

Abstract
Objective
To improve the quality of organs from brain-dead donors by assessing the influence of alternative strategies on the early behavior of kidneys after transplantation into unmodified hosts.
Summary Background Data
Kidneys transplanted from living donors perform consistently better than those from cadaver sources. The authors have recently shown that donor brain death produces inflammatory changes in peripheral organs within hours, amplifies coincident ischemia–reperfusion injury, and accelerates acute and chronic rejection. Normalization of the graft by donor hormone treatment has hitherto been unsuccessful.
Methods
A standardized rat model of brain death was used. Experimental groups included recipients of allogeneic grafts from living and brain-dead donors (F344→LEW). Donors were treated immediately after induction of brain death either with intravenous steroids, which block inflammatory cytokine release, or a soluble P-selectin glycoprotein ligand (sPSGL), which blocks initial selectin-mediated cellular adhesion. Kidney grafts were examined serially up to 10 days by morphology, immmunohistology, and reverse transcriptase–polymerase chain reaction.
Results
Overall survival of ummodified recipients of kidneys from brain-dead donors was significantly reduced versus living donors. Animals with organs from brain-dead donors that had received steroids or sPSGL survived significantly longer than those from untreated brain-dead donors. The intensity of ischemia–reperfusion injury and of acute rejection was reduced. Cellular infiltration and transcription of mRNA of representative proinflammatory mediators were diminished.
Conclusions
Treatment of organ donors at the time of brain death markedly improves organ quality after kidney transplantation, upgrading it to that from a living donor.
The catastrophic central injury of brain death (BD) has been shown to perturb significantly the function and structure of somatic organs in situ by rapid upregulation of inflammatory mediators and other acute phase proteins. 1,2 The tempo and intensity of acute rejection of heart, kidney, and liver allografts from such donors are accelerated after transplantation because the involved organs appears to trigger host alloresponsiveness as a continuum between nonspecific injury and specific immunologic activity. 3,4 Clinical data have confirmed that kidney grafts from brain-dead organ donors experience more frequent episodes of acute rejection after transplantation. 5 The lack of suitable donors and the increasing numbers of potential recipients have also resulted in the acceptance of “marginal” brain-dead donors, the organs of whom are at significant risk for primary nonfunction or primary delayed function (PDF) compared with those from living donors. PDF alone is associated with significant potential complications, an increase in retransplantation, and a higher recipient death rate. Such grafts, compromised not only by donor-associated conditions such as diabetes and hypertension, but also from insults surrounding the removal and storage, experience both an increased rate of acute and chronic rejection and significantly diminished survival. 6 Several factors have been implicated in the development of this complication, particularly ischemia–reperfusion (I/R), which appears to be an integral part of the syndrome of BD. 7
Because the state of BD occurs before organ retrieval, it is a potential risk factor for graft outcome after transplantation. As a result, means to improve the organ quality of cadaver donors before transplantation have been considered. Earlier attempts focused on altering the hormonal milieu. 8–10 Because the results of these approaches were disappointing, however, they were never introduced into clinical practice. 11 The objectives of the present study are to reassess whether other strategies of donor treatment at the time of BD improve organ quality by reducing inflammatory changes and subsequent associated acute host immune responsiveness. To prevent the nonspecific changes occurring within peripheral organs after the central catastrophe, two treatments were tested and their effects compared. First, a single dose of steroids was administered with induction of BD to reduce the release of inflammatory cytokines. Second, soluble P-selectin glycoprotein ligand (sPSGL) was given in similar fashion to prevent leukocyte adhesion to the inflamed vascular endothelium of the graft.
METHODS
Brain Death Model
Inbred adult (200–250 g) male Fisher (F344) and Lewis (LEW) rats (Harlan Sprague-Dawley, Indianapolis, IN) acted as kidney donors and recipients. BD was produced in donor animals by gradually increasing intracranial pressure by slow inflation of a No. 3 Fogarty catheter balloon (Fogarty Arterial Embolectomy Catheter: 3F, Baxter Healthcare Co., Irvine, CA) introduced into the intracranial cavity through an occipital bur hole, as described. 12 BD with herniation of the brain stem was confirmed by electroencephalography, apnea, areflexia, and maximally dilated and fixed pupils. All rats were intubated via a tracheostomy using a No. 13 blunt-tipped cannula and mechanically respirated at a rate of 85/min and a tidal volume of 2.0 mL for 6 hours (Rodent ventilator, model 683, Harvard Instruments, South Natick, MA). Intraarterial blood pressure was continuously monitored via a PE50 catheter placed in the left femoral artery and attached to a transducer and recorder. Only animals with stable mean arterial blood pressure (MAP > 80 mm Hg) were accepted as donors in the study to preclude as much as possible the effects of peripheral ischemia secondary to hypotension. After 6 hours, the left kidney was removed for transplantation. Sham-operated rats in which a bur hole was drilled but no Fogarty catheter inserted served as living donor controls. Urine production was determined via a PE50 catheter placed directly into the urinary bladder after a small laparotomy incision. Maintenance anesthesia with pentobarbital (Nembutal, Abbott Laboratories, North Chicago, IL; 40 mg/kg) was administered to the control rats as needed for the 6-hour period before kidney removal. Brain-dead animals received no anesthesia because previous studies had shown it did not influence physiologic parameters. 1
Surgical Techniques
Renal allografts from F344 donors were grafted into unmodified anesthetized LEW recipients using standard microsurgical techniques. After removal of the left native kidney from the host, the donor kidney was transplanted orthotopically to the recipient renal vessels and ureter by end-to-end anastomoses using 10-0 prolene. Right native nephrectomy was performed 6 days later. The time between release of the vascular clamps and return of uniform cortical blood flow was defined as reperfusion time after transplantation.
Experimental Groups
For survival times in group 1 (n = 12), kidneys were placed into unmodified hosts 6 hours after donor BD and mechanical ventilation. Sham-operated and ventilated (6 hours) F344 animals were used as living donor controls (group 2, n = 12). Immediately after induction of BD, the donor treatment groups received either sPSGL (500 μg; group 3, n = 10) or glucocorticosteroids (10 mg/kg; group 4, n = 10) intravenously. The time of death of the unmodified graft recipients resulting from renal failure secondary to complete and irreversible rejection of their allografts was compared between the groups.
The F344→LEW strain combination was chosen as model because it is a weak strain combination in which the acute rejection process, even in unmodified hosts, occurs at a attenuated tempo, emphasizing any donor-associated changes. 3 If a strong strain combination had been used, in which the acute rejection process is generally complete by 7 to 10 days, such differences would be difficult to determine accurately.
Postoperative complications such as hydronephrosis secondary to ureteric obstruction were ruled out by autopsy. Additional grafted organs were analyzed serially by morphology, immunohistology, and polymerase chain reaction (PCR) immediately before transplantation, 6 hours after grafting (a period examined specifically because the early changes were presumably inflammatory), and at 1, 3, and 10 days, as host alloresponsiveness was becoming apparent (n = 3–5 per time point per group).
Histology and Immunohistology
Kidneys were fixed in 10% buffered formalin. Paraffin sections were stained with hematoxylin and eosin and assessed by light microscopy. For immunohistology, cortical segments were immediately snap-frozen in liquid nitrogen. Monoclonal antibodies obtained from Serotec (Wiesbaden, Germany) were directed against CD5+ T cells (OX19), CD4+ T-helper cells (W3/25), CD8+ T cytotoxic/suppressor cells (OX-8), monocyte/macrophages (ED1), and CD25+ cells (activated T and B cells). Additional antibodies were directed against MHC class 2 antigens (OX-3). After specific monoclonal antibody staining, the sections were then treated with alkaline-labeled rabbit antimouse IgG, followed by indicating mouse antialkaline phosphatase complex; sections were counterstained with hematoxylin. The number of labeled cells in 20 consecutive high-power fields (×400 magnification) was determined in three kidneys per group. Expression of MHC class 2 was based on semiquantitative assessment. 13
Polymerase Chain Reaction
Sample Preparation
Kidney tissue biopsy samples were removed at the time points indicated above and immediately snap-frozen in liquid nitrogen and stored at −70°C. Before analysis, the thawing tissues were transferred in 700 μL guanidinium isothiocyanate solution and homogenized using an Ultraturrax tissue homogenizer (Jahnke und Kunkel, Staufen i.Breisgau, Germany). Total RNA was extracted from core biopsies by a standard method (ethanol-phenol precipitation). RNA was reverse-transcribed as previously described. 14 DNase digestion was performed before cDNA synthesis.
Quantification of Cytokine Genes
Cytokine gene expression was measured using PCR on the ABI PRISM 7700 Sequence Detection System (TaqMan, Foster City, CA). Genes encoding for the following products were investigated: CD3, CD25, interleukin-2, Mip-1α, interferon-γ, Fas ligand, INOS, HO-1, interleukin-10, perforin, and tumor necrosis factor-α. The method uses the 5′ nuclease activity of Taq polymerase to cleave a nonextendable dual-labeled fluorogenic hybridization probe during the extension phase of the PCR. One fluorescent dye serves as a reporter (FAM, 6-carboxyfluorescein), and its emission spectrum is quenched by the second fluorescent dye (TAMRA, 6-carboxy-tetramethyl-rhodamine).
The reactions are monitored in real time during the log phase of product accumulation. The increase in the reporter dye fluorescence intensity during PCR is proportional to the amplification of the target sequence. The cycle number at which the amplification plot crosses a fixed threshold above baseline is defined as the threshold cycle (Ct). To control for variation in DNA content across different preparations, all results are related to the concentration of a gene whose expression remains more or less constant, the so-called housekeeping gene. We choose for the housekeeping gene the rat aldolase gene because it did not cross-react with genomic DNA.
Relative quantitation was performed according to the comparative ΔCt method (User Bulletin #2, Perkin-Elmer, 1997; DNA/RNA Real-Time Quantitative PCR, Perkin-Elmer, 1998 Applied Biosystems, Weiterstadt, Germany). The difference between the mean Ctcytokine and the mean Ctaldolase was calculated to normalize for different amounts of DNA (ΔCt). The result for the cytokine gene expression after versus before treatment was given by a unitless value through the formula 2–ΔCt.
The PCR reaction was performed in a final volume of 25 μL containing 1 μL cDNA, 12.5 μL Master Mix (TaqMan Universal PCR Master Mix, Perkin Elmer), 200 nmol/L hybridization probe, 50 to 900 nmol/L of each primer, and 5.5 μL distilled water. After an initial step of 2 minutes at 50°C involving activation of uracyl-n-glycosylase and degradation of any preexisting contaminating RNA sequences, a denaturation and a hot start for AmpliTaq Gold DNA polymerase was performed at 95°C for 10 minutes. The amplification took place in a two-step PCR (40 cycles; 15 seconds denaturation step [95°C] and 1 minute annealing/extension step [60°C]). The mean Ct values for aldolase and the cytokines were calculated from double determinations.
Statistical Analysis
Statistical significance was ascertained using the Student t test, log-rank-sum test, and Mann-Whitney test. The results are expressed as mean ± SD and were considered significant at P < .05. Graft survival was expressed graphically using the Kaplan-Meier survival curve.
RESULTS
Physiologic Changes After Brain Death and Ischemia–Reperfusion
During inflation of the Fogarty balloon and the gradual onset of BD, the animals reacted with sharply increased arterial blood pressure for 15 to 30 minutes (average MAP = 206 ± 38 mm Hg at 10 minutes vs. MAP = 102 ± 15 before injury, n = 40, P < .0001). After this period of autonomic storm, most of the rats then maintained stable blood pressure (average MAP 80–100 mm Hg) until the kidney was removed for transplantation after the 6-hour ventilation period. Hypotensive animals were excluded. Living donor control animals were consistently normotensive (average MAP 80–100 mm Hg, n = 20). Electroencephalography showed flat-line tracings in brain-dead animals compared with continued physiologic activity in the controls. The average urine production of control animals during this pretransplant period was 0.9 ± 0.3 mL per 6 hours (n = 20); the brain-dead rats showed some diuresis (1.2 ± 0.3 mL per 6 hours, n = 20, P < .005), indicating diabetes insipidus, a common phenomena in brain-dead organ donors.
The cold ischemic time necessary for the transplant procedure did not differ appreciably between the groups (23 ± 2 minutes for group 1, 20 ± 2 minutes for group 2, 21 ± 4 minutes for groups 3 and 4). However, there were significant differences in the period of reperfusion between the release of the vascular clamps and complete cortical perfusion of the grafts (group 1, 18 ± 5 minutes vs. group 2, 4 ± 2 minutes, P < .0001; group 3, 8 ± 4 minutes, P < .001; group 4, 7 ± 5 minutes, P < .001; group 3 vs. group 4, NS).
Allograft Survival
Recipients of kidney allografts from brain-dead donors experienced an accelerated rate of acute rejection and died of renal failure significantly earlier than controls (P = .01, Fig. 1). Recipients of treated kidneys from groups 3 and 4 showed survival times comparable to living donor controls (group 2, NS) and significantly better survival times versus the group 1 animals (P < .01).
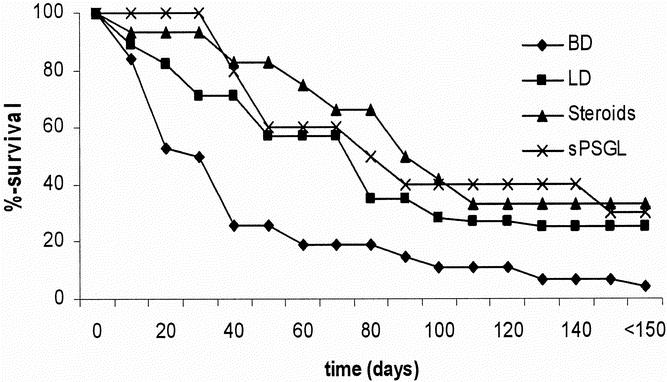
Figure 1. The tempo of acute rejection and the resultant survival of unmodified recipients of renal allografts are shown (Kaplan-Meier survival curve). Survival of animals with grafts from brain-dead donors is significantly shorter than in animals with grafts from living donor or treated animals (all groups P < .01 vs. brain-dead donors).
Histology
Although characteristic morphologic changes associated with immunologic rejection were observed in the transplanted allografts of all groups, the tempo and intensity of the process were strikingly different. Within the first 24 hours, margination and pooling of neutrophils were obvious in the microvasculature and glomeruli of kidneys from the untreated brain-dead donor animals in group 1. By day 3, relatively large numbers of mononuclear cells had scattered throughout the graft substance and had formed clumps around vessels and tubules. Minimal cellular activity was noted during this initial interval in the grafts of recipients in groups 2, 3, and 4. By day 10, histologic changes in group 1 allografts included dense cellular infiltrates, increasing interstitial inflammation, loss of tubules, and endotheliitis. The grafts of animals dying acutely of renal failure in this group exhibited massive cellular infiltration, interstitial edema, and necrosis. In contrast, cellular activity was significantly diminished in the kidneys of living donor controls and in the animals in groups 3 and 4; differences in the intensity or tempo of the acute phenomenon between these groups were minor.
Immunopathology
Immunolabeled infiltrating cells occurred in relatively small numbers before transplantation in the kidneys of living donor controls and treated brain-dead donor groups. In recipients of organs from untreated brain-dead donors, however, the number of ED1+, CD4+, and CD8+ cells was significantly greater (P < .05), and they were located predominantly around vessels and in direct contact with tubules. Cellular infiltration increased quickly within 6 hours of transplantation, and by 24 hours macrophages and T-cell populations had become increasingly obvious (Table 1, Fig. 2). Cellular infiltrates were relatively few in the kidneys from recipients in groups 2, 3, and 4 at this time point. Although all leukocyte populations increased slightly up to 10 days in these latter groups, they remained consistently lower than in group 1. Numbers of B cells did not vary significantly between the groups and contributed less than 5% of graft cellularity at any point (data not shown).
Table 1. INFILTRATING CELL POPULATIONS

*P < .05.
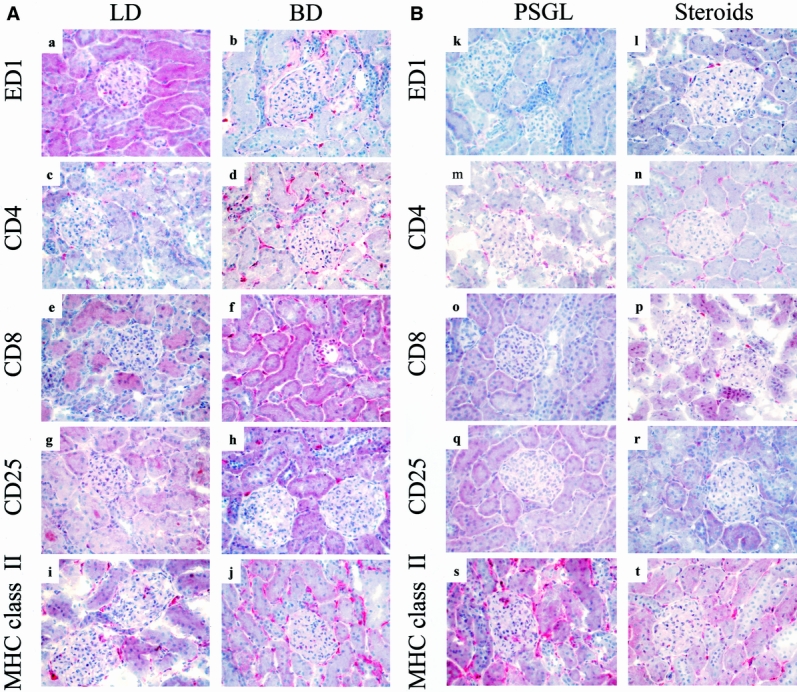
Figure 2. Effects of brain death on intragraft events 24 hours after transplantation. Numbers of ED1+, CD4+, CD8+, and CD25+ cells present in control kidneys are minimal (a–h). Donor brain death, in contrast, markedly enhanced the infiltration and accumulation of these leukocyte subtypes within perivascular and periglomerular areas (k–t). Cellular infiltrates were absent at 24 hours in the soluble P-selectin glycoprotein ligand (sPSGL) donor treatment group and in living donor kidneys, although their presence was diminished less by steroid treatment. Intragraft expression of MHC class 2 was increased and was particularly evident in interstitial macrophages in kidneys from untreated brain-dead donors; it was expressed only moderately in controls and after steroid treatment (i, j). sPSGL treatment did not affect MHC class 2 expression, which remained comparable to brain-dead organs (a–t). (Serial cryostat sections, representative of 4 animals per group per time-point; hematoxylin counterstain, ×400)
Expression of MHC class 2 became apparent on infiltrating mononuclear cells (10–20%) before transplantation and within a few hours thereafter on endothelial cells (focal) in group 1 kidneys, in contrast to those in groups 2 and 4 (no staining). Interestingly, the infiltrating cells in grafts from donors treated with sPSGL (group 3) expressed MHC class 2 antigens in a pattern comparable to group 1 before and after transplantation. By 6 and 24 hours, induction of MHC class 2 was even more obvious on leukocytes, interstitial dendritic cells, and endothelial cells in both untreated brain-dead donor kidneys (at 24 hours, interstitial dendritic cells 10–20%, endothelial cells diffuse, mononuclear cells 20–50%) and after treatment with sPSGL (at 24 hours, interstitial dendritic cells 10–20%, endothelial cells diffuse, mononuclear cells 10–20%; see Fig. 2). Although subsequent MHC antigen expression in groups 2 and 4 was consistently present, it remained less than in groups 1 and 3 at all time points.
Polymerase Chain Reaction
mRNA expression in kidneys of brain-dead and living donors was assessed by PCR before transplantation and serially thereafter for the representative inflammatory cytokines tumor necrosis factor-α, interferon-γ, MIP-1α, perforin, and Fas ligand. These were significantly (P < .05) upregulated 6 hours after BD in kidneys from group 1 hosts versus controls (n = 4 per group;Fig. 3). Donor treatment with steroids limited the transcription of these proinflammatory mediators after reperfusion (P < .05 at 6 hours after transplantation), but sPSGL treatment had less effect. Differences between the groups became less obvious at 3 and 10 days, although organs from untreated brain-dead donors in most instances showed the highest and living donor organs the lowest transcription rates. There were no statistically significant differences in the transcription of CD25, CD3, interleukin-2, and interleukin-10 as markers and products of infiltrating cells between the groups before transplantation, although untreated organs in group 1 showed increased expression after transplantation (P < .05). The expression of CD25 and CD3 was reduced more by treatment with sPSGL than with steroids (P < .05). HO-1 transcription was significantly increased after treatment in both groups at the early time points (P < .05 vs. brain-dead and living donors) but returned to baseline levels after 3 days and 10 days. There were no significant differences between brain-dead and living donors during the observation period. Donor treatment with sPSGL did not upregulate the transcription of INOS, in contrast to treatment with steroids, which reduced INOS transcription significantly at 0 hours, 6 hours, and 3 days (P < .05) and kept it at a lower level at day 10. At all time points, the transcription of INOS was highest in the untreated BD group; little expression was noted in controls.

Figure 3. mRNA expression in kidney tissue in brain-dead and control grafts before transplantation (0 hours) and up to 10 days after transplantation.
DISCUSSION
The syndrome of BD is characterized by rapid and chaotic swings in blood pressure, an early hypertensive phase followed by later normotension or hypotension. Intense catecholamine release from sudden sympathetic stimulation either from direct neural activity or from excessive production of endogenous catecholamines (“autonomic storm”) elevates peripheral vascular resistance and alters tissue perfusion. 15–17 The vasoconstriction is presumably so severe that flow to peripheral organs may be significantly reduced despite markedly increased perfusion pressure. 18 These circulatory changes may produce severe ischemia of donor organs before their removal, a damaging insult that is worsened by reperfusion after transplantation. As a result, the incidence of acute tubular necrosis leading to delayed graft function is often increased in grafted kidneys from brain-dead donors. 19,20 The unstable period of “autonomic storm” during and after BD also produces intense inflammatory changes in the prospective graft. 21 Recent experimental examination of the relation between explosive BD and the activation of peripheral organs has shown rapid infiltration of leukocyte populations with upregulation of their associated cytokines and other products. 1 Changes in the organ secondary to the nonspecific, antigen-independent insult of BD and associated I/R appear to trigger host alloresponsiveness, which in time accelerates and intensifies the process of both acute and chronic rejection.
Attempts to modulate systemic activation after BD have been problematic. The effects of treatment of the brain-dead donor by hormonal substitution therapy have been conflicting: benefit to the quality of the organ to be transplanted has been found only sporadically. 8,22 The present study investigated strategies involving both nonspecific blocking of proinflammatory mediators using steroids and the more selective inhibition of infiltrating leukocytes with sPSGL. The antiinflammatory actions of steroids presumably inhibit the production of cytokines and other cell products. sPSGL has been shown to be effective in preventing I/R injury in experimental models. 23,24 P-and E-selectin are early adhesion molecules that mediate rolling and initial attachment of circulating leukocytes to vascular endothelium in inflammatory states. 25,26 A high-affinity ligand for these molecules, PSGL-1 (CD162) is a sulfated, sialyl Lewis X-bearing glycoprotein expressed on cell surfaces and essential for their diapedesis into rejecting grafts. 27,28 Binding of the selectins to their ligand is important in both in BD and I/R injury. 3,29 Because selectins mediate the initial events of leukocyte adhesion, it is reasonable to postulate that selectin-directed blockade would reduce cell-mediated damage associated with BD in similar fashion to its effects in I/R. 23 Indeed, our results indicate that sPSGL inhibits the infiltration of various leukocyte populations and expression of MHC class 2 antigens in the kidney both after donor BD and after transplantation. It has been shown both clinically and experimentally that impaired early function secondary to initial I/R injury influences later graft behavior and is a risk factor for chronic graft dysfunction. 30 In the present study, sPSGL treatment of the donor led to delayed occurrence of acute rejection in the unmodified hosts, resulting in survival times comparable to those of living donor grafts. Treatment of the donor with steroids showed similar effects, with reduction of cellular infiltration within 24 hours after transplantation. Steroids were also more effective than sPSGL in decreasing the transcription of various leukocyte and endothelial cell-derived cytokines and mediators. I/R injury is diminished by this treatment, indicated by downregulated activity of proinflammatory cytokines and Fas ligand and perforin, factors involved in cell death after injury. INOS expression, a potent vasodilator, decreased in organs from treated brain-dead donors. Nevertheless, there were significant differences in the return of uniform cortical blood flow, which was defined as reperfusion time after transplantation. This suggests that both drugs reduced reperfusion injury and could influence significantly the catecholamine-mediated vasoconstriction and microcirculatory disturbance occurring rapidly after BD.
The upregulation of cytokines and other cell products after 24 hours may be due to the transient effects of these drugs. Cellular infiltrates were still present in the steroid-treated group, to a lesser extent than in the organs in group 1, but greater than those occurring after sPSGL treatment and in living donor kidneys. By day 3, the presence of the proinflammatory lymphocyte-associated cytokines tumor necrosis factor-α and interferon-γ were associated with MHC class 2 expression in the kidneys from group 1 more than in those of the treated groups 3 and 4.
Upregulation of MHC class 2 molecules in peripheral tissue and infiltrating leukocytes is an important effect of BD and other nonspecific endothelial injuries, including I/R. 31 It is important that in group 1 organs, MHC class 2 was expressed before the organ was removed, in contrast to its absence or significantly reduced expression in the other groups. The presence of MHC class 2 antigens increases graft immunogenicity via the T-cell recognition process and acts as an important trigger for host alloresponsiveness. As a result, early suppression of its expression contributes to improved graft survival.
These experiments show that intervention early after the induction of BD either with steroids or sPSGL inhibits the effects of this central catastrophe in peripheral tissue. Thus, this neurologic state should not be considered a static condition, but a dynamic process that influences significantly the quality of peripheral organs. Although the kinetics of inflammatory changes in the human donors and the optimal time point for therapeutic intervention before harvesting remain unanswered, reduction of the initial BD-mediated inflammatory immunologic changes in this reproducible rat model at least can minimize sequential cellular activity and modulate I/R injury and may prevent eventual renal dysfunction. In the context of allotransplantation, a more inert brain-dead donor organ, which resembles one from a living donor, does not trigger immediate host immunity and may attenuate and delay the initial effects of acute rejection and the later changes of the chronic process. This observation may have important clinical application.
Discussion
Prof. L. Fernandez-Cruz: Thank you, Dr. Pratschke, for your excellent presentation. I believe you and the group of Berlin should be congratulated for this excellent work. It seems that the brain death of the donor is a significant risk factor for organ survival after transplantation. It is very interesting to see that you decrease the inflammatory response after brain death with an early intervention. A relationship between brain death and graft performance after transplantation is of outstanding interest for transplant surgeons and biologists. In this context I have a few questions.
You did your investigations in the kidney; I am wondering if the state of brain death causes inflammation of other organs besides the kidney. My second question is, is there a relationship between brain death and ischemia perfusion? I think this is interesting. Do you think they evoke similar mechanism of injury? Can you explain further how early no specific injury can produce long-term changes?
Thank you.
Sir P. Morris: I too enjoyed this paper. It is a further study in the series from Nick Tilney’s group, and you yourself have been involved in most of them. The paper that you presented elaborates on this intriguing concept of the injury recurring after, or during, brain death. I have one comment and one quick question. You did state in your presentation that the survival of spouse transplants, where the matching of donor and recipient is usually poor, leads to a comparable outcome of a kidney transplant as that in parent-to-child transplants. This is not strictly so, in that within the European collaborative study Opelz has demonstrated a major influence of matching on the outcome of spouse transplants, so that transplants between the better-matched spouses do considerably better than between poorly matched spouses. This is now confirmed in the UNOS data. Thus, I think it is important to realize that even in the spouse transplants without brain death, there is an immunologic influence of matching.
The question I want to ask is, I think, of considerable clinical relevance, and that is: What is the effect of steroids given after brain death has been established? In your model, for example, if you gave steroids 3 hours after establishing a brain death, would you see an influence similar to that as demonstrated by the administration of steroids at the time brain death is induced? I do not know whether you have done those experiments or not, but I would be very interested to hear your comments.
Prof. P. Bell: I enjoyed your paper and would like to ask a question about nonheart-beating donors in this context. If you are correct, then if you take biopsies from nonheart-beating donors, they should have little or no class 2 antigens on their kidney and they should do better, because the donor has not had any upregulation of their immune system. Have you tried that? If not, maybe you should.
It is my impression, from our nonheart-beating donor cases, that they do better after recovery from their initial ATN. They seem to have less rejection episodes and do fine long term with good renal function.
Dr. J. Pratschke (closing): Thank you very much for the questions. First I would like to address the question of Prof. Fernández-Cruz. We have shown proinflammatory activation in our experimental setups in the kidney as well as in the heart immediately after brain death. Beside this, we also observed for both organs an accelerated course of rejection after allogeneic transplantation. There is some evidence of similar perturbations in the liver, shown by Van der Hoeven and coworkers in a similar experimental approach. In large animals rendered brain-dead there are only few data available, demonstrating patches of myocyte necrosis and delayed organ function after heart transplantation. These investigations have been done in the 1980s by Reichart, Novitzky, and Cooper at Cape Town. In humans the only data available are from Koo, Welsh, McLaren, and coworkers published in Kidney International showing in human kidneys significant differences in terms of ischemia/reperfusion injury and rejection episodes between cadaver and living donors.
The second question is a very important one as there is indeed a close relationship between brain death and ischemia/reperfusion. At the moment brain injury is induced, the animals react uniformly with sharply increased mean arterial blood pressure and vascular resistance of peripheral organs. These phenomena are well known and summarized in the literature as “autonomic storm.” As a consequence, the perfusion of peripheral organs is reduced during and after brain death induction due to microcirculatory disturbances. Our hypothesis is that organ ischemia starts at the moment of brain death, despite increased mean arterial blood pressure, and persists on different levels until organ harvesting. It is likely that early-onset ischemia leads to deteriorated reperfusion injury, which we observed experimentally after transplantation of organs from brain-dead donors.
Early nonspecific injuries, as for example the brain death of the donor, surgical manipulations, ischemia, and following reperfusion injury, have a common pathway: these injuries result in unspecific inflammatory activation of the prospective graft before transplantation and lead to an enhanced interaction with specific allogeneic factors after transplantation. The higher the activation level of the organ before grafting, the worse the injury afterward, leading to reduced organ mass and early onset of fibrosis, arteriosclerosis, and in kidneys glomerulosclerosis. Our data show clearly that early inflammatory changes can be interrupted with steroids and in a more specific approach with sPSGL. Interestingly, without further immunosuppressive treatment, rebound phenomena were observed over time in our series. We hypothesize that by diminishing the initial injury as much as possible, we will see beneficial effects not only shortly after transplantation, but even over the long term.
To address the question of Sir Peter Morris: Until now we did not define a resection kinetics with different time points of therapeutical intervention. In our experiments we gave the drug at the moment of brain death to show principally that there is a chance to stop brain death-induced proinflammatory changes. This is of course not completely comparable to the human situation, where there is usually a period of intensive care between brain death and organ harvesting. I agree that it would be very interesting to examine later interventions and the outcome thereafter.
Prof. Bell’s question about the nonheart-beating donor situation is hard to answer and I am afraid I can only speculate. Recently Dr. Tilney and his fellow I. Laskowski performed a few experiments with nonheart-beating donors, showing that some of the nonheart-beating donors are comparable or even better grafts than those from cadaver sources. To answer this question sufficiently, still a lot of experiments have to be done.
Thank you very much for the discussion.
Footnotes
Drs. Pratschke and Wilhelm are recipients of a Research Fellowship award from the Deutsche Forschungsgemeinschaft (DFG)(Pr 578/2-2 and Wi 1677/1-1), Germany. This work was supported by USPHS grant 5 RO 1 DK 4490-28.
Correspondence: Johann Pratschke, MD, Department of General, Visceral and Transplantation Surgery, Humboldt University Berlin, Charité, Campus Virchow, Berlin, Augustenburgerplatz 1, 13353 Berlin, Germany.
E-mail: johann.pratschke@charite.de
Accepted for publication April 2001.
References
- 1.Takada M, Nadeau KC, Hancock WW, et al. Effects of explosive brain death on cytokine activation of peripheral organs in the rat. Transplantation 1998; 65: 1533–1542. [DOI] [PubMed] [Google Scholar]
- 2.Van der Hoeven JA, Ploeg RJ, Postema F, et al. Induction of organ dysfunction and up-regulation of inflammatory markers in the liver and kidneys of hypotensive brain dead rats: a model to study marginal organ donors. Transplantation 1999; 68: 1884–1890. [DOI] [PubMed] [Google Scholar]
- 3.Pratschke J, Wilhelm MJ, Kusaka M, et al. Accelerated rejection of rat renal allografts from brain-dead donors. Ann Surg 2000; 232: 263–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wilhelm MJ, Pratschke J, Beato F, et al. Activation of the heart by donor brain death accelerates acute rejection after transplantation. Circulation 2000; 102: 2426–2433. [DOI] [PubMed] [Google Scholar]
- 5.Koo DD, Welsh KI, McLaren AJ, et al. Cadaver versus living donor kidneys: impact of donor factors on antigen induction before transplantation. Kidney Int 1999; 56: 1551–1559. [DOI] [PubMed] [Google Scholar]
- 6.Tullius SG, Nieminen M, Bechstein WO, et al. Contribution of early acute rejection episodes to chronic rejection in a rat kidney retransplantation model. Kidney Int 1998; 53: 465–472. [DOI] [PubMed] [Google Scholar]
- 7.Kusaka M, Pratschke J, Wilhelm MJ, et al. Activation of proinflammatory mediators in rat renal isografts by donor brain death. Transplantation 2000; 69: 405–410. [DOI] [PubMed] [Google Scholar]
- 8.Novitzky D, Cooper DKC, Reichart B, et al. Hemodynamic and metabolic response to hormonal therapy in brain-dead potential organ donors. Transplantation 1987; 43: 852–854. [PubMed] [Google Scholar]
- 9.Pratschke J, Wilhelm MJ, Kusaka M, et al. Brain death and its influence on donor organ quality and outcome after transplantation. Transplantation 1999; 67: 343–348. [DOI] [PubMed] [Google Scholar]
- 10.van der Hoeven JA, Ter Host GT, Molema G, et al. Effects of brain death and hemodynamic status on function and immunological activation of the potential donor liver in the rat. Ann Surg 2000; 232: 804–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gramm HJ, Meinhold H, Bickel U, et al. Acute endocrine failure after brain death? Transplantation 1992; 54: 851–857. [DOI] [PubMed] [Google Scholar]
- 12.Pratschke J, Wilhelm MJ, Kusaka M, et al. A model of gradual onset brain death for transplant-associated studies in rats. Transplantation 2000; 69: 427–430. [DOI] [PubMed] [Google Scholar]
- 13.Hancock WW, Whitley WD, Tullius SG, et al. Cytokines, adhesion molecules, and the pathogenesis of chronic rejection of rat renal allografts. Transplantation 1993; 56: 643–650. [DOI] [PubMed] [Google Scholar]
- 14.Murphy GM, Jia XC, Yu AC, et al. Reverse transcription and polymerase chain reaction technique for quantification of mRNA in primary astrocyte Cultures. J Neurosci Res 1993; 15: 643–651. [DOI] [PubMed] [Google Scholar]
- 15.Mertes PM. Physiology of brain death. In: Tilney NL, Strom TB, Paul LC, eds. Transplantation biology: Cellular and molecular aspects. Philadelphia: Lippincott; 1996: 275–289.
- 16.Shivalkar B, Van Loon J, Wieland W, et al. Variable effects of explosive or gradual increase of intracranial pressure on myocardial structure and function. Circulation 1992; 87: 230–239. [DOI] [PubMed] [Google Scholar]
- 17.Bittner HB, Kendall SW, Campell KA, et al. A valid experimental brain death organ donor model. J Heart Lung Transplant 1995; 14: 308–317. [PubMed] [Google Scholar]
- 18.Herijgers P, Leunens V, Tjandra-Maga TB, et al. Changes in organ perfusion after brain death in the rat and its relation to circulating catecholamines. Transplantation 1996; 62: 330–335. [DOI] [PubMed] [Google Scholar]
- 19.Lagiewska B, Pacholczyk M, Szostek M, et al. Hemodynamic and metabolic disturbances observed in brain-dead organ donors. Transplant Proc 1996; 28: 165–166. [PubMed] [Google Scholar]
- 20.Nagareda T, Kinoshita Y, Tanaka A, et al. Clinicopathology of kidneys from brain dead patients treated with vasopressin and epinephrine. Kidney Int 1993; 43: 1363–1370. [DOI] [PubMed] [Google Scholar]
- 21.Wilhelm MJ, Pratschke J, Laskowski IA, et al. Brain death and its impact on the donor heart-lessons from animal models. J Heart Lung Transplant 2000; 1: 414–418. [DOI] [PubMed] [Google Scholar]
- 22.Harms J, Isemer FE, Kolenda H. Hormonal alteration and pituitary function during course of brain stem death in potential organ donors. Transplantation 1993; 56: 363. [PubMed] [Google Scholar]
- 23.Dulkanchainun TS, Goss JA, Imagawa DK, et al. Reduction of hepatic ischemia/reperfusion injury by a soluble P-selectin glycoprotein ligand-1. Ann Surg 1998; 227: 832–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Takada M, Nadeau KC, Shaw GD, et al. The cytokine-adhesion molecule cascade in ischemia/reperfusion injury of the rat kidney. J Clin Invest 1997; 99: 2682–2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McEver RP, Moor KL, Cumminigs RD. Leukocyte trafficking mediated by selectin carbohydrate interactions. J Biol Chem 1995; 270: 11025–11028. [DOI] [PubMed] [Google Scholar]
- 26.Ley K, Tedder TF. Leukocyte interactions with vascular endothelium. J Immunol 2001; 1995: 525–528. [PubMed] [Google Scholar]
- 27.Sako D, Chang XJ, Barone KM. Expression cloning of a functional glycoprotein ligand for P-selectin. Cell 1993; 75: 1179–1186. [DOI] [PubMed] [Google Scholar]
- 28.Turunen JP, Paavonen T, Mujuri ML. Sialyl Lewis X- and L-selectin-dependent site-specific lymphocyte extravasation into renal transplants during acute rejection. Eur J Immunol 1994; 24: 1130–1136. [DOI] [PubMed] [Google Scholar]
- 29.Han KT, Sharar SR, Philips ML, et al. Sialyl Lewis X oligosaccharide reduces ischemia-reperfusion injury in the rabbit ear. J Immunol 1995; 155: 4011–4015. [PubMed] [Google Scholar]
- 30.Takada M, Nadeau KC, Shaw GD, et al. Prevention of late renal changes after initial ischemia/reperfusion injury by blocking early selectin binding. Transplantation 1997; 64: 1520–1525. [DOI] [PubMed] [Google Scholar]
- 31.Shoskes DA, Parfrey NA, Halloran PF. Increased major histocompatibility complex antigen expression in unilateral ischemic acute tubular necrosis in the mouse. Transplantation 1990; 49: 201–207. [DOI] [PubMed] [Google Scholar]