model that you referred to uses a carcinogen called BOP. That has been probably one of the most extensively studied animal models of pancreatic cancer that we have. The originator of that model, Parvis Pour from Omaha, believes it is of islet cell origin and has been spending the last 10 years trying to prove that this is a different kind of cancer. In fact, he is trying to prove that all human cancer originates from the islet cells rather than from the ducts, so that story is a little bit off. But it probably is at least an acinar cell carcinoma, in the opinion of most, so it may not be comparable completely.
The one question I can answer: are the changes local or general as induced by this carcinogen? They are definitely local. It is not seen in the rest of the pancreas or elsewhere in the main pancreatic duct.
The final question was why aren’t these changes permanent? I’d love to know. Maybe I will be back another year. Thank you very much.
Footnotes
Presented at the 112th Annual Meeting of the Southern Surgical Association, December 4–6, 2000, Palm Beach, Florida.
Correspondence: Carlos Fernández-del Castillo, MD, Department of Surgery, Massachusetts General Hospital, WACC 336, Boston, MA, 02114.
Accepted for publication December 2000.
References
- 1.Z’graggen K, Rivera JA, Compton CC, et al. Prevalence of activating K-ras mutations in the evolutionary stages of neoplasia in intraductal papillary mucinous tumors of the pancreas. Ann Surg 1997; 226: 491–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jimenez RE, Warshaw AL, Z’graggen K, et al. Sequential accumulation of K-ras mutations and p53 overexpression in the progression of pancreatic mucinous cystic neoplasms to malignancy. Ann Surg 1999; 230: 501–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rivera JA, Graeme-Cook F, Werner J, et al. A rat model of pancreatic ductal adenocarcinoma: targeting chemical carcinogens. Surgery 1997; 122: 82–90. [DOI] [PubMed] [Google Scholar]
- 4.Jimenez RE, Z’graggen K, Hartwig W, et al. Immunohistochemical characterization of pancreatic tumors induced by dimethylbenzanthracene in rats. Am J Pathol 1999; 154: 1223–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Silverman DT, Swanson CA, Gridley G, et al. Dietary and nutritional factors and pancreatic cancer: a case-control study based on direct interviews. J Natl Cancer Inst 1998; 90: 1710–1719. [DOI] [PubMed] [Google Scholar]
- 6.Bockman DE, Black O, Mills LR, et al. Origin of tubular complexes developing during induction of pancreatic adenocarcinoma by 7,12-dimethylbenz(a)anthracene. Am J Pathol 1978; 90: 645–658. [PMC free article] [PubMed] [Google Scholar]
- 7.Izzotti A, Camoirano A, Cartiglia C, et al. Patterns of DNA adduct formation in liver and mammary epithelial cells of rats treated with 7,12-dimethylbenz(a)anthracene, and selective effects of chemopreventive agents. Cancer Res 1999; 59: 4285–4290. [PubMed] [Google Scholar]
- 8.Turteltaub KW, Frantz CE, Creek MR, et al. DNA adducts in model systems and humans. J Cell Biochem (Suppl) 1993; 17F: 138–148. [DOI] [PubMed] [Google Scholar]
- 9.Wang M, Abbruzzese JL, Friess H, et al. DNA adducts in human pancreatic tissues and their potential role in carcinogenesis. Cancer Res 1998; 58: 38–41. [PubMed] [Google Scholar]
- 10.Xue L, Yang K, Newmark H, et al. Epithelial cell hyperproliferation induced in the exocrine pancreas of mice by a Western-style diet. J Natl Cancer Inst 1996; 88: 1586–1590. [DOI] [PubMed] [Google Scholar]
- 11.Chowdhury P, Nishikawa M, Blevins GW Jr, et al. Response of rat exocrine pancreas to high-fat and high-carbohydrate diets. Proc Soc Exp Biol Med 2000; 223: 310–315. [DOI] [PubMed] [Google Scholar]
- 12.Birt DF, Julius AD, White LT, Pour PM. Enhancement of pancreatic carcinogenesis in hamsters fed a high-fat diet ad libitum and at a controlled calorie intake. Cancer Res 1989; 49: 5848–5851. [PubMed] [Google Scholar]
- 13.Chow WH, Gridley G, Fraumeni JF, et al. Obesity, hypertension, and the risk of kidney cancer in men. N Engl J Med 2000; 343: 1305–1311. [DOI] [PubMed] [Google Scholar]
- 14.Albanes D. Total calories, body weight, and tumor incidence in mice. Cancer Res 1987; 47: 1987–1992. [PubMed] [Google Scholar]
- 15.Guthrie N, Carroll KK. Specific versus nonspecific effects of dietary fat on carcinogenesis. Prog Lipid Res 1999; 38: 261–271. [DOI] [PubMed] [Google Scholar]
- 16.Kritchevsky D. The effect of over- and undernutrition on cancer. Eur J Cancer Prev 1995; 4: 445–451. [DOI] [PubMed] [Google Scholar]
- 17.Kim YI, Shirwadkar S, Choi SW, et al. Effects of dietary folate on DNA strand breaks within mutation-prone exons of the p53 gene in rat colon. Gastroenterology 2000; 119: 151–161. [DOI] [PubMed] [Google Scholar]
- 18.Guo Z, Heydari A, Richardson A. Nucleotide excision repair of actively transcribed versus nontranscribed DNA in rat hepatocytes: effect of age and dietary restriction. Exp Cell Res 1998; 245: 228–238. [DOI] [PubMed] [Google Scholar]
- 19.Singh J, Hamid R, Reddy BS. Dietary fat and colon cancer: modulating effect of types and amount of dietary fat on ras-p21 function during promotion and progression stages of colon cancer. Cancer Res 1997; 57: 253–258. [PubMed] [Google Scholar]
- 20.Singh J, Hamid R, Reddy BS. Dietary fat and colon cancer: modulation of cyclooxygenase-2 by types and amount of dietary fat during the postinitiation stage of colon carcinogenesis. Cancer Res 1997; 57: 3465–3470. [PubMed] [Google Scholar]
- 21.Davidson LA, Lupton JR, Jiang YH, et al. Carcinogen and dietary lipid regulate ras expression and localization in rat colon without affecting farnesylation kinetics. Carcinogenesis 1999; 20: 785–791. [DOI] [PubMed] [Google Scholar]