

Abstract
Objective
To examine the cellular mechanisms involved in the pathogenesis of necrotizing enterocolitis (NEC).
Summary Background Data
Necrotizing enterocolitis is a major cause of death and complications in neonates; the cellular mechanisms responsible for NEC are unknown. The inducible form of cyclooxygenase (i.e., COX-2) is activated by the transcription factor nuclear factor (NF)-κB and is thought to play a role in inflammation.
Methods
Segments of perforated and adjacent uninvolved small intestine from neonates with NEC were analyzed for COX-2 expression by immunohistochemistry. NEC was induced in weanling (18 days old) rats by occlusion of superior mesenteric vessels for 1 hour and intraluminal injection of platelet activating factor (50 μg/kg). Small intestine was harvested for protein extraction. Western immunoblot was performed to determine expression of COX-2. Gel shift assays were performed to assess NF-κB binding activity.
Results
Immunohistochemical analysis showed increased COX-2 protein expression in the perforated intestinal sections of all 36 neonates but not in adjacent normal intestine. Increased expression of COX-2 protein and NF-κB binding activity was noted in the small intestine of weanling rats at 0 and 3 hours after induction of NEC.
Conclusions
Increased COX-2 expression was identified in all neonatal intestinal segments resected for perforated NEC. In addition, a coordinate induction of COX-2 expression and NF-κB binding was noted in a rodent model of NEC. These findings suggest that the COX-2/NF-κB pathway may play a role in the pathogenesis of NEC. Therapeutic agents that target this pathway may prove useful in the treatment or possible prevention of NEC.
Necrotizing enterocolitis (NEC), characterized by edema, ischemia, and intestinal necrosis, most commonly involves the terminal ileum and the proximal colon and is a major cause of death and complications in neonates. 1,2 NEC is predominately a disease of premature infants; in recent years, its incidence has become more prevalent with the increasing survival of low-birthweight premature infants. It remains the most frequently encountered gastrointestinal emergency in newborns, with an incidence ranging from 1 to 2.4 cases per 1,000 live births; the case fatality rate ranges from 20% to 40%. 3,4 In addition to prematurity, additional risk factors such as perinatal stress, decreased splanchnic perfusion, hypoxia, patent ductus arteriosus, and hyperosmolar enteral feedings have been described in association with NEC. 5–7 Although several risk factors for NEC have been identified, the exact cellular mechanisms involved in its pathogenesis are unknown.
Cyclooxygenase (COX) catalyzes the rate-limiting step of arachidonic acid metabolism into prostaglandins, leukotrienes, and thromboxanes. 8 Two isoforms of the COX enzyme have been identified. COX-1 is constitutively expressed in many tissues, including the gastrointestinal mucosa. 9 The inducible form, COX-2, is normally undetectable in most tissues; however, increased expression of COX-2 has been shown in inflammatory conditions of the gastrointestinal tract (e.g., inflammatory bowel disease). 10 COX-2 expression is increased by proinflammatory cytokines such as interleukin 1, interleukin 6, and tumor necrosis factor-α. 11–13 In addition, the proinflammatory transcription factor nuclear factor-κB (NF-κB) plays an important role in the induction of COX-2 gene transcription. 14
NF-κB is an important protein for the activation of many inflammatory mediators and cytokines. 15,16 The NF-κB proteins, p50 and p65, are expressed in all cell types as either heterodimer or homodimer subunits. They are normally sequestered in the cytoplasm bound to the inhibitory protein IκB. On activation, IκB is rapidly phosphorylated and degraded by proteasomes. This degradation of IκB releases NF-κB, allowing it to translocate into the nucleus, where it binds to its consensus sequence within the promoter region of various target genes. 15,16 The activation of NF-κB is known to be involved in several inflammatory conditions, such as inflammatory bowel disease 17 and pancreatitis. 18 However, the role of NF-κB in the pathogenesis of NEC is unknown.
The purpose of our study was to discern the molecular mechanisms contributing to NEC by examining the potential role of COX-2 and NF-κB in this disease process. We evaluated paired intestinal samples from neonates with NEC and other noninflammatory conditions of the small bowel for expression of COX-2. In addition, we used a well-characterized rodent model of NEC to extend our clinical findings.
METHODS
Materials
Platelet activating factor was purchased from Calbiochem Corp. (La Jolla, CA). Monoclonal antibodies specific for COX-2 and COX-1 were from Cayman Chemical (Ann Arbor, MI). Antibodies for IκB and β-actin were purchased from Santa Cruz Biotechnology (Santa Cruz, CA) and Sigma (St. Louis, MO), respectively. The NF-κB oligonucleotide was purchased from Promega (Madison, WI). For immunohistochemical staining of COX-2 and COX-1, a mouse ABC Staining System (Santa Cruz, CA) was used. The enhanced chemiluminescence system for Western immunoblot analysis was purchased from Amersham (Arlington Heights, IL). All other reagents were of molecular biology grade and were purchased from either Sigma or Amresco (Solon, OH).
Human Intestinal Sections
Paraffin-embedded blocks of intestinal sections from 36 infants who underwent resection for NEC at The University of Texas Medical Branch and the University of Alabama at Birmingham were analyzed. Multiple sections (4-μm thick) from each specimen, representing the areas of perforation or necrosis from NEC as well as adjacent uninvolved segments, were prepared for staining with hematoxylin and eosin and immunohistochemical studies using specific antibodies. Additional sections of intestine from six infants who required resection for noninflammatory conditions of the gastrointestinal tract (e.g., intestinal atresia, intussusception, colonic biopsy) were also analyzed. In one patient, sections of intestine were analyzed from the areas of perforated NEC and compared with a segment obtained at the time of ileostomy closure after recovery from NEC.
Animal Study
Weanling Swiss Webster rats (Charles River Breeding Laboratories, Inc., Wilmington, MA) were acclimated in an environment of controlled temperature (23°C), humidity, and lighting (12 hours light/12 hours dark) for 1 week. At day 18 of age, rats (∼25 g weight) were randomized to receive either sham operation (n = 12) or NEC (n = 12). All rats were anesthetized using pentobarbital (Nembutal; 40 mg/kg given intraperitoneally), and the peritoneal cavity was entered through a midline incision. In the sham (control) group, the superior mesenteric vessels were exposed but not occluded, and the vehicle for platelet activating factor (0.25% bovine serum albumin) was injected into the proximal jejunum. In the NEC group, intestinal injury was induced by transient (60 minutes) clamping of the superior mesenteric vessels with atraumatic microvascular clamps and intraluminal injection of platelet activating factor (50 μg/kg) into the proximal jejunum. The midline abdominal wound was closed in one layer with 3-0 silk after instillation of normal saline (20 mL/kg) into the peritoneal cavity. Rats were killed at 0 and 3 hours after removal of the vascular clamp and reperfusion. Full-thickness small intestine (from the ligament of Treitz to the ileocecal valve) was harvested and divided into equal lengths, with the proximal half designated jejunum and the distal half designated ileum. Portions of small intestine were fixed in 10% formalin for morphologic studies. The remaining sections of the jejunum and ileum were rapidly frozen in liquid N2 and stored at −70°C for protein extraction.
Preparation of Nuclear Protein Extracts
For the purification of nuclei, tissue samples were homogenized in Buffer A (50 mmol/L HEPES [pH 7.4], 10 mmol/L KCl, 1 mmol/L EDTA, 1 mmol/L EGTA, 1 mmol/L dithiothreitol, 0.1 μg/mL phenylmethylsulfonyl fluoride, 1 μg/mL pepstatin A, 1 μg/mL leupeptin, 10 μg/mL soybean trypsin inhibitor, 10 μg/mL aprotinin, and 0.5% NP-40). After 10 minutes on ice, the lysates were centrifuged at 4,000 g for 4 minutes at 4°C. After discarding the supernatant, the nuclear pellet was resuspended in Buffer B (Buffer A + 1.7 mol/L sucrose) and centrifuged at 15,000 g for 30 minutes at 4°C. The purified nuclear pellet was then incubated in Buffer C (same as Buffer A except 400 mmol/L KCl and without NP-40) with frequent vortexing for 30 minutes at 4°C. After centrifugation at 15,000 g for 5 minutes at 4°C, the supernatant was saved for nuclear extract. Protein concentrations of nuclear and cytosolic extracts were measured as described by Bradford. 19
Western Immunoblot Analysis
Protein extract (100 μg) was resolved by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and electroblotted to Immobilon-P nylon membranes as previously described. 20 Filters were then incubated for 3 hours at room temperature with 1:500 dilution of primary antibody in blocking solution, washed three times, and incubated with a horseradish peroxidase-labeled secondary antibody (1:1,000 dilution) for 1 hour at room temperature. The immune complexes were visualized using the enhanced chemiluminescence detection method.
Immunohistochemical Analysis
Paraffin-embedded sections were deparaffinized in three xylene washes followed by a graded alcohol series. Slides were incubated with 0.01% H2O2 solution for 10 minutes to remove any endogenous peroxidase activity. Primary antibody was added for 30 minutes at room temperature, washed with three changes of phosphate-buffered saline, and incubated with secondary antibody for 30 minutes at room temperature. The sections were then incubated with AB enzyme reagent for 30 minutes and washed with phosphate-buffered saline. The peroxidase substrate was added for stain development. The sections were dehydrated, permanently mounted with mounting medium, and observed by light microscopy.
Electrophoretic Mobility Shift Assay
A single strand of the NF-κB consensus sequence oligonucleotide was end-labeled with [γ-32P] ATP and T4 polynucleotide kinase and then annealed to its complementary nonradioactive oligonucleotide, as described previously. 21 Electrophoretic mobility shift assay reaction mixture contained 45,000 cpm 32P-end-labeled oligonucleotide and 25 μg crude nuclear extract in a final volume of 20 μL with 0.5 μg poly (dI·dC) as a nonspecific competitor. The reaction was incubated at room temperature for 20 minutes. Competition binding experiments were performed by adding unlabeled oligonucleotide, in molar excess, with the protein extract and binding buffer for 5 minutes at room temperature. The labeled probe was then added and incubation was continued for 20 minutes at room temperature. Reaction mixtures were loaded on a 4% nondenaturing polyacrylamide gel and resolved by electrophoresis at 200V at 2 hours. The gels were then dried and autoradiographed at −70°C.
RESULTS
To determine whether COX-2 is altered in NEC, intestinal sections from 36 infants with perforated NEC were examined by immunohistochemistry. COX-2 protein expression was markedly increased in the perforated sections from all 36 infants compared with adjacent uninvolved intestine;Figure 1 shows representative sections from 3 of the 36 neonates. COX-2 protein expression was distributed throughout the crypts and villi of the intestinal epithelium. In addition, increased COX-2 expression was noted in the submucosa and muscularis layers, which may represent COX-2 induction within inflammatory cells in these layers.

Figure 1. Cyclooxygenase-2 (COX-2) expression in necrotizing enterocolitis (NEC). Three representative histologic sections of intestine from infants with NEC show marked COX-2 protein expression by immunohistochemical staining compared with adjacent uninvolved segments (×100). Brown staining represents COX-2 protein expression in the intestinal epithelium, as assessed by a masked pathologist.
To determine whether induction of COX-2 was specific for NEC or simply a nonspecific finding in diseased intestine, small bowel resected for noninflammatory conditions from six infants was analyzed for COX-2 expression. Expression of COX-2 was not found in these noninflammatory conditions of the bowel (data not shown), thus suggesting that increased COX-2 expression is specific to inflammatory conditions of the intestine and may contribute to the pathogenesis of NEC. In addition, COX-2 expression was analyzed in a neonate with perforated NEC treated with resection and ileostomy and compared with a segment obtained at the time of ileostomy closure 6 weeks later (Fig. 2). Similar to our findings in the intestinal segments from the 36 neonates, COX-2 was strongly expressed in the perforated segment, but expression was not detected in the intestinal end resected at ileostomy closure.

Figure 2. Cyclooxygenase-2 (COX-2) expression in necrotizing enterocolitis (NEC) and ileostomy stoma. Histologic sections of immunohistochemical staining with COX-2 antibody in NEC intestine are compared with a segment obtained at ileostomy closure after recovery from NEC (×100). Brown staining denotes COX-2 expression.
To elucidate the molecular mechanisms contributing to NEC, a model using 18-day-old weanling rats was used to determine the expression of COX-2. 22 Consistent with our findings in neonates, increased COX-2 protein levels were noted at 0 and 3 hours in both the jejunum and ileum of weanling rats with NEC (Fig. 3). In contrast, COX-1 protein was expressed at relatively high levels in the sham (control) animals and did not increase with NEC. The blot was stripped and reprobed with β-actin, which indicated intact and relatively equal loading of the protein samples. These results corroborate our findings in neonates and further suggest a role for the COX-2 isoform in this disease process.

Figure 3. Induction of cyclooxygenase-2 (COX-2) in experimental necrotizing enterocolitis (NEC). Western blot analysis of protein (100 μg) from full-thickness small bowel (J, jejunum; I, ileum) at 0 and 3 hours after NEC treatment or sham operation in weanling rats. COX-2 protein levels were increased in the NEC group at both time points. In contrast, COX-1 expression was constitutively expressed and not altered with injury. The blot was stripped and reprobed for β-actin to control for protein loading. (Representative data of four experiments.)
The transcription factor NF-κB is important for the activation of COX-2 after inflammation. 14 Therefore, we next determined whether NF-κB binding activity was induced with NEC. Using nuclear protein extracted from the small bowel of weanling rats, electrophoretic mobility shift assays were performed to determine the changes in NF-κB binding activity (Fig. 4). NF-κB binding activity was increased in both the jejunum and ileum at 0 and 3 hours after intestinal injury compared with sham controls. The presence of multiple bands suggested activation of different NF-κB subunit proteins (e.g., p65 and p50). To ensure binding specificity, a molar excess of the unlabeled NF-κB oligonucleotide was used in competition experiments showing complete inhibition of protein binding to the labeled probe (lane 9, Fig. 4).

Figure 4. Nuclear factor (NF)-κB binding activity. Small bowel (J, jejunum; I, ileum) was harvested at 0 and 3 hours after necrotizing enterocolitis or sham operation. Nuclear protein (25 μg) was assayed for NF-κB activity with a labeled oligonucleotide containing the NF-κB consensus sequence. Competition experiments were performed with 100-fold molar excess of unlabeled NF-κB oligonucleotide (lane 9). (Representative data of four experiments.)
NF-κB binding activity is regulated by its inhibitory protein IκB in the cytoplasm. 15,16 On activation, IκB rapidly degrades and releases NF-κB to translocate into nucleus to regulate transcription of target genes. We next determined IκB expression in protein extracts from intestine of rats with NEC and sham controls. In both the jejunum and ileum of rats with experimental NEC, a rapid degradation of IκB protein levels was noted at 0 and 3 hours after bowel reperfusion, which correlates inversely with NF-κB activation (Fig. 5). Equal loading of protein was confirmed by reblotting with β-actin. Therefore, these findings further suggest that with NEC degradation of IκB occurs, resulting in the activation of NF-κB binding, which may then activate downstream inflammatory target genes such as COX-2.
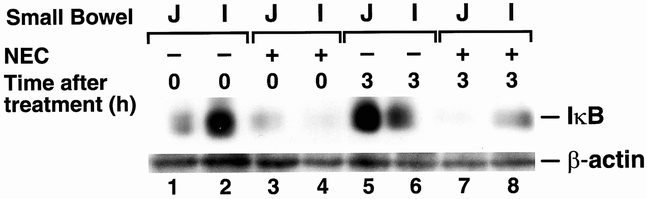
Figure 5. Expression of IκB. Western blot analysis of protein lysate (100 μg) extracted from small bowel (J, jejunum; I, ileum) after necrotizing enterocolitis or sham operation. The blot was sequentially probed with antibodies to IκB and β-actin. (Representative data of four experiments.)
DISCUSSION
Cyclooxygenase, the enzyme that catalyzes the first two steps in the biosynthesis of the prostaglandins from arachidonic acid, exists in two isoforms. 8,23 The inducible form, COX-2, is barely detectable during normal physiologic conditions; however, in response to various proinflammatory stimuli, it is rapidly induced. An increased expression of COX-2 has been found in the intestinal epithelial cells from patients with inflammatory bowel disease and has been implicated in its pathogenesis. 10 In the current study, we showed a marked increase in COX-2 protein expression in intestinal epithelium from infants with NEC. This increase was consistently noted in all 36 specimens of NEC with a variable spectrum of intestinal injury. Although we could not correlate the abundance of COX-2 expression with the severity of intestinal injury, induction of COX-2 was clearly shown in these pathologic segments of intestine when compared with adjacent uninvolved segments or the resected intestine from patients with noninflammatory gastrointestinal conditions. We also noted a resolution of COX-2 induction when an intestinal segment of NEC was compared with recovered intestine in an infant. In a similar fashion, we found that COX-2 expression was increased in the small bowel of rats using an experimental model of NEC. In contrast, COX-1 expression was not altered with NEC, which further supports the role of the COX-2 isoform in the pathogenesis of NEC.
A potential role for the COX/prostaglandin pathway in NEC has been suggested in previous studies, although there is no consensus regarding the exact role that this pathway plays in the disease process. Prostaglandin E1 is a potent vasodilator of the splanchnic bed 24 and stimulates crypt cell proliferation. 25 The low incidence of NEC in infants fed breast milk has been attributed in part to the presence of prostaglandin E1 in mother’s breast milk. 26 In contrast, thromboxane, another byproduct of COX, is a potent vasoconstrictor and mediates many of the deleterious responses associated with inflammation. 23 Indomethacin, a nonspecific inhibitor of both COX-1 and COX-2, causes vasoconstriction and has been used effectively in infants to medically close patent ductus arteriosus. 26 Unfortunately, it has also been associated with an increased incidence of NEC because of its effect on mesenteric hypoperfusion, which leads to mucosal ischemia and subsequent bacterial invasion. 6 In our study, the levels of COX-1 did not change in NEC. However, it is possible that the underlying mechanism of indomethacin-induced NEC may be due to nonspecific inhibition of constitutively expressed prostaglandins that play a protective role in the gastrointestinal tract. Grosfeld et al 6 reported a cytoprotective role for prostaglandin E1 in a rat bowel ischemic model, showing increased survival and a decreased rate of bowel necrosis and perforation. Future studies using specific inhibitors of COX-1 and COX-2 are required to define the role of the COX/prostaglandin pathway in the pathogenesis of NEC.
In the current study, we showed a causal association of increased COX-2 expression in NEC intestinal sections, which suggests a possible role for COX-2 in the inflammatory changes associated with NEC. However, another possibility is that induction of COX-2 may be a protective mechanism in the inflamed intestine. For example, recent studies have shown that COX-2 can accelerate the healing of gastric ulceration 27 and play a role in reducing epithelial injury in dextran sodium sulfate-induced colitis. 28 Prostaglandins produced by COX-2 may prevent intestinal epithelial injury and stimulate crypt cell survival. Therefore, the induction of COX-2 in NEC, as shown in our study, could represent a reparative response to injury as opposed to actually producing the deleterious effects. Specific inhibitors of COX-2 may improve or worsen intestinal injury, depending on the tissue-specific actions of COX-2 during NEC. Studies are in progress in our laboratory to address these issues using selective COX-2 inhibitors and COX-2 knockout mice.
To discern the mechanisms involved in the regulation of COX-2 in NEC, we next evaluated NF-κB, an important regulator of many target genes (e.g., cytokines, COX-2) that are crucial in the inflammatory responses of various tissues. 15 There are two NF-κB consensus sites in the promoter region of the human COX-2 gene, which play an integral role in COX-2 induction by cytokines and lipopolysaccharide. 29,30 We found that induction of NF-κB binding is an early event in the small intestine of rats using an experimental model of NEC. Various cytokines (e.g., tumor necrosis factor-α, interleukin 1, interleukin 6), which are found in increased amounts in infants with NEC, 31 may be responsible for the induction of NF-κB activation in NEC. Although it is beyond the scope of this study to speculate that NF-κB regulates COX-2 expression in NEC, the coordinate induction of COX-2 protein expression and NF-κB binding activity suggests a role for NF-κB in the inflammatory response of NEC. Recent studies have shown that proteasome inhibitors, which interfere with NF-κB activation, protect mice against lethal shock induced by lipopolysaccharide or tumor necrosis factor-α. 32 May et al 33 have also recently shown that selective inhibition of NF-κB ameliorated inflammatory responses in two experimental murine models of acute inflammation. We postulate that NF-κB may potentially be an even more attractive molecular target for the treatment of inflammatory intestinal conditions because it is upstream of COX-2 and other proinflammatory cytokines.
In conclusion, we have shown a marked induction of COX-2 protein expression in severely injured intestinal segments of NEC but not in intestine resected for noninflammatory conditions. This induction of COX-2 was also noted in an experimental model of NEC in weanling rats, suggesting an important role of the COX-2 pathway in the pathogenesis of NEC. We have also shown coordinate induction of NF-κB binding activity associated with degradation of IκB in NEC, suggesting that NF-κB may also be contributing to the inflammation associated with NEC. Therapeutic agents that target either NF-κB or COX-2 may ameliorate intestinal injury as a result of NEC.
The authors thank Wan Qin Hu and Jell Hsieh for their technical support and Eileen Figueroa and Karen Martin for manuscript preparation.
Discussion
DR. MAX R. LANGHAM, JR. (Gainesville, Florida): Dai Chung and his colleagues from the University of Texas Medical Branch have clearly shown us that cyclooxygenase 2 (COX-2) expression is increased in the intestinal segments obtained from babies with perforated NEC but not in children with atresias or in patients that have NEC but a healthy portion of bowel. This observation in humans is very important since it adds to a growing body of literature that improves our insights into the pathophysiology of NEC, and also the biology of the cyclooxygenase system and its functions in the normal and diseased gut.
Other investigators have shown that nonselective COX inhibition increases intestinal permeability after ischemia/reperfusion, while selective COX-2 inhibition can cause a pathologic immune response to luminal protein antigens. It can limit intestinal smooth muscle cell proliferation after LPS challenge and delay return of gut motility after a normal laparotomy. So selective COX-2 inhibition can cause some deleterious effects in a number of animal models.
All of this data put together, though, makes it appear that the COX-2 system functions as a proximal controller of a whole variety of responses in normal and injured intestine. I have one comment and several questions.
The weanling rat model that is used here, in my opinion, is not a clinically relevant model of necrotizing enterocolitis. In fact, we do not know what the pathophysiology of necrotizing enterocolitis is, and there is no clinically relevant model. In human beings, weaning normally occurs between 6 months and 2 years of age, and children that achieve weaning status even when they develop overwhelming sepsis from gut-derived organisms, as occurs in third-world countries, commonly do not generally have intestinal necrosis as part of their pathology. Nevertheless, it is reasonable to assume that using a model such as Dr. Chung has shown us will help improve our understanding of the molecular controllers of the gut. And this improved understanding of these controllers may lead to better ability to diagnose and treat NEC.
My questions are: COX-2 induction appears nearly complete immediately after 1 hour of ischemia. And when you think about this, that means that NF-κB has to be activated, transferred to the nucleus, that transcription has to occur and protein expression be complete. And that happens within an hour at time point zero. In fact, there is no evidence that there is any increase at 3 hours time point. I was wondering if the authors had any information for us regarding the duration of ischemia or the rate response for this. Because in trying to determine the kinetics of this event, it would be very helpful to have some interim data where the event was not complete.
Brad Warner’s lab in Cincinnati has shown decreased salivary, serum, and urine levels of epidermal growth factor in infants with NEC. Have you looked in your model at whether or not epidermal growth factor affects the COX-dependent inflammatory response to ischemia reperfusion?
Similarly, Dan Ledbeatter and Sunny Juul, while they were in Gainesville, showed that babies who were being treated with erythropoietin were much less likely to develop necrotizing enterocolitis than case-matched controls. Subsequently, they demonstrated the presence of intact human EpO in breast milk, and EpO receptors on gut mucosa. None of us know what that means. Do the authors have any insight, based on their understanding of the COX-2 system in the gut, which would allow them to speculate as to a possible mechanism that might link these two?
I would like to thank Dr. Chung, Dr. Evers, and their colleagues for allowing me to discuss their exciting work and for furnishing me a copy of their manuscript in advance of the meeting. Sue and I would also like to express our appreciation to the membership of the Association for including us in your fellowship.
DR. MARTIN J. HESLIN (Birmingham, Alabama): Dr. Aust, Dr. Townsend, members, and guests. I rise to congratulate Dr. Chung and Dr. Evers on their really important work.
Here, a clinician recognizes a problem, evaluates molecular markers in a clinical specimen, takes it back to the laboratory, creates a model that, for whatever shortcomings it has, has a lot of the similarities of necrotizing enterocolitis, and looks at similar mediators in an effort to exploit molecular pathways. This is not a trivial undertaking.
The questions that I have relate to COX-2 pathways, and with my background being in malignancies and cancer, we look at COX-2 overexpression as something that is bad. And with the markers or the products of COX-2 overexpression, prostaglandins and thromboxanes, as you mentioned, there are some that are viewed as helpful and some that are deleterious. Prostaglandins can be measured in tissue levels, and it would be important to look at these markers. I was wondering if you had or planned to look at the products of COX-2 overexpression and the individual prostaglandins that are produced in necrotizing enterocolitis and the small intestine of the animal model.
Secondly, in your manuscript in your conclusion you mentioned COX-2 inhibition as potentially being protective. With COX-2 in malignancy, we look at it as an induction of a number of processes that allow cells to escape but are also involved in normal processes in inflammation. COX-2 is believed to be an upstream regulator of metalloprotease, which is involved in the normal process of tissue remodeling and healing. Angiogenesis, again, which is involved in normal inflammation, the ingrowth of new vessels in inflamed tissue and then subsequent resolution. As many children with necrotizing enterocolitis don’t progress to perforation and subsequently heal, do you believe or do you have any evidence that COX-2 inhibition would be protective or ultimately deleterious?
I would like to thank the Association for allowing me to discuss this manuscript.
DR. DAVID MERCER (Houston, Texas): Dr. Aust, Dr. Townsend, members, and guests. I’d like to congratulate the authors on an excellent paper nicely demonstrating that COX-2 immunoreactivity is increased, not only in patients with necrotizing enterocolitis but also in a rat model where enterocolitis is experimentally induced.
It would seem imperative to me, and I’m echoing some of the comments made already by others, since the COX-2 enzyme can have both beneficial and deleterious effects, do the authors have any preliminary data or do they plan to do any work with COX-2 inhibitors that are selected for the COX-2 enzyme? Or do you have any data with COX-2 deficient knockout mice?
I was somewhat surprised to see changes in the COX-2 so early on in this model, too, at only 60 minutes of ischemia. It also begged the question what happens with ischemia alone, as well as platelet activating factor alone, as I do not see those controls in the manuscript. The reason I say that, I’m not too surprised that the IκB degradation and NF-κB got turned on so rapidly, but it seems like a short period of time for the NF-κB to translocate and then to turn on transcription, translate that into protein, and then have protein that is functional in making prostanoids. So I would ask the question there, are you surprised by the rapidity of the response in your model?
And then, along those lines, have you looked at any earlier time points with ischemia as for IκB degradation and NF-κB translocation? When does it occur? Is it 5 minutes, 15 minutes, 30 minutes, or does it have to be 60 minutes for the IκB degradation to occur?
Again, like Dr. Heslin noted, the COX enzyme produces a number of prostanoids, PGE2, prostacyclin, thromboxanes. Have you looked at any of these prostanoids in your model, either in the necrotizing enterocolitis patients or in the experimentally induced enterocolitis, to find out which is the principal prostanoid involved, as it may help to ascertain whether these are beneficial or deleterious prostanoids being produced?
Lastly, with regards to NF-κB, as the authors have acknowledged in their manuscript, there are a number of subunits involved here, p50, p65, C-rel. Have you done any supershift assays to determine which of the NF-κB subunits is pertinent in your model? And along those lines, do you really think that it would be clinically relevant to use NF-κB inhibitors in patients when, while they may turn on a lot of bad actors, they probably also turn on a lot of good actors that help to combat information?
I’d like to thank the Society for the privilege of the floor.
DR. DAI H. CHUNG (Galveston, Texas): I would like to thank all the discussants for their insightful comments and questions.
Dr. Langham asked whether we have any information regarding the duration of ischemia on COX-2 induction or the rate of response for earlier timepoints. We learned that our present model produces a severe lethal injury of NEC. All of the rats died approximately 4 to 5 hours after induction of NEC. Therefore, we are currently performing additional experiments to determine the expression of COX-2/NF-κB in the gut after variable duration of ischemic insult (shorter than one hour of vessel clamping) as well as a prolonged reperfusion period of up to 24 hours.
We have not studied the specific effects of EGF or COX/NF-κB responses during NEC in our rodent model. However, we plan to investigate not only EGF but other mitogenic gut hormones, such bombesin and neurotensin, for their potential protective responses in this NEC model.
We do not know of any specific relationship between human EpO and COX/prostaglandin pathway in the gut during NEC.
Dr. Heslin and Dr. Mercer asked whether we measured specific levels of individual prostaglandins that are produced in NEC. We did not measure tissue prostaglandin levels in the small intestine, but we plan to do so in our animal model.
Regarding the question of whether COX-2 induction in NEC is a protective or deleterious mechanism, the data from our present study do not support a cause and effect relationship of COX-2 induction in NEC. However, in recent literature induction of COX-2 has been shown to play a role in both pro- and anti-inflammatory responses in the gut. Therefore, we are conducting experiments to further elucidate a role for COX-2 induction in NEC by studying the effect of specific COX-2 inhibitors as well as the use of COX-2 knockout mice on intestinal injury during NEC; however, we have no preliminary data as yet.
Dr. Mercer, we have looked at the effect of inhibiting NF-κB, an upstream regulator of COX-2, in our rodent model using a specific inhibitor of NF-κB. Our preliminary data suggest that rats receiving this NF-κB inhibitor show increased survival beyond 6 to 8 hours after induction of NEC; however, we have not yet assessed COX-2/prostaglandin levels in small intestinal tissues.
We have not examined the expression of COX-2 protein and NF-κB binding activity after ischemia-reperfusion injury alone or administration of platelet activating factor alone, but we plan to do studies to further discern the role of these insults on induction of the COX/NF-κB pathway.
We agree that it is important to perform a time-course study to further define NF-κB activation during NEC and we are currently conducting experiments to examine expression of COX-2 and activation of NF-κB during the early, as well as late, period after induction of NEC. We did not perform supershift assays to identify which subunits of NF-κB are involved during NEC; however, we plan to do so.
Footnotes
Presented at the 112th Annual Session of the Southern Surgical Association, December 4-6, 2000, Palm Beach, Florida.
Supported by grants from National Institutes of Health (PO1 DK35608, RO1 DK48498, T32 DK07639), and Shriners Hospitals for Children (#8250). R.T.E. is a recipient of a National Alpha Omega Alpha Student Research Fellowship and a McLaughlin Fellowship Award.
Correspondence: Dr. Dai H. Chung, Department of Surgery, The University of Texas Medical Branch, 301 University Blvd., Children’s Hospital, Room 3.220, Galveston, TX 77555-0353.
E-mail: dhchung@utmb.edu
Accepted for publication December 2000.
References
- 1.Kliegman RM, Fanaroff AA. Necrotizing enterocolitis. N Engl J Med 1984; 310: 1093–1103. [DOI] [PubMed] [Google Scholar]
- 2.Hsueh W, Caplan MS, Tan X, et al. Necrotizing enterocolitis of the newborn: pathogenetic concepts in perspective. Pediatr Dev Pathol 1998; 1: 2–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ryder RW, Shelton JD, Guinan ME. Necrotizing enterocolitis: a prospective multicenter investigation. Am J Epidemiol 1980; 112: 113–123. [DOI] [PubMed] [Google Scholar]
- 4.Kosloske AM. Epidemiology of necrotizing enterocolitis. Acta Paediatr Suppl 1994; 396: 2–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Martinez-Tallo E, Claure N, Bancalari E. Necrotizing enterocolitis in full-term or near-term infants: risk factors. Biol Neonate 1997; 71: 292–298. [DOI] [PubMed] [Google Scholar]
- 6.Grosfeld JL, Chaet M, Molinari F, et al. Increased risk of necrotizing enterocolitis in premature infants with patent ductus arteriosus treated with indomethacin. Ann Surg 1996; 224: 350–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Foglia RP. Necrotizing enterocolitis. Curr Probl Surg 1995; 32: 757–823. [DOI] [PubMed] [Google Scholar]
- 8.Needleman P, Turk J, Jakschik BA, et al. Arachidonic acid metabolism. Ann Rev Biochem 1986; 55: 69–102. [DOI] [PubMed] [Google Scholar]
- 9.Williams CS, Dubois RN. Prostaglandin endoperoxide synthase: why two isoforms? Am J Physiol 1996; 270: G393–400. [DOI] [PubMed] [Google Scholar]
- 10.Singer II, Kawka DW, Schloemann S, et al. Cyclooxygenase 2 is induced in colonic epithelial cells in inflammatory bowel disease. Gastroenterology 1998; 115: 297–306. [DOI] [PubMed] [Google Scholar]
- 11.Serou MJ, DeCoster MA, Bazan NG. Interleukin-1 beta activates expression of cyclooxygenase-2 and inducible nitric oxide synthase in primary hippocampal neuronal culture: platelet-activating factor as a preferential mediator of cyclooxygenase-2 expression. J Neurosci Res 1999; 58: 593–598. [PubMed] [Google Scholar]
- 12.Guan Z, Buckman SY, Miller BW, et al. Interleukin-1 beta-induced cyclooxygenase-2 expression requires activation of both c-Jun NH2-terminal kinase and p38 MAPK signal pathways in rat renal mesangial cells. J Biol Chem 1998; 273: 28670–28676. [DOI] [PubMed] [Google Scholar]
- 13.Diaz A, Chepenik KP, Korn JH, et al. Differential regulation of cyclooxygenase 1 and 2 by interleukin-1 beta, tumor necrosis factor-alpha, and transforming growth factor-beta 1 in human lung fibroblasts. Exp Cell Res 1998; 241: 222–229. [DOI] [PubMed] [Google Scholar]
- 14.Appleby SB, Ristimaki A, Neilson K, et al. Structure of the human cyclooxygenase-2 gene. Biochem J 1994; 302: 723–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thanos D, Maniatis T. NF-κB: a lesson in family values. Cell 1995; 80: 529–532. [DOI] [PubMed] [Google Scholar]
- 16.Mercurio F, Manning AM. NF-κB as a primary regulator of the stress response. Oncogene 1999; 18: 6163–6171. [DOI] [PubMed] [Google Scholar]
- 17.Schreiber S, Nikolaus S, Hampe J. Activation of nuclear factor κB in inflammatory bowel disease. Gut 1998; 42: 477–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Steinle AU, Weidenbach H, Wagner M, et al. NF-κB/Rel activation in cerulein pancreatitis. Gastroenterology 1999; 116: 420–430. [DOI] [PubMed] [Google Scholar]
- 19.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 1976; 72: 248–254. [DOI] [PubMed] [Google Scholar]
- 20.Ethridge RT, Hellmich MR, DuBois RN, et al. Inhibition of heat-shock protein 70 induction in intestinal cells overexpressing cyclooxygenase 2. Gastroenterology 1998; 115: 1454–1463. [DOI] [PubMed] [Google Scholar]
- 21.Ehlers RA, Hernandez A, Bloemendal LS, et al. Mitochondrial DNA damage and altered membrane potential (delta psi) in pancreatic acinar cells induced by reactive oxygen species. Surgery 1999; 126: 148–155. [PubMed] [Google Scholar]
- 22.Musemeche CA, Baker JL, Feddersen RM. A model of intestinal ischemia in the neonatal rat utilizing superior mesenteric artery occlusion and intraluminal platelet-activating factor. J Surg Res 1995; 58: 724–727. [DOI] [PubMed] [Google Scholar]
- 23.Vane JR, Bakhle YS, Botting RM. Cyclooxygenase 1 and 2. Annu Rev Pharmacol Toxicol 1998; 38: 97–120. [DOI] [PubMed] [Google Scholar]
- 24.Gustafson T, Hedner P, Ingemansson S. Prostaglandin E1 effect on human splanchnic circulation at different dose levels. Invest Radiol 1982; 17: 249–253. [PubMed] [Google Scholar]
- 25.Cohn SM, Schloemann S, Tessner T, et al. Crypt stem cell survival in the mouse intestinal epithelium is regulated by prostaglandins synthesized through cyclooxygenase-1. J Clin Invest 1997; 99: 1367–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reid B, Smith H, Friedman Z. Prostaglandins in human milk. Pediatrics 1980; 66: 870–872. [PubMed] [Google Scholar]
- 27.Wallace JL, Reuter BK, McKnight W, et al. Selective inhibitors of cyclooxygenase-2: are they really effective, selective, and GI-safe? J Clin Gastroenterol 1998; 27: S28–34. [DOI] [PubMed] [Google Scholar]
- 28.Morteau O, Morham SG, Sellon R, et al. Impaired mucosal defense to acute colonic injury in mice lacking cyclooxygenase-1 or cyclooxygenase-2. J Clin Invest 2000; 105: 469–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schmedtje JF Jr., Ji YS, Liu WL, et al. Hypoxia induces cyclooxygenase-2 via the NF-κB p65 transcription factor in human vascular endothelial cells. J Biol Chem 1997; 272: 601–608. [DOI] [PubMed] [Google Scholar]
- 30.Lo CJ, Fu M, Lo FR, Cryer HG. Cyclooxygenase 2 (COX-2) gene activation is regulated by cyclic adenosine monophosphate. Shock 2000; 13: 41–45. [DOI] [PubMed] [Google Scholar]
- 31.Ford HR, Sorrells DL, Knisely AS. Inflammatory cytokines, nitric oxide, and necrotizing enterocolitis. Sem Pediatr Surg 1996; 5: 155–159. [PubMed] [Google Scholar]
- 32.Lauzurica P, Martinez-Martinez S, Marazuela M, et al. Pyrrolidine dithiocarbamate protects mice from lethal shock induced by LPS or TNF-α. Eur J Immunol 1999; 29: 1890–1900. [DOI] [PubMed] [Google Scholar]
- 33.May MJ, D’Acquisto F, Madge LA, et al. Selective inhibition of NF-κB activation by a peptide that blocks the interaction of NEMO with the IκB kinase complex. Science 2000; 289: 1550–1554. [DOI] [PubMed] [Google Scholar]