

Abstract
Objective
To screen a library of small chemicals for compounds that activate the DPC4 signal transduction pathway in a human pancreatic cancer cell line.
Summary Background Data
Various tumor-suppressor genes are mutated in all human cancers. Specifically, DPC4 (deleted in pancreatic carcinoma, locus 4 or MADH4/SMAD4) is a tumor-suppressor gene mutated in approximately 50% of human pancreatic adenocarcinomas. DPC4 plays an important role in the well-studied transforming growth factor-β (TGFβ) signaling pathway. It would be useful to identify therapies that augment or restore the downstream functions of this critical signal transduction pathway, in hopes that such therapy would have a rational role in anticancer therapy.
Methods
Using a commercially available plasmid vector with a luciferase reporter gene already incorporated, a DPC4-specific reporter construct was genetically engineered. This was done by inserting six copies of the palindromic Smad binding element (6SBE), which is a DNA binding element specific for DPC4, in front of the minimal promoter in the plasmid. This construct was then stably integrated into the genome of a human pancreatic cancer cell line (PANC-1) that has wild-type DPC4. Several stably transfected clones were tested for basal luciferase expression and inducibility with TGFβ, which is known to activate the DPC4 signal transduction pathway. A single transfected clone was chosen for the drug screen based on basal luciferase (reporter) expression and TGFβ inducibility. A systematic screen of the chemical library was then performed, using luciferase activity to detect DPC4 activity and induction of the signaling pathway.
Results
A high-throughput system based on this stably integrated reporter system was used to screen a library of 16,320 random compounds to identify agents that conferred robust augmentation of the DPC4 signal transduction pathway. Of the 16,320 compounds screened, 11 were associated with a 2- to 5-fold induction of luciferase activity, and one with a 12-fold activation. The latter compound was shown to be a novel histone deacetylase inhibitor and was further characterized.
Conclusions
These results confirm the feasibility of a specific high-throughput reporter system to screen a large compound library in human cells efficiently. The screening identified several compounds capable of augmenting DPC4-specific luciferase reporter activity, and a specific mechanism for one compound was identified. The discovery of such agents will aid our understanding of complex tumor-suppressive signaling pathways and may identify other potential therapeutic targets within this critical signaling pathway. In addition, random drug screening provides an unbiased method for identifying drugs or lead compounds for potential therapeutic use.
Various tumor-suppressor genes are mutated in all human cancers. DPC4 (deleted in pancreatic carcinoma, locus 4), also known as MADH4/SMAD4, is a tumor-suppressor gene located on chromosome 18q21.1. It is known to be inactivated in approximately 50% of human pancreatic adenocarcinomas, 1,2 as well as in many biliary 3,4 and colorectal cancers. 5,6 The DPC4 protein belongs to the evolutionarily conserved family of Smad proteins that serve as crucial intracellular mediators of the transforming growth factor-β (TGFβ) signaling pathway. The pathway is summarized in Figure 1. All members of the TGFβ superfamily of cytokines signal through membrane-bound serine threonine kinase receptor complexes. TGFβ and related ligands bind to their cell membrane receptors, which in turn phosphorylate the receptor-regulated Smad proteins, Smads 1 to 3. These phosphorylated Smads then form heteromeric complexes with DPC4 (specifically Smad2 and Smad3). These heteromeric complexes are translocated to the nucleus, where DPC4 binds a specific DNA sequence, the Smad-binding element (SBE), functioning as a transcriptional regulator. 7–10
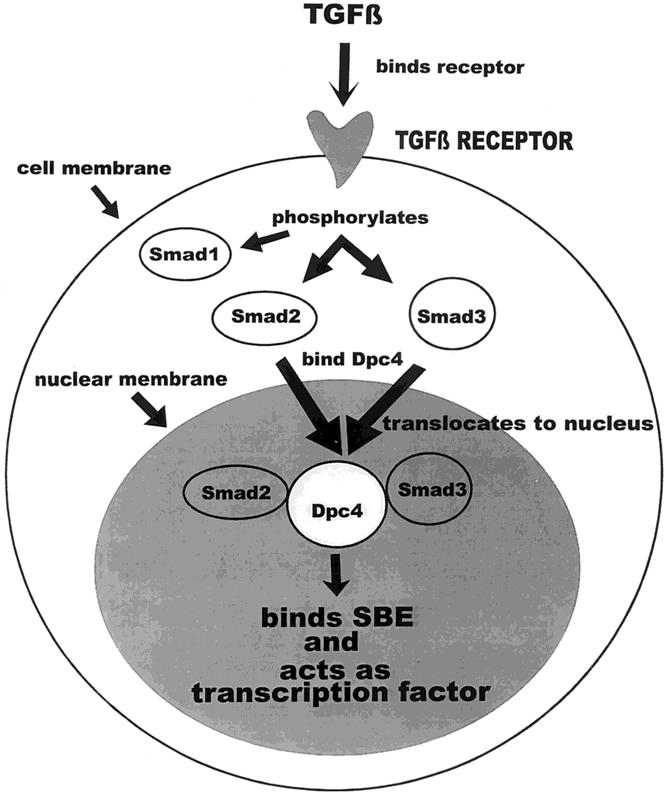
Figure 1. The transforming growth factor-β (TGFβ)/DPC4 pathway. TGFβ binds to its cell surface receptor, a serine threonine kinase, which then phosphorylates Smads 1 to 3. Once phosphorylated, Smad2 and Smad3 form heteromeric complexes with DPC4, which are translocated to the nucleus. There the complex binds the Smad-binding element (SBE) and acts as a transcription factor.
TGFβ can play complex roles in tumorigenesis, suppressing carcinogenesis in the early stages and acting as a tumor promoter in later stages. TGFβ is a potent inhibitor of epithelial cell growth, whose downstream functions include induction of apoptosis and cell-cycle arrest. Loss of DPC4 during tumor progression relieves this inhibition through a variety of mechanisms. 11
Reexpression of the DPC4 protein in vitro 12–16 and in vivo 11 has been shown to restore TGFβ signaling, induce apoptosis, and lead to growth inhibition. Therefore, it would be useful to identify therapies that augment or restore the downstream functions of this critical signal transduction pathway. The current methods commonly used to elucidate signal transduction pathways involve physical interactions (two-hybrid analysis or protein affinity chromatography), experimental genetic means (yeast or bacterial mutagenesis), and observations of natural genetic variance (human tumors or inherited disease susceptibility). Each of these methods has its limitations and strengths. Screening of random chemical libraries can encompass all the benefits of current methodologies. Such screening is both unbiased and high-throughput, meaning it can be performed efficiently on large numbers of compounds. Moreover, it can be readily applied to probe complex biologic systems in mammalian cells, making it applicable to human disease. 17 In addition, random screening of chemical libraries may identify drugs or lead compounds for potential therapeutic use in human cancers. This report confirms the feasibility of a specific high-throughput reporter system to screen a large compound library in human pancreatic cancer cells. The screening identified several chemicals capable of augmenting and inhibiting DPC4-specific luciferase reporter activity.
METHODS
Development of Reporter Constructs
The pGL3-promoter vector (Promega, Madison, WI) is a commercially available 5.0-kb plasmid vector containing an SV40 minimal viral promoter linked to a luciferase reporter gene. A luciferase reporter construct (p6SBE-luc) was genetically engineered by inserting six copies of the palindromic SBE (a sequence-specific DNA-binding element for DPC4) in front of the minimal promoter. The design and a map of the final construct are shown in Figure 2. The construct was confirmed by direct sequencing. The pGL3-control vector (Promega), which has a strong viral promoter and constitutively expresses luciferase at high levels when transfected into cells, was used to test the specificity of inhibitors. A p53-specific reporter was designed in similar fashion using the same parent vector.

Figure 2. The 6SBE-luc reporter construct. The pGL3-promoter vector was cut by two restriction enzymes, KpnI and SmaI. Six copies of the Smad-binding element (SBE) with KpnI and SmaI compatible (“sticky”) ends were then inserted in this site, in front of the SV40 minimal promoter and luciferase reporter gene. (Construct reported in Dai JL, Turnacioglu K, Schutte M, et al. Dpc4 transcriptional activation and dysfunction in cancer cells. Cancer Res 1998; 58:4592–4597.)
Development of Stably Transfected Cell Lines
The p6SBE-luc reporter construct was then stably integrated into the genome of a human pancreatic cancer cell line (PANC-1, which is wild-type for the DPC4 gene). The PANC-1 cell line was purchased from American Type Culture Collection (Manassas, VA). Stable transfectants were generated by cotransfection of pcDNA3.1 (Invitrogen, Carlsbad, CA) and p6SBE-luc into PANC-1 cells with Lipofectamine (Life Technologies, Inc.). pcDNA3.1 is a commercially available vector containing a G418 antibiotic resistance gene used in selection of clones. Transfection was performed according to the Lipofectamine protocol. Transfected cells were diluted and selected in multiple 96-well plates in the presence of 0.5 mg/mL G418 (Life Technologies, Inc.). Single clones were identified and expanded, then tested for basal luciferase expression and TGFβ inducibility. A single clone for use in drug screening was chosen on the basis of high (six- to eightfold) induction of luciferase activity by 0.5 ng/mL TGFβ.
Stable transfectants of the p53 reporter were made in similar fashion. Hs766T, a pancreatic cancer cell line that is wild-type for p53, was used.
Compound Screening
Each of the 16,320 compounds in the commercially available library (DIVERSet, ChemBridge, San Diego, CA) was dissolved and diluted in DMSO at 1 mg/mL and aliquoted into 96-well plates. Cells were then plated into 96-well cluster plates and incubated with each compound after further dilution in culture medium to a final concentration of 2 μg/mL. For each 96-well plate of compounds, three corresponding plates of cells were prepared (Fig. 3). On the first, the cells were treated with library compounds alone to identify any potential DPC4 agonists in the drug library. On the second plate, all wells contained cells plus TGFβ at low concentration (0.06 ng/mL), providing a two- to threefold increase in luciferase activity. This was done to identify any compounds acting synergistically with TGFβ to increase DPC4 activity. All wells on the third plate contained cells plus TGFβ at 0.50 ng/mL, providing a six- to eightfold increase in luciferase activity. This allowed screening for compounds that would inhibit DPC4 activity. Luciferase activity was measured 24 hours after the treatment of cells with compounds in the library. Steady-Glo luciferase substrate (Promega) was used to assay luciferase activity. Up to 16 96-well plates could be assembled in a Wallac (Gaithersburg, MD) Trilux photodetector for measurement. All of the readouts from each experiment were compared with control wells (untreated), and a number reflecting the relative increase in the luciferase activity was calculated for each chemical and stored in an Excel spreadsheet (Microsoft, Redmond, WA). Potentially promising agonists or synergists (twofold or greater activation) and inhibitors (less than 0.5-fold inhibition) were retested and confirmed.
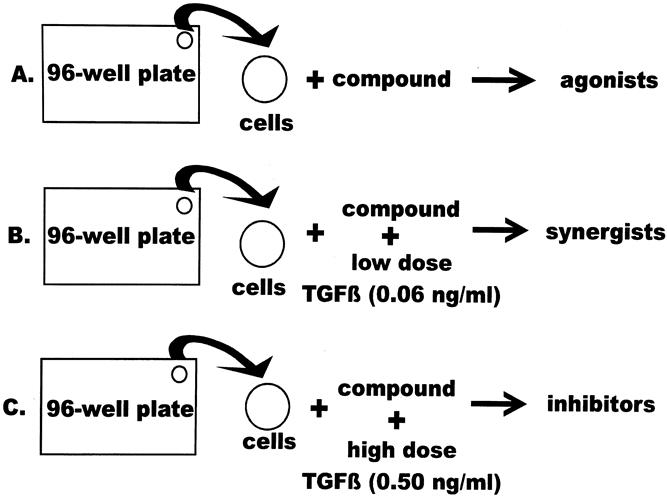
Figure 3. Compound screening procedure. Cells were plated on 96-well plates. A single drug from the library was added to a single well on each of three 96-well plates. The three plates contained (A) cells alone plus compound, to identify agonists, (B) cells alone plus compound plus low-dose transforming growth factor-β (TGFβ; 0.06 ng/mL), to identify synergists, and (C) cells alone plus compound plus high-dose TGFβ (0.5 ng/mL), to stimulate the reporter maximally and identify inhibitors.
RESULTS
Six copies of the palindromic SBE were successfully cloned into the pGL3-promoter vector and confirmed by direct sequencing. After confirming the TGFβ responsiveness in transient transfections in PANC-1 cells, stable transfectants were generated as described. A single clone was chosen for compound screening.
A high-throughput system based on this stably integrated DPC4-specific reporter system was used to screen the library of 16,320 compounds to identify compounds that conferred augmentation of the DPC4 signal transduction pathway (either alone or synergistically with TGFβ) or inhibited the DPC4 pathway.
Identifying Agonists
Of the 16,320 compounds screened, 12 were found to increase luciferase activity in the DPC4 reporter system. These 12 compounds were termed agonists. Five of the 12 compounds showed 3.0- to 5.0-fold activation, 6 compounds showed 2.0-to 2.9-fold activation, and 1 compound showed greater than 10.0-fold activation (12.0- to 18.0-fold).
This latter compound (6-(1,3–dioxo-1 H-3 H-benzo[de]isoquinolin-2-yl)hexanoic acid hydroxyamide) was further characterized and identified as a novel histone deacetylase inhibitor, termed Scriptaid, and reported previously. 17 This compound is a general transcriptional activator and was generally applicable to exogenous gene constructs, including viral and cellular promoters, different cell lines and reporter genes, as well as stably integrated and transiently introduced sequences. Using the Chemfinder software (Cambridge Soft, Cambridge, MA), we were able to identify structurally similar compounds within the library. One such compound, termed Nullscript, conferred no increased transcriptional activity. By comparing the structures of Scriptaid, Nullscript, and Trichostatin A (a known histone deacetylase inhibitor), we were able to determine a structure–function relationship, identifying the 5-carbon aliphatic chain as the functional component (Fig. 4).
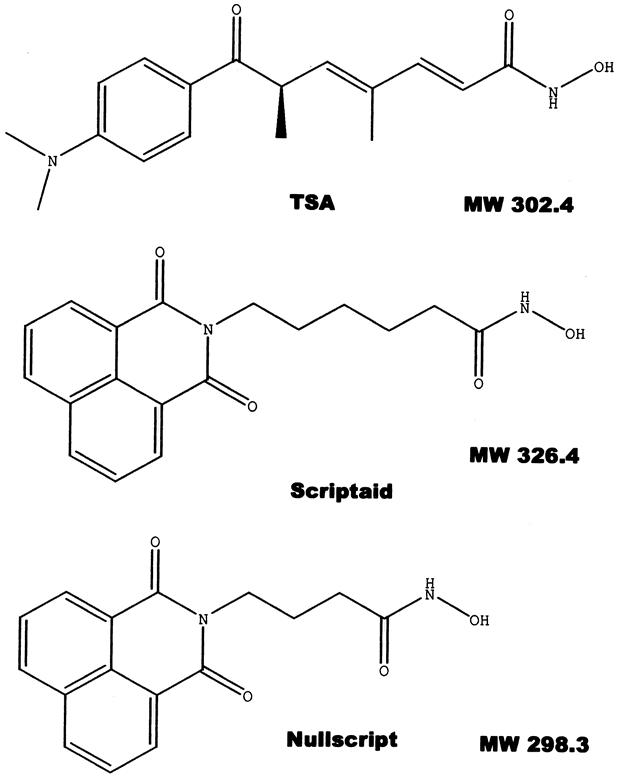
Figure 4. Structural similarities of Trichostatin A (TSA), Scriptaid, and Nullscript. They possess the same hydroxamic acid group, an aliphatic chain, and an aromatic cap at the other end. The aliphatic linkers of TSA and Scriptaid are 5-carbon in length, whereas the linker is only 3-carbon in Nullscript.
Two compounds with three- to fivefold induction of luciferase activity had similar chemical structures (acridin-9-yl-amines), differing only by a methyl group, and conferred similar augmentation of luciferase activity. Two additional compounds with three- to fivefold induction of the reporter were structurally alike, having benzo[g]quinolin-4-yl groups and conferring similar augmentation in reporter activity. The remaining compound in the 3.0- to 5.0-fold group and the six compounds with 2.0- to 2.9-fold activity were not structurally similar. The two acridin-9-yl-amines, as well as a structurally dissimilar agonist, are shown in Figure 5. Titration curves were performed to determine the concentration of maximal reporter activation for each agonist. The concentration for maximal activation ranged from 1 to 10 μg/mL. At levels above the maximal level of reporter activation for a given compound, cell death was observed.
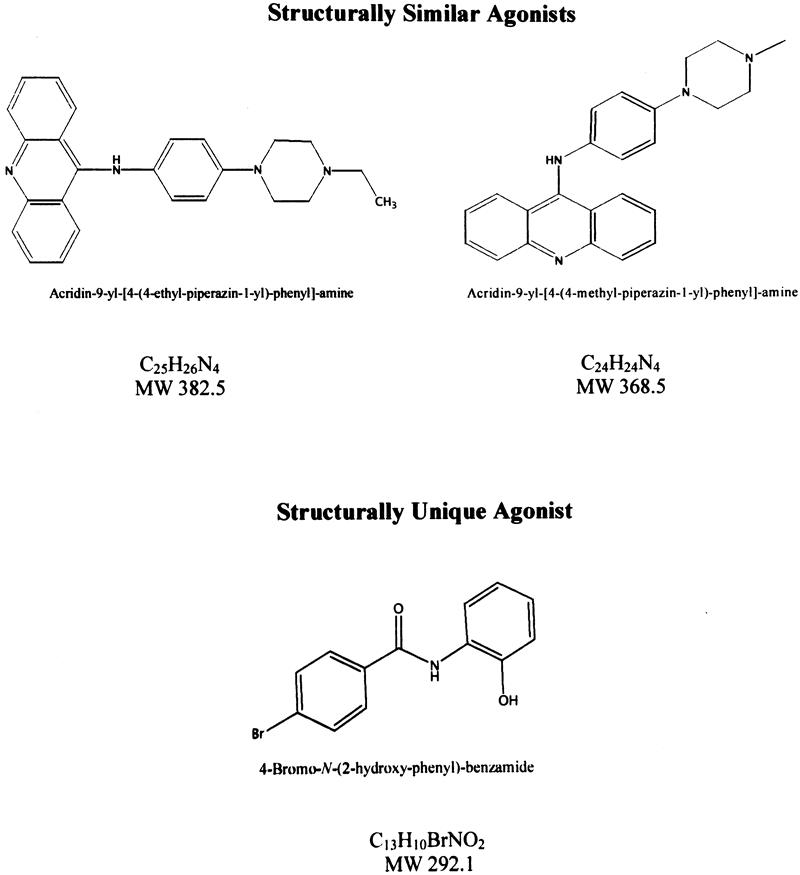
Figure 5. The chemical structures, formulas, and molecular weights (MW) of two structurally similar DPC4 agonists and one structurally unique agonist.
Identification of Synergists
Within the library, eight compounds were identified as synergists. Alone, these compounds did not significantly augment DPC4-reporter activity, but in conjunction with low-dose TGFβ (see Fig. 3), they showed two- to fourfold activation. These compounds showed no structural similarity to TGFβ or the other agonists identified.
Identification of Inhibitors
Eight inhibitors were identified during the drug screening. All were confirmed and retested in a cell line that constitutively expresses luciferase. In this system all the inhibitors showed decreased reporter activity, identifying them as general transcriptional inhibitors.
Screening in a Different Reporter System: The p53 System
The 12 agonists and 8 synergists were screened using a similar system but with a different reporter construct (T.A.S. and S.E.K., data not published). The reporter construct in this system was also engineered from the pGL3-promoter vector but contained an enhancer element specific for p53. A pancreatic cancer cell line that had wild-type p53 was used to develop stable transfectants. In this system, 5 of the 12 agonists identified in the DPC4 system showed no augmentation of reporter activity, suggesting that these compounds were acting specifically in the TGFβ/DPC4 pathway, thus identifying them as the compounds most interesting for further testing. The remaining seven agonists, including Scriptaid, augmented luciferase activity in both the DPC4-specific and p53-specific reporter systems, suggesting that these compounds were acting as general transcriptional activators (i.e., activating the general transcriptional machinery of the cell). Of note, all eight potential synergists showed no augmentation of reporter activity in the p53-specific system. This, in conjunction with their dependence on the presence of TGFβ for reporter induction, suggests that these compounds were specific for the DPC4 pathway being studied.
DISCUSSION
The true activation of a complex biologic signaling pathway can be difficult to evaluate and quantify. The detection of downstream transcriptional events using a specific reporter construct is a useful method for studying such pathways. This type of manipulation requires a basic understanding of interactions between members of the pathway but does not require complete and precise knowledge of the pathway. We implemented a high-throughput compound screen to identify reliable and important regulators of a well-studied tumor-suppressive pathway. The TGFβ pathway comprises a number of tumor-suppressor genes including DPC4. 1,4,18 Our reporter construct contains six copies of the SBE, allowing us to measure processes that result in the nuclear localization of DPC4. 19 The discovery of agents that interact with or bypass deficits in the TGFβ pathway by augmenting the downstream pathway of DPC4 would aid in understanding this critical tumor-suppressive pathway while providing lead compounds for further testing or perhaps therapeutic use.
The random screening of large libraries of random chemical compounds encompasses many of the benefits of current methodologies used to dissect complex biologic systems. It has been shown to be unbiased, efficient, and feasible when used to probe complex biologic systems. 17,20,21 Its utility when applied to mammalian cells makes it readily applicable to human disease.
Our results confirm the feasibility of using a specific high-throughput reporter system to screen efficiently a large compound library in human cells to dissect a complex biologic pathway. Showing the feasibility of such an approach opens the doors to study any complex biologic system in this manner. In this case, we studied the TGFβ/DPC4 tumor-suppressive pathway. The initial screen of 16,320 compounds identified 20 lead compounds (12 agonists, 8 synergists) that are promising for further investigation. Several compounds identified in the screen have structural similarities. By searching chemical databases for compounds with similar chemical structures, we may be provided with more compounds to study for potential pathway activation, presumably from an enriched pool.
One compound, Scriptaid, was further classified as a histone deacetylase inhibitor, which acts as a general transcriptional activator. 17 The remaining compounds are now promising chemicals for further investigation. Further studies should include testing the compounds in transient transfections with a mutant SBE luciferase reporter known not to bind DPC4. Using such a system would enable us to identify the specificity of the compounds identified and to classify drugs further as general transcriptional activators or drugs acting specifically to alter the DPC4 pathway.
The five agonists and eight synergists that did not activate the p53-reporter system are especially interesting compounds for further study. Their lack of activity in the p53-reporter system suggests that these 13 compounds are specifically augmenting the DPC4 tumor-suppressive pathway. By identifying where in the pathway these compounds are acting, we may further define the pathway. Once their role in the pathway is understood, these compounds may be candidates for therapeutic intervention. For instance, a chemical that is found to augment DPC4 activity by increasing DPC4 translocation to the nucleus would require the presence of functional DPC4 protein to be effective and may serve as a useful chemotherapeutic agent in tumors that have wild-type DPC4. If, instead, a compound bypasses the need for the DPC4 protein by augmenting the binding of other Smad proteins to DNA, this compound might be useful in tumors lacking functional DPC4. Reexpression of the DPC4 protein in vitro 12–16 and in vivo 11 has been shown to restore TGFβ signaling, induce apoptosis, and lead to growth inhibition, suggesting that the restoration of this critical pathway could be a means of rational therapy.
A recently developed immunohistochemical technique has led to extremely sensitive and specific labeling for the DPC4 gene product. 22–25 DPC4 inactivation appears to be a late event in pancreatic tumorigenesis, with an increased incidence being identified along the precursor progression model. For example, the DPC4 protein is present in flat, papillary, or atypical papillary intraductal neoplasms (PanIN-1A, 1B, and 2) but is inactivated in approximately 30% of severely atypical intraductal neoplasms (PanIN-3), as shown by immunohistochemical labeling to identify the genetic status of DPC4. 22 Further, the DPC4 gene is inactivated in 50% of pancreatic adenocarcinomas. 1,2
Using these current techniques, 22–25 one could identify the DPC4 status of a resected or biopsied pancreatic cancer. Potentially, this classification could be used to guide rational therapy. For example, an agent found to augment the downstream action of DPC4 might be therapeutic in wild-type tumors, whereas agents that bypass deficits in the pathway may restore the function of the pathway in tumors that are deficient in DPC4.
With advances in immunohistochemistry and drug screening specific for tumor-suppressive pathways, one can begin to think about rational therapy for pancreatic and other cancers. Similar screens can be performed for other tumor-suppressor pathways (e.g., p16, p53, BRCA2, MKK4), and compounds that could bypass or augment these pathways may be identified. By knowing the genetic status of a patient’s tumor, therapy could then be tailored to include compounds that augment intact pathways and restore dysfunctional pathways critical in tumor suppression.
Discussion
DR. B. MARK EVERS (Galveston, Texas): I would like to congratulate the authors on a nicely presented and well-done study. The authors have used a high-throughput system to rapidly screen the potential effectiveness of over 16,000 compounds to activate the DPC4 tumor suppressor in a human pancreatic cancer cell line. Using this novel and rapid screening procedure, they have identified potential agents that look promising in the activation of the DPC4 protein, which is an intracellular regulator of the TGF-beta signalling pathway. The restoration or activation of the DPC4 protein in pancreatic cancers may provide for a novel approach to treat this disease of which there are no effective treatments. I would like to ask Dr. Sohn three questions regarding this study.
First, as you know, this is a highly artificial system where you have used a vector with not one Smad binding element but 6 copies of this site placed in front of luciferase. I certainly can understand and appreciate the need to do this in your system but then worry that the cutoff that you have used to assume potential biological relevance is only a two-fold stimulation of luciferase activity. Obviously, you have to start somewhere, but could you comment on how you chose this fold stimulation as your cutoff point. As a corollary to this question, since you have only measured the ability of these compounds to induce activity of a reporter gene, have you assessed whether the compounds that stimulate luciferase activity can actually increase the expression of the wild type DPC4 gene and protein in the Panc-1 cell line? This is obviously a critical experiment and would determine whether this rapid screening method can provide you with the information that you are seeking; that is, do these compounds activate DPC4 in vivo?
My second question has to do with the potential compounds that you have identified in your study. The chemical scriptaid produced the most marked induction of luciferase activity, but this also occurred using the p53 reporter system, suggesting that this may not be specific for DPC4. However, you found that five of the agonists that stimulated DPC4 activity did not stimulate P53 activity. Have you had the opportunity to better assess these five compounds in terms of their chemical actions and possible utility as therapeutic agents?
Finally, one of the major objectives of this study is to determine whether these chemical agents can restore the function of DPC4 in pancreatic cancer cells in which 50% of these cancers are known to have DPC4 mutation. If that is the case, why did you use a wild-type Smad binding element? Why not use a mutated Smad binding element to determine whether your compounds induce the activity of the mutated promoter element?
I enjoyed the presentation and the paper. I think that the system that you describe has the possibility of greatly accelerating by light years the ability to discover potential compounds that could be used in the adjuvant treatment of cancers. One could even envision using this screening method to better ‘customize‘ chemothereapy based upon the individual tumor genetic makeup and the ability to respond to chemotherapeutic compounds.
I thank you again for the opportunity to comment on this paper.
DR. MARSHALL M. URIST (Birmingham, Alabama): Drs. Yeo, Sohn, and colleagues have presented an elegant and sophisticated technique to screen large numbers of agents for their ability to enhance or suppress the growth of pancreatic cancer cells. More than simply a biological observation, re-expression of the DPC4 protein has been shown in other studies to re-establish TGF-beta signaling, induce apoptosis, and result in growth inhibition.
I would like to know if you have shown any phenotypic changes in your cell culture population to indicate that the drugs detected do have activity in the manner indicated by the assay.
How specific are these transcriptional activators? One you have described as a general transcriptional activator. Are there also adverse consequences of this property? Are there additional tumor suppressor pathways that can be measured by this technique and how many do you postulate you would have to measure in order to apply a treatment protocol?
The authors have prepared an excellent manuscript that I recommend to everyone. It clearly describes the complexity of their technique and the potential for its application. And I would like to thank them for the opportunity to review their manuscript and comment on their work.
DR. TAYLOR A. SOHN (Baltimore, Maryland): I’d like to thank Dr. Evers and Dr. Urist for their comments. I will handle Dr. Evers’ questions first.
With regard to choosing the 6SBE or the 6Smad binding elements, when the initial construct was made, it was made in blue script, and we made multiple concatemers of different lengths. We found that the six copies worked best in our reporter system. It is admittedly an artificial system, but we had to start somewhere, and we picked two-fold as our cutoff.
If you look at our screen and our normalization numbers, there are many, many compounds that activate 1.2, 1.3-fold, but 2-fold seemed to be a reasonable cutoff, as we looked at all the data that we obtained on the 16,000 drugs.
We have not assessed our protein expression of DPC4. And part of the reason for not doing that is that it is not necessarily the levels of DPC4 protein that matter. There have been experiments that have been done that showed that sequestering normal levels of DPC4 in the cytoplasm has the same effect as not having DPC4. So it may in fact just be translocation to the nucleus that is important and not necessarily overall DPC4 levels. It would be simple enough in the future to do experiments assessing that and to further develop a mechanism.
You mentioned the five agonists that were specific for DPC4, and you also mentioned Scriptaid, which was a general transcriptional activator. Scriptaid we have a mechanism for. It’s a histamine deacetylase inhibitor and we know that is a general transcriptional activator.
And to handle Dr. Urist’s question, yes, there are adverse effects of using a general transcriptional activator, and you would worry about doing that in treatment of cancer. That’s why TSA and other histamine deacetylase inhibitors are not used.
We have not further characterized the 5 agonists. Actually, the compounds that are more interesting to me are probably the 8 synergists, because they not only are specific – they don’t show up in our p53 screen– but they also work in conjunction with DPC4, suggesting that they are very specific for the pathway.
The reason we chose a wild-type SBE element is it is that we want findings that bind to the wild-type element. That is, they are binding to DNA in the nucleus to initiate transcription. It is the DPC4 protein that is mutated, not the binding element, so we wanted to start screening there.
Dr. Urist asked about phenotypic changes in our cells. We have looked at the cells only in the sense of doing killing curves or concentration curves. Once we have identified compounds, we have tested them over a range of concentrations. And, as you would expect, at very high concentrations the cells die and at lower concentrations the activation wears off. But we have not further characterized any of these compounds extensively.
The beauty of this is that it can be applied to any complex biological system. We have, in fact, on the same screen in p53, we used a different pancreatic cancer cell line that was wild type for p53 and performed a similar screen, and actually, that screen is even more interesting than this one. I think the p53 pathway is more ubiquitous and more widespread, and we captured a lot more interesting compounds in that screen.
In terms of further applications, we can certainly envision being able to do this for a number of oncogenic and tumor suppressive pathways. And then, in combination with our immunohistochemical techniques and our molecular genetic techniques, we can characterize or get a molecular fingerprint, per se, of a patient’s tumor. And we can then choose from this list of compounds. We can choose to augment pathways that we know are wild type in a patient’s tumor or to choose something that bypasses a pathway in a system that we know is mutant.
I’d like to thank the Association for the privilege of closing the paper.
Footnotes
Presented at the 112th Annual Meeting of the Southern Surgical Association, December 4–6, 2000, Palm Beach, Florida.
Supported in part by grants from the National Institutes of Health SPORE Grant in Gastrointestinal Research (RO1-CA62694), the Niarchos Foundation, and the American Hepato-Pancreato-Biliary Association/Ethicon.
Correspondence: Charles J. Yeo, MD, Professor of Surgery and Oncology, The Johns Hopkins Medical Institutions, Blalock 606, 600 N. Wolfe St., Baltimore, MD 21287-4606.
E-mail: cyeo@jhmi.edu
Accepted for publication December 2000.
References
- 1.Hahn SA, Schutte M, Hoque ATMS, et al. DPC4, a candidate tumor-suppressor gene at 18q21.1. Science 1996; 271: 350–353. [DOI] [PubMed] [Google Scholar]
- 2.Schutte M, Hruban RH, Hedrick L, et al. DPC4 gene in various tumor types. Cancer Res 1996; 56: 2527–30. [PubMed] [Google Scholar]
- 3.Hahn SA, Bartsch D, Schroers A, et al. Mutations of the DPC4/Smad4 gene in biliary tract carcinoma. Cancer Res 1998; 58: 124–1126. [PubMed] [Google Scholar]
- 4.Goggins M, Shekher M, Turnacioglu K, et al. Genetic alterations of the transforming growth factor beta receptor genes in pancreatic and biliary adenocarcinomas. Cancer Res 1998; 58: 5329–5332. [PubMed] [Google Scholar]
- 5.Takagi Y, Kohmura H, Futamura M, et al. Somatic alterations of the DPC4 gene in human colorectal cancers in vivo. Gastroenterology 1996; 111: 1369–1372. [DOI] [PubMed] [Google Scholar]
- 6.Thiagalingam S, Lengauer C, Leach FS, et al. Evaluation of candidate tumour suppressor genes on chromosome 18 in colorectal cancers. Nat Genet 1996; 13: 342–346. [DOI] [PubMed] [Google Scholar]
- 7.Lagna G, Hata A, Hemmati-Brivanalou A, et al. Partnership between DPC4 and SMAD proteins in TGF-β signaling pathways. Nature 1996; 383: 832–836. [DOI] [PubMed] [Google Scholar]
- 8.Derynck R, Feng XH. TGF-beta receptor signaling. Biochem Biophys Acta 1997; 1333: F105–150. [DOI] [PubMed] [Google Scholar]
- 9.Heldin CH, Miyazono K, ten Dijke P. TGF-beta signaling from cell membrane to nucleus through SMAD proteins. Nature 1997; 390: 465–471. [DOI] [PubMed] [Google Scholar]
- 10.Attisano L, Wrana J. Smads as transcriptional co-modulators. Curr Opin Cell Biol 1998; 10: 188–194. [DOI] [PubMed] [Google Scholar]
- 11.Schwarte-Waldhoff I, Volpert OV, Bouck NP, et al. Smad4/DPC4 mediated tumor suppression through suppression of angiogenesis. Proc Natl Acad Sci 2000; 97: 9624–9629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.de Caestecker MP, Hemmati P, Larisch-Bloch S, et al. Characterization of functional domains within Smad4/DPC4. J Biol Chem 1997; 272: 13690–13696. [DOI] [PubMed] [Google Scholar]
- 13.de Winter JP, Roelen BAJ, ten Dijke P, et al. DPC4 (SMAD4) mediates transforming growth factor-beta1 (TGF-beta1) induced growth inhibition and transcriptional response in breast tumour cells. Oncogene 1997; 14: 1891–1899. [DOI] [PubMed] [Google Scholar]
- 14.Grau AM, Zhang L, Wang W, et al. Induction of p21waf1 expression and growth inhibition by transforming growth factor beta involve the tumor suppressor gene DPC4 in human pancreatic adenocarcinoma cells. Cancer Res 1997; 57: 3929–3934. [PubMed] [Google Scholar]
- 15.Dai JL, Bansal RK, Kern SE. G1 cell cycle arrest and apoptosis induction by nuclear Smad4/Dpc4: phenotypes reversed by a tumorigenic mutation. Proc Natl Acad Sci USA 1999; 96: 1427–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Atfi A, Buisine M, Mazars A, et al. Induction of apoptosis by DPC4, a transcriptional factor regulated by transforming growth factor-beta through stress-activated protein kinase/c-Jun N-terminal kinase (SAPK/JNK) signaling pathway. J Biol Chem 1997; 272: 24731–24734. [DOI] [PubMed] [Google Scholar]
- 17.Su GH, Sohn TA, Ryu B, et al. A novel histone deacetylase inhibitor identified by high-throughput transcriptional screening of a compound library. Cancer Res 2000; 60: 3137–3142. [PubMed] [Google Scholar]
- 18.Whitman M. Smads and early developmental signaling by the TGF-β superfamily. Genes Dev 1998; 12: 2445–2462. [DOI] [PubMed] [Google Scholar]
- 19.Dai JL, Turnacioglu K, Schutte M, et al. Dpc4 transcriptional activation and dysfunction in cancer cells. Cancer Res 1998; 58: 4592–4597. [PubMed] [Google Scholar]
- 20.Komarov PG, Komorova EA, Kondratov RV, et al. A chemical inhibitor of p53 that protects mice from the side effects of cancer therapy. Science 1999; 285: 1733–1737. [DOI] [PubMed] [Google Scholar]
- 21.Mayer TU, Kapoor TM, Haggarty SJ, et al. Small molecule inhibitor of mitotic spindle bipolarity identified in a phenotype-based screen. Science 1999; 286: 971–974. [DOI] [PubMed] [Google Scholar]
- 22.Wilentz RE, Iacobuzio-Donahue CA, Aragani P, et al. Loss of expression of DPC4 in pancreatic intraepithelial neoplasia (PanIN): evidence that DPC4 inactivation occurs late in neoplastic progression. Cancer Res 2000; 60: 2002–2006. [PubMed] [Google Scholar]
- 23.Wilentz RE, Su GH, Dai JL, et al. Immunohistochemical labeling for dpc4 mirrors genetic status in pancreatic adenocarcinomas: a new marker for DPC4 inactivation. Am J Pathol 2000; 156: 37–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Iacobuzio-Donahue CA, Klimstra DS, Adsay NV, et al. Dpc-4 protein is expressed in virtually all human intraductal papillary mucinous neoplasms of the pancreas. Comparison with conventional ductal adenocarcinomas. Am J Pathol 2000; 157: 755–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maitra A, Molberg K, Albores-Saavedra J, et al. Loss of Dpc4 expression in colonic adenocarcinomas correlates with the presence of metastatic disease. Am J Pathol 2000; 157: 1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]