

Abstract
Objective
To assess the strategy of combining oncolytic herpes simplex virus (HSV) therapy with immunomodulatory therapy as treatment for experimental colon cancer. The oncolytic HSV recombinant NV1023 and the interleukin 12 (IL-12)-secreting oncolytic NV1042 virus were evaluated in vitro and in vivo with respect to antitumor efficacy.
Summary Background Data
Genetically engineered, replication-conditional, attenuated HSVs have shown oncolytic activity against a wide variety of solid malignancies. Other strategies for treating cancer have involved immunomodulation and cytokine gene transfer using viral vectors. This study has combined both of these strategies by inserting the murine IL-12 gene into a replication-competent HSV. This approach allows oncolytic therapy to replicate selectively within and lyse tumor cells while providing the host immune system with the cytokine stimulus necessary to recruit and activate inflammatory cells needed to enhance the antitumor effect.
Methods
NV1023 is a multimutant HSV based on the wild-type HSV-1 F strain. NV1042 was created by insertion of the mIL-12 gene into NV1023. Cytotoxicity and viral proliferation of both NV1023 and NV1042 within murine CT26 colorectal cancer cells were first shown. Cells infected with NV1042 were then shown to produce significant levels of IL-12. Using an experimental flank model of colon cancer, mice were treated with both high and low doses of NV1023 or NV1042 and were followed up for both cure and reduction in tumor burden.
Results
Both viruses could replicate within and kill CT26 cells in vitro, with 100% cytotoxicity achieved after infection by either virus. Only NV1042 could produce mIL-12. Therapy using high viral doses to treat animals in vivo showed equal efficacy between NV1023 and NV1042, with five of seven cures for each virus. When viral doses were lowered, only the cytokine-producing NV1042 virus could reduce tumor burden and cure animals of their disease.
Conclusions
Both NV1023 and NV1042 have the oncolytic potential to kill colon cancer cells at higher doses. Cytokine production by NV1042 may allow lower doses of viral therapy to be used without losing antitumor efficacy. The combination of oncolytic viral therapy and immunomodulatory strategies should be further investigated as treatment for colon cancer.
Colon cancer continues to be one of the leading causes of cancer death in the United States. 1 Colon cancer develops in more than 130,000 people each year, with almost half expected to die of their disease despite aggressive therapy. 2 Although many patients can undergo primary resection at diagnosis, the high recurrence rate and the high incidence of hepatic and distant metastases both carry a poor prognosis. 3,4 Chemotherapy has provided some options for palliation, but many patients fail to respond to this therapy. 5,6 Novel treatments are needed to improve the outcome of this common and often fatal disease.
Therapy based on oncolytic herpes simplex virus (HSV) has been studied as therapy for many experimental solid malignancies. 7–12 Genetically engineered, replication-competent, oncolytic HSVs have been designed with deletion for one or more strategic genes that are important for viral growth. 7,13–17 It is believed that tumor cells maintain genes that complement these viral deletions and thereby support viral replication, whereas normal host tissue does not. 12,18 Thus, genetic manipulation permits these mutants to target and kill tumor cells selectively. Such oncolytic HSV therapy has reached phase I clinical trials. 19,20
Another novel antitumor strategy involves the use of viral vectors to deliver immunomodulatory agents such as cytokines, chemokines, or costimulatory molecules to sites of tumor growth. 21–24 This strategy seeks to recruit and activate inflammatory cells that have immediate antitumor effects and that may also provide lasting immunity against recurrent disease by stimulating immunologic memory. 25,26 Interleukin 12 (IL-12) is one such cytokine that is a promising agent for cancer gene therapy because of its many potent antitumor effects. 22,25,27 The goals of the current study were to determine whether an oncolytic HSV could be used as a vehicle for cytokine gene transfer, and to determine whether such combination therapy could enhance tumor kill compared with the oncolytic effects alone.
METHODS
Cells and Cell Culture
The murine colorectal cancer cell line CT26 was used for all experiments. This cell line was obtained from the ATCC and maintained in RPMI supplemented with 10% FCS, 5 mmol/L nonessential amino acids, and 1% penicillin and streptomycin. The cells were maintained in a 5% CO2 humidified incubator at 37°C.
Viruses
NV1020 is a nonselected clonal derivative of R7020 and has been previously characterized (Fig. 1). 14 NV1021 and NV1022 are precursor viruses to NV1023 and NV1042 and were derived from NV1020. NV1021 contains the Escherichia coli lacZ gene under control of the ICP47 (US12) promoter. The α4-driven thymidine kinase (TK) gene present in NV1020 and NV1021 was deleted from NV1021 to create the TK-negative virus NV1022. NV1023 was created by repair of the endogenous HSV-1 TK gene and the UL24 promoter in NV1022. The correct recombinants were identified by Southern blot analysis and were subsequently amplified. To create NV1042, murine IL-12 cDNA was inserted into NV1021 and TK-negative recombinants were selected in the presence of 20 μmol/L acyclovir. The endogenous TK gene and UL24 promoter were repaired to generate stocks of NV1042. The structures of NV1042 was confirmed by Southern blot analysis.
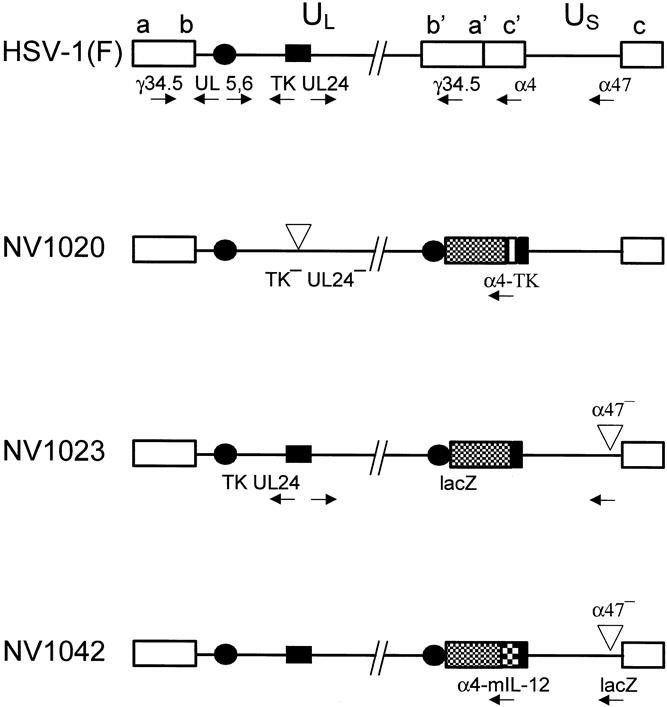
Figure 1. Genomic structures of the wild-type herpes simplex virus (HSV)-1(F) strain parent virus and derivative recombinants. HSV-1 contains both long (UL) and short (US) unique sequences. Locations of thymidine kinase (TK), UL24, UL5/6, γ134.5, α4, and α47 genes are shown. NV1020 has been deleted for the endogenous TK and UL24 genes; there is a duplication of the UL5/6 gene (solid circles), insertion of HSV-2 DNA (hatched box), and an exogenous copy of the TK gene driven by the α4 promoter. 14 NV1023 has been repaired for the endogenous TK/UL24 genes, the exogenous α4-TK gene has been removed, and the Escherichia coli lacZ gene has been inserted at the α47 locus under control of the α47 promoter. The mIL-12 gene has been inserted under control of a hybrid α4-TK promoter.
In Vitro Cytotoxicity and Viral Proliferation Assays
To evaluate the toxicity of each virus in vitro, CT26 cells were plated at 5 × 104 cells/well in 12-well plates (Costar; Corning Inc., Corning, NY). Cells were infected with either NV1023 or NV1042 at multiplicities of infection (MOIs; ratio of virions to cells) of 0, 0.1, 1, and 5. Cell viability was counted during 168 hours after infection at 48-hour intervals using trypan blue exclusion. In a separate assay, 5 × 105 cells/well were plated in six-well plates (Corning Inc.). These cells were infected with NV1023 or NV1042 at an MOI of 0 or 0.1. Cells and supernatants were harvested by cell scraping at 0, 6, 24, 48, 72, 96, and 120 hours after infection, subjected to three cycles of freeze–thaw lysis, and stored at −80°C. Viral titers were determined by standard plaque assay on Vero cell monolayers. All in vitro experiments were performed in triplicate.
Measurement of IL-12 Production
The ability of NV1042-infected CT26 cells to produce IL-12 in vitro was evaluated. CT26 cells were plated at 5 × 104 cells/well in 12-well plates (Corning Inc.) and infected with NV1042 at MOIs of 0, 0.1, 1, and 5. Supernatants from these infected cells were collected during 168 hours, and production of murine IL-12 (p70) was quantified by enzyme-linked immunosorbent assay according to the manufacturer’s instructions (R&D Systems, Minneapolis, MN). Assays were performed in triplicate. As a control, cells were also infected with the noncytokine-bearing parent NV1023 virus at an MOI of 1. These supernatants were also evaluated for IL-12 production by enzyme-linked immunosorbent assay.
Animal Studies
Balb/c male mice (4–6 weeks old) purchased from the National Cancer Institute (Bethesda, MD) were used for all animal experiments. All animal work was approved by the Memorial Sloan-Kettering Institutional Animal Care and Use Committee and performed under strict guidelines. Animals were anesthetized by intraperitoneal injection of ketamine–xylazine for all experiments. Flank tumors were established by subcutaneous injection of 5 × 105 CT26 cells. Animals were followed up daily for tumor growth. Tumor volume was calculated by the formula: ellipsoid volume = (4/3)*(π)*(length/2)*(width/2). When tumors reached an approximate size of 30 to 35 mm3, animals were randomly distributed into five equal groups. Control animals were treated with injection of 50 μL saline. Previous experiments had shown that saline injection is equivalent to treatment by injection of heat-inactivated virus. Animals treated with low-dose viral therapy were injected intratumorally with 1 × 106 plaque-forming units (pfu) of NV1023 or NV1042 in 50 μL virus buffer. Animals treated with high-dose viral therapy were injected intratumorally with 1 × 107 pfu NV1023 or NV1042 in 50 μL virus buffer. Tumors were measured periodically for 21 days. Tumor volumes were calculated using the formula for a prolate spheroid, 4/3(π)ab2, with a as the radius of the long axis and b as the radius of the short axis in millimeters. These measurements were carefully performed with vernier calipers. At this time, control tumors were at risk for ulcerating and the experiment was ended according to animal committee guidelines. Average tumor volume was calculated, with the value for nonpalpable tumors entered as 0.
RESULTS
In Vitro Cytotoxicity and Viral Proliferation Assays
The ability of NV1023 and NV1042 to kill CT26 colorectal cancer cells in vitro was evaluated at three different MOIs (Fig. 2). There was no difference in toxicity between these two viruses at the doses evaluated. MOI = 5 was the most toxic dose and resulted in 100% cell kill by 72 hours after infection. At MOI = 1, 100% toxicity was noted by 120 hours. The lowest MOI (0.1) resulted in 89% ± 17 toxicity at 168 hours for NV1023 and 92% ± 2 toxicity for NV1042. With respect to viral proliferation at MOI = 0.1, there was also no difference between NV1023 and NV1042 with respect to viral growth (Fig. 3). After infection with 50,000 pfu of either NV1023 or NV1042 (at 0 hours), viral uptake and disassembly resulted in a decrease in viral titers as measured at 6 and 24 hours after infection. Viral replication was significantly increased by 48 hours after infection and resulted in titers of 5.1 × 107 pfu NV1023 and 1.2 × 108 pfu NV1042 by 120 hours.

Figure 2. In vitro cytotoxicity of NV1023 and NV1042 against the murine colorectal cancer cell line CT26. Monolayer cell cultures were infected at multiplicities of infection (MOIs) of 0.1, 1, and 5, and viable cells were counted every 24 hours by trypan blue exclusion. Data are presented as mean cell survival (vs. controls) from triplicate wells (± standard deviation), with no difference in cytotoxicity noted between the two viruses.
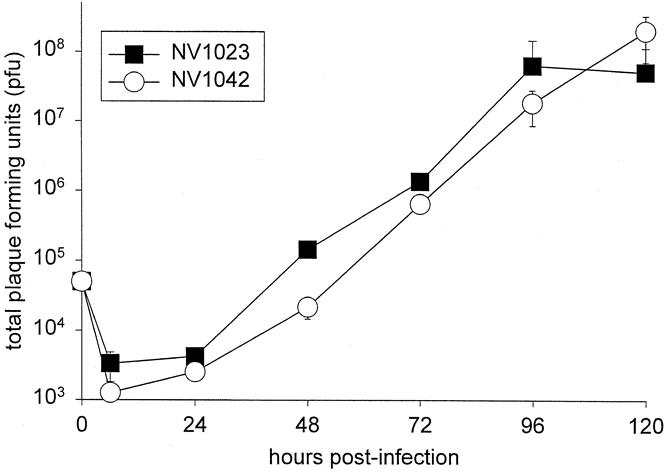
Figure 3. Replication of NV1023 and NV1042 in CT26 cells. In vitro infection was performed with a multiplicity of infection (MOI) of 0.1 and virions were collected every 24 hours for 120 hours. Data represent mean plaque-forming units (± standard deviation) from triplicate samples. There was no difference in the ability of these viruses to replicate in CT26 cells.
IL-12 Production
Production of IL-12 was evaluated in CT26 cells infected with NV1042. Infection at MOI = 5 resulted in a significant amount of IL-12 production in the first 24 hours (10 ng/mL) compared with MOI = 1 (1 ng/mL) and 0.1 (0 ng/mL)(Fig. 4). A plateau of IL-12 production was reached by 72 hours (20–27 ng/mL) at MOI = 5. At this dosage, 100% cytotoxicity was seen and the lack of viable cells capable of producing more IL-12 was responsible for this plateau. At MOI = 1, levels of IL-12 were not attained as rapidly as seen with MOI = 5. At the less-toxic MOI = 1, a higher plateau of IL-12 production was achieved (36–38 ng/mL). However, the 100% toxicity seen by 120 hours killed all viable cells capable of producing IL-12 and resulted in a plateau of cytokine production at this MOI as well. At the lowest MOI of 0.1, minimal IL-12 production was seen within the first 72 hours. As viral replication occurred, IL-12 levels increased significantly from 72 to 168 hours after infection. Because this MOI was not completely cytotoxic, live cells were available to sustain both viral proliferation and IL-12 production. Paradoxically, therefore, the highest levels of IL-12 were seen with the lowest dose of virus.

Figure 4. Production of interleukin 12 (IL-12) was measured in CT26 cells infected with NV1042 at multiplicities of infection (MOIs) of 0.1, 1, and 5. Supernatants from cultures of infected CT26 cells were harvested at varying time points after infection. IL-12 concentrations were assayed by enzyme-linked immunosorbent assay to determine cumulative IL-12 production (mean ± standard deviation). The highest viral titer (MOI = 5) produced the most initial IL-12, but significant toxicity at this dosage caused an early plateau of cytokine production. At the lower MOI = 1, a slight delay in the IL-12 peak was noted before viral replication and IL-12 production occurred. Eventual toxicity at this dose resulted in a plateau as well. The least toxic MOI (0.01) resulted in significant IL-12 production, although at a later time point after viral replication occurred.
Animal Studies
The ability of NV1023 and NV1042 to kill flank tumors in a model of colorectal cancer was evaluated in vivo. The ability of NV1042 to reduce tumor burden and cure more animals than NV1023 was also shown. At 21 days after treatment, control tumors reached a mean volume of 1,230 ± 265 mm3. Tumors treated with 1 × 107 pfu NV1023 measured 35 ± 30 mm3 (P < .005 vs. controls), whereas tumors treated with the same dose of NV1042 measured 115 ± 80 mm3 (P < .005 vs. controls) (Fig. 5). In both the NV1023 and NV1042 groups, five animals per group were completely cured of their tumor. Animals treated with 1 × 106 pfu NV1023 grew tumors to the same size as the control group, with a mean volume of 1,295 ± 235 mm3. Animals treated with 1 × 106 pfu NV1042 showed a significant reduction in mean tumor volume (450 ± 230 mm3). This was significantly different from both control tumors (P < .05) and from tumors treated with low doses of NV1023 (P < .05) Only one animal was cured in the low-dose NV1023 group, whereas four of seven animals were cured of their tumor in the low-dose NV1042 group. All seven control tumors grew throughout the 21-day study.

Figure 5. Comparison between NV1023 and NV1042 as viral therapy for a model of colorectal cancer. Tumor was established in balb/c mice by subcutaneous flank injection of 5 × 105 CT26 murine colorectal cancer cells. When tumors reached a volume of 30 to 35 mm3, mice were treated by direct injection of 1 × 106 plaque-forming units (pfu; low dose) or 1 × 107 pfu (high dose) of NV1023 or NV1042. Control tumors were injected with saline. (A) Single intratumoral injection of 1 × 107 pfu of either NV1023 or NV1042 significantly reduced tumor burden compared with controls (P < .005). (B) Single intratumoral injection of 1 × 106 pfu NV1023 showed no difference in tumor reduction compared with controls. The cytokine-producing NV1042 virus showed significant antitumor efficacy compared with both controls (P < .05) and the parent NV1023 virus (P < .05).
DISCUSSION
This study evaluated the use of oncolytic HSV therapy in conjunction with cytokine gene therapy as treatment for experimental colon cancer. HSVs have been extensively studied in attempts to harness their natural ability to infect and lyse tumor cells. Deletions of strategic herpes genes have resulted in several generations of oncolytic viruses that are safe for in vivo use in experimental models. First-generation genetically engineered HSV vectors were deleted for single genes such as thymidine kinase, ribonucleotide reductase, or uracyl N-glycosylase. 13,15,16,28–30 Second-generation vectors with multiple mutations were then developed to decrease the chance of wild-type reversion, theoretically improving safety margins for in vivo use. G207 and MGH-1 were two such viruses, both of which had the viral ribonucleotide reductase gene inactivated, and these viruses also had both copies of the γ134.5 genes deleted. 7,31 The γ134.5 gene functions as a virulence factor by preventing the shutoff of protein synthesis in the HSV-infected cell, thereby permitting enhanced viral replication. 32,33
Another multimutated HSV that was originally designed as vaccine therapy against HSV-1 and -2 infection is R7020. 14 Recently, this agent has shown success as therapy against experimental cancers resistant to other types of treatment. 10 R7020 has had the joint region of the viral genome deleted, including one of the two γ134.5 neurovirulence genes. 14 Maintenance of one of the γ134.5 genes has been shown to enhance the oncolytic activity of mutant HSV vectors, thereby providing R7020 with increased therapeutic efficacy compared with double γ134.5 mutants. 31,34 The viruses constructed for the current study were based on a nonselected clonal derivative of R7020 called NV1020. Along with deletion of one γ134.5 gene, NV1020 is deleted for UL24, UL5 to 6, and the L/S junction, all of which attenuate virulence. 14 The novel mutant described in this study, NV1023, is restored for UL24 gene, which has been shown to enhance the ability of the herpes virus to replicate. 35,36 NV1023 also has a lacZ marker gene insertion within the ICP47 locus. The ICP47 gene product functions to inhibit major histocompatibility complex (MHC) class I peptide presentation by infected cells in humans. 37 Disabling the ICP47 locus in NV1023 should allow MHC class I peptide statement, thereby increasing immune recognition of the HSV-infected tumor in humans. In theory, therefore, restoring the UL24 gene and disabling the ICP47 locus should enhance the oncolytic potential of NV1023 compared with NV1020.
The promising anticancer potential of IL-12 was the basis for the construction of NV1042. This heterodimeric cytokine is produced by antigen-presenting cells such as B cells, dendritic cells, and macrophages. 25,27 IL-12 has been shown to enhance T-cell, LAK-cell, and NK-cell cytotoxicity, to induce expression of interferon γ from T and NK cells, and to promote the induction and differentiation of T-helper type 1 lymphocytes. 22,25,26,38–40 IL-12 has also been reported to have antiangiogenic effects that inhibit tumor growth. 41 IL-12-induced activation of the immune system provides both early and long-term immune stimulation, leading to both immediate tumor regression and the generation of immunologic memory that may prevent recurrent disease. 25,26
Most previous studies examining local IL-12 delivery have involved purely immunostimulatory strategies that used gene delivery by means of replication-incompetent viral vectors, fibroblasts, and dendritic cells. 42–44 In the current studies, IL-12 was delivered by the replication-competent oncolytic virus NV1042, constructed by insertion of the mIL-12 gene into NV1023. NV1042 is a novel HSV vector that has both oncolytic activity and the ability to deliver a potentially therapeutic cytokine to sites of tumor; it thus provides two separate potential strategies for tumor killing. HSV-mediated oncolysis may produce direct killing of cancer cells, while local expression of IL-12 may produce immunostimulatory anticancer effects by recruitment and activation of immune cells in an environment rich in putative tumor antigens. Only one other study has described the use of a cytokine-secreting oncolytic HSV. 38 In that study, however, an IL-12-secreting HSV repaired for the UL24 gene was examined but was compared with a parent virus that did not contain UL24. Because UL24 gene expression is associated with enhanced viral replication, the improved outcome shown in this other study may be secondary to UL24 gene repair, and not from IL-12 secretion. 36,37 In the current studies, we compared the cytokine-secreting oncolytic virus to a UL24-repaired virus identical in every other way and showed that it is indeed the cytokine secretion that resulted in the enhanced biologic effects.
The in vitro data presented in this investigation show equal oncolytic activity and similar viral growth between NV1023 and NV1042. At all MOIs evaluated, these viruses were toxic to CT26 colon cancer cells, resulting in 100% cell kill at MOIs of 1 and 5. These data confirm the oncolytic activity of both viruses and show that the insertion of mIL-12 did not affect the ability of NV1042 to replicate within and kill CT26 colon cancer cells. The ability of NV1042-infected CT26 cells to express and secrete mIL-12 was also shown. IL-12 production was limited by viral toxicity at higher doses. Lower viral doses resulted in higher cumulative IL-12 production, probably because more viable cells were available to support both viral replication and IL-12 production.
In vivo studies using doses of 107 pfu showed equal antitumor efficacy between NV1023 and NV1042 when virus was injected into tumors. It is likely that the oncolytic effects of these viruses were too effective for the benefits of cytokine secretion to be noted. It is also likely that delivery of 107 pfu resulted in significant and early tumor kill and IL-12 secretion was never achieved. Many animals in the groups treated with 107 pfu were completely cured of their tumors, with no toxicity noted. These results are promising with respect to the oncolytic activity of these viruses in general. At the lower dose of 106 pfu, NV1042 resulted in a significant reduction in tumor burden and several animal cures, whereas NV1023 failed to show any reduction in tumor burden compared with controls. The antitumor effects of IL-12 secretion were apparent only at a viral dose that was too low for sufficient cell kill to occur by oncolysis alone. This combination therapy, therefore, allows lower doses of virus to be used without any significant loss in antitumor efficacy, potentially allowing maximal benefit while minimizing toxicity. The synergistic action of combined oncolytic therapy and immunotherapy seen in these studies is highly encouraging for clinical studies of such combined therapy in humans.
Discussion
DR. B. MARK EVERS (Galveston, Texas): I want to compliment the authors on an elegant and well-done study assessing novel therapies for the potential treatment of colorectal cancer. The authors have assessed both in vitro and in vivo the tumor-killing ability of a replication-competent herpes simplex virus alone or in combination with a plasmid engineered to express the gene for IL-12. In vivo the authors have used an experimental model of colon cancer growth by placement of colon cancers in the flank of mice and allowing them to grow in this subcutaneous location. Either the HSV parental virus or the virus engineered to produce IL-12 were able to significantly reduce tumor burden when given as single agents. The combination therapy was significantly better than the agents alone. Therefore, this study has the potential of providing a novel and exciting therapeutic option in the treatment of colorectal cancers. I have three questions for Dr. Fong.
First, you note tumor suppression with single-agent treatment in vivo. What happens after the completion of therapy to these tumors – that is, does cancer cell growth remain suppressed or do the tumors resume their growth pattern and eventually become as large as the control tumors? In the same regard, you speak in the paper of an actual cure of these tumors with combination treatment. What exactly constitutes a “cure”? Does this mean that there was no histologic evidence of a tumor at the subcutaneous site?
The second question has to do with mechanism of action. I was wondering whether you could shed some light on the actions of the oncolytic virus alone and the virus containing IL-12 – that is, do the cancer cells undergo apoptosis, and if so, are the mechanisms different for the virus alone compared with the virus expressing IL-12?
My final question has to do with the overall clinical applications of this type of therapy. Have you evaluated this treatment in other models such as using colon cancer cells that have been trained to metastasize to the liver when injected into either the spleen or the cecal wall? This type of model may allow you to better assess whether these agents could potentially be useful in the treatment of colorectal cancer metastasis. Along the same lines, you have injected the viruses directly into the tumor in your in vivo model. Could these agents be used systemically or would the toxicity be so high as to prevent this route of delivery?
Once again, I thoroughly enjoyed the presentation of this potentially promising and novel therapy for colorectal cancer.
DR. MICHAEL CHOTI (Baltimore, Maryland): I would like to congratulate Drs. Fong, Brennan, and the other investigators on this excellent and interesting study looking at this novel approach of combining the oncolytic viral therapy with cytokine delivery using IL-12 in this murine colorectal model. I have three questions for Dr. Fong.
First, I am also interested in your thoughts regarding the mechanism of IL-12 action. Do you think this is a synergistic effect? Do you have any data to show synergistic versus an additive effect of these two local cytotoxic therapies, if you will?
And a related question: do you think the IL-12 mechanism is truly immunostimulatory, in that generation of immunologic memory has been achieved? Have you done the studies where, in those animals that have been cured, you subsequently challenge them with wild-type CT-26 to see if tumor grows in that model? And, as you mentioned, IL-12 has been shown in some models to have such an immunologic memory.
A second question relates to the clinical application. The intratumoral injection in this model using oncolytic virus is similar, if you will, to other local ablative therapies, if you will. And perhaps the addition of effective immunostimulatory cytokine, in effect, makes a local ablative therapy potentially clinically useful. Because if you can elicit a systemic immune effect, then this may be an advantageous local ablative therapy compared to some of the other ablative therapies that are currently being used. And similarly, have you looked at other approaches, as was asked, intraarterial and systemic delivery, and have you studied this combination as well as strictly the oncolytic virus in the systemic and arterial approaches?
Finally, have you looked at other cytokines using this model, particularly other cytokines such as GM-CSF or others that are known to stimulate systemic immunologic response, again emphasizing the potential benefit of this therapy, if you will, creating the local oncolytic effect as well as the vaccine effect for its clinical efficacy?
I did enjoy this paper, and I would like to thank the authors and the Association for the opportunity to review the manuscript and discuss it. Thank you.
DR. YUMAN FONG (New York, New York): Thank you, Dr. Evers and Dr. Choti, for your comments. In response to both your comments about what happens long term, we have actually watched some of these animals out 2 to 3 months and never found tumor in the flank, and rechallenged those animals to see what would happen with rechallenge, and in a significant number of animals we have been unable to have tumor regrow. So I believe it is a long-term cure, and I believe that there is immunologic memory against the particular tumors.
In terms of what are the mechanisms of action, for the oncolytic virus it is very simple. The viral life cycle is infection, internalization, replication, and lysis of the cells. And it is quite simple and quite straightforward, and that’s why it is so appealing to us as a gene therapy strategy. And that’s because in most of the other gene therapy strategies that we have been working on for the last decade, we have been trying to be too smart. To try to put in tumor-suppressor genes, to try to put in specific cell cycle regulators works great in animal models, but I am very worried that ultimately by themselves they will not be useful clinically. And that’s why we sort of reverted somewhat to the very simple biologic action of tumor-killing viruses.
And this idea of engineering the viruses so that they are much more specific for tumor cells, I hope, will became an important and future standard therapy for cancers, and that is the basis of the oncolytic therapy.
For the interleukin 12, it is an attempt to stimulate the immune system. It is an attempt to put an immunostimulatory cytokine exactly where the tumor antigen should be after the tumor cells are killed by oncolysis. And the reason that we know that it is an immunostimulatory strategy in mechanism is because we have done blockade experiments, blocking down the CD-4 and CD-8 subsets of cells in mice and have been able to abrogate the effects of this additional gene.
We have been unable to show that there is an additional advantage of having such a gene in nude animals that are immunodeficient, so we believe that part of it is immunostimulatory.
In terms of the clinical implications, what are some of the other models that we have used? In terms of tumor types, we have actually used these types of viruses in many different tumors, and have been successful in many of them. In terms of actual animal models, we have used peritoneal perfusion for gastric cancer. We have used bladder perfusion for bladder cancer. We have used liver perfusion for liver metastases, lung perfusion for lung metastases – a whole wide variety of delivery groups and means.
In terms of delivering it systemically, at a high enough dose, we can produce effect. And the reason that, when we went to a clinical program, we decided to go with a regional approach first is because delivering the virus at the closest proximity to the tumor, we believe, will be the easiest way of showing efficacy and reducing toxicity.
And as a proof of principle, in the first studies that we and others will do in man, we have to aim for the lowest dose of virus that will show us a biologic effect and not have so much toxicity that the entire program will be turned off before it got started. That’s why in our phase I clinical trials that we have just begun oncolytic viruses in the treatment of metastatic colorectal cancer. We plan to treat metastatic colorectal cancer isolated to the liver using virus delivered by intraarterial infusion. I believe that long term we will, and other groups will, look at systemic delivery. In the short term, in phase I trials, I believe that everybody should consider using regional delivery of these agents.
In terms of whether it is synergistic or additive, that’s going to be very difficult to look at, and that’s because in most of the studies looking at chemotherapy synergy, the way the synergy is calculated is that you go look to see, in tissue culture, what the LD50 of a particular drug is, and you try to calculate what the curve would look like if two drugs were added together and then compare it to what the actual is.
For the immunostimulatory agents that we are using, you can’t do that in vitro. And, therefore, whether it is additive or whether it is synergistic, I think it’s going to be very difficult to prove. But as long as it’s more than single agent, I think it shows promise.
And, lastly, in terms of other cytokines, many cytokines have been tested. We have also tested adhesion molecules and antigen-presentation molecules such as B7 and ICAM, again trying to add to the immunostimulatory strategy.
Again, I thank the Society for allowing me to present this paper today.
Footnotes
Presented at the 112th Annual Meeting of the Southern Surgical Association, December 4–6, 2000, Palm Beach, Florida.
Dr. Fong was supported in part by grants RO1CA75416, RO1CA72632, and RO1CA61524 from the National Institutes of Health, and MBC-99366 from the American Cancer Society.
Correspondence: Dr. Yuman Fong, Department of Surgery, Memorial Sloan-Kettering Cancer Center, 1275 York Ave., New York, NY 10021.
E-mail: fongy@mskcc.org
Accepted for publication December 2000.
References
- 1.Greenlee RT, Murray T, Bolden S, et al. Cancer statistics, 2000. CA Cancer J Clin 2000; 50: 7–33. [DOI] [PubMed] [Google Scholar]
- 2.August DA, Ottow RT, Sugarbaker PH. Clinical perspective of human colorectal cancer metastasis. Cancer Metastas Rev 1984; 3: 303. [DOI] [PubMed] [Google Scholar]
- 3.Blumgart LH, Fong Y. Surgical options in the treatment of hepatic metastasis from colorectal cancer. Curr Probl Surg 1995; 32: 333–421. [DOI] [PubMed] [Google Scholar]
- 4.Panis Y, Ribiero J, Chretien Y, et al. Dormant liver metastases, an experimental study. Br J Surg 1994; 19: 221–233. [DOI] [PubMed] [Google Scholar]
- 5.Fong Y, Kemeny N, Paty P, et al. Treatment of colorectal cancer: hepatic metastasis. Semin Surg Oncol 1996; 12: 219–252. [DOI] [PubMed] [Google Scholar]
- 6.Kemeny N, Huang Y, Cohen A, et al. Hepatic arterial infusion of chemotherapy after resection of hepatic metastases from colorectal cancer. N Engl J Med 1999; 341: 2039–2048. [DOI] [PubMed] [Google Scholar]
- 7.Mineta T, Rabkin SD, Yazaki T, et al. Attenuated multi-mutated herpes simplex virus-1 for the treatment of malignant gliomas. Nat Med 1995; 1: 938–943. [DOI] [PubMed] [Google Scholar]
- 8.Bennett J, Kooby D, Delman D, et al. Antitumor efficacy of regional oncolytic viral therapy for peritoneally disseminated cancer. J Mol Med 2000; 78: 166–174. [DOI] [PubMed] [Google Scholar]
- 9.Kooby DA, Carew JF, Halterman MW, et al. Oncolytic viral therapy for human colorectal cancer and liver metastases using a multimutated herpes simplex virus type-1 (G207). FASEB J 1999; 6: 499–504. [DOI] [PubMed] [Google Scholar]
- 10.Advani SJ, Chung S, Yan SY, et al. Replication-competent, nonneuroinvasive genetically engineered herpes virus is highly effective in the treatment of therapy-resistant experimental human tumors. Cancer Res 1999; 59: 2055–2058. [PubMed] [Google Scholar]
- 11.Kucharczuk JC, Randazzo B, Chang MY, et al. Use of a “replication-restricted” herpes virus to treat experimental human malignant mesothelioma. Cancer Res 1997; 57: 466–471. [PubMed] [Google Scholar]
- 12.Yoon SS, Nakamura H, Carroll NM, et al. An oncolytic herpes simplex virus type 1 selectively destroys diffuse liver metastases from colon carcinoma. FASEB J 2000; 14: 301–311. [PubMed] [Google Scholar]
- 13.Pyles RB, Thompson RL. Evidence that the herpes simplex virus type 1 uracil DNA glycosylace is required for efficient viral replication and latency in the murine nervous system. J Virol 1994; 68: 4963–4972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meignier B, Longnecker R, Roizman B. In vivo behavior of genetically engineered herpes simplex viruses R7017 and R7020: construction and evaluation in rodents. J Infect Dis 1988; 158: 602–614. [DOI] [PubMed] [Google Scholar]
- 15.Goldstein DJ, Weller SK. Herpes simplex virus type 1-induced ribonucleotide reductase activity is dispensable for virus growth and DNA synthesis: isolation and characterization of an ICP6 lacZ insertion mutant. J Virol 1988; 62: 196–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goldstein DJ, Weller SK. Factor(s) present in herpes simplex virus type 1-infected cells can compensate for the loss of the large subunit of the viral ribonucleotide reductase: characterization of an ICP6 deletion mutant. Virology 1988; 166: 41–51. [DOI] [PubMed] [Google Scholar]
- 17.Chou J, Kern ER, Whitley RJ, et al. Mapping of herpes simplex virus-1 neurovirulence to gamma 134.5, a gene nonessential for growth in culture. Science 1990; 250: 1262–1266. [DOI] [PubMed] [Google Scholar]
- 18.Carroll NM, Chiocca EA, Takahashi K, et al. Enhancement of gene therapy specificity for diffuse colon carcinoma liver metastases with recombinant herpes simplex virus. Ann Surg 1996; 224: 323–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Markert J, Medlock M, Rabkin S, et al. Conditionally replicating herpes simplex virus mutant, G207 for the treatment of malignant glioma: results of a phase I trial. Gene Ther 2000; 7: 867–874. [DOI] [PubMed] [Google Scholar]
- 20.Rampling R, Cruickshank G, Papanastassiou V, et al. Toxicity evaluation of replication-competent herpes simplex virus (ICP 34.5 null mutant 1716) in patients with recurrent malignant glioma. Gene Ther 2000; 7: 866. [DOI] [PubMed] [Google Scholar]
- 21.D’Angelica M, Fong Y. Cytokine gene therapy for human tumors. Surg Oncol Clin North Am 1998; 7: 537–563. [PubMed] [Google Scholar]
- 22.Siders WM, Wright PW, Hixon JA, et al. T cell- and NK cell-independent inhibition of hepatic metastases by systemic administration of an IL-12-expressing recombinant adenovirus. J Immunol 1998; 160: 5465–5474. [PubMed] [Google Scholar]
- 23.Andreansky S, He B, Cott JV, et al. Treatment of intracranial gliomas in immunocompetent mice using herpes simplex viruses that express murine interleukins. Gene Ther 1998; 5: 121–130. [DOI] [PubMed] [Google Scholar]
- 24.Karpoff HM, D’Angelica M, Blair S, et al. Prevention of hepatic tumor metastases in rats with herpes viral vaccines and gamma-interferon. J Clin Invest 1997; 99: 799–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pham-Nguyen KB, Yang W, Saxena R, et al. Role of NK and T cells in IL-12-induced anti-tumor response against hepatic colon carcinoma. Int J Cancer 1999; 81: 813–819. [DOI] [PubMed] [Google Scholar]
- 26.Tasaki K, Yoshida Y, Maeda T, et al. Protective immunity is induced in murine colon carcinoma cells by the expression of interleukin-12 or interleukin-18, which activate type 1 helper T cells. Cancer Gene Ther 2000; 7: 247–254. [DOI] [PubMed] [Google Scholar]
- 27.Zitvogel L, Lotze MT. Role of interleukin-12 (IL-12) as an anti-tumor agent: experimental biology and clinical application. Res Immunol 1995; 146: 628–638. [DOI] [PubMed] [Google Scholar]
- 28.Martuza RL, Malick A, Markert JM, et al. Experimental therapy of human glioma by means of a genetically engineered virus mutant. Science 1991; 252: 854–856. [DOI] [PubMed] [Google Scholar]
- 29.Jia WW, McDermott M, Goldie J, et al. Selective destruction of gliomas in immunocompetent rats by thymidine kinase-defective herpes simplex virus type 1. J Natl Cancer Inst 1994; 86: 1209–1215. [DOI] [PubMed] [Google Scholar]
- 30.Mineta T, Rabkin SD, Martuza RL. Treatment of malignant gliomas using ganciclovir-hypersensitive, ribonucleotide reductase-deficient herpes simplex viral mutant. Cancer Res 1994; 54: 3963–3966. [PubMed] [Google Scholar]
- 31.Kramm CM, Chase M, Herrlinger U, et al. Therapeutic efficiency and safety of a second-generation replication-conditional HSV1 vector for brain tumor gene therapy. Hum Gene Ther 1997; 8: 2057–2068. [DOI] [PubMed] [Google Scholar]
- 32.Chou J, Chen JJ, Gross M, et al. Association of a M(r) 90,000 phosphoprotein with protein kinase PKR in cells exhibiting enhanced phosphorylation of translation initiation factor eIF-2 alpha and premature shutoff of protein synthesis after infection with gamma 134.5-mutants of herpes simplex virus 1. Proc Natl Acad Sci USA 1995; 92: 10516–10520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chou J, Roizman B. The gamma 34.5 gene of herpes simplex virus 1 precludes neuroblastoma cells from triggering total shutoff of protein synthesis characteristic of programmed cell death in neuronal cells. Proc Natl Acad Sci USA 1992; 89: 3266–3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chung RY, Saeki Y, Chiocca EA. B-myb promoter retargeting of herpes simplex virus gamma 34.5 gene-mediated virulence toward tumor and cycling cells. J Virol 1999; 73: 7556–7564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jacobson JG, Martin SL, Coen DM. A conserved open reading frame that overlaps the herpes simplex virus thymidine kinase gene is important for viral growth in cell culture. J Virol 1989; 63: 1839–1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jacobson JG, Chen SH, Cook WJ, et al. Importance of the herpes simplex virus UL24 gene for productive ganglionic infection in mice. Virology 1998; 242: 161–169. [DOI] [PubMed] [Google Scholar]
- 37.York IA, Roop C, Andrews DW, et al. A cytosolic herpes simplex virus protein inhibits antigen presentation to CD8+ T lymphocytes. Cell 1994; 77: 525–535. [DOI] [PubMed] [Google Scholar]
- 38.Parker JP, Gillespie GY, Love CE, et al. Engineered herpes simplex virus expressing IL-12 in the treatment of experimental murine brain tumors. Proc Natl Acad Sci USA 2000; 97: 2208–2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mehrotra PT, Wu DCJA, Mostowski HS, et al. Effects of IL-12 on the generation of cytotoxic activity in human CD8+ T lymphocytes. J Immunol 1993; 151: 2444. [PubMed] [Google Scholar]
- 40.McKnight AJ, Zimmer GJ, Fogelman I, et al. Effects of IL-12 on helper T cell-dependent immune responses in vivo. J Immunol 1994; 152: 2172–2179. [PubMed] [Google Scholar]
- 41.Duda DG, Sunamura M, Lozonschi L, et al. Direct in vitro evidence and in vivo analysis of the antiangiogensis effects of interleukin 12. Cancer Res 2000; 60: 1111–1116. [PubMed] [Google Scholar]
- 42.Nishioka Y, Hirao M, Robbins PD, et al. Induction of systemic and therapeutic antitumor immunity using intratumoral injection of dendritic cells genetically modified to express interleukin 12. Cancer Res 1999; 59: 4035–4041. [PubMed] [Google Scholar]
- 43.Gambotto A, Tuting T, McVey DL, et al. Induction of antitumor immunity by direct intratumoral injection of a recombinant adenovirus vector expressing interleukin-12. Cancer Gene Ther 1999; 6: 45–53. [DOI] [PubMed] [Google Scholar]
- 44.Tahara H, Lotze MT, Robbins PD, et al. IL-12 gene therapy using direct injection of tumors with genetically engineered autologous fibroblasts. Hum Gene Ther 1995; 6: 1607–1624. [DOI] [PubMed] [Google Scholar]