

Abstract
Objective
To examine the effect of trauma plasma on clonogenic progenitor cultures.
Summary Background Data
Severely injured trauma patients often experience altered hematopoietic functions, manifested by an increased susceptibility to infection and the development of a persistent anemia. Experimental and clinical data suggest that trauma results in the release of cytokines into the plasma that have hematopoietic regulatory function, but few studies have examined human bone marrow.
Methods
Plasma was obtained from 42 severely injured patients admitted to the surgical intensive care unit from days 1 to 15 after injury. Bone marrow and normal plasma were obtained from volunteers. Bone marrow mononuclear cells were isolated and plated for granulocyte-monocyte colony-forming unit (CFU-GM) and erythroid burst-forming unit (BFU-E) growth. Parallel cultures were incubated with 2% (v/v) trauma or normal plasma. Additional cultures were plated with neutralizing concentrations of antibodies to transforming growth factor (TGF)-β1 and MIP-1α. Circulating plasma TGF-β1 was determined by bioassay. mRNA from bone marrow stromal cultures was extracted and probed for TGF-β1 and macrophage inflammatory protein (MIP)-1α.
Results
Trauma plasma suppressed CFU-GM and BFU-E colony growth by 40% to 60% at all time periods after injury compared with cultures incubated with normal plasma. Using a noncontact culture system, the authors showed that this inhibition of BFU-E and CFU-GM colony growth was mediated by bone marrow stroma. The inhibition appeared to be due to soluble plasma-induced bone marrow stromal products that did not require direct cell–cell contact. The addition of anti-TGF-β1 antibodies reversed the suppressive effect of trauma plasma on CFU-GM and BFU-E colony growth during the early but not late time points after injury. Trauma but not normal plasma induced TGF-β1 mRNA in bone marrow stroma.
Conclusions
Trauma plasma inhibits bone marrow BFU-E and CFU-GM colony growth for up to 2 weeks after injury. This inhibition is mediated through the interaction of trauma plasma with bone marrow stroma. TGF-β1 production by bone marrow stroma appears to plays an important role in the early but not late bone marrow suppression after injury.
Bone marrow (BM) dysfunction has been shown to occur in experimental animal models of hemorrhagic shock, hypoxia, soft tissue injury, and thermal injury. 1–5 Whether a similar alteration in BM function occurs in humans after injury is less clear and has been not well studied. Moore et al 6 showed that there was a marked rise in circulating granulocyte-macrophage colony-forming units (CFU-GM) in patients for up to 10 days after blunt torso trauma. Coincident with the release of immature progenitors into the periphery was the fact that peripheral mononuclear cells from trauma patients produced reduced levels of colony-stimulating activity in response to lipopolysaccharide compared with controls. The degree of BM failure also appeared to correlate with the degree of injury. Persistent anemia has long been documented in intensive care unit patients. 7 This anemia has been examined after thermal injury, 8 and most recently Iversen et al 9 reported a decrease in bone marrow erythroid burst-forming units (BFU-E) in patients after spinal cord injury.
Successful hematopoiesis is a complex regulated process in which pluripotential stem cells proliferate and differentiate to generate all necessary mature blood cells. 10 The process of immune and red blood cell formation requires the interaction between BM progenitor and stem cells with their microenvironment, which includes BM stromal cells, extracellular matrix proteins, and other soluble factors. 11–13 BM stroma has previously been reported to influence hematopoiesis by the elaboration of regulatory cytokines, 14 as well as to regulate progenitor cell growth and differentiation through direct cell–cell contact. 15,16 Thus, BM stroma is central in both the maintenance and the regulation of hematopoiesis. Consequently, any disruption of BM hematopoiesis after injury is likely to be at least partly mediated through the interaction of BM stroma with hematopoietic progenitor cells.
Trauma results in the migration of inflammatory cells to the site of injury, and a multitude of cytokines and chemokines have been documented to be secreted into both the local tissue environment and systemic circulation. 17–20 Many of these cytokines and wound fluids have been shown to have an effect on immune cells. For example, Hauser et al 21 showed that fluid surrounding the sites of fracture in trauma patients contained high levels of interleukin (IL)-6 and IL-8. Plasma obtained from rats subjected to hemorrhagic shock has been shown to inhibit CFU-GM formation from BM cells. 2 The hypothesis of these experiments is that plasma obtained from patients after severe injury would inhibit hematopoiesis in both red blood cell (as measured by BFU-E) and white blood cell (as measured by CFU-GM) lineages, and that this inhibition would be mediated through plasma’s interaction with the BM stroma.
METHODS
Reagents and Cytokines
Diethyl pyrocarbonate (DEPC), bovine serum albumin (BSA), hydrocortisone, Ficoll-Hypaque, and 2-mercaptoethanol (2-ME) were purchased from Sigma (St. Louis, MO). Rabbit anti-TGF-β antibodies were purchased from R&D Systems (Minneapolis, MN). Previous work from our laboratory showed that this antibody preferentially neutralized the β-1 isoform. 22 The Immunology Department of the Genetics Institute (Cambridge, MA) generously provided recombinant human granulocyte macrophage-colony stimulating factor (rhGM-CSF), rh-erythropoietin (rhEPO), IL-3, rhMIP-1α, and rabbit antihMIP-1α.
Subjects
This study was reviewed and approved by the Institutional Review Board of UMDNJ-New Jersey Medical School, and informed consent was obtained from each donor or his or her legal representative. Peripheral blood was obtained in heparinized tubes from multiply injured patients admitted to the surgical intensive care unit at University Hospital. Per protocol, only one blood sample was obtained from each patient. Samples were drawn from patients at various times after initial trauma, beginning at 12 hours after admission for up to 15 days. As a control, peripheral blood was obtained from age- and sex-matched healthy donors (three or four per group). Plasma was separated by centrifugation at 10,000 g for 15 minutes. Aliquots of plasma were stored at −80°C in sterile siliconized tubes until use.
Hematopoietic Progenitor Cultures
Bone marrow was obtained from the posterior iliac crest of healthy volunteers. Low-density BM mononuclear cells (BMNCs) were separated by Ficoll-Hypaque density gradient (Pharmacia LKB Biotechnology, Piscataway, NJ) and resuspended in RPMI 1640 (Sigma) containing 10% fetal calf serum (FCS; Hyclone Laboratories, Logan, UT). BMNCs (105 cells) were plated in duplicate in Iscove’s media containing 30% FCS, 2% BSA, 1% methylcellulose, 2 × 10−4 mol/L 2-ME, glutamine (Cellgro; Mediatech, Herndon, VA), and 2% (v/v) of plasma from trauma patients or normal volunteers. Parallel cultures containing only media without any plasma served as controls. CFU-GM cultures were supplemented with 3 U/mL rhGM-CSF and BFU-E cultures with 2 U/mL rhEPO and 6 U/mL rhIL-3. Cultures were incubated at 37°C in 5% CO2. CFU-GM colonies with greater than 20 cells were enumerated at day 10 and BFU-E colonies were counted at day 15 by an observer who was unaware of the treatment.
Bone Marrow Stromal Cultures
Unseparated BM cells (107 cells) were suspended in 5 mL stromal media and added to 25-cm2 Falcon 3109 tissue culture flasks (Becton Dickinson, Franklin Lakes, NJ). Stromal medium consisted of α-MEM (Life Technologies, Grand Island, NY), 12.5% FCS, 12.5% horse serum (Hyclone Laboratories), 0.1 μmol/L hydrocortisone, 0.1 mmol/L 2-ME, and 1.6 mmol/L glutamine. Cultures were incubated at 33°C for 3 days. On day 3, mononuclear cells were isolated from the nonadherent cell population by Ficoll-Hypaque density gradient and then replaced into the original culture flasks. Stromal cultures were reincubated until the adherent cells were confluent. Throughout the culture period, 50% of the stromal media were replaced at weekly intervals.
Noncontact Stromal Cultures
Stromal cultures were prepared as described above with the following modification. Unseparated BM cells (4 × 106) were placed in the outer well of a 24-transwell plate (Corning Costar Co., Cambridge, MA). When the stroma reached confluence, the remaining nonadherent cells were removed and replaced with fresh media. The stroma was then irradiated with 150 Gy delivered with cesium source (mark 1 Model 68-A-3 gamma irradiator; J. L. Shepard, San Fernando, CA), and the plates were reincubated overnight. Low-density BMNCs (105 cells) obtained from another volunteer donor were placed in the inner wells. Plasma at a final concentration of 2% from healthy controls or trauma patients was added to the outer wells. Parallel cultures containing no stroma in the outer wells were performed to control for the presence of stroma. All cultures were incubated at 37°C for 7 days. On day 7, the BMNCs from the inner wells were harvested and then cultured for CFU-GM and BFU-E colony growth as described above. BM cell viability exceeded 95% on day 7 just before initiating the CFU-GM and BFU-E cultures. There were no differences in viability between groups.
Stroma Depletion of Bone Marrow Mononuclear Cells
Bone marrow mononuclear cells were depleted of stromal cells by passage twice through nylon-wool columns (Fenwell Laboratories, Deerfield, IL) as previously described. 23 Briefly, columns were prepared by loosely packing approximately 0.6 to 0.7 g sterile scrubbed nylon wool in a 10-mL syringe. Columns were equilibrated with prewarmed RPMI-1640 containing 5% FCS. BMNCs obtained from healthy volunteers were resuspended in similar medium at 2.5 × 107 cells/mL and then added to the nylon-wool column, and then incubated for 45 minutes at 37°C. At the end of the incubation period, the nonadherent cells were eluted from the column with prewarmed RPMI-1640 containing 5% FCS. The recovered cells were depleted of stromal cells because they did not form stromal monolayers even up to 4 weeks in culture (data not shown). Further, the total number of CD34+ cells added to the column as determined by flow cytometry was similar in the recovered fraction (±0.001%). Stroma-depleted BMNCs eluted from the columns were then plated for CFU-GM and BFU-E progenitor growth as described above.
Determination of Trauma Plasma TGF-β1 Level
TGF-β1 levels in trauma plasma were quantitated using a bioassay based on the growth inhibition of mink lung epithelial cells (CCL 64) by TGF-β1. 22 CCL 64 cells (5 × 103) were added to 24-well tissue culture plates in a total volume of 0.5 mL RPMI-1640 supplemented with 2 mmol/L glutamine, 10% FCS, and 15 mmol/L HEPES. The CCL 64 cells were incubated for 24 hours, after which 25 and 50 μL of plasma samples to be tested were added to the wells. All plasma samples were run in triplicate. After the addition of the plasma, the cultures were incubated for an additional 3 days. The plasma levels of TGF-β1 were determined by a comparison of a standard curve created by assaying CCL 64 cell viability against known concentration of TGF-β1 (0.02–20 pg/mL).
Bone Marrow Progenitor Cultures With Trauma Plasma and Anti-TGF-β and Anti-MIP-1α Antibodies
Bone marrow mononuclear cells obtained from healthy volunteers were cultured for CFU-GM and BFU-E colony growth as described above. Plasma from trauma patients or normal volunteers was added at a concentration of 2% to the BM progenitor cultures in the presence of various concentrations of anti-TGF-β (1–2,000 ng/mL) or anti-MIP-1α (1–1,000 ng/mL) antibodies. Nonimmune rabbit IgG antibodies were used as controls. CFU-GM and BFU-E colonies were enumerated as previously described. The data for the CFU-GM and BFU-E colonies, grown in the presence of trauma plasma, are reported as the percentage of growth compared with colonies grown with normal plasma.
Northern Analysis of TGF-β1 and MIP-1α mRNA
Bone marrow stroma from normal volunteers was grown to confluence. Trauma or normal plasma was added (2% v/v) and the cultures were incubated for 48 hours. Steady-state mRNA for TGF-β and MIP-1α in BM stroma was determined by Northern analysis. Total RNA (10 mg) was separated by electrophoresis on 1.2% agarose and then transferred to nylon membranes. RNA ladders, 0.24 to 9.5 kb and 0.16 to 1.77 kb, were used as molecular weight markers. Membranes were UV cross-linked and baked for 1 hour at 80°C under vacuum and hybridized with cDNA probes for TGF-β1, MIP-1α, and 18S rRNA. Probes were randomly labeled with (a-32P)-dATP, 3,000 Ci/mmol (Dupont/NEN, Boston, MA) using the Prime-IT II random primer labeling kit (Stratagene, La Jolla, CA). After hybridization, membranes were washed twice at room temperature with 2× SSC containing 0.1% SDS. This was followed with another wash for 20 minutes at 37°C with a buffer of similar stringency, followed by exposure to x-ray film as described. 23 Ethidium bromide visualization of rRNA and hybridization with cDNA for rRNA were used as loading controls and for normalization. Densitometric scanning compared the autoradiograms.
Statistical Analysis
All data are expressed as mean ± standard deviation. Statistical analyses were performed using analysis of variance and the Tukey-Kramer multiple comparisons test. P < .05 was considered significant.
RESULTS
Patient Demographics and Characteristics
Plasma was obtained from 42 patients (30 men, 12 women; mean age 36 years [range 18–68]). The mechanism of injury was a motor vehicle crash in 18, pedestrian stuck by a motor vehicle in 10, gunshot wounds in 8, and fall in 6. The mean Injury Severity Score was 28 ± 9. There were eight deaths, all secondary to sepsis and progressive organ failure. The postinjury course was complicated by at least one major infection in 38 of the 42 patients. The patients received an average of 16 ± 11 units of packed red blood cells, 20 ± 15 units of fresh-frozen plasma, and 10 ± 5 units of platelets before the plasma sample was obtained. Data for the complete blood counts, differential, and reticulocyte counts at the time the plasma sample was obtained are shown in the Table.
Table 1. COMPLETE BLOOD COUNTS FOR TRAUMA PATIENTS

Effect of Trauma Plasma on Hematopoietic Progenitors
The numbers of BFU-E and CFU-GM colonies grown with media alone were comparable to those grown in the presence of 1% or 2% normal plasma. All individual plasma samples from trauma patients were inhibitory to both CFU-GM and BFU-E; thus, the data were grouped into three time periods after severe injury: days 1 to 5 (early), 6 to 10 (middle), and 11 to 15 (late) (Fig. 1). We observed significant suppression of both CFU-GM and BFU-E at all three time periods (P < .05), which ranged from 40% to 60% when compared with the normal controls. Trauma plasma at a concentration of 1% also showed significant inhibition at each time point (P < .05), but there was a small increased variability between plates (data not shown). There were no statistically significant differences in CFU-GM or BFU-E colony growth between plasma obtained from different time periods.
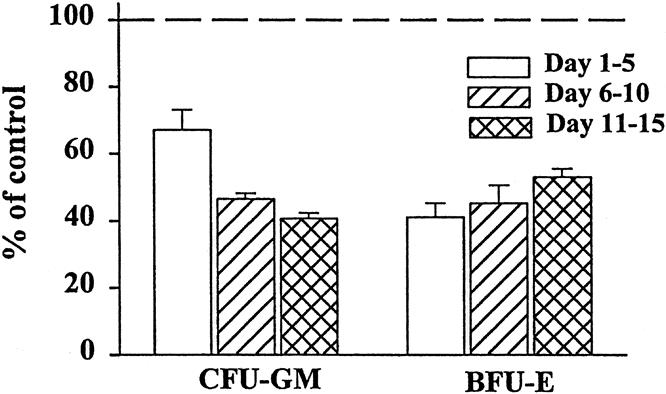
Figure 1. Granulocyte-monocyte colony-forming unit (CFU-GM) and erythroid burst-forming unit (BFU-E) colony growth was significantly inhibited by the addition of 2% plasma from trauma patients. Data for individual trauma plasma time points were combined in 5-day intervals and compared with growth with 2% normal plasma (dashed line). N = 8 to 12 individual plasma samples per time point. All time points P < .05 versus normal plasma.
Effects of Trauma Plasma in Stromal Noncontact Progenitor Cultures
Bone marrow stroma plays a central role in the regulation of hematopoiesis, 5,11 both by direct contact with the progenitor cells 15,16 and through the production of cytokines. 14 To determine whether the inhibition of BM progenitor growth by trauma plasma is mediated by the bone marrow stroma and whether it was due to the production of soluble mediators or required cell–cell contact, we performed a set of experiments using a transwell culture system. In these experiments, the BM stroma was physically separated from BMNCs by a microporous membrane that prevented direct contact between BMNCs and BM stroma but allowed soluble factors to diffuse freely between the two chambers. Under these conditions, trauma plasma added to the plates inhibited BFU-E and CFU-GM (Fig. 2) growth to a similar degree observed when BMNCs were plated under normal conditions (i.e., the stroma was in contact with BMNCs). These results suggest that direct contact between BMNCs and BM stromal cells is not necessary for trauma plasma to inhibit CFU-GM and BFU-E.
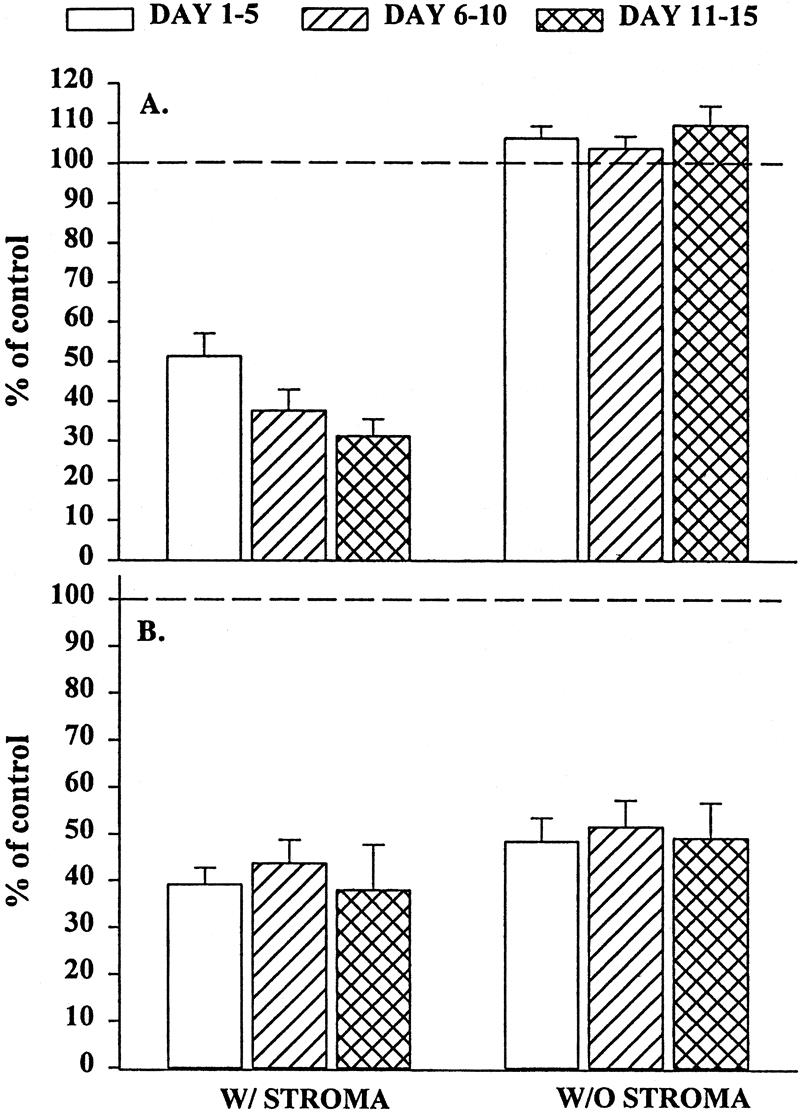
Figure 2. Granulocyte-monocyte colony-forming unit (CFU-GM) colonies (A) and erythroid burst-forming unit (BFU-E) colonies (B) in the presence of 2% trauma plasma grown in transwell cultures with or without stroma. N = 8 to 12 individual trauma plasma samples per time point. Dashed line represents CFU-GM and BFU-E with normal plasma. All time periods with stroma were significantly different from control and the same time period in cultures without stroma (P < .05). Data indicate that inhibition of CFU-GM was clearly mediated through bone marrow stroma.
Elimination of stromal cells from the transwell CFU-GM cultures reversed the inhibition. These findings suggest that trauma plasma inhibits CFU-GM growth by the production of soluble factor(s) that are elaborated by BM stroma and is not due to a direct effect on BM progenitors. In contrast, trauma plasma suppressed BFU-E growth even in the absence of BM stroma. BMNCs obtained from Ficoll-Hypaque separation of BM may contain a small amount of stromal elements, which can potentially form stromal monolayers. Because of this, it remained unclear whether the suppressive effect of trauma plasma on BFU-E growth was a direct effect on the progenitor cells, was mediated by BM stroma, or both.
Effect of Trauma Plasma on Stroma-Depleted Progenitor Cultures
To determine whether the residual stromal cells influenced our BFU-E results, BMNCs were passed through nylon-wool columns, after which the stromal-depleted BMNCs were cultured for BFU-E and CFU-GM in the presence of 2% trauma or normal plasma. BMNCs obtained after passage through nylon wool are incapable of forming stromal monolayers. After stromal depletion, trauma plasma had no effect on CFU-GM colony growth (Fig. 3), and the growth was similar to the numbers of colonies formed in the transwell cultures. In contrast, BFU-E cultures showed consistent and significant improvement in growth over that seen in the transwell cultures.

Figure 3. Granulocyte-monocyte colony-forming unit (CFU-GM) colonies (A) and erythroid burst-forming unit (BFU-E) colonies (B) after passage through nylon wool incubated with 2% trauma plasma. N = 8 to 12 individual plasma samples per time point. Dashed line represents CFU-GM and BFU-E growth with normal plasma. Stromal depletion restored CFU-GM and BFU-E growth to control values.
Circulating Plasma TGF-β1 Level
The mean TGF-β1 level in all samples was 15 ± 5 ng/mL. There were no significant differences in the plasma concentration of TGF-β1 between the three time periods (Fig. 4). This concentration was significantly greater than that in normal controls (<0.5 ng/mL) and indicates that severely injured patients who require intensive care have a significant and persistent elevation in plasma TGF-β1. The average level of TGF-β in 2% trauma plasma was approximately 0.3 pg/mL.
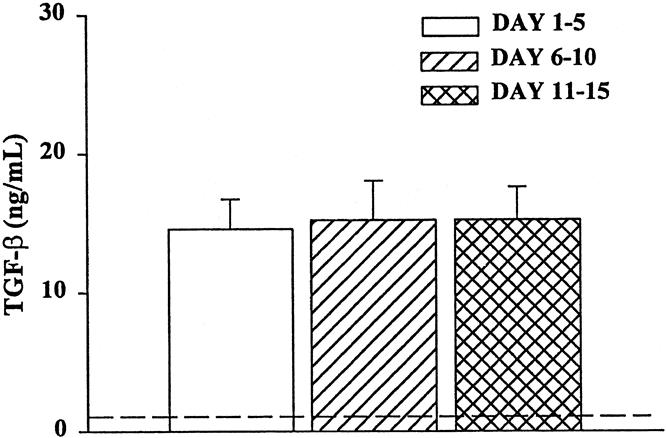
Figure 4. Plasma TGF-β levels from trauma patients. TGF-β was quantitated with or without neutralizing anti-TGF-β using a bioassay with CCL 64 cells. N = 8 to 12 individual plasma samples per time point. All time points were significantly elevated (P < .05) compared with normal volunteers (dashed line).
Effect of Anti-TGF-β and Anti-MIP-1α Neutralizing Antibodies on the Inhibitory Effect of Trauma Plasma
Because BM stroma is a rich source of TGF-β1 and MIP-1α, we postulated that trauma plasma might induce the production of these cytokines in BMNC cultures, which would then inhibit CFU-GM and BFU-E growth. To explore this possibility, different concentrations of anti-TGF-β1 and anti-MIP-1α were incubated with BMNCs and 2% trauma plasma. At 200 to 600 ng/mL, anti-TGF-β1 abrogated either completely (days 1–5 and 6–10) or partially (days 11–15) the suppressive effect of trauma plasma on CFU-GM and BFU-E (Fig. 5). Incubation with anti-MIP-1α or nonimmune IgG had no effect on BFU-E or CFU-GM growth at all time periods (data not shown). These data suggest that a significant degree of inhibition of BFU-E and CFU-GM colony growth for the first 10 days after injury is mediated through the production of TGF-β1 by BM stroma, whereas other factors are involved late after injury.

Figure 5. Granulocyte-monocyte colony-forming unit (CFU-GM) (A) and erythroid burst-forming unit (BFU-E) (B) growth in the presence of 2% trauma plasma and neutralizing anti-TGF-β. N = 8 to 12 individual plasma samples per time point. The numbers of CFU-GM and BFU-E were comparable in cultures with normal plasma or equivalent concentrations of antibodies, represented by the dashed line. *P < .05 versus control.
Trauma Plasma Induces TGF-β1 mRNA
The small quantity of TGF-β1 present in the plasma cannot fully explain the results of the above experiments, which showed increased CFU-GM and BFU-E colony growth after neutralization of TGF-β1. Thus, we assessed whether trauma plasma could induce TGF-β1 production by BM stromal cells. Normal BM stroma was incubated with trauma plasma, after which steady-state TGF-β1 mRNA concentrations were measured. A representative Northern blot is shown in Figure 6 and clearly shows a marked induction of TGF-β1 mRNA when BM stroma is exposed to trauma plasma. Plasma from all time points (three or four per time point) induced TGF-β1. No detectable levels of MIP-1α mRNA were observed after incubation of BM stroma with trauma plasma (data not shown).
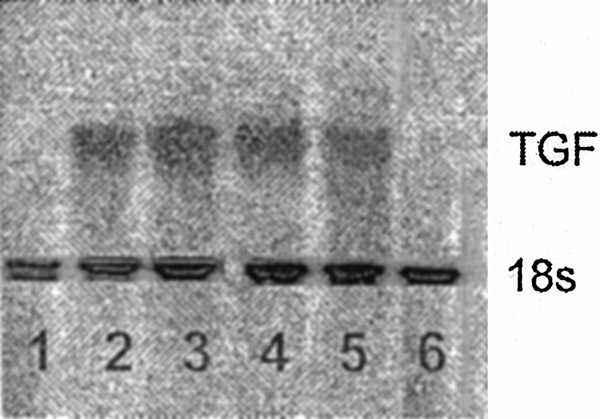
Figure 6. Representative Northern analysis for TGF-β1 in normal bone marrow stroma incubated for 48 hours with 2% (v/v) normal (lanes 1 and 6) or trauma plasma (lanes 2–5). Bands in lanes 2 to 5 clearly show that trauma plasma resulted in an increase steady state TGF-β1 mRNA.
DISCUSSION
Hematopoietic suppression has been shown after injury in both animal models 1–4,24 and humans. 6,25 The primary focus of these investigations has been on the altered growth of myeloid progenitor cells after injury and its relationship to subsequent infection. None of these studies examined human bone marrow. Because plasma obtained from trauma patients has been found to contain increased levels of cytokines and chemokines, 19,20 many of which are known to regulate hematopoiesis, 26,27 it is likely that plasma obtained from trauma patients would have an effect on BM hematopoiesis. The data presented here clearly show that plasma from severely injured patients inhibits both BFU-E and CFU-GM growth for at least 15 days after injury. The degree of inhibition in these experiments with human trauma plasma was similar to the magnitude of CFU-GM growth inhibition we previously observed when plasma obtained from rats subjected to hemorrhagic shock was plated with normal rodent BM. 2
Because all plasma samples tested in these experiments were inhibitory to CFU-GM and BFU-E proliferation, we grouped the data into three time points based on the clinical course of the patients. The early time period (≤ day 5) was the immediate postresuscitation/postoperative phase after injury and represented the effects of soft tissue injury, shock, tissue ischemia, and reperfusion. Infectious complications occurred rarely and were observed in only two patients before day 5. Both patients had pneumonia, diagnosed on day 5. Samples from patients during the late period (≥day 11) were all from patients sustaining one or more infectious complications and/or organ dysfunction. The middle time period represented a more heterogenous group, including patients whose severe injuries kept them in the intensive care unit but who had not developed infectious complications yet, and patients with infectious and other complications. Thus, we postulated that although all samples were inhibitory to hematopoietic progenitor growth, there might be a change in the degree or mechanism of the inhibition based on the timing of the samples and the clinical course of the patients. This was borne out by the results using anti-TGF-β antibodies. Samples from the late time period continued to be inhibitory to CFU-GM and BFU-E colony growth compared with plasma obtained before day 10. Thus, the presence of ongoing infection appears to elaborate other factors that play a role in the persistence of BM suppression.
Although the mechanism of trauma plasma’s inhibition of BM progenitor growth remains to be determined, data from experimental models point to the interaction between plasma and BM stroma as a likely etiology. 28 Further, BM stroma is an integral component in the control of hematopoiesis, and stromal cells within the BM microenvironment are known to regulate hematopoiesis through the elaboration of hematopoietic factors, direct cell–cell contact, and the production of extracellular matrix glycoproteins. 11,14,29 Thus, any regulatory effect on CFU-GM and BFU-E growth by plasma from trauma patients would likely involve the bone marrow stroma. The results from our transwell cultures clearly showed that cell–cell contact was not necessary for trauma plasma to inhibit CFU-GM and BFU-E colony formation. Consistent with the hypothesis that the effect of trauma plasma was mediated through its interaction with BM stroma was the finding that depletion of stromal elements significantly reversed the inhibition of CFU-GM and BFU-E (see Figs. 2 and 3). Data from our transwell cultures also pointed out a potential difference in the inhibition of BFU-E and CFU-GM colony growth by trauma plasma. CFU-GM growth returned to control values after either Ficoll-Hypaque or nylon-wool separation of BMNC; thus, it appears that trauma plasma does not have a direct inhibitory effect on CFU-GM cells. Conversely, BFU-E growth never returned to baseline. Although the 15% to 20% inhibition in the BFU-E numbers after passage through nylon wool was not statistically significant compared with control, we cannot rule out a possible type 2 statistical error, and consequently we cannot definitively exclude whether trauma plasma may directly inhibit BFU-E cells as well as regulating their growth via BM stroma. A difference in the mechanism of the inhibition of BFU-E growth by trauma plasma compared with CFU-GM may explain the clinical observation that persistent anemia (i.e., quantitative red cell defect) appears more profound and long-lasting than a white cell defect after injury.
The above findings suggested that some factor or factors were either present in trauma plasma or was produced by other cells within the BM in response to the trauma plasma. Although many potential antiproliferative cytokines exist, Meert et al 17 showed that TGF-β levels were increased in a rat model of femur fracture, and that the increase in TGF-β correlated with immunosuppression. TGF-β has been shown by others to suppress erythroid and myeloid progenitors. 30,31 Elevated TGF-β levels have also been found in the cerebrospinal fluid of patients with traumatic brain injuries;32 however, scant information is available regarding the temporal pattern of TGF-β levels in humans after severe injury. The data from the experiments reported here showed that plasma levels of TGF-β1 were elevated for up to 2 weeks after severe trauma. Although the small amounts of TGF-β1 (<0.5 ng) that were present in the 2% plasma added to the culture plates have been shown to inhibit BFU-E and CFU-GM under specific culture conditions, 33 depletion of stromal cells reversed this inhibition. Therefore, we believe that the quantity of TGF-β1 present in the plasma of trauma patients was insufficient to inhibit colony growth in the absence of stroma. Thus, BM stroma appears to mediate the inhibition of BFU-E and CFU-GM growth by trauma plasma. These findings are similar to what was observed in rats exposed to hypoxia. 28
Although circulating levels of TGF-β1 could not explain the inhibition observed, the production of TGF-β1 by bone marrow stroma in response to trauma plasma remained a possibility. This hypothesis is supported by our finding that trauma, but not normal, plasma induced TGF-β1 mRNA expression in BM stromal monolayers. However, the addition of anti-TGF-β1 antibodies reversed the inhibition of CFU-GM and BFU-E growth for only the first 10 days after injury. Plasma obtained after day 10 remained inhibitory despite the addition of anti-TGF-β1 antibodies. Thus, hematopoietic inhibition secondary to trauma plasma, although it appears constant, comprises at least two distinct mechanisms that either change over time after injury or are influenced by infection or organ failure. The need for seriously injured patients to remain in an intensive care unit for more than 10 days is usually due to the development of infectious complications and/or organ failure. All of the late trauma plasma came from patients who sustained at least one infectious complication. Infection after injury has been shown to increase the plasma concentration of several antiproliferative cytokines in addition to TGF-β such as interferon-γ, IL-6, and IL-10. 21 Thus, these and other potential mediators of late hematopoietic inhibition potentially present in trauma plasma obtained after day 10 remain to be evaluated.
In summary, trauma plasma inhibited both BFU-E and CFU-GM progenitor growth. This inhibition was present immediately after injury and persisted for at least 2 weeks after admission In the case of CFU-GM cells, this inhibitory effect appeared to be mediated entirely through the interaction between trauma plasma and BM stroma. For BFU-E cells, the inhibitory effect of plasma appears to be mediated through its interaction with BM stroma, although trauma plasma may also exert a direct effect on the BFU-E growth. In addition, antiproliferative levels of TGF-β1 are clearly present in the plasma of these patients for up to 2 weeks after injury, and trauma plasma can induce the production of TGF-β1 in normal BM stroma. Whereas TGF-β1 plays a significant role in the inhibition of CFU-GM and BFU-E for the first 10 days after injury, other mechanisms appear responsible for the ongoing hematopoietic dysfunction observed after that time point. We conclude that the inhibition of hematopoiesis by trauma plasma significantly contributes to both the clinical problems of persistent anemia and the increased susceptibility to infection observed in this group of seriously injured patients. Further studies are needed to elucidate the molecular mechanism of BM failure after injury.
Footnotes
Supported in part by National Institutes of Health grants HL-57675, HL-54973, HL-63097, and the Ruth Estrin Goldberg Memorial for Cancer Research.
Correspondence: David H. Livingston, MD, University Hospital E-245, 150 Bergen St., Newark, NJ 07103.
E-mail: livingst@umdnj.edu
Accepted for publication October 10, 2000.
References
- 1.Gamelli R, He L, Liu H. Marrow granulocyte-macrophage progenitor cell response to burn injury as modified by endotoxin and indomethacin. J Trauma 1994; 37: 339–346. [DOI] [PubMed] [Google Scholar]
- 2.Livingston DH, Gentile PS, Malangoni MA. Bone marrow failure after hemorrhagic shock. Circ Shock 1990; 30: 255–263. [PubMed] [Google Scholar]
- 3.Raff G, Livingston D, Wang M, et al. Hemorrhagic shock abolishes the myelopoietic response to turpentine-induced soft tissue injury. J Surg Res 1995; 59: 75–79. [DOI] [PubMed] [Google Scholar]
- 4.Fontes B, Moore FA, Moore EE, et al. Gut ischemia induces bone marrow failure and increases risk of infection. J Surg Res 1994; 57: 505–509. [DOI] [PubMed] [Google Scholar]
- 5.Mohr AM, Upperman JS, Taneja R, et al. The differential effects of acute hypoxia and endotoxin on the secretion and expression of bone marrow interleukin-1 and interleukin-6. Shock 1997; 7: 324–331. [DOI] [PubMed] [Google Scholar]
- 6.Moore FA, Peterson VM, Moore EE, et al. Inadequate granulopoiesis after major torso trauma: a hematopoietic regulatory paradox. Surgery 1990; 108: 667–675. [PubMed] [Google Scholar]
- 7.Corwin H, Gettinger A, Rodriguez R, et al. Efficacy of recombinant human erythropoietin in the critically ill patient: a randomized, double-blind, placebo-controlled trial. Crit Care Med 1999; 27: 2346–2350. [DOI] [PubMed] [Google Scholar]
- 8.Deitch EA, Sittig KM. A serial study of the erythropoietic response to thermal injury. Ann Surg 1993; 217: 293–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Iversen P, Hjeltnes N, Holm B, et al. Depressed immunity and impaired proliferation of hematopoietic progenitor cells in patients with complete spinal cord injury. Blood 2000; 96: 2081–2083. [PubMed] [Google Scholar]
- 10.Lord B, Dexter T. Which are hematopoietic stem cells? Exp Hematol 1995; 23: 1237–1240. [PubMed] [Google Scholar]
- 11.Clark B, Keating A. Biology of bone marrow stroma. Ann NY Acad Sci 1995; 770: 70–78. [DOI] [PubMed] [Google Scholar]
- 12.Sachs L. The molecular control of blood cell development. Science 1987; 238: 1374. [DOI] [PubMed] [Google Scholar]
- 13.Zon L. Developmental biology of hematopoiesis. Blood 1995; 86: 2876–2882. [PubMed] [Google Scholar]
- 14.Verfaillie C. Soluble factor(s) produced by human bone marrow stroma increase cytokine-induced proliferation and maturation of primitive hematopoietic progenitors while preventing their terminal differentiation. Blood 1993; 82: 2045–2053. [PubMed] [Google Scholar]
- 15.Verfaillie C, Catanzaro P. Direct contact with stroma inhibits proliferation of human long-term culture initiating cells. Leukemia 1996; 10: 498–504. [PubMed] [Google Scholar]
- 16.Hurley R, McCarthy J, Verfaillie C. Direct adhesion to bone marrow stroma via fibronectin receptors inhibits hematopoietic progenitor proliferation. J Clin Invest 1995; 96: 511–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meert K, Ofenstein J, Genyea C, et al. Elevated transforming growth factor-beta concentration correlates with posttrauma immunosuppression. J Trauma 1996; 40: 901–906. [DOI] [PubMed] [Google Scholar]
- 18.Jiang J, Tian K, Chen H, et al. Plasma cytokines and endotoxin levels in patients with severe injury and their relationship with organ damage. Injury 1997; 28: 509–513. [DOI] [PubMed] [Google Scholar]
- 19.Hoch R, Rodriguez R, Manning T, et al. Effects of accidental trauma on cytokine and endotoxin production. Crit Care Med 1993; 21: 839–845. [DOI] [PubMed] [Google Scholar]
- 20.Nast-Kolb D, Waydhas C, Gippner-Steppert C, et al. Indicators of the posttraumatic inflammatory response correlate with organ failure in patients with multiple injuries. J Trauma 1997; 42: 446–454. [DOI] [PubMed] [Google Scholar]
- 21.Hauser C, Zhou X, Joshi P, et al. The immune microenvironment of human fracture/soft-tissue hematomas and its relationship to systemic immunity. J Trauma 1997; 42: 895–904. [DOI] [PubMed] [Google Scholar]
- 22.Rameshwar P, Denny T, Stein D, et al. Monocyte adhesion in patients with bone marrow fibrosis is required for the production of fibrogenic cytokines. Potential role for interleukin-1 and TGF-beta. J Immunol 1994; 153: 2819–2830. [PubMed] [Google Scholar]
- 23.Rameshwar P, Gascon P. Induction of negative hematopoietic regulators by neurokinin-A in bone marrow stroma. Blood 1996; 88: 98–106. [PubMed] [Google Scholar]
- 24.Livingston DH. Interferon-gamma reverses bone marrow inhibition following hemorrhagic shock. Arch Surg 1991; 126: 100–103. [DOI] [PubMed] [Google Scholar]
- 25.Peterson V, Hansbrough J, Buerk C, et al. Regulation of granulopoiesis following severe thermal injury. J Trauma 1983; 23: 19–24. [DOI] [PubMed] [Google Scholar]
- 26.Keller J, Bartelmez S, Sitnicka E, et al. Distinct and overlapping direct effects of macrophage inflammatory protein-1 alpha and transforming growth factor beta on hematopoietic progenitor/stem cell growth. Blood 1994; 84: 2175–2181. [PubMed] [Google Scholar]
- 27.Mayani H, Little M, Dragowska W, et al. Differential effects of the hematopoietic inhibitors MIP-1 alpha, TGF-beta, and TNF-alpha on cytokine-induced proliferation of subpopulations of CD34+ cells purified from cord blood and fetal liver. Exp Hematol 1995; 23: 422–427. [PubMed] [Google Scholar]
- 28.Taneja R, Rameshwar P, Upperman J, et al. Effects of hypoxia on granulocyte-monocyte progenitors in rats. Am J Hematol 2000; 64: 20–25. [DOI] [PubMed] [Google Scholar]
- 29.Jacobsen K, Osmond D. Microenvironment organization and stromal cell associations of B lymphocyte precursors cells in mouse bone marrow. Eur J Immunol 1990; 20: 2395–2404. [DOI] [PubMed] [Google Scholar]
- 30.Garbe A, Spyridonidis A, Mobest D, et al. Transforming growth factor-beta 1 delays formation of granulocyte-macrophage colony-forming cells, but spares more primitive progenitors during ex vivo expansion of CD34+ haemopoietic progenitor cells. Br J Haematol 1997; 99: 951–958. [DOI] [PubMed] [Google Scholar]
- 31.Murohashi I, Endho K, Nishida S, et al. Differential effects of TGF-beta 1 on normal and leukemic human hematopoietic cell proliferation. Exp Hematol 1995; 23: 970–979. [PubMed] [Google Scholar]
- 32.Morganit-Kossman M, Hans V, Lenzlinger P, et al. TGF-β is elevated in the CSF of patients with severe traumatic brain injuries and parallels blood-brain barrier function. J Neurotrauma 1999; 16: 617–628. [DOI] [PubMed] [Google Scholar]
- 33.Dybedal I, Jacobsen S. Transforming growth factor beta (TGF-beta), a potent inhibitor of erythropoiesis: neutralizing TGF-beta antibodies show erythropoietin as a potent stimulator of murine burst-forming unit erythroid colony formation in the absence of a burst-promoting activity. Blood 1995; 86: 949–957. [PubMed] [Google Scholar]