

Abstract
Objective
To provide an introduction to the concept of DNA methylation and its function in normal cells, and to explain the possible mechanisms as to how abnormalities in this phenomenon can relate to carcinogenesis. The clinical implications with reference to common malignancies encountered in surgical practice are discussed.
Summary Background Data
Methylation of DNA is a heritable, enzyme-induced modification to DNA structure without alteration of the specific sequence of the base pairs responsible for encoding the genome. DNA methylation can both directly inhibit the expression of genes and also increase the probability that affected genes undergo a mutational event. Although DNA methylation plays an essential role in normal biologic processes, distinct and abnormal patterns of methylation are observed in cancers. In particular, there has been increased documentation that methylation of the promoter regions of several genes, including known tumor suppressor genes, results in the subsequent failure to express their functional proteins. Consequently, DNA methylation may represent an early and fundamental step in the pathway by which normal tissue undergoes neoplastic transformation. Further, an assessment of the methylation profiles within neoplastic tissues may provide key information in enhancing the diagnosis, predicting the clinical behavior, and designing specific treatment plans for individual patients.
Methods
Published literature from 1925 to 2000 contributing to an understanding of the purpose of DNA methylation and how pathology of this phenomenon could contribute to cancer are reviewed. Theories on these issues and the evidence that led to them are described. The present status of the subject in a clinical context is discussed.
Results
Gene expression can be significantly modulated by alterations in DNA methylation patterns. Methylation within the promoter regions of tumor suppressor genes causes their silencing, and methylation within the gene itself can induce mutational events. These mechanisms may play a fundamental role in precipitating the development of a large and diverse number of human cancers.
Conclusions
DNA methylation is an important factor in the development of cancer. A greater understanding of the relationship between DNA methylation events at the molecular level and its interaction in the clinical context may provide the basis for future advances in the surgical and pharmacologic management of malignant diseases.
GENE EXPRESSION, CELL BEHAVIOR, AND PATHWAYS TO CANCER
The expression of genetic information within an individual cell dictates how that cell subsequently behaves. Events at the molecular level can influence the expression of certain genes and thereby adversely affect cellular function to such a degree as to initiate a pathologic process. Cancer cells, for example, must undergo a number of molecular events that allow them to acquire several distinct but pathologic behavioral properties. These properties result in deleterious clinical consequences for the patient in whom the cancer cell resides, but paradoxically empowers that abnormal cell and its progeny with a survival advantage over normal cells.
The fundamental properties required to generate the characteristic malignant attributes associated with cancer cells are the ability to replicate without limitations, indifference to positive growth signals, disregard for growth inhibitory factors, evasion of programmed cell death (apoptosis), sustained angiogenesis, and the ability to invade and metastasize. 1 Each of these traits is influenced by a gene or set of genes. Failure to express the gene correctly and produce functional regulatory proteins leads to the uncontrolled patterns of cell behavior observed in a typical neoplasm.
Basic models describing the evolution of normal cells into cancer cells imply that a series of distinct proliferative changes are undergone, which in some cancers can be classified into distinct histopathologic lesions. Common examples are the adenoma-to-carcinoma sequence for colorectal cancer and the metaplasia-to-dysplasia-to-carcinoma sequence for esophageal adenocarcinoma. At each stage, the cell acquires some but not all the attributes of a cancer cell as defined at the molecular level, and each one of these changes is secondary to a discrete molecular event. Knudson in 1971 2 proposed the “two-hit” hypothesis and used it to explain this phenomenon in retinoblastoma. The suggestion was that to transform the normal cell into a malignant cell, two discrete “hits” or molecular events to both alleles of a gene involved in the control of cell proliferation—in effect a tumor suppressor gene—are necessary. Intermediate steps in this overall multistage process may result in limited gene inactivation and the formation of the premalignant lesions that are sometimes observed preceding fully invasive cancer.
Much of the focus of molecular biologic research has concentrated on investigating the role of genetic changes—that is, direct alterations of DNA base sequence through mutation, deletion, or insertion, and their effect on subsequent gene expression and cell behavior. Recently, alternative mechanisms of gene modulation have been coming under scrutiny that, without disrupting the actual sequence of a gene, affect its expression and remain preserved after cell division. The inheritance of information on the basis of gene expression rather than base sequence is termed epigenetics. The methylation of DNA is recognized as a key mechanism in the regulation of gene expression in this way, and evidence for its role in the development of a wide variety of cancers is accumulating. 3
METHYLATION OF DNA: A NORMAL CELLULAR FUNCTION
DNA Methylation and Gene Inactivation
In 1953, a model for the structure of DNA was outlined by Watson and Crick. 4 This consists of two complementary strands of nitrogen containing base pairs joined to a 5-carbon sugar, deoxyribose. These link together to form a polymer with phosphodiesterase bonds, creating a sugar–phosphate backbone to which the bases are attached. The four bases consist of two pyrimidine groups, cytosine (C) and thymine (T), and two purine groups, guanine (G) and adenine (A). Their sequential combination constitutes the code for each gene. The complementary strands give rise to a double helix geometry as a result of the hydrogen bonds linking C with G, and A with T (Fig. 1).

Figure 1. DNA double helix and CpG dinucleotide pairs. DNA structure with opposing base pairs arranged on a double helix sugar–phosphate backbone. CpG dinucleotide pair units that are the sites for possible methylation are outlined.
Transcription is the process by which a messenger ribonucleic acid (mRNA) template is formed from the DNA sequence of a gene. RNA polymerase, which is responsible for generating the manufacture of mRNA from a DNA strand, recognizes and binds, before transcribing the gene, to a promoter region located upstream from the gene itself. The mRNA then forms the framework for the assembly of amino acids, where each triple sequence of base pairs represents the code for a certain amino acid. This second process, translation, concludes with the formation of cell proteins that regulate or support functions of the cell. This transcription–translation pathway from DNA to protein refers to the so-called central dogma in the molecular biology of the cell, and successful gene expression relates to the transformation of a specific genetic sequence into a properly functioning cellular protein.
DNA methylation is an enzyme-induced chemical modification to the DNA structure. A methyl (−CH3) group is covalently bonded to the 5-carbon on the cytosine base. This process is mediated by one or more of a group of enzymes known as DNA methyltransferases. The methyl group is provided by S-adenosyl methionine (SAM), and this is converted to S-adenosyl homocysteine (SAH) in the process. This is recycled back to SAM in a folate- and cobalamin-dependant pathway (Fig. 2). Biologic methylation in vertebrates occurs only on the cytosine bases, and further only on those linked directly to a guanine by the phosphodiesterase link, forming a CpG dinucleotide pair (see Fig. 1).
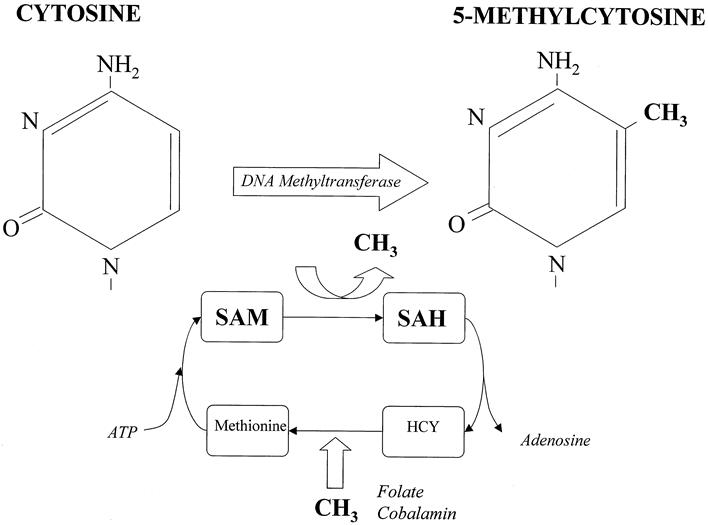
Figure 2. The methylation cycle. Methylation of the 5-carbon on the cytosine residue is executed by the DNA methyltransferase enzyme, which uses a methyl group from S-adenyl methionine (SAM). This is converted to S-adenyl homocysteine (SAH), which is then broken down to homocysteine (HCY) and adenosine. SAM is reconstituted from HCY by methionine. Folate and cobalamin are required for and provide the methyl groups for this reaction.
Early descriptions documented that the presence of DNA methylation roughly correlated with a decrease in genetic expression. It is now established that this inverse relationship between DNA methylation and gene expression is specific for when methylation occurs at the promoter regions of the gene. The process was initially thought to be due simply to the physical effect of the methyl group protruding from the DNA and interfering with the mechanics of transcription. 5 At present the inhibitory mechanism is thought to occur through the binding of specific proteins to the methylated DNA sequences. These proteins, such as MeCP2, belong to a family of proteins that contain a methyl-CpG binding domain (MBD) that recognizes and binds preferentially to methylated CpG groups irrespective of gene sequence. The protein also contains a transcriptional repression domain (TRD), which forms a complex with a variety of corepressor molecules (e.g., mSin3A) and histone deacetylase proteins (e.g., HDAC1, HDAC2). When this protein complex binds to methylated DNA, the histone proteins around which the DNA strands are wrapped to form chromatin become deacetylated. This causes changes in the chromatin structure, making it more condensed and the DNA less accessible, preventing active transcription from taking place (Fig. 3). Reversal of this effect can be achieved using an inhibitor of histone deacetylase, trichostatin A (TSA), which overrides this transcriptional silencing. 6–8
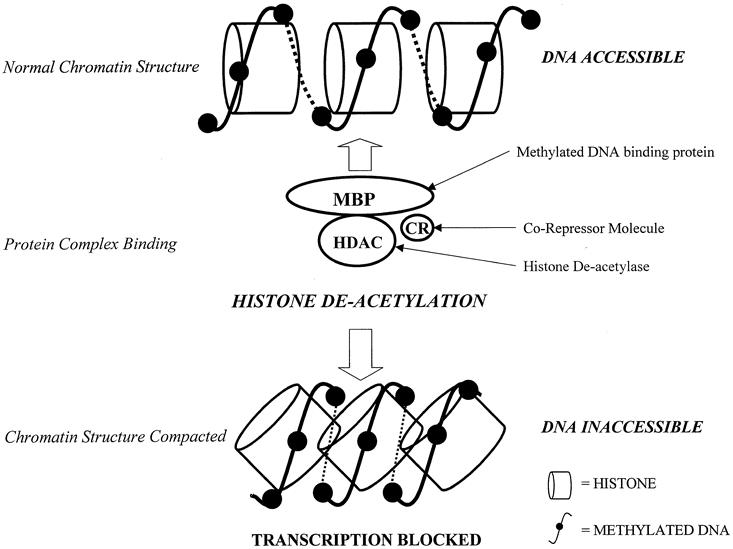
Figure 3. Transcriptional repression resulting from alteration of chromatin structure. Methylated DNA binds to a protein complex consisting of a methyl binding protein (MBP), which has a methyl-binding domain and a transcriptional repression domain, a corepressor molecule (CR), and a histone deacetylase (HDAC). After binding of this complex to the methylated DNA, the histones around which the DNA is wrapped become deacetylated, resulting in a compression and compaction of the chromatin structure. This makes the DNA inaccessible, and thus functional transcription is no longer possible.
Function of Methylated DNA
The possible existence of methylated cytosines within DNA has been known since the early part of the last century. They were initially described within the DNA from the tubercle bacillus 9 and subsequently were extracted from calf thymus, where they were known as epi-cytosine, producing a different chromotographic profile from normal cytosine. 10 Their purpose was not defined until relatively recently. It was speculated that they acted as a primitive host defense mechanism to silence invading DNA from viral organisms and provided an explanation for the latency of certain viral infections and how such agents can escape detection. 11,12
Today, DNA methylation is known to be an essential and normal component of mammalian embryogenesis. Targeted mutations of DNA methyltransferase genes in mice knock out or drastically reduce enzyme production and methylation activity and result in early embryonic lethality. 13 Several different genes encoding DNA methyltransferases have been identified. These enzymes exhibit two distinct functions but vary in their ability to perform one or the other. The functions are known as maintenance methyltransferase activity and de novo methyltransferase activity. Maintenance methyltransferase activity is responsible for methylation of newly synthesized strands of DNA based on the template of a single parent strand. Maintenance methyltransferase activity recognizes the hemimethylated pattern of the parent strand and then faithfully reproduces this on the daughter strand, allowing this feature to be heritable after DNA replication and cell division. The second function, de novo methyltransferase activity, is thought to be responsible for new methylation (methylation of previously unmethylated bases). This activity occurs extensively in the early stages of embryo development after implantation. Before implantation, the fertilized egg undergoes a wave of demethylation that deletes most of the preexisting patterns of methylation inherited from parental DNA, paving the way for a new pattern of methylation in the embryo. 14
A moderately decreased level of the methyltransferase results in less extensive disruption of methylation patterns and is compatible with fetal survival. In humans, mutations of one of the genes coding for a methyltransferase, DNMT3B, thought to be responsible for specific methylation of centromeric satellite repeat sequences, results in the rare ICF syndrome (immunodeficiency, centromeric heterochromatin instability, and facial anomalies). 15–17 Once a new methylation pattern has been established throughout the genome in the embryo, it remains constant and is maintained throughout life. This function provides the means by which the cell can regulate its own activity by switching off the expression of certain genes when not required. Examples of this phenomenon in normal cells are the global silencing of genes on the additional X-chromosome in females, preventing the double expression of genetic information compared with males, 18 and the imprinting of genes (i.e., the inactivation of one set of parental genes out of the two alternative copies available). 19
METHYLATION OF DNA: A ROLE IN CARCINOGENESIS
Models of Tumorigenesis
Altered methylation patterns are known to occur in the DNA of cancer cells. Two patterns have been observed: wide areas of global hypomethylation along the genome, and localized areas of hypermethylation at certain specific sites, the CpG islands, within the gene promoter regions. 20,21
Based on these patterns, several theories have emerged to implicate DNA methylation mechanisms in carcinogenesis. 22 In fundamental genetic models of cancer, the amplification of protooncogenes, or the silencing of tumor suppressor genes, disrupts the balance that normally controls cell proliferation and drives it through the succession of events leading to full malignant status. Thus, in theory, decreased methylation, and hence relief of transcriptional silencing, may allow the expression of previously quiescent protooncogenes to become active and induce the cell proliferation events (Fig. 4). Alternatively, increased methylation at previously unmethylated sites, such as the promoter regions of a tumor suppressor gene, may result in their silencing through inhibition of transcription and their inability to suppress cell proliferation.

Figure 4. Mechanisms of carcinogenesis induced by methylation events. (A) Activation of previously silent protooncogenes after hypomethylation. (B) Silencing of tumor suppressor genes after methylation of gene promoter region.
Abnormal methylation patterns can also indirectly affect gene activity with the disruption of the transcription–translation process by increasing the probability for a mutational event to take place and reducing overall chromosomal stability, resulting in the manufacture of a dysfunctional protein product. Methylated cytosine has a greater propensity to undergo spontaneous deamination and the formation of thymine. If this does occurs on a tumor suppressor gene, then a point mutation develops and loss of control of cell proliferation can occur (Fig. 5).
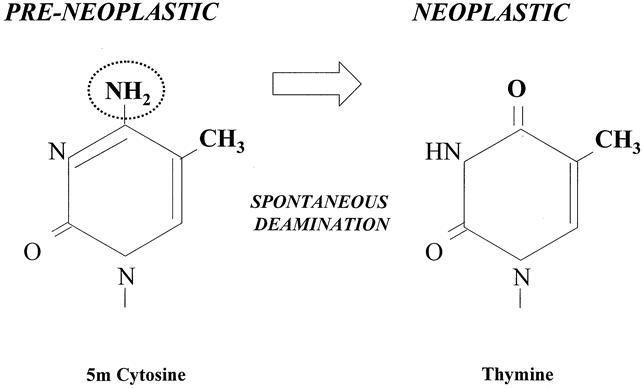
Figure 5. Methylation precipitating a point mutation. Cytosine-to-thymine point mutation after deamination of methylated cytosine.
Hypomethylation and Gene Activation
Despite considerable early interest, 23 investigators have failed consistently to implicate a definitive role for hypomethylation and the subsequent activation of protooncogenes (see Fig. 4). An example of such a relationship was noted in the BCL-2 gene and human B-cell chronic lymphocytic leukemia, 24 but further support for hypomethylation and resultant activation of protooncogenes in this manner has not been forthcoming. Although reduced levels of methylation of genes including C-MYC in human tumors have also been reported, it has not been possible to show convincingly that this is indeed responsible for increased levels of gene expression rather than merely a secondary characteristic observed in cancer cells. 25
Hypermethylation and Gene Silencing
In the human genome, the overall prevalence of CpG dinucleotide pairs is significantly less than would be expected statistically from the possible combinations available. The actual prevalence is only about 1% as opposed to the expected 6% (1/16). However, localized high-density concentrations of CpG repeat sequences between several hundred to a few thousand base pairs are noted to exist as islands in the promoter regions of many common genes, and in particular genes associated with tumor suppression. 26 These are normally unmethylated, but should these regions become methylated, failure to transcribe the downstream gene occurs, causing silencing of that gene (see Fig. 4).
This hypothesis can be confirmed if the reduction of DNA methyltransferase function through pharmacologic inhibition or gene inactivation results in reexpression of previously silenced genes. Substantive evidence that localized hypermethylation is responsible for rather than secondary to tumor-related gene inactivation has been accumulating from data derived from experimental animal models and cell culture technology, and observations in human tumor tissues.
For example, mice with heterozygous, multiple intestinal neoplasia mutations of the adenomatous polyposis coli (Apc) gene (Min mice) develop a condition that resembles human familial adenomatous polyposis coli. These mice develop multiple (>100) intestinal polyps in the first 6 months of life. Reduction of DNA methyltransferase activity in Min mice can be achieved through cross-breeding with mice heterozygous for a DNA methyltransferase gene that express lower levels of endogenous DNA methyltransferase. Further inhibition of the enzyme activity can be achieved pharmacologically with the addition of 5-aza-2′-deoxycytidine (5-aza-dC), potentiating the genetic effect. Through this combination of genetic and pharmacologic disruption of enzyme function, polyp formation can be drastically reduced by more than fifty-fold in these mice. 27
Another example is the hypermethylation of the CpG promoter region of the mismatch repair gene MLH1 observed in a subgroup of human colorectal cancers that show microsatellite instability. Microsatellite sequences are polymorphic, short, repeating segments of DNA between 1 and 4 base pairs distributed across the genome, and alterations to their pattern frequently occur if there is a deficiency in the cells’ ability to repair defects in DNA. The methylation of MLH1 results in failure to produce a functional protein and impairs the ability of the cell to repair mismatches that occur in the genome during proliferation, resulting in an increased mutation rate some 100 times greater than that in normal cells. Microsatellite instability is noted in approximately 13% of all sporadic cases of colorectal cancer and in nearly all patients with hereditary nonpolyposis colorectal cancer, which in turn is linked to mutations of the mismatch repair genes hMLH1 and hMSH2. In a significant proportion of tumors positive for microsatellite instability, no mutational abnormality can be shown, but hypermethylation and loss of hMLH1 protein expression does occur. This phenomenon is also observed in colorectal cancer cell lines, in which pharmacologic reversal of methylation events with 5-aza-dC restores both expression of hMLH1 protein and mismatch repair capacity. 28
In a similar manner, the inactivation of the CDKN2A (p16) tumor suppressor gene, located on the short arm of chromosome 9, has been reported in several human cancers. The p16 product protein is a cyclin-dependant kinase inhibitor and interferes with the cell cycle, thereby preventing cellular proliferation. Genetic alterations to it through point mutations and deletions have been reported in a wide variety of human cancers. 29 Loss of heterozygosity of p16 is commonly found in both esophageal adenocarcinoma and in Barrett’s metaplastic epithelium, the premalignant epithelium from which adenocarcinoma can evolve. 30 However, further mutations or deletions of the remaining copy of the p16 gene are not common in this cancer. 31 The explanation for adequate total gene inactivation in this situation was unclear. Recent demonstrations have shown that the promoter region of p16 is hypermethylated in patients with esophageal adenocarcinoma, suggesting that this epigenetic mechanism can act in concert with a genetic event to inactivate the gene fully. 32 The observation that this pattern is also observed in the metaplastic Barrett’s epithelium, particularly when associated with the more advanced dysplastic lesions 33 but without the presence of frank malignancy, indicates that the loss of p16 function as a result of mutation and methylation is an early molecular event in the evolution of adenocarcinoma. Hypermethylation of the p16 promoter region is now the most widely reported epigenetic event to occur in the development of human cancers. 34 Pharmacologic inhibition of methylation with 5-aza-dC in bladder cancer cell lines that also show methylation of the p16 promoter region can allow gene reactivation and transcription of the p16 product to recur. 35
Disruption of imprinting or parental specific gene inactivation, which is a normal function of DNA methylation within the cell, is seen in patients with Wilms tumors. Chromosome 11 contains the genes IGF2, which has characteristics of an autocrine growth factor, and H19, which appears to acts as a tumor suppressor gene to control cell proliferation. These genes are reciprocally expressed, with only paternal IGF2 and maternal H19 being active, to balance cellular growth. Loss of imprinting of these genes can occur in Wilms tumors. 36 The maternal chromosome reverts to a paternal pattern of gene expression, with methylation at normally unmethylated locations upstream to the H19 gene, switching off H19 expression. 37
The catalogue of genes involved in the control of cell proliferation and whose promoter regions are methylated in specific cancers is rapidly increasing. Documented reports of these associations are summarized in the Table.
Table. EXAMPLES OF GENES INVOLVED IN CELL PROLIFERATION AND METHYLATED IN CANCER

It appears that gene suppression can be achieved with relatively small amounts of methylation at specific defined sites in the promoter regions. 35 Some degree of selectivity in this process is therefore implied. This is illustrated by the observation that hypermethylation of the p15 promoter, observed in leukemia, and hypermethylation of the p16 promoter occur independently but rarely simultaneously in the common cancers, despite their close proximity to each other at location 9p21. 45 The increased occurrence of methylation of genes from normal tissues with aging, itself a risk factor for cancer, again suggests that methylation may be an early event in carcinogenesis. 39,53
This complements our present understanding of the genetic abnormalities that are already documented in certain neoplasms but which alone cannot fully explain the complete picture of the molecular events leading to malignant transformation. Methylation could act as an epigenetic means of inactivating one genetic copy. This, in combination with an independent genetic event or a second methylation event, can provide sufficient suppression of gene expression and failure to produce functional proteins to permit carcinogenesis (Fig. 6).
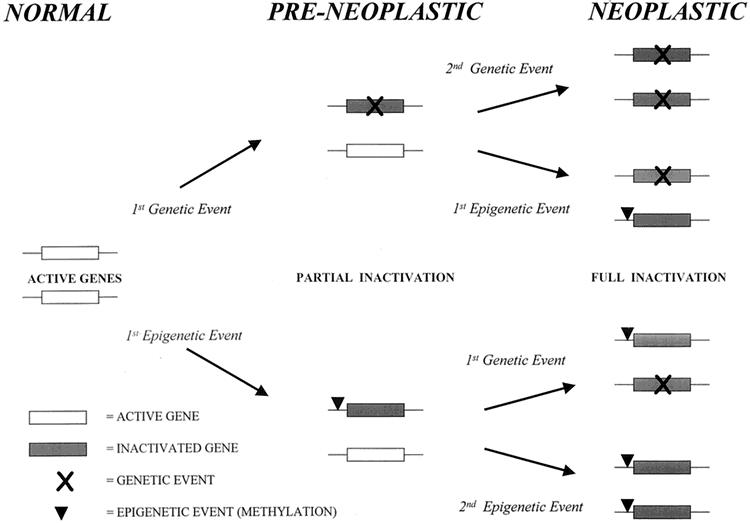
Figure 6. Alternative pathways to cancer. Combination of independent genetic (mutation, deletion, insertion) and epigenetic (methylation) events leading to complete gene inactivation through different routes and molecular heterogeneity within cancers. Genetic events may also be precipitated by an initial methylation event.
This revised model remains consistent with Knudson’s hypothesis and provides another explanation for the differing clinical characteristics of individual tumors based on heterogeneity at the molecular level. It is no coincidence that methylation is now being reported at several genetic locations in which mutation of only one copy has been strongly associated with a cancer in the past. These include RB and retinoblastoma, 59APC and colorectal cancer, 60 and VHL (von Hippel-Lindau) and renal cancers. 61 Mutation of the BRCA1 gene is closely associated with familial breast and ovarian cancers but is less common in sporadic cases. Methylation of BRCA1 has been reported in sporadic breast and ovarian tumors, 62 particularly if there is concurrent loss of heterozygosity at the BRCA1 locus. 63 Medullary and mucinous breast tumors, more common in familial breast cancers, show a greater degree of BRCA1 methylation than ductal tumors. 63
The combination of genetic and epigenetic events in cancer now provides a mechanism for complete inactivation of both allelic locations. Classification of tumors into categories based on molecular characteristics and whether they display certain methylation patterns is possible. Tumors can be termed as displaying a CpG island methylator phenotype, or CpG island methylator phenotype positive (CIMP+), if simultaneous methylation at multiple gene promoter regions, such as p16, hMLH1, or THBS1, occurs. This has been reported in some gastrointestinal tract cancers, and it is postulated that the pathway to cancer may be different depending on the CIMP status of the tumor. 64 For example, CIMP+ tumors show a higher degree of microsatellite instability and an association with a high mutation rate on certain genes such as K-RAS, but a low association with TP53 (p53) mutation. CIMP-negative tumors, in contrast, have a greater association with mutations at the p53 tumor suppressor gene but few other locations. 65 Whether distinguishing between molecularly heterogeneous groups of tumors of the same origin in this way correlates with clinically useful parameters remains to be determined. Both hypermethylation of p16 and mutation of p53 are recognized as early and fundamental events in carcinogenesis, and they may represent the first steps in alternative routes to the same goal. It is also possible that the classification of tumors in this manner may correlate with their clinical behavior and account for different outcomes or responses to treatment.
We do not know what induces methylation to occur at previously unmethylated locations in incipient cancer cells or, indeed, whether methylation is the primary event responsible for genetic silencing or merely a secondary event. Theoretically, abnormal methylation patterns could arise as a result of an overactivity of a “methylating” factor or the loss of a “demethylating” factor. The observation that DNA methyltransferase levels are increased in cancers 66 would tend to suggest this concept, but current investigations indicate that the elevation is likely to be a secondary effect of increased cell proliferation rather than a causal mechanism. 67 Further, in human colon cancer cell lines, abolition of DNMT1 activity, normally lethal in embryonic mouse cells, does not alter the methylation pattern either at normally methylated locations or at abnormally hypermethylated areas such as the p16 promoter region. This suggests a role for DNA methyltransferases other than DNMT1 or, alternatively, that as yet unelucidated factors are involved in maintaining malignant patterns of methylation. 68 Putative proteins with demethylating activity have been reported, but implication in this process remains inconclusive. 69 The actual initiation and maintenance of tumor methylation patterns will no doubt be an area of active research during the next few years. For the present it is accepted that promoter region methylation of genes involved in the control of cell proliferation results in their inactivation, and this is a fundamental event in the pathway to carcinogenesis.
Methylation-Related Mutational Events
Mutation of the p53 tumor suppressor gene is thought to play a key role in the development of many cancers. 70 The gene contains 513 base pairs, of which 42 are part of a CpG dinucleotide pair. In normal cells these base pairs show ubiquitous methylation, and the methylated cytosine residues show a high propensity to undergo deamination to form thymine. 71 In this way, a C-to-T point mutational event occurs. Because thymine is a normal component of human DNA, this mutation may not be correctly recognized by the DNA repair mechanisms. Instead of repairing the mutated thymine, the complementary strand guanine may be substituted for adenine to form the normal T–A opposition. Hence, a G-to-A point mutation occurs.
The transformation of C to T can occur either through spontaneous deamination of 5mC or by an enzyme-mediated mechanism where methyltransferase binding results in deamination before the methyl transfer to form uracil, which is then substituted by thymine after two rounds of DNA replication. This second reaction is observed in bacterial cells and is increased when the availability of the methyl donor SAM is deficient. This may be an explanation for the increased levels of carcinogenesis seen in mice fed a methyl group-deficient diet, 72,73 and the inverse relationship between dietary folate, essential in the SAM metabolism cycle, and colorectal tumors in humans. 74 There is, however, no evidence as yet that this particular SAM deficient mechanism plays a significant role in human cells. The major cause of the high mutation rate at CpG dinucleotides is likely to be spontaneous deamination of 5mC. 75 The vast majority of the mutational hot spots of the p53 gene occur at the CpG sites, although these sites represent less than a tenth of the total p53 genetic code, and further the C-to-T and G-to-A transitions are among the most abundant recorded mutations. 70 This event would be consistent with a methylation-induced genetic effect, and similar patterns have been noted in other genes such as the factor IX gene. 76
Some other theories for mutational induction secondary to DNA methylation are:
Failure to produce O6-methylguanine-DNA methyltransferase (MGMT), a DNA repair protein that normally protects from mutations occurring at guanine bases, results in increased G-to-A mutations. 77
Damage to DNA effected by oxygen free radicals has the potential to cause adenine mutations during tumor progression. This effect is limited by glutathionine S transferase and DNA repair protein encoded from the GTSP1 gene. Hypermethylation of the promoter region of the GSTP1, with subsequent gene inactivation, has been documented in breast and prostate tumors, and it is speculated that these tumors may be predisposed to increased oxidative DNA damage by this mechanism. 78
The hypomethylation observed along the genome in cancers may also predispose to increased mutational rates by contributing to overall chromosomal instability. 79
These observations show that abnormal methylation patterns contribute to the evolution of cancer by several different potential pathways.
CLINICAL RELEVANCE OF DNA METHYLATION
An understanding of the molecular events that lead to the evolution of cancer and are responsible for the heterogeneity of tumors in individual patients can be of benefit to the clinician for at least three reasons: it can improve the accuracy and timing of the diagnosis of cancer, it can provide prognostic information about the cancer, and it can offer a potential means for cancer therapy.
Diagnosis of Early Cancers
One key to improving the clinical outcome in patients with cancer is the urgent need to diagnose the disease at its earliest possible stage, which translates into a survival benefit for the patient. If diagnosis is possible before extensive local invasion, lymph node spread, or disseminated disease, then the surgical resection can be less radical, with fewer complications and side effects. This forms the rationale for population screening and the surveillance of high-risk patients. However, the investigative methods used in these cancer prevention programs, such as serum tumor markers, radiologic procedures, and endoscopic examinations, all have major drawbacks, such as limited sensitivity and specificity, expense, and patient compliance. Further, the diagnosis of equivocal lesions in asymptomatic patients creates a new clinical dilemma, in which further investigations and surgery are conducted without proof that they are necessary or beneficial. For example, ductal carcinoma in situ is often found incidentally in screening mammography; similarly, Barrett’s epithelium may be noted during upper gastrointestinal endoscopy. Lesions such as these have a risk of developing into invasive cancer, but it is impossible to predict with any certainty which patients will have malignant disease and require surgery, versus which will remain quiescent, making potentially hazardous intervention less justifiable.
If methylation of gene promoter regions does prove to be a consistent and early event in the incipient cancer cells, perhaps with specific gene combinations for different cancers, a potential tool for diagnosing premalignant lesions could become available. This may provide the means for a more accurate screening and surveillance rationale by identifying higher-risk patients on a molecular basis. It would also provide justification for more definitive treatment of patients who have molecular but not yet all the typical pathologic or microscopic features associated with frank malignancy. Prevention of a subsequent cancer in such patients may be accomplished by the focal resection or ablation of the involved tissue. Surgical management of such potential cancer would be done at a stage before invasion and metastasis, allowing the use of minimally invasive procedures that are associated with fewer complications. Application of this rationale to the growing number of patients currently requiring regular surveillance endoscopy to monitor Barrett’s metaplasia in the esophagus would be ideal. Barrett’s esophagus is a premalignant lesion, but esophageal adenocarcinoma develops in approximately 10% of patients. 80,81 The incidence of esophageal adenocarcinoma is increasing, 82 and thus selective surveillance of patients with Barrett’s esophagus is essential. Categorization of patients to receive intense endoscopic surveillance while relieving others from this need may be an achievable goal using DNA methylation profiles in Barrett’s tissue biopsy specimens.
Predicting Outcome and Monitoring Progress
Staging of tumors based on the levels and pattern distribution of DNA methylation may provide a convenient way to assess a tumor’s biologic aggressiveness and to predict patient outcome. It would seem likely that the methylation and inactivation of genes essential to specific vital cell proliferation events would be associated with a particular behavioral trait of a tumor, such as the ability to form distant metastases. Based on the principle that mutations of key genes such as p53 in certain cancers have been linked with a poor prognosis, 83 methylation profiles may be able to augment current staging classifications.
The presence of free tumor DNA in the serum of patients has been recognized as a potential means of monitoring the efficacy of cancer therapy. 84 Although genetic defects in the DNA specific to the tumor of origin can be identified and sometimes correlated with clinical parameters, 85 the process is expensive and time-consuming and may not be a reliable reflection of the state of the disease. 86,87 Abnormal gene promoter region methylation patterns within circulating serum tumor DNA from patients with breast, 87 lung, 88 liver, 89 and head and neck 90,91 tumors have recently been identified. This provides a rapid, quantitative, and less expensive biologic marker. Subsequent clinical correlation will determine whether this approach has the sensitivity and sensitivity to be a useful molecular serum marker. If so, methylation patterns of circulating DNA released by the tumor may provide a means for monitoring the progress of a tumor and its response to therapy.
Potential Tailored Therapeutic Options
Information about how a cancer develops through molecular events could allow a clinician to predict more accurately how such a cancer is likely to respond to specific chemotherapeutic agents. In this way, a regimen tailored to the individual patient and based on knowledge of the tumor’s chemosensitivity could be designed. A simple example is with tumors that increase their proliferation in response to steroid hormones. Breast and prostate cancers, some of which are trophic to circulating estrogens and androgens, respectively, can respond to endocrine therapy. Some tumors, however, become insensitive to hormonal blockade, proliferating independently, and are unlikely to respond to trials of antisteroid hormone medication. Breast tumors that express the estrogen receptor (ER+) are usually sensitive to antiestrogen treatment, particularly if the progesterone receptor (PR) is also expressed, whereas those lacking ER receptors (ER-) do not normally respond to this form of treatment. 92 Neither mutations of the ER gene nor the PR gene in breast cancers lacking expression of these receptors have been identified to explain this form of molecular heterogeneity. 93,94 Methylated promoters to the ER gene and the PR gene do, however, correlate with the failure of expression of these receptors in human breast tissues. 95 In cell culture models, treatment of breast cancer cell lines with 5-aza-dC demethylates the ER promoter and results in ER reexpression. 96,97 In this instance, direct heterogeneity in the methylation profile between two tumors can predict potential response to a treatment modality, and a theoretical means of altering the tumor phenotype (and hence its response) is a possibility. Similar paradigms may be proven for other tumors and growth factors such as prostate cancers and androgen receptors, and may also explain the sexual dimorphism associated with certain cancers.
As our understanding of tumor biology increases, the genes involved in different intracellular biochemical reactions specific to individual cancers will be identified. The methylation profile of such genes could be used to predict the efficacy of therapy designed to interrupt these pathways. Reversal of abnormal methylation patterns would seem an attractive and logical therapeutic means of arresting cancer growth or spread or even obliterating a neoplasm. Several molecular study groups are investigating the transcriptional failure that accompanies DNA methylation, and with this understanding will come several potential targets for engineering novel pharmacologic weapons against cancer. Patients with leukemia have already been treated with 5-aza-dC in a clinical setting. 97 The drug primarily interferes with the DNA methyltransferase function, and though initial results have been promising, the effect may be from general cellular toxicity rather than targeted strikes against methylated DNA segments. 98 Combination of lower doses of 5-aza-dC with a histone deacetylase or other inhibitor of the methylation–transcription blockade complex may be another option in this area. 99
The science of DNA methylation has experienced rapid growth and interest during the past decade, and its implications relate directly to diseases in the realm of surgical oncology. The current concepts regarding methylation complement our present understanding of the genetic basis of malignancy. The union of epigenetic and genetic information pertaining to specific cancers in individual patients will provide a more precise definition of the exact nature of the malignancy. This information is likely to affect the nature of surgical therapy by a greater use of limited-access technology in early disease and the more appropriate application of extensive resection in advanced disease.
Footnotes
Correspondence: Tom R. DeMeester, MD, Department of Surgery, University of Southern California School of Medicine, 1510 San Pablo St. HCC 514, Los Angeles, CA 90033.
E-mail: demeester@surgery.hsc.usc.edu
Accepted for publication September 29, 2000.
References
- 1.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000; 100: 57–70. [DOI] [PubMed] [Google Scholar]
- 2.Knudson AGJ. Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci USA 1971; 68: 820–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jones PA, Laird PW. Cancer epigenetics comes of age. Nat Genet 1999; 21: 163–167. [DOI] [PubMed] [Google Scholar]
- 4.Watson JD, Crick FHC. Genetic implications of the structure of deoxyribonucleic acid. Nature 1953; 171: 964–967. [DOI] [PubMed] [Google Scholar]
- 5.Razin A, Riggs AD. DNA methylation and gene function. Science 1980; 210: 604–610. [DOI] [PubMed] [Google Scholar]
- 6.Nan X, Campoy FJ, Bird A. MeCP2 is a transcriptional repressor with abundant binding sites in genomic chromatin. Cell 1997; 88: 471–481. [DOI] [PubMed] [Google Scholar]
- 7.Jones PL, Veenstra GJ, Wade PA, et al. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat Genet 1998; 19: 187–191. [DOI] [PubMed] [Google Scholar]
- 8.Bestor TH. Gene silencing. Methylation meets acetylation. Nature 1998; 393: 311–312. [DOI] [PubMed] [Google Scholar]
- 9.Johnson TB, Coghill RD. The discovery of 5-methyl-cytosine in tuberculinic acid, the nucleic acid of the tubercle bacilus. J Am Chem Soc 1925; 47: 2838–2844. [Google Scholar]
- 10.Hotchkiss RD. The quantitative separation of purines, pyrimidines, and nucleosides by paper chromatography. J Biol Chem 1948; 175: 315–332. [PubMed] [Google Scholar]
- 11.Laird PW, Jaenisch R. The role of DNA methylation in cancer genetic and epigenetics. Annu Rev Genet 1996; 30: 441–464. [DOI] [PubMed] [Google Scholar]
- 12.Robertson KD, Ambinder RF. Methylation of the Epstein-Barr virus genome in normal lymphocytes. Blood 1997; 90: 4480–4484. [PubMed] [Google Scholar]
- 13.Li E, Bestor TH, Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell 1992; 69: 915–926. [DOI] [PubMed] [Google Scholar]
- 14.Razin A, Shemer R. DNA methylation in early development. Hum Mol Genet 1995; 4: 1751–1755. [DOI] [PubMed] [Google Scholar]
- 15.Jeanpierre M, Turleau C, Aurias A, et al. An embryonic-like methylation pattern of classical satellite DNA is observed in ICF syndrome. Hum Mol Genet 1993; 2: 731–735. [DOI] [PubMed] [Google Scholar]
- 16.Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 1999; 99: 247–257. [DOI] [PubMed] [Google Scholar]
- 17.Xu GL, Bestor TH, Bourchis D, et al. Chromosome instability and immunodeficiency syndrome caused by mutations in a DNA methyltransferase gene. Nature 1999; 402: 187–191. [DOI] [PubMed] [Google Scholar]
- 18.Grant SG, Chapman VM. Mechanisms of X-chromosome regulation. Annu Rev Genet 1988; 22: 199–233. [DOI] [PubMed] [Google Scholar]
- 19.Razin A, Cedar H. DNA methylation and genomic imprinting. Cell 1994; 77: 473–476. [DOI] [PubMed] [Google Scholar]
- 20.Feinberg AP, Vogelstein B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature 1983; 301: 89–92. [DOI] [PubMed] [Google Scholar]
- 21.Baylin SB, Hoppener JW de B, Steenbergh PH, et al. DNA methylation patterns of the calcitonin gene in human lung cancers and lymphomas. Cancer Res 1986; 46:2917–2922. [PubMed]
- 22.Laird PW. Oncogenic mechanisms mediated by DNA methylation. Mol Med Today 1997; 3: 223–229. [DOI] [PubMed] [Google Scholar]
- 23.Goelz SE, Vogelstein B, Hamilton SR, Feinberg AP. Hypomethylation of DNA from benign and malignant human colon neoplasms. Science 1985; 228: 187–190. [DOI] [PubMed] [Google Scholar]
- 24.Hanada M, Delia D, Aiello A, et al. bcl-2 gene hypomethylation and high-level expression in B-cell chronic lymphocytic leukemia. Blood 1993; 82: 1820–1828. [PubMed] [Google Scholar]
- 25.Sharrard RM, Royds JA, Rogers S, Shorthouse AJ. Patterns of methylation of the c-myc gene in human colorectal cancer progression. Br J Cancer 1992; 65: 667–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bird AP. CpG-rich islands and the function of DNA methylation. Nature 1986; 321: 209–213. [DOI] [PubMed] [Google Scholar]
- 27.Laird PW, Jackson-Grusby L, Fazeli A, et al. Suppression of intestinal neoplasia by DNA hypomethylation. Cell 1995; 81: 197–205. [DOI] [PubMed] [Google Scholar]
- 28.Herman JG, Umar A, Polyak K, et al. Incidence and functional consequences of hMLH1 promoter hypermethylation in colorectal carcinoma. Proc Natl Acad Sci USA 1998; 95: 6870–6875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kamb A, Gruis NA, Weaver-Feldhaus J, et al. A cell cycle regulator potentially involved in genesis of many tumor types. Science 1994; 264: 436–440. [DOI] [PubMed] [Google Scholar]
- 30.Barrett MT, Sanchez CA, Galipeau PC, et al. Allelic loss of 9p21 and mutation of the CDKN2/ p16 gene develop as early lesions during neoplastic progression in Barrett’s esophagus. Oncogene 1996; 13: 1867–1873. [PubMed] [Google Scholar]
- 31.Cairns P, Mao L, Merlo A, et al. Rates of p16 (MTS1) mutations with 9p loss. Science 1994; 265: 415–416. [DOI] [PubMed] [Google Scholar]
- 32.Wong DJ, Barrett MT, Stoger R, et al. p16 INK4a promoter is hypermethylated at a high frequency in esophageal adenocarcinomas. Cancer Res 1997; 57: 2619–2622. [PubMed] [Google Scholar]
- 33.Klump B, Hsieh CJ, Holzmann K, et al. Hypermethylation of the CDKN2/p16 promoter during neoplastic progression in Barrett’s esophagus. Gastroenterology 1998; 115: 1381–1386. [DOI] [PubMed] [Google Scholar]
- 34.Herman JG, Merlo A, Mao L, et al. Inactivation of the CDKN2/ p16/MTS1 gene is frequently associated with aberrant DNA methylation in all common human cancers. Cancer Res 1995; 55: 4525–4530. [PubMed] [Google Scholar]
- 35.Gonzalgo ML, Hayashida T, Bender CM, et al. The role of DNA methylation in expression of the p19/ p16 locus in human bladder cancer cell lines. Cancer Res 1998; 58: 1245–1252. [PubMed] [Google Scholar]
- 36.Rainier S, Johnson LA, Dobry CJ, et al. Relaxation of imprinted genes in human cancer. Nature 1993; 362: 747–749. [DOI] [PubMed] [Google Scholar]
- 37.Steenman MJ, Rainier S, Dobry CJ, et al. Loss of imprinting of IGF2 is linked to reduced expression and abnormal methylation of H19 in Wilms’ tumour [published erratum appears in Nat Genet 1994 Oct;8(2): 203]. Nat Genet 1994; 7: 433–439. [DOI] [PubMed] [Google Scholar]
- 38.Toyota M, Ahuja N, Suzuki H, et al. Aberrant methylation in gastric cancer associated with the CpG island methylator phenotype. Cancer Res 1999; 59: 5438–5442. [PubMed] [Google Scholar]
- 39.Ahuja N, Li Q, Mohan AL, et al. Aging and DNA methylation in colorectal mucosa and cancer. Cancer Res 1998; 58: 5489–5494. [PubMed] [Google Scholar]
- 40.Schutte M, Hruban RH, Geradts J, et al. Abrogation of the Rb/ p16 tumor-suppressive pathway in virtually all pancreatic carcinomas. Cancer Res 1997; 57: 3126–3130. [PubMed] [Google Scholar]
- 41.Esteller M, Levine R, Baylin SB, et al. MLH1 promoter hypermethylation is associated with the microsatellite instability phenotype in sporadic endometrial carcinomasAberrant methylation of p16 (INK4a) is an early event in lung cancer and a potential biomarker for early diagnosis. Oncogene 1998; 17: 2413–2417. [DOI] [PubMed] [Google Scholar]
- 42.McCluskey LL, Chen C, Delgadillo E, et al. Differences in p16 gene methylation and expression in benign and malignant ovarian tumors. Gynecol Oncol 1999; 72: 87–92. [DOI] [PubMed] [Google Scholar]
- 43.Gonzalgo ML, Bender CM, You EH, et al. Low frequency of p16/CDKN2A methylation in sporadic melanoma: comparative approaches for methylation analysis of primary tumors. Cancer Res 1997; 57: 5336–5347. [PubMed] [Google Scholar]
- 44.Cameron EE, Baylin SB, Herman JG. p15 (INK4B) CpG island methylation in primary acute leukemia is heterogeneous and suggests density as a critical factor for transcriptional silencing. Blood 1999; 94: 2445–2451. [PubMed] [Google Scholar]
- 45.Herman JG, Civin CI, Issa JP, et al. Distinct patterns of inactivation of p15 INK4B and p16 INK4A characterize the major types of hematological malignancies. Cancer Res 1997; 57: 837–841. [PubMed] [Google Scholar]
- 46.Fleisher AS, Esteller M, Wang S, et al. Hypermethylation of the hMLH1 gene promoter in human gastric cancers with microsatellite instability. Cancer Res 1999; 59: 1090–1095. [PubMed] [Google Scholar]
- 47.Strathdee G, MacKean MJ, Illand M, Brown R. A role for methylation of the hMLH1 promoter in loss of hMLH1 expression and drug resistance in ovarian cancer. Oncogene 1999; 18: 2335–2341. [DOI] [PubMed] [Google Scholar]
- 48.Ahuja N, Mohan AL, Li Q, et al. Association between CpG island methylation and microsatellite instability in colorectal cancer. Cancer Res 1997; 57: 3370–3374. [PubMed] [Google Scholar]
- 49.Hiraguri S, Godfrey T, Nakamura H, et al. Mechanisms of inactivation of E-cadherin in breast cancer cell lines. Cancer Res 1998; 58: 1972–1977. [PubMed] [Google Scholar]
- 50.Graff JR, Herman JG, Lapidus RG, et al. E-cadherin expression is silenced by DNA hypermethylation in human breast and prostate carcinomas. Cancer Res 1995; 55: 5195–5199. [PubMed] [Google Scholar]
- 51.Graff JR, Greenberg VE, Herman JG, et al. Distinct patterns of E-cadherin CpG island methylation in papillary, follicular, Hurthle’s cell, and poorly differentiated human thyroid carcinoma. Cancer Res 1998; 58: 2063–2066. [PubMed] [Google Scholar]
- 52.Bachman KE, Herman JG, Corn PG, et al. Methylation-associated silencing of the tissue inhibitor of metalloproteinase-3 gene suggests a suppressor role in kidney, brain, and other human cancers. Cancer Res 1999; 59: 798–802. [PubMed] [Google Scholar]
- 53.Issa JP, Ottaviano YL, Celano P, et al. Methylation of the oestrogen receptor CpG island links ageing and neoplasia in human colon. Nat Genet 1994; 7: 536–540. [DOI] [PubMed] [Google Scholar]
- 54.Ottaviano YL, Issa JP, Parl FF, et al. Methylation of the estrogen receptor gene CpG island marks loss of estrogen receptor expression in human breast cancer cells. Cancer Res 1994; 54: 2552–2555. [PubMed] [Google Scholar]
- 55.Issa JP, Baylin SB, Belinsky SA. Methylation of the estrogen receptor CpG island in lung tumors is related to the specific type of carcinogen exposure. Cancer Res 1997; 56: 3655–3658. [PubMed] [Google Scholar]
- 56.Issa JP, Zehnbauer BA, Civin CI, et al. The estrogen receptor CpG island is methylated in most hematopoietic neoplasms. Cancer Res 1996; 56: 973–977. [PubMed] [Google Scholar]
- 57.Li LC, Chui R, Nakajima K, Oh BR, et al. Frequent methylation of estrogen receptor in prostate cancer: correlation with tumor progression. Cancer Res 2000; 60: 702–706. [PubMed] [Google Scholar]
- 58.Jarrard DF, Kinoshita H, Shi Y, et al. Methylation of the androgen receptor promoter CpG island is associated with loss of androgen receptor expression in prostate cancer cells. Cancer Res 1998; 58: 5310–5314. [PubMed] [Google Scholar]
- 59.Ohtani-Fujita N, Dryja TP, Rapaport JM, et al. Hypermethylation in the retinoblastoma gene is associated with unilateral, sporadic retinoblastoma. Cancer Genet Cytogenet 1997; 98: 43–49. [DOI] [PubMed] [Google Scholar]
- 60.Hiltunen MO, Alhonen L, Koistinaho J, et al. Hypermethylation of the APC (adenomatous polyposis coli) gene promoter region in human colorectal carcinoma. Int J Cancer 1997; 70: 644–648. [DOI] [PubMed] [Google Scholar]
- 61.Herman JG, Latif F, Weng Y, et al. Silencing of the VHL tumor-suppressor gene by DNA methylation in renal carcinoma. Proc Natl Acad Sci USA 1994; 91: 9700–9704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Catteau A, Harris WH, Xu CF, Solomon E. Methylation of the BRCA1 promoter region in sporadic breast and ovarian cancer: correlation with disease characteristics. Oncogene 1999; 18: 1957–1965. [DOI] [PubMed] [Google Scholar]
- 63.Esteller M, Silva JM, Dominguez G, et al. Promoter hypermethylation and BRCA1 inactivation in sporadic breast and ovarian tumors. J Natl Cancer Inst 2000; 92: 515–517. [DOI] [PubMed] [Google Scholar]
- 64.Toyota M, Ahuja N, Ohe-Toyota M, et al. CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci USA 1999; 96: 8681–8686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Toyota M, Ohe-Toyota M, Ahuja N, Issa JP. Distinct genetic profiles in colorectal tumors with or without the CpG island methylator phenotype. Proc Natl Acad Sci USA 2000; 97: 710–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.el-Deiry WS, Nelkin BD, Celano P, et al. High expression of the DNA methyltransferase gene characterizes human neoplastic cells and progression stages of colon cancer. Proc Natl Acad Sci USA 1991; 88: 3470–3474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Eads CA, Danenberg KD, Kawakami K, et al. CpG island hypermethylation in human colorectal tumors is not associated with DNA methyltransferase over expression. Cancer Res 1999; 59: 2302–2306. [PubMed] [Google Scholar]
- 68.Rhee I, Jair KW, Yen RW, et al. CpG methylation is maintained in human cancer cells lacking DNMT1. Nature 2000; 404: 1003–1007. [DOI] [PubMed] [Google Scholar]
- 69.Bhattacharya SK, Ramchandi S, Cervoni N, et al. A mammalian protein with specific demethylase activity for mCpG DNA. Nature 1999; 397: 579–583. [DOI] [PubMed] [Google Scholar]
- 70.Greenblatt MS, Bennett WP, Hollstein M, Harris CC. Mutations in the p53 tumor suppressor gene: clues to cancer etiology and molecular pathogenesis. Cancer Res 1994; 54: 4855–4878. [PubMed] [Google Scholar]
- 71.Tornaletti S, Pfeifer GP. Complete and tissue-independent methylation of CpG sites in the p53 gene: implications for mutations in human cancers. Oncogene 1995; 10: 1493–1499. [PubMed] [Google Scholar]
- 72.Mikol YB, Hoover KL, Creasia D, Poirier LA. Hepatocarcinogenesis in rats fed methyl-deficient, amino acid-defined diets. Carcinogenesis 1983; 4: 1619–1629. [DOI] [PubMed] [Google Scholar]
- 73.Cravo ML, Mason JB, Dayal Y, et al. Folate deficiency enhances the development of colonic neoplasia in dimethylhydrazine-treated rats. Cancer Res 1992; 52: 5002–5006. [PubMed] [Google Scholar]
- 74.Giovannucci E, Rimm EB, Ascherio A, et al. Alcohol, low-methionine–low-folate diets, and risk of colon cancer in men. J Natl Cancer Inst 1995; 87: 265–273. [DOI] [PubMed] [Google Scholar]
- 75.Schmutte C, Yang AS, Nguyen TT, et al. Mechanisms for the involvement of DNA methylation in colon carcinogenesis. Cancer Res 1996; 56: 2375–2381. [PubMed] [Google Scholar]
- 76.Bottema CD, Ketterling RP, Vielhaber E, et al. The pattern of spontaneous germ-line mutation: relative rates of mutation at or near CpG dinucleotides in the factor IX gene. Hum Genet 1993; 91: 496–503. [DOI] [PubMed] [Google Scholar]
- 77.Esteller M, Toyota M, Sanchez-Cespedes M, et al. Inactivation of the DNA repair gene O6-methylguanine-DNA methyltransferase by promoter hypermethylation is associated with G to A mutations in K-ras in colorectal tumorigenesis. Cancer Res 2000; 60: 2368–2371. [PubMed] [Google Scholar]
- 78.Esteller M, Corn PG, Urena JM, et al. Inactivation of glutathione S-transferase P1 gene by promoter hypermethylation in human neoplasia. Cancer Res 1998; 58: 4515–4518. [PubMed] [Google Scholar]
- 79.Chen RZ, Pettersson U, Beard C, et al. DNA hypomethylation leads to elevated mutation rates. Nature 1998; 395: 89–93. [DOI] [PubMed] [Google Scholar]
- 80.Cameron AJ, Ott BJ, Payne WS. The incidence of adenocarcinoma in columnar-lined (Barrett’s) esophagus. N Engl J Med 1985; 313: 857–859. [DOI] [PubMed] [Google Scholar]
- 81.Sarr MG, Hamilton SR, Marrone GC, et al. Barrett’s esophagus: its prevalence and association with adenocarcinoma in patients with symptoms of gastroesophageal reflux. Am J Surg 1985; 149: 187–193. [DOI] [PubMed] [Google Scholar]
- 82.Pera M, Cameron AJ, Trastek VF, et al. Increasing incidence of adenocarcinoma of the esophagus and esophagogastric junction. Gastroenterology 1993; 104: 510–513. [DOI] [PubMed] [Google Scholar]
- 83.Hamelin R, Laurent-Puig P, Olschwang S, et al. Association of p53 mutations with short survival in colorectal cancer. Gastroenterology 1994; 106: 42–48. [DOI] [PubMed] [Google Scholar]
- 84.Leon SA, Shapiro B, Sklaroff DM, et al. Free DNA in the serum of cancer patients and the effect of therapy. Cancer Res 1977; 37: 646–650. [PubMed] [Google Scholar]
- 85.Castells A, Puig P, Mora J, et al. K-ras mutations in DNA extracted from the plasma of patients with pancreatic carcinoma: diagnostic utility and prognostic significance. J Clin Oncol 1999; 17: 578–584. [DOI] [PubMed] [Google Scholar]
- 86.Hibi K, Robinson CR, Booker S, et al. Molecular detection of genetic alterations in the serum of colorectal cancer patients. Cancer Res 1998; 58: 1405–1407. [PubMed] [Google Scholar]
- 87.Silva JM, Dominguez G, Garcia JM, et al. Presence of tumor DNA in plasma of breast cancer patients: clinicopathological correlations. Cancer Res 1999; 59: 3251–3256. [PubMed] [Google Scholar]
- 88.Esteller M, Sanchez-Cespedes M, Rosell R, et al. Detection of aberrant promoter hypermethylation of tumor suppressor genes in serum DNA from non-small cell lung cancer patients. Cancer Res 1999; 59: 67–70. [PubMed] [Google Scholar]
- 89.Wong IH, Lo YM, Zhang J, et al. Detection of aberrant p16 methylation in the plasma and serum of liver cancer patients. Cancer Res 1999; 59: 71–73. [PubMed] [Google Scholar]
- 90.Sanchez-Cespedes M, Esteller M, Wu L, et al. Gene promoter hypermethylation in tumors and serum of head and neck cancer patients. Cancer Res 2000; 60: 892–895. [PubMed] [Google Scholar]
- 91.McGuire WL. Hormone receptors: their role in predicting prognosis and response to endocrine therapy. Semin Oncol 1978; 5: 428–433. [PubMed] [Google Scholar]
- 92.Roodi N, Bailey LR, Kao WY, et al. Estrogen receptor gene analysis in estrogen receptor-positive and receptor-negative primary breast cancer. J Natl Cancer Inst 1995; 87: 446–451. [DOI] [PubMed] [Google Scholar]
- 93.Fuqua SA, Hill SM, Chamness GC, et al. Progesterone receptor gene restriction fragment length polymorphisms in human breast tumors. J Natl Cancer Inst 1991; 83: 1157–1160. [DOI] [PubMed] [Google Scholar]
- 94.Lapidus RG, Ferguson AT, Ottaviano YL, et al. Methylation of estrogen and progesterone receptor gene 5′ CpG islands correlates with lack of estrogen and progesterone receptor gene expression in breast tumors. Clin Cancer Res 1996; 2: 805–810. [PubMed] [Google Scholar]
- 95.Jones PA. Altering gene expression with 5-azacytidine. Cell 1985; 40: 485–486. [DOI] [PubMed] [Google Scholar]
- 96.Ferguson AT, Lapidus RG, Baylin SB, Davidson NE. Demethylation of the estrogen receptor gene in estrogen receptor-negative breast cancer cells can reactivate estrogen receptor gene expression. Cancer Res 1995; 55: 2279–2283. [PubMed] [Google Scholar]
- 97.Momparler RL, Cote S, Eliopoulos N. Pharmacological approach for optimization of the dose schedule of 5-Aza-2′-deoxycytidine (Decitabine) for the therapy of leukemia. Leukemia 1997; 11 (suppl 1): S1–6. [PubMed] [Google Scholar]
- 98.Juttermann R, Li E, Jaenisch R. Toxicity of 5-aza-2′-deoxycytidine to mammalian cells is mediated primarily by covalent trapping of DNA methyltransferase rather than DNA demethylation. Proc Natl Acad Sci USA 1994; 91: 11797–11801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Cameron EE, Bachman KE, Myohanen S, et al. Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nat Genet 1999; 21: 103–107. [DOI] [PubMed] [Google Scholar]