

Abstract
The repair mechanisms acting on DNA interstrand crosslinks (ICLs) in eukaryotes are poorly understood. Here, we provide evidence for a pathway of ICL processing that uses components from both nucleotide excision repair (NER) and translesion synthesis (TLS) and predominates during the G1 phase of the yeast cell cycle. Our results suggest that repair is initiated by the NER apparatus and is followed by a thwarted attempt at gap-filling by the replicative Polymerase δ, which likely stalls at the site of the remaining crosslinked oligonucleotide. This in turn leads to ubiquitination of PCNA and recruitment of the damage-tolerant Polymerase ζ that can perform TLS. The ICL repair factor Pso2 acts downstream of the incision step and is not required for Polymerase ζ activation. We show that this combination of NER and TLS is the only pathway of ICL repair available to the cell in G1 phase and is essential for viability in the presence of DNA crosslinks.
Keywords: DNA interstrand crosslinks, nucleotide excision repair, PCNA, translesion synthesis
Introduction
Covalent DNA interstrand crosslinks (ICLs) are produced by several key anticancer drugs, and are also likely to represent a particularly toxic form of spontaneous DNA damage (reviewed by Dronkert and Kanaar, 2001; McHugh et al, 2001).
Numerous ICL repair models have been proposed to account for the limited observations made in yeast and higher eukaryotes to date (Dronkert and Kanaar, 2001; McHugh et al, 2001; Niedernhofer et al, 2004; Saffran et al, 2004; Barber et al, 2005). These models are heavily influenced by those proposed for ICL repair in bacteria, which are the product of genetic investigation coupled to detailed biochemical reconstitution (Cole and Sinden, 1975; Van Houten et al, 1986; Sladek et al, 1989). Together, the Escherichia coli data suggest that dual incisions flanking the ICL, but on one strand only, are catalysed by the nucleotide excision repair (NER) apparatus and this is followed by recombination-mediated gap-filling utilising an intact sister chromatid. The newly assimilated strand provides a good substrate for NER and resynthesis on the second strand, eliminating the remaining crosslinked oligonucleotide.
In G1 phase haploid yeast cells, a homologous recombination substrate is not available. Furthermore, rad52 (recombination deficient) mutant cells display near wild-type sensitivity to ICL-inducing agents when grown to stationary phase (McHugh et al, 2000) indicating that nonrecombinational pathways must operate. We, and others, have previously reported an increase in sensitivity in rev3 mutants in stationary phase culture when compared with exponentially growing cultures (Henriques and Moustacchi, 1980; McHugh et al, 2000), suggesting that polymerase ζ (a heterodimer of the products of the REV3 and REV7 genes) could play an important role in ICL repair in G0/G1 haploid cells.
Yeast cells defective for either rev3 or rev7 demonstrate reduced mutability to a variety of genotoxic agents (Lemontt, 1971; Lawrence and Christensen, 1976; Ruhland and Brendel, 1979; Lawrence et al, 1985). Pol ζ is a B-family polymerase, highly conserved in eukaryotes (Gibbs et al, 1998; Xiao et al, 1998). In genetic and biochemical studies, it has been shown to replicate past UV-induced photoproducts (Nelson et al, 1996; Gibbs et al, 2005). Furthermore, Pol ζ very efficiently extends a range of mismatch primed 3′ ends, likely created during attempts at bypass by other polymerases (Lawrence and Hinkle, 1996; Johnson et al, 2000; Lawrence et al, 2000; Prakash and Prakash, 2002). Yeast cells defective in the Y-family polymerase, Rev1, show a phenotype very similar to those deficient in Rev3 and Rev7, including reduced mutability and sensitivity to damaging agents (Lawrence, 2002). Several recent studies suggest that the activity of translesion synthesis (TLS) polymerases, including Pol ζ, at replication forks stalled by DNA damage is dependent on a specific conserved lysine residue in PCNA (K164) that can be monoubiquitinated in Rad6–Rad18-dependent reaction to control TLS (Hoege et al, 2002; Stelter and Ulrich, 2003; Kannouche et al, 2004; Watanabe et al, 2004).
Here, we show that Pol ζ is required during ICL repair in the G1 phase of the cell cycle. Our observations suggest the predominance of an ICL repair pathway consisting of an NER step followed by TLS past the incised intermediate. Our data are also consistent with Pol δ activity and PCNA modification playing a role in the recruitment of Pol ζ to incised ICL intermediates.
Results
Polymerase ζ, but not Pol η, is required for resistance to crosslinking drugs in G1 cells and controls a mutagenic repair pathway
We examined the role of Pol ζ in ICL repair throughout the cell cycle by treating cells synchronised in G1, S and G2 phases with nitrogen mustard (HN2). A wild-type strain maintains good viability regardless of the cell cycle phase in which it is treated (Figure 1A). In contrast, an isogenic rev3 strain shows much greater sensitivity when treated in G1 than when released into S phase or treated in G2 (Figure 1B), suggesting a key role for Pol ζ during ICL repair in G1 cells. Since Pol ζ is a heterodimer of Rev3 and Rev7, which also interacts with Rev1, we also examined isogenic rev7 and rev1 disruptants for sensitivity in G1, S and G2, and found that these behave indistinguishably from the rev3 strain in these assays (data not shown).
Figure 1.
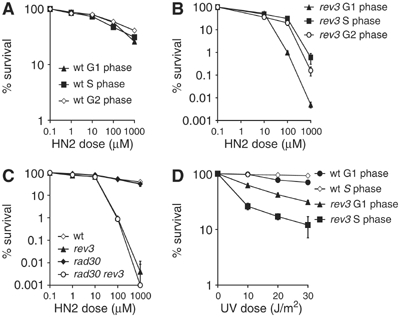
Cells lacking Pol ζ (rev3) are most sensitive to nitrogen mustard (HN2) in the G1 phase of the cell cycle. (A) Wild-type cells treated with increasing doses of HN2 for 30 min in the G1, S and G2 phases of cell cycle. (B) rev3 cells synchronised in G1, S and G2 treated with increasing doses of HN2. (C) rev3, rad30 and rev3 rad30 double mutants treated with HN2 in G1. (D) Wild-type and rev3 cells treated with different doses of UV irradiation in G1 and S phases. Results are the mean of a minimum of three repeats.
The second major translesion polymerase in yeast is encoded by the RAD30 gene (Pol η) (Johnson et al, 1999). Although this factor has previously reported to be dispensable for ICL repair in exponentially growing cells (Grossmann et al, 2001), we wished to examine the possibility of a specific role in G1 cells, including a redundant one with Pol ζ. We created rad30 and rev3 rad30 disruptants and examined their sensitivity to HN2 in G1 (Figure 1C). The rad30 single mutant shows no increase in sensitivity relative to the wild type, and furthermore the rev3 rad30 double mutant is no more sensitive than the rev3 single mutant indicating that there is no significant role for Pol η in the response to ICLs in G1 cells.
Several groups, including our own, have previously reported that drugs that induce ICLs are mutagenic (Ruhland and Brendel, 1979; McHugh et al, 1999). We therefore measured the rate of forward mutation at the CAN1 locus in repair-competent, wild-type and rev3 disruptants either in G1 or the exponential growth phase (Table I). Treatment with 100 μM HN2 induces an elevated forward mutation frequency in wild-type cells, and the majority of HN2-induced forward mutations are rev3-dependent in both growth phases. Therefore, we conclude that if a Pol ζ-dependent pathway predominates in G1, then it is likely that ICL repair reactions in G1 cells significantly contribute to the overall mutagenic effects of ICLs. As expected, a proportion of spontaneous (0 μM HN2) mutations are also rev3-dependent in both growth phases.
Table 1.
Forward mutation frequency (Canr) of wt and rev3 strains treated with HN2
| Exp. (0 μM) | Exp. (100 μM) | G1 (0 μM) | G1 (100 μM) | |
|---|---|---|---|---|
| Wt | 330 (±101) | 5659 × 108 (±575) | 160 (±30) | 6200 × 108 (±565) |
|
rev3 |
88 |
1145 × 108 (±474) |
50 (±24) |
375 × 108 (±35) |
| HN2, nitrogen mustard. | ||||
Most models for the action of TLS polymerases invoke a role for these enzymes in filling postreplication gaps remaining at lesions (Friedberg et al, 2005) and would, therefore, be expected to be most crucial in cells passing through S phase. Consistent with this notion, rev3 cells are much more sensitive to UV irradiation in S phase than those treated in G1, whereas wild-type cells are most sensitive in G1 phase (Figure 1D), indicating that the passage through S phase enhances resistance to this form of DNA damage in a Pol ζ-dependent manner. These results stand in marked contrast to the enhanced sensitivity of rev3 cells towards HN2 in G1 (Figure 1B), suggesting a requirement for Pol ζ in G1 cells during the resolution of ICLs, as opposed to bulky lesions that involve only one DNA strand.
A genetic test to confirm the importance of Pol ζ in ICL repair during G1 phase
We clearly observe sensitivity (Figure 1B) and a G1–S cell cycle transition delay (Figure 2A) in G1-arrested rev3-defective cells treated with HN2. However, from this data we cannot exclude the possibility that ICL repair events are deferred until the cells are released from G1 (e.g. upon plating in the sensitivity experiments) and that the Rev3-dependent pathway operates mainly in S phase. To try and definitively determine if the Pol ζ pathway is operational in G1, we exploited a temperature-sensitive allele of the CDC4 gene, cdc4-1, and its well-characterised suppressor sic1 (Schwob et al, 1994; Verma et al, 1997). At the G1/S transition, Cdc4, with Skp1 and Cdc53 (forming the SCFCdc4 complex), directs ubiquitination and destruction of Sic1, a Clb/Cdk1 inhibitor, permitting entry into S phase (Verma et al, 1997). Therefore, following synchronization with α-factor and incubation at the restrictive temperature of 37°C, we were able to maintain cdc4-1 cells in G1 for extended periods with only a minimal loss of viability (data not shown). In contrast, when sic1 is also deleted in these cells, G1 arrest is suppressed and cells enter S phase precociously. We combined these mutations with disruptions in the Rev7 subunit of Pol ζ creating isogenic cdc4-1 rev7 cells and cdc4-1 sic1 rev7 cells. The experimental strategy is shown in Figure 2B. As expected, double mutant cdc4-1 rev7 cells remain in G1 for the entire duration of the experiment when held at the restrictive temperature (Figure 2E). They have a low level of viability when plated that does not alter over time (2C). In contrast, cdc4-1 sic1 rev7 cells are forced rapidly and precociously into S phase at the restrictive temperature as monitored by FACS analysis (Figure 2E), entering S phase in under 30 min following HN2 treatment. These HN2 treated cdc4-1 sic1 rev7 cells also start to recover viability rapidly (Figure 2C), concomitant with entry into S phase and further progression through the cell cycle (Figure 2E). Therefore, functional Pol ζ is required to tolerate ICLs in G1 phase cells, and artificially forcing cells to enter S phase and beyond helps relieve this requirement, presumably by allowing utilisation of additional pathways available in other phases of the cell cycle. This is a strong evidence that the Pol ζ-dependent ICL repair pathway is the primary repair option for G1 cells. The pathway likely remains functional in other cell cycle phases, but here the ICL repair intermediates can be channeled into alternative pathways, which might be preferred. Repair competent cdc4-1 mutants and cdc4-1 sic1 double mutants exhibit around 70% viability throughout these experiments, at either the restrictive or permissive temperature, at the HN2 dose used (100 μM, data not shown). The NER-defective cdc4-1 rad14 sic1 cells demonstrate the same low viability as the cdc4-1 rad14 strain at the restrictive- (and also permissive-) temperature consistent with a vital role for NER in the initiation of all ICL repair pathways irrespective of cell cycle phase (Figure 2C and D).
Figure 2.
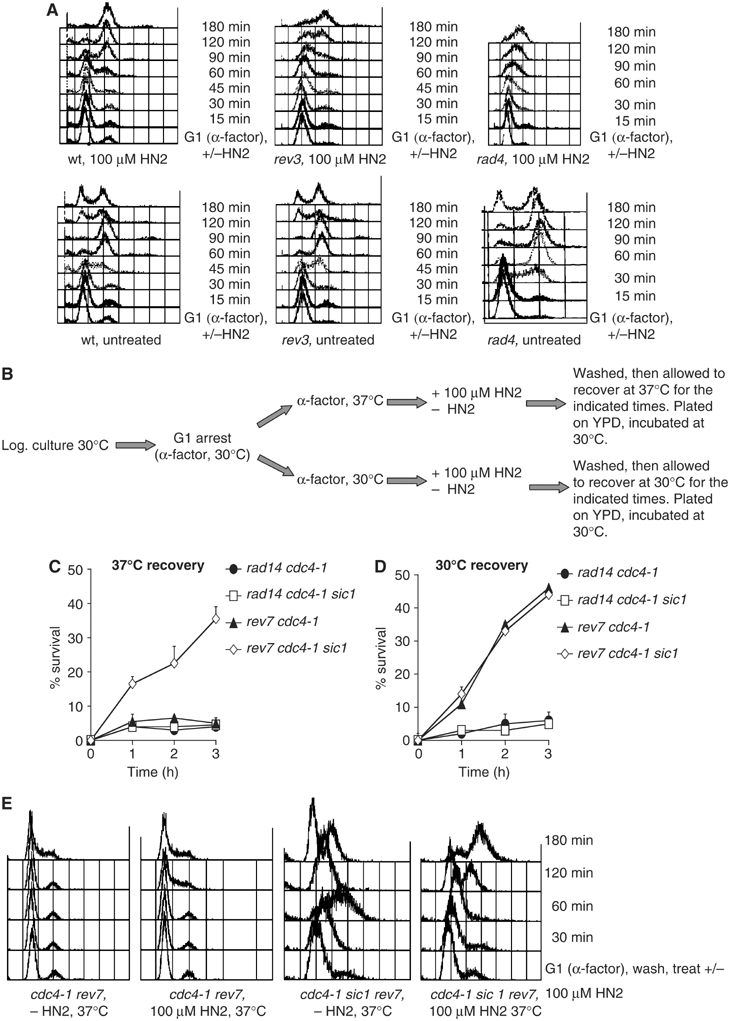
Release of rev3 cells from G1 enhances viability following HN2 treatment. (A) FACS analysis of wild-type, rev3 and rad4 cells arrested in G1 then mock-treated or treated with 100 μM HN2 for 30 min and allowed to enter the cell cycle. (B) Experimental scheme employing cdc4-1, cdc4-1 sic1 cells and their cognate double/multiple rad14 and rev7 mutants. Cells were grown to log phase and G1 arrested with α-factor. The culture was divided into two, one half at 37° and the other at 30°C, held with α-factor and either treated or mock-treated with 100 μM HN2. Cells were washed twice and allowed to recover either at 37 or 30°C in fresh YPD with samples taken, diluted and plated after 60, 120 and 180 min. (C, D) Percentage survival of rad14 cdc4-1, rad14 cdc4-1 sic1, rev7 cdc4-1 and rev7 cdc4-1 sic1 cells held at the restrictive (37°C) and permissive temperature (30°C) over time. (E) FACS analysis of cdc4-1 rev7, cdc4-1 sic1 rev7 arrested in G1 and either untreated or treated with 100 μM HN2 at the restrictive temperature (37°C).
NER, Pso2 and Pol ζ constitute the basic members of the G1 ICL repair pathway
Numerous models presented to date have suggested that ICL repair is initiated by NER incision and that the gap created postincision could be a substrate for gap-filling reactions catalysed by TLS polymerases (Berardini et al, 1999; McHugh et al, 2000; Dronkert and Kanaar, 2001). We wished to test these models more directly, and have used a combined genetic and biochemical approach. First, we constructed strains disrupted for NER genes and rev3, for which we tested G1 phase sensitivity to HN2 (Figure 3A). The NER (rad4) mutant is only slightly more sensitive than the rev3 strain to HN2, but the double mutant is no more sensitive than the rad4 single mutant. This suggests epistasis and, therefore, that these two factors likely act in a common biochemical pathway. Another factor that plays a crucial role in ICL repair in G1 cells is Pso2/Snm1 (Henriques and Moustacchi, 1980; Barber et al, 2005). The role of this factor in ICL repair remains a mystery although recent genetic and biochemical evidence suggest its role might be nucleolytic (Barber et al, 2005; Li et al, 2005). PSO2 also shows epistasis with rev3 in G1 cells and, indeed, a pso2 rev3 rad4 triple mutant is no more sensitive than the rad4 single mutant (Figure 3B). Therefore, these factors collectively appear to represent the basic components of an ICL repair pathway that predominates in G1 cells.
Figure 3.
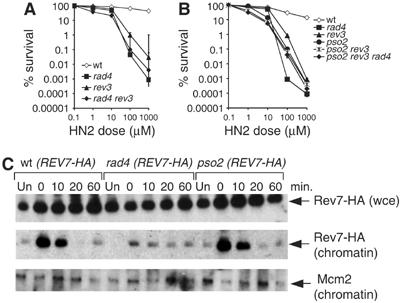
Nitrogen mustard (HN2) sensitivity of rev3, rad4 and pso2 strains, and their cognate double and triple mutants, in G1 phase. (A) HN2 sensitivity of wild-type, rad4, rev3 and double mutant rad4 rev3 cells. (B) HN2 sensitivity of wt, rad4, rev3, pso2 and their cognate double and triple mutants, pso2 rev3 and pso2 rev3 rad4. (C) Chromatin recruitment of Pol ζ in G1 phase is dependent upon ICL damage and NER (rad4), but not functional Pso2. Mcm2 is shown as a loading control since it is tightly chromatin associated in G1 cells. Mock-treated cells are marked ‘Un' and the time of treatment with 100 μM HN2 is shown in minutes.
NER activity is required to recruit Pol ζ to chromatin during the G1 repair pathway
The fundamental prediction of the models described above is that incision of ICLs by NER likely precedes TLS by Pol ζ, and the epistasis of NER genes and REV3 support this. To test this more directly, we performed chromatin recruitment assays using the method devised by Liang and Stillman (1997), whereby fractions representing whole-cell extract/protein (wce), as well as fractions containing pelleted solubilised polynucleosomes, which is chromatin-bound material, are collected. In order to detect Pol ζ in these fractions, we inserted codons for a HA-tag at the 3′-end/C-terminus of the endogenous REV7 gene. Cells were synchronised in G1, treated with HN2 and samples were taken after 0, 10, 20 and 60 min for analysis of the levels of total (wce) Rev7-HA and the proportion of this protein present in the chromatin fraction (Figure 3C). Mcm2 immunoblots were also performed on the chromatin-bound fractions as a positive control, since this protein is tightly associated with chromatin in G1 cells. In untreated, G1-phase wild-type cells, Rev7-HA is not detected in chromatin-bound fractions but is abundant in the wce (Figure 3C). In contrast, immediately after a 30-min treatment with HN2, a greatly increased chromatin-bound fraction is observed, while the wce levels of Rev7-HA are unchanged. This chromatin-bound fraction gradually declines and is not detectable at 60 min despite the fact that the wce levels remain constant for this time. This is also the point at which wild-type cells begin to exit G1 (Figure 2A). Therefore, Pol ζ is strongly associated with chromatin following ICL induction in G1 phase. In contrast, in a rad4 disruptant, Rev7-HA is not actively recruited to the chromatin fraction in G1 cells treated with HN2 (Figure 3C), despite the fact that total levels of Rev7-HA in the wce remain unaffected by the loss of NER. Together, these observations show that Pol ζ is recruited to chromatin in G1 cells bearing crosslinked DNA, and that these ICLs must first be processed by NER in order for this recruitment to occur. Using the same chromatin recruitment assay, we assessed whether Pso2 is also required for recruitment of Pol ζ. Rev7-HA is recruited to chromatin in the pso2 disruptant with kinetics indistinguishable from wild-type cells (Figure 3C). This result places the action of Pso2 in ICL repair concomitant with, or following, the recruitment of Pol ζ to ICL repair intermediates, and suggests that Pso2 is not required to generate the initial substrate for Pol ζ action at ICLs.
Monoubiquitination of PCNA is required for the Pol ζ-dependent step of ICL repair
Several recent genetic studies suggest that the activity of TLS polymerases, including Pol ζ, at replication forks stalled by DNA damage is dependent on a specific conserved lysine residue in PCNA (K164) that can be either: (i) monoubiquitinated in a Rad6–Rad18-dependent reaction to control TLS in response to DNA damage (Hoege et al, 2002; Stelter and Ulrich, 2003); (ii) polyubiquitinated with lysine-63-linked chains in a Ubc13-Mms2- and Rad5-dependent reaction which controls an error-free tolerance pathway (Hoege et al, 2002) or (iii) modified by SUMO in a Siz1-dependent reaction which appears to be required to recruit Srs2 as a step to prevent unwanted recombination and is apparently confined to S phase (Papouli et al, 2005; Pfander et al, 2005). Further, there is evidence that modification of PCNA by SUMO at a second, less well-conserved lysine (K127) acts additively with K164 modification in the SRS2 pathway (Papouli et al, 2005; Pfander et al, 2005). We wanted to investigate whether the Pol ζ activity we observe in G1 cells upon ICL induction is also dependent upon PCNA modification. From Figure 4A it is evident that the PCNA mutant pol30-K164R, which can no longer be ubiquitinated, demonstrates a high level of sensitivity to HN2 in G1 phase and that this is epistatic with rev3, implying an involvement in the same pathway. From the observation that mutation of a second lysine in PCNA, K127, did not affect the sensitivity to crosslinking agents in G1 significantly (Figure 4B), we conclude that SUMO modification of PCNA is irrelevant to the activation of Pol ζ during G1.
Figure 4.
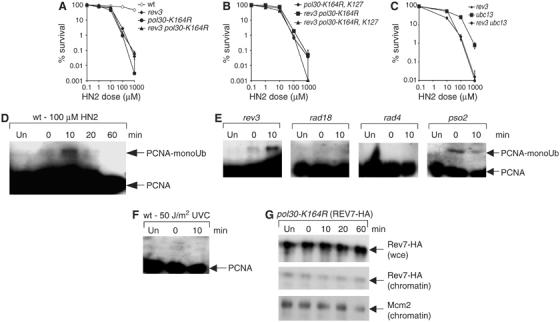
Monoubiquitination of PCNA at K164 residue is required for the response to ICL damage in G1 cells. (A, B) HN2 sensitivities of wt, rev3 and PCNA mutants in G1 cells. (C) Sensitivity of ubc13, rev3 and ubc13 rev3 cells to HN2. (D) Monoubiquitination of PCNA in wild-type cells detected using anti-PCNA antibodies after HISPCNA pull-down from G1 arrested cells mock-treated or treated with 100 μM HN2. Mock-treated cells are marked ‘Un' and the time of treatment with HN2 is shown in minutes. (E) HISPCNA pull-down of rad18, rev3, rad4 and pso2 cells treated with 100 μM HN2, or mock-treated, and probed with anti-PCNA antibody. (F) Monoubiquitination of PCNA in wild-type cells detected using anti-PCNA antibodies after HISPCNA pull-down from G1-arrested cells mock-treated or treated with 50 J/m2 UVC. (G) Recruitment of Rev7-HA to chromatin-bound fractions from pol30-K164R cells either untreated (Un) or treated with 100 μM HN2.
In order to distinguish between a contribution of PCNA mono- versus polyubiquitination in Pol ζ-dependent ICL repair, we determined the G1-phase sensitivity of an ubc13 disruptant, which is defective in PCNA polyubiquitination. We find that it shows only mild sensitivity (Figure 4C), and loss of ubc13 does not modify the sensitivity of rev3 mutants. These genetic data suggest that monoubiquitination of PCNA is the key modification influencing Pol ζ activity at ICLs in G1 cells. In order to detect PCNA monoubiquitination directly, we employed G1-arrested cells harbouring a chromosomally integrated PCNA-His-tagged construct and then HN2-treated or mock-treated them in fresh YPD media taking samples immediately post-treatment, and 10, 20 and 60 min later. From the HISPCNA pull-downs shown in Figure 4D it is clear that ICL damage immediately induces modification of PCNA in G1 cells, which peaks around 10 min after treatment and is short-lived, disappearing within 30 min of treatment. This modified form is also recognised by anti-ubiquitin antibodies, confirming that it is a ubiquitinated species (data not shown). Further support for this notion is provided by the fact that the modification is apparent in wild-type cells (Figure 4D) and siz1 cells (data not shown), but absent from HN2-treated rad18 mutants (see Figure 4E). PCNA ubiquitination in G1 is an ICL-specific reaction, since we did not observe any PCNA modification in G1 wild type cells treated with UVC (Figure 4F), but did observe the reaction following treatment with another crosslinking drug mitomycin C (data not shown). Consistent with the genetic data presented in Figure 4B, we at no point observed any bands with a size consistent with Sumo modification of PCNA, or observed any form of PCNA that could be detected using an anti-Smt3 (budding yeast Sumo) antibody. Therefore, we have shown that PCNA can, in fact, be monoubiqutinated outside of S phase, and this modification is induced by ICLs.
Nucleotide excision repair is required for activation of Pol ζ by PCNA modification
Since the data we presented in Figure 3 places NER upstream of Pol ζ recruitment, but Pso2 action concomitant or after NER, we also assessed PCNA monoubiquitination in rad4 and pso2 cells (Figure 4E). Strikingly, the NER (rad4) mutant fails to monoubiquitinate PCNA, whereas the pso2 cells were competent to do so. Interestingly, the monoubiquitinated form of HISPCNA was strongest immediately following HN2 treatment in pso2 cells in contrast to wild-type cells where this occurs after a further 10 min, perhaps indicating differences in processing of an intermediate that triggers post-translational modification of PCNA. Additional time points were examined in the rad4 strain (up to 60 min), which show that monoubiquitination event is not simply delayed (data not shown). When combined with the findings presented in Figure 3C, these data indicate that NER processing of ICLs is upstream of both PCNA ubiquitination and Pol ζ recruitment to chromatin. Since it has been suggested that the monoubiquitination initiates the recruitment of damage-tolerant polymerases (Stelter and Ulrich, 2003; Kannouche et al, 2004; Watanabe et al, 2004), we asked whether rev3 (Pol ζ)-defective cells are able to monoubiquitinate PCNA (Figure 4E). This is clearly observed, showing that active Pol ζ is not a prerequisite for PCNA modification. Finally, the recruitment of Rev7-HA to chromatin following HN2 treatment was defective in a PCNA K164 (pol30-K164R) mutant (Figure 4G), indicating that monoubiquitination specifically at this conserved residue is required for Pol ζ recruitment to ICLs. Taken together with the chromatin recruitment data presented in Figure 3, we can place NER prior to PCNA modification and Pol ζ recruitment, since Pol ζ is not required for PCNA modification but NER is. We therefore propose that NER incisions are required for the modification of PCNA, which is followed by Pol ζ recruitment to the ICL repair intermediate.
Evidence that the Pol δ subunit Pol32 acts upstream of Pol ζ recruitment at incised ICLs
The current consensus is that PCNA ubiquitination acts as a signal for the switch from a replicative polymerase, upon encountering damage, to a TLS polymerase (Friedberg et al, 2005). Since conventional NER incisions are followed by gap-filling reactions catalysed by the major replicative polymerases (Pol δ and Pol ɛ) in yeast (Wu et al, 2001), we considered it possible that, following NER incisions at ICLs, the normal gap-filling process is initiated. It could, therefore, be that gap-filling is attempted but thwarted by the crosslinked oligonucleotide still tethered to the opposing strand. This replication block could lead to a switch from the normal gap-filling polymerase to Pol ζ, which is able to copy past the tethered adduct. In order to test this hypothesis, we performed four tests. First, we evaluated the HN2 sensitivity of strains lacking a subunit of Pol δ (pol32 mutant) and a temperature-sensitive Pol ɛ (pol2-18) mutant in G1 cells. While the pol2-18 mutant was only marginally sensitive at the restrictive temperature (Figure 5A), the pol32 mutant was sensitive in all cell cycle phases, but most sensitive in G1. In fact, the level of sensitivity is similar to that of rev3 cells, and analysis indicated that pol32 and rev3 are epistatic in G1 (Figure 5B). Second, consistent with a function of Pol δ in the gap-filling reaction following the incision of ICLs, the Pol δ subunit Pol32 is recruited to chromatin only following ICL processing by the NER apparatus during G1 (Figure 5C) since it is greatly reduced in rad4 cells. The recruitment of Pol32 is normal in rev3 cells, supporting the notion that Pol32 recruitment to incised ICLs is not dependent upon Pol ζ (Figure 5C). Third, we examined the recruitment of Rev7-HA to chromatin in the absence of Pol32 (Figure 5D) in G1 cells. This was eliminated, similar to what was observed in the NER-defective strain (Figure 3C), and implies that the action of Pol δ precedes the recruitment of Pol ζ at ICLs. Finally, PCNA ubiquitination was not observed in pol32 strains following HN2 treatment in G1 (Figure 5E), again arguing that Pol δ acts upstream of PCNA modification and Pol ζ recruitment to chromatin. This suggests that a TLS event precipitated by the stalling of Pol δ might indeed occur during the processing of crosslinks.
Figure 5.
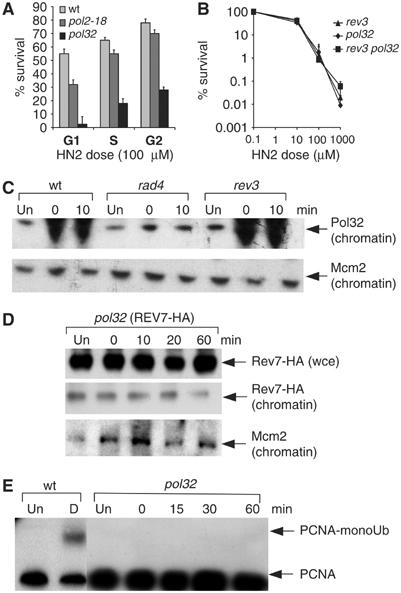
Pol δ is involved in the G1 ICL repair pathway. (A) Sensitivities of wt, pol2-18 (Pol ɛ mutant) and pol32 (Pol δ mutant) cells in G1, S and G2 phases of cell cycle in response to 100 μM HN2. (B) HN2 sensitivities of rev3, pol32 and rev3 pol32 double mutants. (C) Recruitment of Pol32 to chromatin-bound fractions from wt, rad4 and rev3 cells either untreated or treated with 100 μM HN2. (D) Recruitment of Rev7-HA to chromatin-bound fractions from pol32 cells either untreated or treated with 100 μM HN2. (E) Monoubiquitination of PCNA in wt and pol32 cells detected using anti-PCNA antibodies after HISPCNA pull-down from G1-arrested cells either mock-treated (Un) or following HN2 damaging treated (D) with 100 μM HN2 and postincubated for up to 60 min.
Discussion
The mechanisms of eukaryotic ICL repair have remained elusive, but clues are emerging from genetic and biochemical studies in a variety of systems. Here, we have explored, in greater detail, our previous hypothesis that a nonrecombinogenic ICL repair pathway operates in nondividing yeast cells (McHugh et al, 2000). Since double-strand breaks (DSBs) are not involved in this pathway, we also anticipated that elucidation of a pathway for repair of ICLs in G1 might be more facile than in cells passing through S phase, where DSBs arise as a secondary lesion, adding complexity to the interpretation of results. A model, based on the results presented here, for the predominant G1 ICL repair pathway operating in yeast is proposed in Figure 6. The features of our data, and that already in the literature, which support this model are discussed below.
Figure 6.
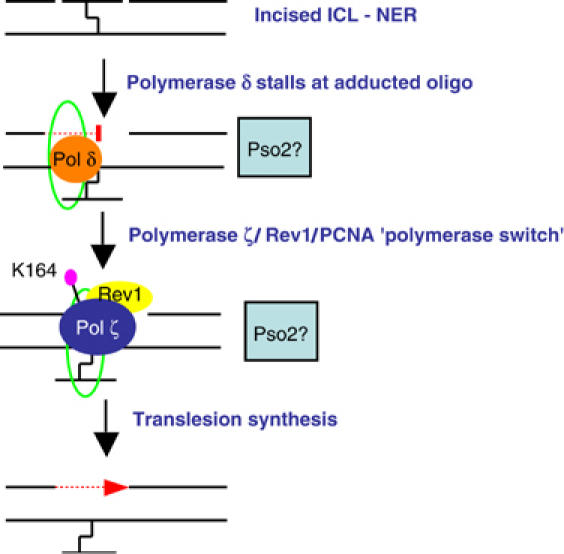
Proposed model, incorporating the roles of NER, Pol δ, PCNA modification and Pol ζ, for the repair of ICLs in G1 phase of the cell cycle.
Previous work has indicated that the first step in ICL repair in yeast involves incision by the NER apparatus (Jachymczyk et al, 1981; Miller et al, 1982). In this regard, there could be some differences between yeast and mammalian systems, since Saccharomyces cerevisiae strains mutated in each of the genes essential for NER (rad1, rad2, rad3, rad4, rad10, rad14, rad25) demonstrate essentially equivalent sensitivity to crosslinking drugs (McHugh et al, 1999), whereas mammalian cells defective for XPF-ERCC1 (RAD1-RAD10 equivalent) often demonstrate much higher sensitivity to crosslinking agents than cells deficient in other NER components (Hoy et al, 1985; Andersson et al, 1996; Damia et al, 1996; Kuraoka et al, 2000). Moreover, observations made in both cellular and biochemical assays implicate XPF and ERCC1 in a specialised ICL incision pathway (De Silva et al, 2000; Kuraoka et al, 2000; Mu et al, 2000), although it is important to note that reactions employing the ‘complete' NER apparatus have also been described (Bessho et al, 1997). It is therefore possible that subpathways of ICL repair operate in mammalian cells triggered by different initial incision events (Zheng et al, 2003). The biochemical reaction mediated by the yeast NER apparatus at ICLs remains uncharacterised, but a considerable body of work, employing sucrose gradients and Southern blotting techniques, indicates that the ICL is released by these NER incisions. It has been shown that this reaction is equally efficient in all phases of the cell cycle (Jachymczyk et al, 1981; Miller et al, 1982; Meniel et al, 1997). Our recent results employing NER-defective cells support this assertion, since these cells show a similar level of sensitivity regardless of whether they are treated in G1-, S- or G2 phase (Barber et al, 2005).
It appears reasonable to assume that a NER reaction initiates ICL repair in all cell cycle phases. In E. coli, two scenarios have been proposed for the repair events downstream of incision by NER. As outlined in the Introduction, when a sister chromatid is available, RecA-mediated transfer into the resulting gap (which has been resected and enlarged by the exonuclease activity of DNA Pol I) could provide a way of providing the substrate for a second round of incision by the NER apparatus. However, a study by Berardini et al (1999) raised the possibility of a nonrecombinogenic ICL repair pathway in bacteria. They demonstrated that repair of a plasmid-borne site-specific HN2 crosslink is repaired by an NER and DNA Pol II-dependent pathway. This appears to be distinct from the NER–RecA pathway. Supporting a combined pathway consisting of incision by NER followed by Pol II TLS, they found that an uvrA Δpolb double mutant is epistatic to a uvrA mutant for HN2 sensitivity.
The work presented here indicates that an analogous pathway is operational in eukaryotes during G1. Our study establishes the order of events in this repair pathway, placing ICL incision by NER prior to PCNA activation that, in turn, precedes TLS by Pol ζ. We can surmise this from: (i) the epistasis of rev3 and NER mutants (Figure 3), as well rev3 with PCNA K164R mutants (Figure 4) in G1 cells treated with HN2, placing these factors within a single repair pathway; (ii) the requirement for an intact NER apparatus for PCNA monoubiquitination in HN2-damaged G1 cells (Figure 4), placing NER upstream of Rad18-dependent PCNA modification; (iii) the requirement for NER and an intact PCNA K164 residue for Pol ζ recruitment to chromatin (Figures 3C and 4G, respectively), placing NER and PCNA monoubiquitination upstream of TLS and (iv) the lack of a requirement for Pol ζ for PCNA ubiquitination, suggesting that PCNA modification occurs prior to TLS. Furthermore, the Pol ζ-dependent ICL repair pathway is apparently most relevant during the G1 phase of the cell cycle since entry of cells into S phase is associated with an increase in resistance to HN2 in Pol ζ-deficient cells, but not NER mutants. We have demonstrated this through use of the cdc4-1 and cdc4-1 sic1 mutations, which allow direct comparison of G1 cells and those forced to enter S phase precociously under the same conditions (Schwob et al, 1994; Verma et al, 1997).
A G1-phase TLS-requiring pathway superficially runs contrary to the known primary role for TLS polymerases, which are generally credited with completing replication past unrepaired lesions during or after S phase in a manner tightly linked to the replicative apparatus (Kannouche et al, 2003; Watanabe et al, 2004; Friedberg et al, 2005; Prakash et al, 2005). Indeed, this has been demonstrated compellingly for Pol η in human cells in response to DNA damage in vivo (Kannouche et al, 2003; Watanabe et al, 2004). We have found that for UV-induced damage, which afflicts only one DNA strand, this is also likely the case for yeast Pol ζ since rev3 cells are more sensitive in S phase than G1. The situation with ICLs is, however, conceptually different in that TLS action is part of a multistep repair/tolerance pathway and not a ‘classical' postreplication tolerance mechanism. In this regard the involvement of PCNA is striking, and its modification by ubiquitin in cells outside of S phase is a novel observation. It has been proposed that for the tolerance of damage during S-phase monoubiquitination of PCNA acts as a molecular switch, exchanging the replicative polymerase for a TLS polymerase when damage is encountered (Stelter and Ulrich, 2003; Kannouche et al, 2004; Watanabe et al, 2004). We therefore explored the possibility that, following incision of the ICL by NER, normal gap-filling synthesis is attempted; however, when the polymerase involved (Pol δ) encounters the still tethered crosslinked oligonucleotide, it sends a signal equivalent to that occurring when replication forks stall. This signals to the Rad6 pathway and ultimately results in PCNA monoubiquitination at K164 precipitating exchange of Pol δ for Pol ζ, possibly in cooperation with Rev1. Our data shows that (i) recruitment of the Pol δ subunit Pol32 to chromatin is NER-dependent, but Pol ζ independent; (ii) Pol32 is required to recruit Rev7-HA (Pol ζ) to chromatin following ICL induction in G1 and (iii) PCNA fails to be monoubiquitinated in pol32 mutants. Taken together, these results suggest that stalled Pol δ is a candidate for triggering PCNA monoubiquitination and subsequent Pol ζ recruitment at incised ICL repair intermediates. Further, our data are consistent with two recent genetic studies that indicate Pol32 plays a cooperative role in TLS catalysed by Pol ζ (Gibbs et al, 2005; Minesinger and Jinks-Robertson, 2005). In addition, PCNA appears to stimulate the TLS activity of Pol ζ (Garg et al, 2005), and although PCNA monoubiquitination does not further increase Pol ζ activity it is capable enhancing Rev1 action, thereby stimulating TLS (Garg and Burgers, 2005).
Our results place the Pso2/Snm1 repair factor at a post-ICL incision step and suggest that it might act concomitantly with or after Pol ζ. The epistasis of pso2 with rev3 in G1 cells treated with crosslinking drugs mirrors that previously reported in stationary phase cells (Henriques and Moustacchi, 1980). Recent genetic and biochemical results suggest that Pso2 might be a nuclease (Barber et al, 2005; Li et al, 2005), and plays an overlapping role with Exo1 in the processing of ICLs and spontaneous damage during S phase (Barber et al, 2005). Although the relevant cellular substrate for Pso2 remains a mystery, our results suggest that structures postulated to arise relatively late during ICL repair pathway would be worth examining in biochemical assays.
We hope that it will be eventually possible to reconstitute the individual steps of this reaction and elucidate the fine details of a eukaryotic ICL repair pathway for the first time. The components of this pathway are well conserved in higher eukaryotes and we suggest that this pathway, or a similar one, is likely to operate in mammals. Recent reports consistent with the involvement of vertebrate and mammalian Pol ζ and other TLS polymerases in ICL repair support this assertion (Wang et al, 2001; Sonoda et al, 2003; Zheng et al, 2003; Zander and Bemark, 2004; Okada et al, 2005).
Materials and methods
Strains and media
Strains used in this study are listed in Supplementary Table I. Deletion strains were constructed using PCR-generated disruption cassettes derived from plasmids pFA6a-KanMX6 (conferring G418 resistance) and pAG32C (conferring hygromycin resistance), respectively, as described by (Longtine et al, 1998). The endogenous REV7 locus of strain DBY747 was tagged at its C-terminal with a 3xHA epitope-tag using cassette conferring G418 resistance generated by PCR from plasmids pFA6a-3HA-KanMx6, as described in Longtine et al (1998). The strain bearing REV7-HA behaves like wild type both in regard to its sensitivity to DNA-damaging agents and also induced mutagenesis. Since rev3 and rev7 mutants phenocopy one another in the ICL repair pathway, genetic manipulations that could not be readily achieved at the REV3 locus in some strain backgrounds were performed by modifying REV7 in certain cases. For detection of modified PCNA, a His-tagged allele of POL30 was integrated into the LEU2 locus using YIp128-P30-His6-POL30, and the endogenous gene was deleted using a URA3 cassette (Stelter and Ulrich, 2003).
Cell synchronization and sensitivity assays
Cells were synchronized in G1 by treatment of exponential YPD cultures with 5 μg/ml of α-mating factor for 2 h. Several strains are of α-mating type, these were synchronised using a mating factor (Sigma) instead of α-factor. After microscopic confirmation of growth arrest, cells were washed with PBS and treated with indicated doses of HN2 for 30 min at 30°C unless indicated otherwise, and appropriate dilutions plated out for cell survival. S-phase cell populations were generated by synchronising in G1, washing off α-mating factor followed by release into YPD. Cells were monitored microscopically for bud formation. When 50% of cells exhibited small buds (30 min from release), they were treated with the indicated dose of HN2, washed and plated. Exponential cultures of cells were synchronised into G2 cells with 10 μg/ml nocodazole treatment for 1.5 h and assessed by microscopic observation. All cell cycle synchronization protocols were also confirmed by a flow-cytometric (FACS) analysis using a Becton-Dickinson FACScan.
Chromatin binding assay and Western blotting of whole-cell extracts
Protein chromatin binding assays were performed according to Liang and Stillman (1997) with the following minor modifications. G1-arrested cells (50 ml at 2 × 108 cells/ml) were used for each tagged strain at each time point. Zymolase (ICN) at 1 mg/ml was used to generate spheroplasts at 30°C for 40 min, and digestion checked by lysis with 10% SDS microscopically. Tightly bound chromatin fractions and wces were run on 4–12% NuSieve Gradient gel (Invitrogen) and the HA epitope detected using a Mouse Anti-HA monoclonal antibody (clone 12CA5) from Roche. Mcm2 was detected using an affinity-purified goat polyclonal antibody raised to the N-terminus of the protein (Santa Cruz Biotech). Immunoaffinity-purified polyclonal antibodies to Pol 32 were a kind gift of Dr Peter Burgers (Gerik et al, 1998).
PCNA modification assays
These were performed as described previously (Papouli et al, 2005).
Supplementary Material
Supplementary Table 1
Acknowledgments
We are grateful to Ian Hickson and Leonard Wu for helpful comments on the manuscript, and all members of the CR-UK DNA Damage and Repair and Genome Integrity Research Groups for help throughout this work. We thank Wei Xiao, Hannah Klein, Zhigang Wang and Maria-Pia Longhese for kindly sharing yeast strains with us, and Peter Burgers for the kind gift of anti-Pol32 antibodies. This work was supported by Cancer Research UK.
References
- Andersson BS, Sadeghi T, Siciliano MJ, Legerski R, Murray D (1996) Nucleotide excision repair genes as determinants of cellular sensitivity to cyclophosphamide analogs. Cancer Chemother Pharmacol 38: 406–416 [DOI] [PubMed] [Google Scholar]
- Barber LJ, Ward TA, Hartley JA, McHugh PJ (2005) DNA interstrand cross-link repair in the Saccharomyces cerevisiae cell cycle: overlapping roles for PSO2 (SNM1) with MutS factors and EXO1 during S phase. Mol Cell Biol 25: 2297–2309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berardini M, Foster PL, Loechler EL (1999) DNA polymerase II (polB) is involved in a new DNA repair pathway for DNA interstrand cross-links in Escherichia coli. J Bacteriol 181: 2878–2882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bessho T, Mu D, Sancar A (1997) Initiation of DNA interstrand cross-link repair in humans: the nucleotide excision repair system makes dual incisions 5′ to the cross-linked base and removes a 22- to 28-nucleotide-long damage-free strand. Mol Cell Biol 17: 6822–6830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole RS, Sinden RR (1975) Repair of cross-linked DNA in Escherichia coli. Basic Life Sci 5B: 487–495 [DOI] [PubMed] [Google Scholar]
- Damia G, Imperatori L, Stefanini M, D'Incalci M (1996) Sensitivity of CHO mutant cell lines with specific defects in nucleotide excision repair to different anti-cancer agents. Int J Cancer 66: 779–783 [DOI] [PubMed] [Google Scholar]
- De Silva IU, McHugh PJ, Clingen PH, Hartley JA (2000) Defining the roles of nucleotide excision repair and recombination in the repair of DNA interstrand cross-links in mammalian cells. Mol Cell Biol 20: 7980–7990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dronkert ML, Kanaar R (2001) Repair of DNA interstrand cross-links. Mutat Res 486: 217–247 [DOI] [PubMed] [Google Scholar]
- Friedberg EC, Lehmann AR, Fuchs RP (2005) Trading places: how do DNA polymerases switch during translesion DNA synthesis? Mol Cell 18: 499–505 [DOI] [PubMed] [Google Scholar]
- Garg P, Burgers PM (2005) Ubiquitinated proliferating cell nuclear antigen activates translesion DNA polymerases eta and REV1. Proc Natl Acad Sci USA 102: 18361–18366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg P, Stith CM, Majka J, Burgers PM (2005) Proliferating cell nuclear antigen promotes translesion synthesis by DNA polymerase zeta. J Biol Chem 280: 23446–23450 [DOI] [PubMed] [Google Scholar]
- Gerik KJ, Li X, Pautz A, Burgers PM (1998) Characterization of the two small subunits of Saccharomyces cerevisiae DNA polymerase delta. J Biol Chem 273: 19747–19755 [DOI] [PubMed] [Google Scholar]
- Gibbs PE, McDonald J, Woodgate R, Lawrence CW (2005) The relative roles in vivo of Saccharomyces cerevisiae Pol eta, Pol zeta, Rev1 protein and Pol32 in the bypass and mutation induction of an abasic site, T-T (6-4) photoadduct and T-T cis-syn cyclobutane dimer. Genetics 169: 575–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs PE, McGregor WG, Maher VM, Nisson P, Lawrence CW (1998) A human homolog of the Saccharomyces cerevisiae REV3 gene, which encodes the catalytic subunit of DNA polymerase zeta. Proc Natl Acad Sci USA 95: 6876–6880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossmann KF, Ward AM, Matkovic ME, Folias AE, Moses RE (2001) S. cerevisiae has three pathways for DNA interstrand crosslink repair. Mutat Res 487: 73–83 [DOI] [PubMed] [Google Scholar]
- Henriques JA, Moustacchi E (1980) Isolation and characterization of pso mutants sensitive to photo-addition of psoralen derivatives in Saccharomyces cerevisiae. Genetics 95: 273–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoege C, Pfander B, Moldovan GL, Pyrowolakis G, Jentsch S (2002) RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature 419: 135–141 [DOI] [PubMed] [Google Scholar]
- Hoy CA, Thompson LH, Mooney CL, Salazar EP (1985) Defective DNA cross-link removal in Chinese hamster cell mutants hypersensitive to bifunctional alkylating agents. Cancer Res 45: 1737–1743 [PubMed] [Google Scholar]
- Jachymczyk WJ, von Borstel RC, Mowat MR, Hastings PJ (1981) Repair of interstrand cross-links in DNA of Saccharomyces cerevisiae requires two systems for DNA repair: the RAD3 system and the RAD51 system. Mol Gen Genet 182: 196–205 [DOI] [PubMed] [Google Scholar]
- Johnson RE, Prakash S, Prakash L (1999) Efficient bypass of a thymine–thymine dimer by yeast DNA polymerase, Poleta. Science 283: 1001–1004 [DOI] [PubMed] [Google Scholar]
- Johnson RE, Washington MT, Haracska L, Prakash S, Prakash L (2000) Eukaryotic polymerases iota and zeta act sequentially to bypass DNA lesions. Nature 406: 1015–1019 [DOI] [PubMed] [Google Scholar]
- Kannouche P, Fernandez de Henestrosa AR, Coull B, Vidal AE, Gray C, Zicha D, Woodgate R, Lehmann AR (2003) Localization of DNA polymerases eta and iota to the replication machinery is tightly co-ordinated in human cells. EMBO J 22: 1223–1233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannouche PL, Wing J, Lehmann AR (2004) Interaction of human DNA polymerase eta with monoubiquitinated PCNA: a possible mechanism for the polymerase switch in response to DNA damage. Mol Cell 14: 491–500 [DOI] [PubMed] [Google Scholar]
- Kuraoka I, Kobertz WR, Ariza RR, Biggerstaff M, Essigmann JM, Wood RD (2000) Repair of an interstrand DNA cross-link initiated by ERCC1-XPF repair/recombination nuclease. J Biol Chem 275: 26632–26636 [DOI] [PubMed] [Google Scholar]
- Lawrence CW (2002) Cellular roles of DNA polymerase zeta and Rev1 protein. DNA Repair (Amst) 1: 425–435 [DOI] [PubMed] [Google Scholar]
- Lawrence CW, Christensen R (1976) UV mutagenesis in radiation-sensitive strains of yeast. Genetics 82: 207–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence CW, Das G, Christensen RB (1985) REV7, a new gene concerned with UV mutagenesis in yeast. Mol Gen Genet 200: 80–85 [DOI] [PubMed] [Google Scholar]
- Lawrence CW, Gibbs PE, Murante RS, Wang XD, Li Z, McManus TP, McGregor WG, Nelson JR, Hinkle DC, Maher VM (2000) Roles of DNA polymerase zeta and Rev1 protein in eukaryotic mutagenesis and translesion replication. Cold Spring Harb Symp Quant Biol 65: 61–69 [DOI] [PubMed] [Google Scholar]
- Lawrence CW, Hinkle DC (1996) DNA polymerase zeta and the control of DNA damage induced mutagenesis in eukaryotes. Cancer Surv 28: 21–31 [PubMed] [Google Scholar]
- Lemontt JF (1971) Mutants of yeast defective in mutation induced by ultraviolet light. Genetics 68: 21–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Hejna J, Moses RE (2005) The yeast Snm1 protein is a DNA 5′-exonuclease. DNA Repair (Amst) 4: 163–170 [DOI] [PubMed] [Google Scholar]
- Liang C, Stillman B (1997) Persistent initiation of DNA replication and chromatin-bound MCM proteins during the cell cycle in cdc6 mutants. Genes Dev 11: 3375–3386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longtine MS, McKenzie A III, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR (1998) Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14: 953–961 [DOI] [PubMed] [Google Scholar]
- McHugh PJ, Gill RD, Waters R, Hartley JA (1999) Excision repair of nitrogen mustard-DNA adducts in Saccharomyces cerevisiae. Nucleic Acids Res 27: 3259–3266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHugh PJ, Sones WR, Hartley JA (2000) Repair of intermediate structures produced at DNA interstrand cross-links in Saccharomyces cerevisiae. Mol Cell Biol 20: 3425–3433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHugh PJ, Spanswick VJ, Hartley JA (2001) Repair of DNA interstrand crosslinks: molecular mechanisms and clinical relevance. Lancet Oncol 2: 483–490 [DOI] [PubMed] [Google Scholar]
- Meniel V, Magana-Schwencke N, Averbeck D, Waters R (1997) Preferential incision of interstrand crosslinks induced by 8-methoxypsoralen plus UVA in yeast during the cell cycle. Mutat Res 384: 23–32 [DOI] [PubMed] [Google Scholar]
- Miller RD, Prakash L, Prakash S (1982) Genetic control of excision of Saccharomyces cerevisiae interstrand DNA cross-links induced by psoralen plus near-UV light. Mol Cell Biol 2: 939–948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minesinger BK, Jinks-Robertson S (2005) Roles of RAD6 epistasis group members in spontaneous Pol ζ-dependent translesion synthesis in Saccharomyces cerevisiae. Genetics 169: 1939–1955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu D, Bessho T, Nechev LV, Chen DJ, Harris TM, Hearst JE, Sancar A (2000) DNA interstrand cross-links induce futile repair synthesis in mammalian cell extracts. Mol Cell Biol 20: 2446–2454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson JR, Lawrence CW, Hinkle DC (1996) Thymine–thymine dimer bypass by yeast DNA polymerase zeta. Science 272: 1646–1649 [DOI] [PubMed] [Google Scholar]
- Niedernhofer LJ, Odijk H, Budzowska M, van Drunen E, Maas A, Theil AF, de Wit J, Jaspers NG, Beverloo HB, Hoeijmakers JH, Kanaar R (2004) The structure-specific endonuclease Ercc1-Xpf is required to resolve DNA interstrand cross-link-induced double-strand breaks. Mol Cell Biol 24: 5776–5787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada T, Sonoda E, Yoshimura M, Kawano Y, Saya H, Kohzaki M, Takeda S (2005) Multiple roles of vertebrate REV genes in DNA repair and recombination. Mol Cell Biol 25: 6103–6111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papouli E, Chen S, Davies AA, Huttner D, Krejci L, Sung P, Ulrich HD (2005) Crosstalk between SUMO and ubiquitin on PCNA is mediated by recruitment of the helicase Srs2p. Mol Cell 19: 123–133 [DOI] [PubMed] [Google Scholar]
- Pfander B, Moldovan GL, Sacher M, Hoege C, Jentsch S (2005) SUMO-modified PCNA recruits Srs2 to prevent recombination during S phase. Nature 436: 428–433 [DOI] [PubMed] [Google Scholar]
- Prakash S, Johnson RE, Prakash L (2005) Eukaryotic translesion synthesis DNA polymerases: specificity of structure and function. Annu Rev Biochem 74: 317–353 [DOI] [PubMed] [Google Scholar]
- Prakash S, Prakash L (2002) Translesion DNA synthesis in eukaryotes: a one- or two-polymerase affair. Genes Dev 16: 1872–1883 [DOI] [PubMed] [Google Scholar]
- Ruhland A, Brendel M (1979) Mutagenesis by cytostatic alkylating agents in yeast strains of differing repair capacities. Genetics 92: 83–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saffran WA, Ahmed S, Bellevue S, Pereira G, Patrick T, Sanchez W, Thomas S, Alberti M, Hearst JE (2004) DNA repair defects channel interstrand DNA cross-links into alternate recombinational and error-prone repair pathways. J Biol Chem 279: 36462–36469 [DOI] [PubMed] [Google Scholar]
- Schwob E, Bohm T, Mendenhall MD, Nasmyth K (1994) The B-type cyclin kinase inhibitor p40SIC1 controls the G1 to S transition in S cerevisiae. Cell 79: 233–244 [DOI] [PubMed] [Google Scholar]
- Sladek FM, Munn MM, Rupp WD, Howard-Flanders P (1989) In vitro repair of psoralen-DNA cross-links by RecA, UvrABC, and the 5′-exonuclease of DNA polymerase I. J Biol Chem 264: 6755–6765 [PubMed] [Google Scholar]
- Sonoda E, Okada T, Zhao GY, Tateishi S, Araki K, Yamaizumi M, Yagi T, Verkaik NS, van Gent DC, Takata M, Takeda S (2003) Multiple roles of Rev3, the catalytic subunit of pol zeta in maintaining genome stability in vertebrates. EMBO J 22: 3188–3197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stelter P, Ulrich HD (2003) Control of spontaneous and damage-induced mutagenesis by SUMO and ubiquitin conjugation. Nature 425: 188–191 [DOI] [PubMed] [Google Scholar]
- Van Houten B, Gamper H, Holbrook SR, Hearst JE, Sancar A (1986) Action mechanism of ABC excision nuclease on a DNA substrate containing a psoralen crosslink at a defined position. Proc Natl Acad Sci USA 83: 8077–8081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma R, Annan RS, Huddleston MJ, Carr SA, Reynard G, Deshaies RJ (1997) Phosphorylation of Sic1p by G1 Cdk required for its degradation and entry into S phase. Science 278: 455–460 [DOI] [PubMed] [Google Scholar]
- Wang X, Peterson CA, Zheng H, Nairn RS, Legerski RJ, Li L (2001) Involvement of nucleotide excision repair in a recombination-independent and error-prone pathway of DNA interstrand cross-link repair. Mol Cell Biol 21: 713–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe K, Tateishi S, Kawasuji M, Tsurimoto T, Inoue H, Yamaizumi M (2004) Rad18 guides pol eta to replication stalling sites through physical interaction and PCNA monoubiquitination. EMBO J 23: 3886–3896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Guo D, Yuan F, Wang Z (2001) Accessibility of DNA polymerases to repair synthesis during nucleotide excision repair in yeast cell-free extracts. Nucleic Acids Res 29: 3123–3130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao W, Lechler T, Chow BL, Fontanie T, Agustus M, Carter KC, Wei YF (1998) Identification, chromosomal mapping and tissue-specific expression of hREV3 encoding a putative human DNA polymerase zeta. Carcinogenesis 19: 945–949 [DOI] [PubMed] [Google Scholar]
- Zander L, Bemark M (2004) Immortalized mouse cell lines that lack a functional Rev3 gene are hypersensitive to UV irradiation and cisplatin treatment. DNA Repair (Amst) 3: 743–752 [DOI] [PubMed] [Google Scholar]
- Zheng H, Wang X, Warren AJ, Legerski RJ, Nairn RS, Hamilton JW, Li L (2003) Nucleotide excision repair- and polymerase eta-mediated error-prone removal of mitomycin C interstrand cross-links. Mol Cell Biol 23: 754–761 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1