

Abstract
A screening for intracellular interactors of the p75 neurotrophin receptor (p75NTR) identified brain-expressed X-linked 1 (Bex1), a small adaptor-like protein of unknown function. Bex1 levels oscillated during the cell cycle, and preventing the normal cycling and downregulation of Bex1 in PC12 cells sustained cell proliferation under conditions of growth arrest, and inhibited neuronal differentiation in response to nerve growth factor (NGF). Neuronal differentiation of precursors isolated from the brain subventricular zone was also reduced by ectopic Bex1. In PC12 cells, Bex1 overexpression inhibited the induction of NF-κB activity by NGF without affecting activation of Erk1/2 and AKT, while Bex1 knockdown accelerated neuronal differentiation and potentiated NF-κB activity in response to NGF. Bex1 competed with RIP2 for binding to the p75NTR intracellular domain, and elevating RIP2 levels restored the ability of cells overexpressing Bex1 to differentiate in response to NGF. Together, these data establish Bex1 as a novel link between neurotrophin signaling, the cell cycle, and neuronal differentiation, and suggest that Bex1 may function by coordinating internal cellular states with the ability of cells to respond to external signals.
Keywords: cell differentiation, growth arrest, NGF, TrkA, PC12 cells
Introduction
Nerve growth factor (NGF) and other members of the neurotrophin family mediate survival, growth and differentiation of neuronal and glial cells by binding to two different types of cell surface receptors, the Trk tyrosine kinases—TrkA, TrkB and TrkC—and the p75 neurotrophin receptor (p75NTR). p75NTR resembles other members of the tumor necrosis factor receptor superfamily in the organization of its extracellular domain and in the presence of a small globular domain in the intracellular region, the so-called death domain (Liepinsh et al, 1997). p75NTR signaling can contribute to neurotrophin-mediated survival, differentiation and neurite outgrowth in a variety of neuronal subpopulations (Roux and Barker, 2002). On the other hand, p75NTR can also mediate cell death by neurotrophins, proneurotrophins and various amyloid peptides, as well as inhibit axonal growth and regeneration in its capacity of signaling receptor for myelin-inhibitory components (Teng and Hempstead, 2004). Thus, the physiological consequences of p75NTR signaling depend on the cellular context and the nature of the activating ligands (Bronfman and Fainzilber, 2004). Mice lacking p75NTR have a multifaceted phenotype, characterized by both positive and negative effects (Lee et al, 1992; von Schack et al, 2001; Naumann et al, 2002), reflecting the complexity of p75NTR signaling.
Trk receptors transmit intracellular signals through several of the canonical pathways activated by other receptor tyrosine kinases, including the Ras/MAP kinase and phosphatidylinositol-3 kinase (PI3K)/AKT signaling pathways (Kaplan and Miller, 2000; Huang and Reichardt, 2003). In contrast, the signaling mechanisms used by p75NTR have remained elusive. Lacking intrinsic catalytic activity, p75NTR signaling is dependent on the ability of this receptor to interact with components of intracellular signaling pathways. Several different p75NTR interacting molecules, with and without catalytic activity, have been identified to date (Gentry et al, 2004). Noncatalytic interactors include a series of scaffolding- and adaptor-like molecules, such as caveolin-1 (Bilderback et al, 1997), Bex3/NADE (Mukai et al, 2000) and TRAF6 (Khursigara et al, 1999; Ye et al, 1999); larger proteins containing zinc-finger domains with some degree of nuclear localization, such as NRIF1/2 (Casademunt et al, 1999) and SC-1 (Chittka and Chao, 1999); and members of the MAGE homology domain family, such as NRAGE (Salehi et al, 2000) and necdin (Tcherpakov et al, 2002), with proposed roles in the regulation of apoptosis. p75NTR interactors with catalytic activity include serine–threonine kinases involved in interleukin and NF-κB signaling, such as IRAK (Mamidipudi et al, 2002) and RIP2 (Khursigara et al, 2001); a protein tyrosine phosphatase (FAP-1) (Irie et al, 1999); and the small GTPase RhoA (Yamashita et al, 1999). How these p75NTR-interacting proteins connect to downstream signaling pathways and cellular responses is less clear, however. Some of the principal downstream events characterized in p75NTR signaling include ceramide production (Dobrowsky et al, 1994), and activation of the transcription factor NF-κB (Carter et al, 1996) and the c-Jun kinases JNK1–3 (Casaccia-Bonnefil et al, 1996; Friedman, 2000; Harrington et al, 2002).
The rat pheochromocytoma cell line PC12 expresses both TrkA and p75NTR and has become a principal model to study NGF action. In response to NGF, PC12 cells exit the cell cycle, extend neurites and differentiate into a neuronal cell type that resembles sympathetic neurons (Greene and Tischler, 1976). An extensive body of evidence supports the fundamental role of TrkA signaling in PC12 cell differentiation in response to NGF (Huang and Reichardt, 2003). More recently, the contribution of p75NTR to this process has also begun to be appreciated (Foehr et al, 2000; Wooten et al, 2001; Hosomi et al, 2003; Sole et al, 2004). NGF signaling in PC12 cells is regulated by the cell cycle, leading to cell differentiation during G1 but to cell cycle progression during other cycle phases (Rudkin et al, 1989). NGF causes an accumulation of cells in the G1 phase as a result of a block in the transition from G1 to S phase (van Grunsven et al, 1996). Interestingly, NGF receptors present a cyclical expression at the extracellular surface of exponentially growing PC12 cells, with high levels of TrkA during M and early G1, and of p75NTR during late G1, S and G2 (Urdiales et al, 1998). The molecular mechanisms linking NGF receptor signaling to the cell cycle, however, remain poorly understood (Lopez-Sanchez and Frade, 2002).
In the present study, we set out to identify novel intracellular interactors of p75NTR using a functional screening method based on T7 phage display, and identified the brain-expressed X-linked 1 (Bex1) protein as a new intracellular interactor of p75NTR. Our results indicate that Bex1 may represent a distinct class of upstream intracellular modulators of neurotrophin receptors, linking neurotrophin signaling to the cell cycle.
Results
Identification of Bex1 as an intracellular interactor of p75NTR
A C-terminal fragment of the rat ortholog of a human protein originally named Bex1 (Brown and Kay, 1999) was isolated in a T7 phage display screen for interacting partners of the intracellular domain of p75NTR (Figure 1A). Up to six paralog Bex genes have been identified in rodents and humans (Alvarez et al, 2005; Koo et al, 2005). Bex1 and Bex2 are very similar in protein sequence (87% identity), while Bex3/NADE—a previously characterized interactor of the p75NTR death domain (Mukai et al, 2000)—is only 30% identical to either Bex1 or Bex2 and thus represents a more divergent member of this family (Figure 1A). Despite their low similarity, the main region in Bex3/NADE implicated in its interaction with p75NTR was included in the Bex1 cDNA fragment isolated by phage display (Figure 1A), suggesting the existence of a conserved p75NTR-binding interface among Bex proteins.
Figure 1.
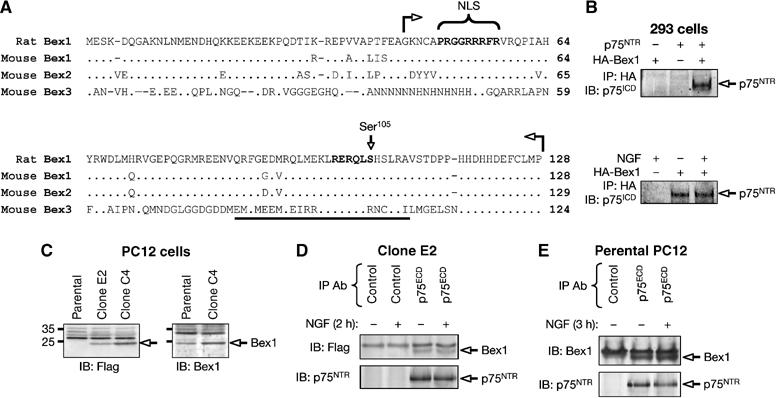
Identification of Bex1 as a novel intracellular interactor of p75NTR. (A) Alignment of primary amino-acid sequences of rat Bex1 and mouse Bex1, Bex2 and Bex3 performed with ClustalX (Jeanmougin et al, 1998). The region of rat Bex1 identified by T7 phage display is indicated between arrows. The p75NTR-binding site in Bex3/NADE is underlined (Mukai et al, 2000). NLS and Ser105 are indicated. (B) Interaction between HA-tagged Bex1 (HA-Bex1) and p75NTR overexpressed in 293 cells analyzed by IP with anti-HA antibodies and Western IB with antibodies against the intracellular domain of p75NTR (p75ICD). In the lower panel, transfected cells were treated for 12 h with NGF prior to lysis and IP as indicated. (C) IB of cell lysates (50 μg protein) from parental PC12 cells and clones C4 and E2 overexpressing Flag-Bex1 using anti-Flag (left) or anti-Bex1 antibodies. (D) Interaction between endogenous p75NTR and Flag-Bex1 in clone E2 in the presence or absence of NGF analyzed by IP with control antibodies or antibodies against the extracellular domain of p75NTR (p75ECD), followed by IB with anti-Flag antibodies. (E) Interaction between endogenous Bex1 and p75NTR in parental PC12 cells analyzed by IP with control or anti-p75ECD antibodies, followed by IB with anti-Bex1 antibodies.
p75NTR could be specifically co-immunoprecipitated with hemagglutinin (HA)-tagged Bex1 in transfected HEK293 T (293) cells (Figure 1B). Flag-tagged Bex1 (Flag-Bex1) could also be co-immunoprecipitated with endogenous p75NTR in two PC12 cell clones—termed E2 and C4—generated by stable transfection (Figure 1C and D). Using anti-Bex1 antibodies, endogenous Bex1 could be specifically co-immunoprecipitated with endogenous p75NTR in parental PC12 cells (Figure 1E), indicating that the two proteins are able to interact when expressed at physiological levels. Association between endogenous Bex1 and p75NTR was also observed in lysates of primary Schwann cells (data not shown). NGF treatment did not affect the interaction between Bex1 and p75NTR—either transfected or endogenous (Figure 1B, D and E).
Overlapping expression of Bex1 and p75NTR mRNAs in developing mesenchyme and vascular and nervous tissues
Expression of Bex1, Bex2 and Bex3 mRNAs could be detected by RT–PCR in PC12 cells and primary cultures of embryonic day (E) 14.5 rat dorsal root ganglion (DRG) cells (Figure 2A). Only Bex1 and Bex3 mRNAs could be found in Schwann cells isolated from the newborn rat sciatic nerve and in E17.5 hippocampal cultures (Figure 2A). mRNAs encoding all three Bex isoforms could also be detected in newborn (P1) cerebral cortex, hippocampus and olfactory bulb (Figure 2B). Bex1 mRNA expression could be localized more precisely by in situ hybridization within specific regions of the developing rat embryo (Figure 2C). At E13.5, Bex1 mRNA was widely expressed throughout the developing nervous system and in vascular and mesenchymal structures. In particular, prominent Bex1 mRNA expression could be observed in the somitic mesenchyme, heart, aorta, dorsal root ganglia, sympathetic ganglia and the lateral motor columns of the developing spinal cord, all tissues known to express high levels of p75NTR mRNA at this stage (Ernfors et al, 1988; Huber and Chao, 1995; Cotrina et al, 2000; von Schack et al, 2001). At later stages of brain development, Bex1 mRNA was also observed in structures known to express p75NTR mRNA, including the olfactory bulb, striatum, thalamus, cerebral cortex and hippocampus (Figure 2C). In the cortex, Bex1 mRNA was detected in the subplate and cortical plate, where cells expressing p75NTR mRNA are known to reside at this stage (DeFreitas et al, 2001; Kendall et al, 2003).
Figure 2.
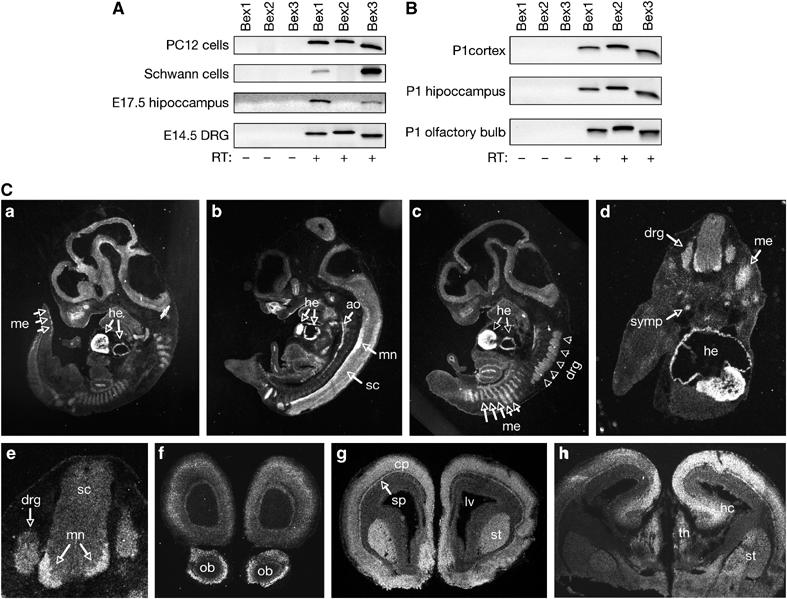
Overlapping expression of Bex1 and p75NTR mRNAs in developing mesenchyme and vascular and nervous tissues. (A) RT–PCR analysis of Bex1, Bex2 and Bex3 mRNA expression in cultures of PC12 cells, Schwann cells, E17.5 hippocampus and E14.5 dorsal root ganglia (DRG). RT, reverse transcriptase. (B) RT–PCR analysis of Bex1, Bex2 and Bex3 mRNA expression in P1 cerebral cortex, hippocampus and olfactory bulb. (C) Bex1 mRNA expression analyzed by in situ hybridization in sagittal (a–c) and transverse (d, e) sections of E13.5 rat embryos, and in coronal sections of E19.5 rat brain (f–h). me, mesenchyme; he, heart; ao, aorta; sc, spinal cord; mn, motorneurons; drg, dorsal root ganglia; symp, sympathetic ganglia; ob, olfactory bulb; st, striatum; cp, cortical plate; sp, subplate; lv, lateral ventricle; hc hippocampus; th, thalamus.
Dynamic nucleocytoplasmic trafficking of Bex1 in response to NGF in p75NTR-expressing cells
The subcellular localization of Bex1 was visualized by introducing a Bex1 construct tagged with a green fluorescent protein moiety (GFP-Bex1) into 293, PC12 and Schwann cells. GFP fluorescence was present in both nucleus and cytoplasm of all these cell types, with 80–85% of cells displaying brighter fluorescence in the nucleus, and 15–20% in the cytoplasm (Figure 3A, B and G). A putative nuclear localization signal (NLS) was identified in the N-terminal half of Bex1 (Figure 1A). Mutation of the Arg53–Arg54–Arg55 triplet in this motif into Ala–Ala–Ala (GFP-Bex1mutNLS) excluded GFP-Bex1 from the nucleus of 293 cells (Figure 3A), indicating that Bex1 harbors a functional NLS. In cells cotransfected with Bex1 and p75NTR, NGF produced a redistribution of GFP-Bex1 from the nucleus to the cytoplasm (Figure 3A), suggesting that NGF may be able to regulate the nuclear export of Bex1 in cells expressing p75NTR.
Figure 3.
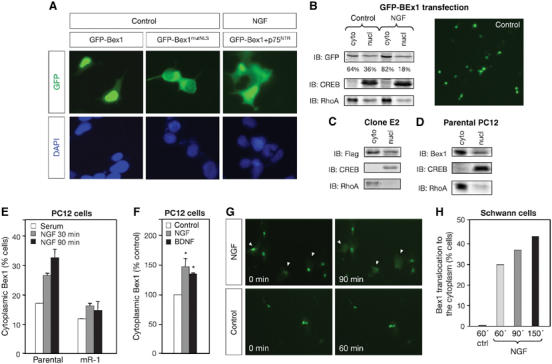
Dynamic nucleocytoplasmic trafficking of Bex1 in response to NGF. (A) 293 cells were transfected with GFP-Bex1, GFP-Bex1mutNLS or GFP-Bex1 and p75NTR, and treated with NGF for 2 h as indicated. (B) Subcellular localization of GFP-Bex1 in transiently transfected PC12 cells—before or after 90 min with NGF—was assessed by fractionation and IB with GFP antibodies. CREB and RhoA were used as markers for nuclear and cytoplasmic compartments, respectively. Percentage in each fraction is indicated. A micrograph of cells expressing GFP-Bex1 grown under control conditions is shown to the right. (C) Subcellular localization of Flag-Bex1 in stable transfected PC12 cells (clone E2). (D) Subcellular localization of endogenous Bex1 in naïve PC12 cells. (E) GFP-Bex1 was transiently transfected in parental or mR-1 PC12 cells and the percentage of cells displaying GFP-Bex1 in a predominant cytoplasmic localization following NGF treatment during the indicated times was quantified. A minimum of 900 cells were counted per condition. Results are average of three independent experiments±s.e.m. (F) Translocation of GFP-Bex1 after NGF or BDNF treatment in PC12 cells. *P<0.05 versus control. (G) GFP-Bex1 was transiently transfected in Schwann cells and photographed at different times after NGF or control treatments. Arrowheads indicate cells showing cytoplasmic translocation of Bex1 in response to NGF treatment. (H) The number of Schwann cells showing translocation of Bex1 from the nucleus to the cytoplasm after the indicated treatments and times was quantified. Results are percentage of cells showing Bex1 translocation at each time point. In all, 70 cells were observed in each case.
Cell fractionation and immunoblotting (IB) showed that 60–70% of all GFP-Bex1 was present in the cytoplasm of transiently transfected PC12 cells (Figure 3B). Similar proportions were also observed in stably transfected (Figure 3C) and parental PC12 cells (Figure 3D), showing that both ectopic and endogenous Bex1 have a similar subcellular distribution. The apparent discrepancy between IB and GFP fluorescence is likely due to the compact size and shape of the nucleus, which makes the fluorescence signal appear to be more concentrated in this compartment (e.g. Figure 3B). The proportion of PC12 cells that showed a higher concentration of GFP-Bex1 fluorescence in the cytoplasm—normally 15–20%—increased over time upon NGF treatment to 30–35% (Figure 3E), indicating the ability of NGF to regulate Bex1 translocation. NGF had no effect in PC12 cell lines that lack expression of full-length p75NTR, such as mR-1 cells (Benedetti et al, 1993) (Figure 3E) or NRA5 cells (Rabizadeh et al, 1994) (data not shown), suggesting a role for p75NTR in the regulation of Bex1 nucleocytoplasmic trafficking. In agreement with this, BDNF—which can interact with p75NTR but not TrkA—was also able to increase the proportion of PC12 cells showing GFP-Bex1 predominantly in the cytoplasm (Figure 3F). Similar to PC12 cells, nuclear GFP-Bex1 relocalized to the cytoplasm in 30% of Schwann cells within 60 min of NGF treatment (Figure 3G and H). Since Schwann cells do not express TrkA, these results indicated that nucleocytoplasmic trafficking of Bex1 in response to NGF must have been mediated by p75NTR signaling.
Dynamic regulation of Bex1 protein levels during the cell cycle and upon neuronal differentiation
Prolonged serum withdrawal—which arrests PC12 cells in the G1 phase of the cell cycle (Rudkin et al, 1989; Urdiales et al, 1998)—drastically affected the endogenous levels of Bex1 protein in PC12 cells, with little or no Bex1 detected after 24 h in serum-free medium (Figure 4A). Bex1 expression recovered in arrested cells after prolonged NGF treatment and concomitantly with postmitotic neuronal differentiation (Figure 4A), in agreement with the abundant Bex1 expression observed in postmitotic neurons in vivo (Figure 2C). In order to examine the regulation of Bex1 levels during the cycle, cell cultures were synchronized in G1 by 24 h serum starvation, after which serum was added back to initiate cell cycle re-entry. Bex1 levels were largely restored 12 h after serum re-addition, as the cells entered S phase, and then decreased again 36–48 h thereafter, as cells returned to G1 (Figure 4B). A second peak in Bex1 was observed as cells re-entered S phase 72 h after serum addition (Figure 4B). Thus, the levels of endogenous Bex1 oscillated during the cell cycle in a pattern that resembled that of p75NTR expression in the membrane of PC12 cells, which had previously been found to be maximal during S phase and lowest during early G1 (Urdiales et al, 1998).
Figure 4.
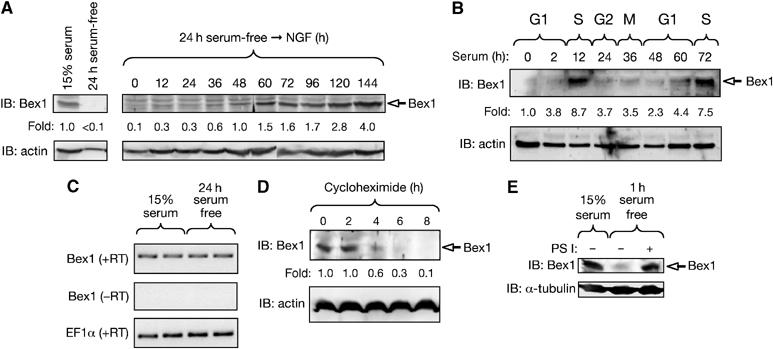
Dynamic regulation of Bex1 protein levels during the cell cycle and upon neuronal differentiation. (A) PC12 cell cultures were serum-starved for 24 h and then treated with NGF plus 1.5% serum for up to 6 days. Expression of endogenous Bex1 was analyzed by IB. Re-probing for actin was used as loading control. Fold change relative to actin is indicated. Similar results were obtained in three independent experiments. (B) PC12 cell cultures were serum-starved for 24 h and then re-exposed to serum for up to 3 days. Expression of endogenous Bex1 was analyzed by IB. Fold change relative to actin is indicated. Similar results were obtained in three independent experiments. (C) RT–PCR analysis of Bex1 mRNA levels in exponentially growing and serum-starved PC12 cells. RT–PCR for EF-1α was used as loading control. (D) Exponentially growing cultures of PC12 cells were treated with cycloheximide for the indicated periods of time and subsequently analyzed for endogenous expression of Bex1 by IB. (E) Endogenous Bex1 was analyzed by IB in PC12 cells after 1 h serum-starving in the presence or absence of PS I. Levels of Bex1 in exponentially growing cells (15% serum) are shown as control. Reprobing for tubulin was used as loading control.
Serum starvation of PC12 cells did not affect the steady-state level of Bex1 mRNA (Figure 4C), indicating a post-transcriptional regulation of Bex1 levels. Using the protein synthesis inhibitor cycloheximide, the half-life of Bex1 was estimated at approximately 5 h in exponentially growing cells (Figure 4D). Upon serum withdrawal, however, Bex1 declined even more rapidly, falling below detection levels between 30 and 90 min after serum deprivation (Figure 4E and data not shown). Interestingly, this decline could be prevented by treatment with the proteasome inhibitor PS I (Figure 4E), confirming that Bex1 levels are regulated by protein degradation.
Phosphorylation of Bex1 in Ser105 by the serine–threonine kinase AKT stabilizes Bex1 and protects it from proteasomal degradation
A motif in the C-terminal portion of the protein (100RERQLS105) matched the consensus site for phosphorylation by the serine–threonine kinase AKT (RxRxxS/T) (Figure 5A). After metabolic labeling of 293 cells with 32P-orthophosphate, incorporation of 32P was observed in wild-type Bex1, but not in a mutant with a Ser105 to Ala substitution (Figure 5B), indicating that Ser105 is an endogenous phosphorylation site in this protein. In addition, no phosphorylated Bex1 could be detected after treatment with LY294002, an inhibitor of PI3K, the upstream activator of AKT (Figure 5C). Moreover, reduction of endogenous AKT activity by serum withdrawal also attenuated Bex1 phosphorylation (Figure 5C). In in vitro kinase assays, wild type, but not mutant, Bex1 was readily phosphorylated by wild-type AKT, while kinase-dead AKT had no effect (Figure 5D), indicating that Bex1 is a direct target of AKT in vitro. Using antibodies that recognize a phosphorylated consensus substrate site for AKT, an increase in the phosphorylation of a band corresponding to Bex1 could be observed in PC12 cells treated with NGF, a stimulus that is known to acutely activate AKT in these cells (Figure 5E). Bex1S105A was consistently expressed at levels lower than wild-type Bex1 in exponentially growing cells (e.g. Figure 5B and F), suggesting a role for Ser105 phosphorylation in regulating the stability of the protein. In agreement with this, treatment with the proteasomal inhibitor PS I produced a significant increase in the levels of Bex1S105A (Figure 5F). Using PS I, we could also confirm that the reduced phosphorylation of the S105A mutant observed in 293 cells was not due to its otherwise low level of expression (Figure 5B, right panel). Taken together, these results indicated that Bex1 can be phosphorylated in Ser105 by AKT, and that this event regulates Bex1 protein turnover.
Figure 5.
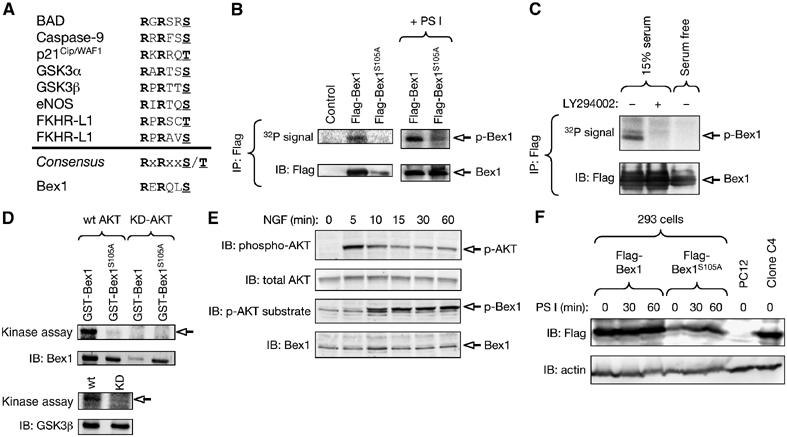
Phosphorylation of Bex1 in Ser105 by the serine–threonine kinase AKT. (A) Consensus sites for phosphorylation in a subset of known AKT substrates shown together with the corresponding site in the Bex1 sequence. (B) Analysis of 32P incorporation in HEK293 T cells that had been metabolically labeled with 32P-orthophosphate after transfection with Flag-tagged versions of wild-type Bex1 or a mutant Bex1 with a Ser105 to Ala substitution (Flag-Bex1S105A). An autoradiogram of anti-Flag immunoprecipitates and its corresponding control IB with anti-Flag antibodies is shown. The panel to the right shows the same experiment performed in cells treated with the proteasomal inhibitor PS I, showing that the reduced phosphorylation of the S105A mutant was not due to its otherwise low level of expression. (C) Bex1 phosphorylation analyzed by metabolic labeling of transfected HEK293 T cells following 2 h treatment with the PI3K inhibitor LY294002 or after 24 h serum withdrawal. (D) Kinase assay of bacterially produced GST-tagged Bex1 constructs incubated with lysates of HEK293 T cells transiently transfected with wild-type or kinase-dead versions of AKT. An autoradiogram of GST pull-downs and its corresponding control IB with anti-GST antibodies is shown. GSK3β was used as positive control (lower panels). Arrows denote phosphorylated products. (E) Analysis of AKT activation and Bex1 phosphorylation in PC12 cells treated with NGF. Immunoblots of phospho-AKT, total AKT, phospho-AKT substrates and Bex1 are shown. Phospho-AKT and phospho-Bex1 are indicated. (F) Steady-state levels of Flag-tagged wild type and S105A Bex1 in HEK293 T cells in the presence and absence of the proteasomal inhibitor PS I analyzed by IB with anti-Flag antibodies. Lysates of parental and clone C4 PC12 cells were used as negative and positive controls, respectively.
Deregulated Bex1 expression prevents growth arrest and neuronal differentiation of PC12 cells
We could not observe any significant increase in cell death after transient transfection of GFP-Bex1 in PC12 cells (Supplementary Figure 1), nor in stable PC12 clones overexpressing Bex1 (data not shown). Parental PC12 cells and clones overexpressing Bex1 showed no differences in cell cycle progression during exponential growth (Figure 6A and data not shown). However, upon 24 h serum withdrawal, a larger proportion of cells in clones C4 and E2 were in S phase compared to parental cells (Figure 6A and Supplementary Figure 2), suggesting that Bex1 overexpression may have interfered with cell cycle arrest. Incorporation of BrdU was examined under two growth-arrest conditions, that is, serum withdrawal and NGF treatment. Strikingly, cells overexpressing Bex1 continued proliferating at normal levels under either condition (Figure 6B), indicating a failure to exit the cell cycle as a result of deregulated Bex1 expression. Following 5-day treatment with NGF in low serum, clones overexpressing Bex1—as well as transiently transfected cells—failed to differentiate, and showed only incipient signs of neuronal differentiation (Figure 6C and data not shown). Taken together, these results indicated a role for Bex1 in cell cycle progression and neuronal differentiation.
Figure 6.
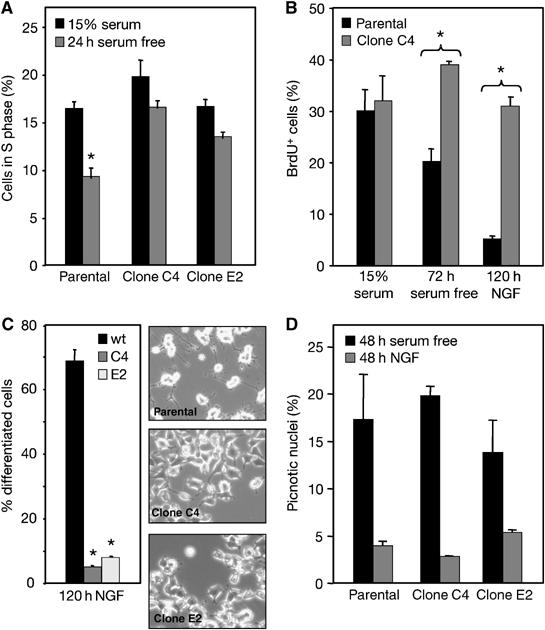
Deregulated Bex1 expression prevents growth arrest and neuronal differentiation of PC12 cells. (A) The percentage of cells in S phase was determined by flow cytometry analysis of exponentially growing and serum-starved cultures of parental PC12 and clones C4 and E2 overexpressing Bex1. Results shown are the mean±s.e.m. of three independent experiments, each performed in triplicate. *P<0.01 versus 15% serum. (B) The percentage of BrdU-positive cells was determined by immunofluorescence analysis of BrdU incorporation in exponentially growing (15% serum), serum-starved and NGF-treated cultures of parental PC12 and clone C4 cells. Counterstaining of nuclei was done with DAPI. Results are shown as average±s.e.m. of three independent experiments each performed in triplicate. *P<0.01. (C) The percentage of differentiated cells (i.e. bearing neurites longer than two-cell body diameters) was quantified in parental PC12 and clones C4 and E2 5 days after NGF treatment. Results are shown as average±s.e.m. of three independent experiments each performed in triplicate. *P<0.01 versus parental PC12 cells (wt). Representative micrographs of NGF-treated cultures are shown. (D) Percentage of dead cells in parental PC12 cells and clones C4 and E2 after 48 h serum starvation in the presence or absence of NGF. Results are shown as average±s.e.m. of three independent experiments each performed in triplicate.
Bex1 modulates proximal signaling by p75NTR, but not TrkA
Although PC12 clones overexpressing Bex1 were refractory to NGF-induced growth arrest and differentiation, they showed survival responses to this factor that were comparable to those of parental PC12 cells (Figure 6D). In agreement with this, NGF stimulation of parental PC12 cells or clones overexpressing Bex1 resulted in comparable activation of the Ras/MAPK and the PI3K/AKT pathways, the two major signaling pathways activated downstream of TrkA (Figure 7A), suggesting that Bex1 overexpression did not interfere with proximal TrkA signaling.
Figure 7.
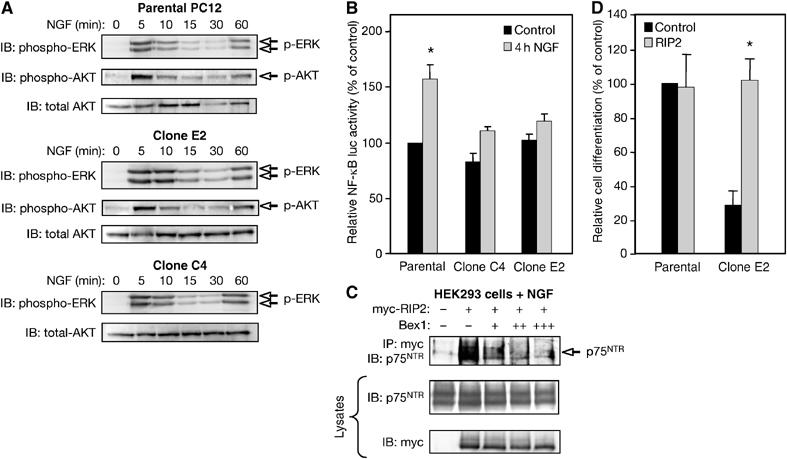
Bex1 modulates proximal signaling by p75NTR but not TrkA. (A) NGF-mediated phosphorylation of ERK and AKT was determined in parental PC12 cells and clones overexpressing Bex1 by IB of cell lysates using phospho-specific antibodies. Reprobing of filters with total AKT was used as loading control. (B) NF-κB activity was assessed using a firefly luciferase reporter gene in parental PC12 cells and clones overexpressing Bex1 before or after a 4 h treatment with NGF. Luciferase activity was normalized to that of a control construct carrying a Renilla luciferase gene. Results are expressed as average±s.e.m. of three independent experiments each performed in quadruplicate. *P<0.05 versus control. (C) HEK293 T cells were cotransfected with p75NTR, myc-tagged RIP2 and increasing amounts of Bex1. After a 4 h treatment with NGF, RIP2 was immunoprecipitated from cell lysates with anti-myc antibodies and filters were probed with anti-p75NTR antibodies. Aliquots of total cell lysates were directly probed with anti-myc and anti-p75NTR antibodies as control. (D) RIP2 was transiently transfected in parental PC12 cells and clone E2 together with a GFP expression plasmid and then stimulated with NGF. After 2 days, the fraction of differentiated cells was counted in each condition. Results were normalized to the extent of cell differentiation observed in control parental cells (i.e. 25–32% of transfected cells), and are expressed as average±s.e.m. of three independent experiments each performed in triplicate. *P<0.05 versus control.
The first pathway to be characterized downstream of p75NTR leads to activation of the NF-κB transcription factor (Carter et al, 1996), and has been implicated in survival (Wood, 1995; Yoon et al, 1998; Hamanoue et al, 1999) and neuronal differentiation (Foehr et al, 2000; Wooten et al, 2001) of PC12 cells and other cell types. Although similar levels of basal NF-κB activity could be observed in parental cells and clones overexpressing Bex1, NGF was able to stimulate this pathway only in parental cells (Figure 7B), indicating that Bex1 can modulate NF-κB activity in response to NGF in these cells. Although TrkA has also been implicated in the induction of NF-κB activity by NGF in PC12 cells (Foehr et al, 2000), the fact that the two most likely candidate pathways for NF-κB activation by TrkA—that is, Ras/MAPK and PI3K/AKT—were unaffected by Bex1 overexpression, suggested interference with p75NTR-derived signals.
The serine–threonine kinase RIP2 has been shown to be required for NF-κB activation by p75NTR in response to NGF, and interacts directly with the death domain of this receptor upon NGF stimulation (Khursigara et al, 2001). A progressive reduction in the association between RIP2 and p75NTR was observed in cells that received increasing amounts of Bex1 (Figure 7C), suggesting that Bex1 and RIP2 competed for an overlapping binding site in p75NTR. In agreement with this, RIP2 transfection in cells overexpressing Bex1 completely rescued neuronal differentiation in response to NGF (Figure 7D).
Bex1 knockdown potentiates NF-κB activity and accelerates PC12 cell differentiation in response to NGF
An siRNA construct directed to the rat Bex1 mRNA sequence significantly reduced—but did not completely abolish—endogenous Bex1 mRNA levels (Figure 8A). A small increase in Bex2 mRNA could also be observed (Figure 8A). Cotransfection of Bex1 siRNA and Flag-Bex1 indicated a reduction of 60–70% in Bex1 protein levels in siRNA-treated cells (Figure 8B). In agreement with a role for Bex1 in the regulation of NF-κB activity, Bex1 knockdown potentiated NF-κB activity and produced a more robust response to NGF (Figure 8C). In addition, a significant potentiation of the differentiation response was observed after 12 h of NGF in synchronized PC12 cells expressing Bex1 siRNA (Figure 8D, see Supplementary data for details). Interestingly, such levels of differentiation would not normally be observed in naïve cells until after 24–36 h of NGF treatment, that is, roughly the time it takes for cells to move from S to G1 and endogenous Bex1 to disappear. In agreement with this notion, the differentiation advantage of siRNA-treated cells waned over time, such that no difference could be seen between the two groups by 60 h of NGF treatment (Figure 8D).
Figure 8.
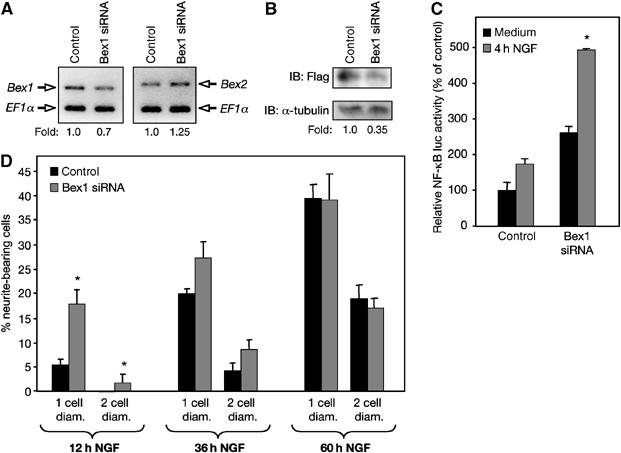
Bex1 knockdown potentiates NF-κB activity and accelerates PC12 cell differentiation in response to NGF. (A) Bex1 and Bex2 mRNA levels were analyzed by RT–PCR in PC12 cells transfected with a Bex1 siRNA or a control construct. EF1α is shown as loading control. Fold changes relative to control are indicated. (B) Bex1 protein levels were analyzed by IB in PC12 cells transfected with Bex1 siRNA or a control construct together with Flag-Bex1. α-Tubulin is shown as loading control. Fold change relative to control is indicated. (C) The effects of Bex1 knockdown on NF-κB activity were tested in PC12 cells that also received small amounts of ectopic RIP2 (which in our hands enhances NF-κB responses to NGF without altering basal levels), before (medium) or after a 4 h treatment with NGF. Results are expressed as average±s.e.m. of three experiments performed in quadruplicate. *P<0.05 versus control. (D) Neuronal differentiation was assessed in synchronized in S phase, PC12 cells transfected with Bex1 siRNA or a control construct as the percentage of cells displaying neurites longer than one or two cell diameters, as indicated (see Supplementary data for details). Results are shown as average±s.d. of a representative experiment performed in triplicate. *P<0.05 versus control. Similar results were obtained in two additional experiments.
Bex1 inhibits neuronal differentiation of precursors from the brain subventricular zone
The general ability of Bex1 to regulate neuronal differentiation was tested in neural precursors derived from the brain subventricular zone (SVZ). Proliferating SVZ precursors expressed Bex1, Bex2 and Bex3 mRNA as assessed by RT–PCR (Figure 9A). Upon differentiation, however, the expression levels of all three genes declined significantly (Figure 9A). We transfected Bex1 into proliferating SVZ precursors which, after an additional round of neurosphere culture, were induced to differentiate by mitogen withdrawal. 9B). Ectopic Bex1 interfered with neuronal differentiation—assessed by coexpression of GFP and βIII-tubulin—of neurosphere-amplified SVZ precursors, as only half as many cells overexpressing Bex1 were able to differentiate into neurons compared to control (Figure 9B and C).
Figure 9.
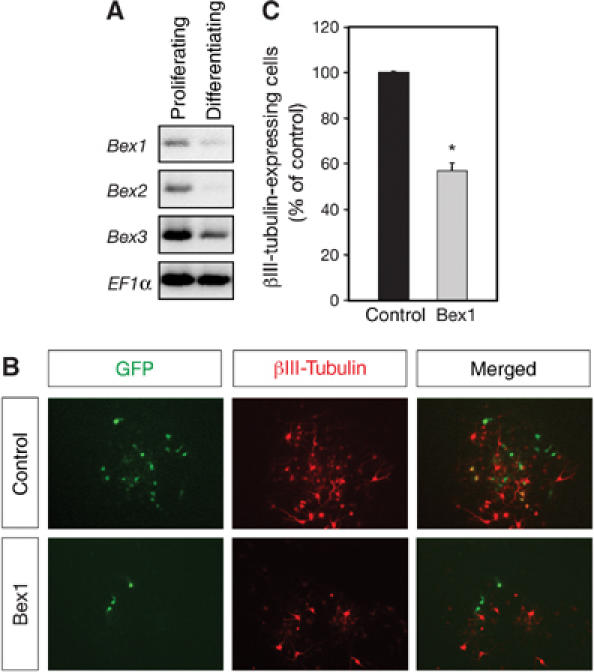
Bex1 inhibits neuronal differentiation of precursors from the brain SVZ. (A) Bex1, Bex2 and Bex3 mRNA levels were analyzed by RT–PCR in floating neurospheres derived from the postnatal SVZ (proliferating) or differentiating neural precursors after 5 days of mitogen withdrawal. EF1α is shown as loading control. (B) Neural precursors transfected with GFP (control) or GFP-Bex1 (Bex1) were allowed to differentiate after mitogen withdrawal. Micrographs show GFP fluorescence, βIII-tubulin immunocytochemistry (in red) and their overlay (merged). In the examples shown, double-positive cells (yellow) are observed in control, but not Bex1-transfected cells. (C) Quantification of neuronal differentiation of SVZ precursors as the percentage of βIII-tubulin-positive cells among the GFP-expressing cell population. Under control conditions (set here to 100%) 20–30% of the cells expressed βIII-tubulin and extended neurites. Average results of three independent experiments are shown ±s.d. *P<0.05 versus control.
Discussion
The search for intracellular interacting partners of p75NTR has opened multiple inroads into the signaling mechanisms of this receptor. Candidate p75NTR interactors have been identified through either educated guesses (Khursigara et al, 1999, 2001; Mamidipudi et al, 2002) or the yeast-two-hybrid technique (Casademunt et al, 1999; Chittka and Chao, 1999; Yamashita et al, 1999; Mukai et al, 2000; Salehi et al, 2000; Tcherpakov et al, 2002). Using T7 phage display, we have identified the Bex1 protein as an intracellular interactor of p75NTR. The expression of Bex1 during embryonic development showed a remarkable overlap with that of p75NTR throughout the developing nervous system and in vascular and mesenchymal structures. In transverse sections of E13.5 rat embryos, for example, the predominant pattern of Bex1 mRNA expression—that is, DRG, sympathetic and motorneurons, somitic mesenchyme and heart—was essentially indistinguishable from that of p75NTR at the same developmental stage (Ernfors et al, 1988). This is unlike many other intracellular interactors of p75NTR which, for the most part, display a much broader pattern of expression than the receptor (Kendall et al, 2002, 2003). The significance of this observation is unclear at present, but given the emerging roles of p75NTR in stem cell renewal and neuronal and glial differentiation, and the ability of Bex1 to modulate similar processes, it suggests that Bex1 may play an important role in coupling p75NTR signaling to the intracellular machinery controlling cell renewal, cell cycle exit and differentiation. The expression of Bex1 in many postmitotic neurons of the peripheral and central nervous systems suggests other functions for this protein—unrelated to cell cycle and growth control—which remain to be explored.
Bex1 belongs to a growing family of small proteins of unknown function, but with several features suggesting roles as adaptors or modulators of intracellular signaling pathways. Bex1 interacted directly with the intracellular domain of p75NTR, but this interaction was not affected by NGF binding to the receptor, suggesting that Bex1 does not function as a downstream effector of p75NTR signaling, at least not in the conventional sense. On the other hand, the dynamic regulation of Bex1 levels during cell cycle, growth arrest and cell differentiation indicated that other signaling inputs or cellular states may regulate Bex1 binding to p75NTR. Thus, for example, no Bex1 complexed with p75NTR could be recovered in cells synchronized in the G1 phase of the cycle by serum starvation. Together with the ability of Bex1 to modulate ligand-dependent recruitment of bonafide downstream effectors, such as the adaptor and serine–threonine kinase RIP2, these observations suggest that Bex1 may represent a novel class of upstream intracellular regulators of p75NTR function.
Bex1 showed a dynamic nucleocytoplasmic distribution in cells expressing p75NTR following stimulation with NGF, which resulted in rapid relocalization of Bex1 from the nucleus to the cytoplasm. This is also in contrast to several other known interactors of p75NTR which translocate to the nucleus upon NGF binding (Casademunt et al, 1999; Chittka and Chao, 1999). NGF could induce nuclear export of Bex1 in cells expressing p75NTR but not TrkA, suggesting a two-way communication between the interacting proteins, that is, p75NTR signaling regulates Bex1 localization, and Bex1 levels regulate p75NTR signaling. Phosphorylation of Ser-105 had no effect on the subcellular distribution of Bex1 (M Vilar, unpublished observations), so the mechanisms controlling nucleocytoplasmic trafficking of Bex1 remain to be elucidated. Intriguingly, although NGF induced a partial redistribution of Bex1 protein to the cytoplasm in all cell types tested, this did not translate into a detectable increase in its interaction with membrane-bound p75NTR, perhaps due to the particular internalization kinetics of this receptor (Bronfman et al, 2003) or due to a distinct subcellular compartmentalization. Interestingly, the intracellular domain of p75NTR has been observed to undergo endoproteolytic cleavage and nuclear translocation (Jung et al, 2003; Kanning et al, 2003; Frade, 2005), although the studies reported so far differ as to the role of NGF in those processes.
In addition to p75NTR signaling, other stimuli are likely to regulate Bex1 function, as demonstrated by the ability of serum to induce Bex1 phosphorylation in Ser105 via the PI3K/AKT pathway. In the absence of serum, Bex1 was dephosphorylated—presumably due to low AKT activity—and rapidly degraded by the proteasome. The functional importance of the rapid Bex1 turnover is supported by at least two sets of observations: (i) endogenous Bex1 levels oscillated during the cell cycle, being lowest at G1 and highest at S phase; (ii) preventing the normal cycling and downregulation of Bex1 had dramatic effects on cell proliferation and the ability of cells to exit the cell cycle and differentiate. The oscillation of Bex1 levels during the cell cycle could conceivably be controlled by cycles of phosphorylation and de-phosphorylation at Ser105. However, whether AKT activity normally oscillates with the cycle in dividing cells has not yet been investigated. Likewise, following NGF addition, AKT only remains phosphorylated for 30–60 min, after which Bex1 may be dephosphorylated and degraded.
Bex1 overexpression caused PC12 cells to become resistant to growth arrest induced by serum withdrawal or NGF treatment, suggesting that Bex1 levels need to be downregulated at G1 in order for cells to exit the cycle and differentiate. In agreement with this, Bex1 knockdown accelerated cell cycle exit and neuronal differentiation. In PC12 cells, Bex1 overexpression interferred with cell cycle exit but not progression, suggesting that Bex1 is not an intrinsic component of the cell cycle machinery but may rather function as a gate-keeper of growth arrest and cell differentiation, preventing cell cycle exit during S, G2 and M phases. In contrast to Bex1, several previously identified downstream effectors of p75NTR have been shown to promote mitotic cycle arrest (Lopez-Sanchez and Frade, 2002; Chittka et al, 2004). Together with its ability to regulate p75NTR function, this suggests that Bex1 could be part of a feedback mechanism to gate p75NTR activity according to the phase of the cell cycle. In line with its effects in PC12 cells, Bex1 was downregulated upon differentiation of SVZ precursors, and its overexpression affected the ability of those cells to undergo neuronal differentiation upon mitogen withdrawal, indicating a broader role for Bex1 in the control of neuronal differentiation. Interestingly, neurospheres grown from the early postnatal SVZ have been shown to express p75NTR, and lack of p75NTR expression was reported to decrease their capacity to undergo neuronal differentiation in response to BDNF (Hosomi et al, 2003).
The mechanisms by which Bex1 prevents cell cycle exit are at present unclear, but could involve interference with signaling pathways activated by growth arrest and differentiation signals. Despite the recognized importance of TrkA-derived signals in cell cycle exit and neuronal differentiation of PC12 cells, Bex1 overexpression did not interfere with the activation of two primary targets of TrkA signaling, that is, Erk1/2 and AKT. Additional studies will be required to establish whether Bex1 may affect further downstream steps of TrkA-activated pathways. On the other hand, elevated Bex1 expression prevented —and reduced expression enhanced— the induction of NF-κB activity by NGF. Upon NGF treatment, PC12 clones overexpressing Bex1 exhibited only very short neurites and failed to fully differentiate, a phenotype that resembled that obtained after inhibition of NF-κB activity in these cells (Foehr et al, 2000). In agreement with the importance of the interplay between Bex1 and RIP2 in this process, elevating RIP2 levels relieved the differentiation block of Bex1-overexpressing cells, suggesting that Bex1 may modulate p75NTR signaling by antagonizing the recruitment and activation of adaptor proteins such as RIP2.
In conclusion, our studies establish Bex1 as a novel link between the cell cycle and neurotrophic factor signaling. Unlike many other receptor-interacting molecules, Bex may function in part as an upstream modulator of receptor signaling, coordinating biological responses to external signals with internal cellular states. These findings open interesting possibilities for exploring Bex1 function in the control of neural stem cell renewal and neurogenesis in the peripheral and central nervous systems.
Materials and methods
T7 phage display
A T7 phage display cDNA library was prepared from differentiated PC12 cells according to manufacturer's instructions (Novagen). The intracellular domain of p75NTR (p75ICD) was produced and purified as described previously (Liepinsh et al, 1997). Purified p75ICD was biotinylated using the EZ-link reagent (Pierce), and immobilized onto streptavidin-coated paramagnetic beads (Promega). Biopanning was performed using standard procedures (see Supplementary data).
RT–PCR and in situ hybridization
For RT–PCR studies, total RNA purification and first-strand cDNA synthesis were performed with kits from Invitrogen and Stratagene, respectively. Primer sequences for rat Bex1, Bex2 and Bex3 and RT–PCR conditions are available upon request. In situ hybridization was performed using standard protocols (see Supplementary data).
Cell culture and transfection
PC12, HEK293 T and Schwann cells were cultured using standard protocols (see Supplementary data). Synchronized PC12 cultures were obtained after 24 h incubation in serum-free medium. After serum re-addition, cell cycle phases were confirmed by flow cytometry. Cultures of SVZ-derived precursors were established from postnatal day 9 rat SVZ using standard protocols (see Supplementary data). Cell transfection of PC12, HEK293 T and Schwann cells was carried out using polyethylenimine (PEI, 25 kDa, Sigma-Aldrich) as described (Scott et al, 2005). Neural precursors were transfected using Fugene-6 (Roche) after one neurosphere passage, then allowed to grow in suspension for 5 days before induction of neuronal differentiation.
Immunoprecipitation, IB and metabolic labeling
Total cell lysates, immunoprecipitation (IP) and metabolic labeling were prepared and performed using standard protocols (see Supplementary data). Antibodies were obtained from various sources as indicated in the Supplementary data. Polyclonal anti-Bex1 antibodies were prepared by immunizing rabbits with a peptide derived from rat Bex1 (KNCAPRGGRRRFRVRQPI) conjugated to keyhole limpet hemocyanin.
Pharmacological treatments and siRNA
NGF (Alomone Labs) was used at 100 ng/ml in low serum containing medium (typically 1.5% FCS) unless otherwise indicated. Cycloheximide (Sigma) was used at 40 μg/ml, PS I (Calbiochem) at 25 μM and LY294002 (Calbiochem) at 50 μM. An siRNA construct targeting the rat Bex1 mRNA sequence was assembled in the TOPO vector (Invitrogen) using the mouse U6 RNA Pol III promoter upstream of a hairpin DNA sequence derived from Bex1 (see Supplementary data). An siRNA construct directed to Wnt3 was used as negative control.
Flow cytometry, BrdU incorporation, cell death and gene reporter assays
Cell cycle phases were analyzed by flow cytometry in cells stained with propidium iodine as described previously (Rudkin et al, 1989; Urdiales et al, 1998). Cell proliferation was assessed by measuring BrdU incorporation using standard procedures (see Supplementary data). Cell death was quantified as the number of picnotic nuclei in cultures stained with DAPI. For cell differentiation assays, the proportion of cells bearing neurites longer than one- or two-cell diameters was quantified. Gene reporter assays were performed using standard protocols (see Supplementary data).
Supplementary Material
Supplementary Information
Acknowledgments
We thank Moses Chao for anti-p75NTR antibodies and mR-1 cells, and various lab members for help and support. We also thank Wilma Friedman and Mike Fainzilber for comments on the manuscript, and Xiaoli-Li and Carolina Svensson for secretarial help. This work was supported by grants from the Swedish Foundation for Strategic Research, the Swedish Research Council (33X-10908-10A), the Swedish Cancer Society (3474-B97-05XBC), the Vth Framework Program of the European Union (QLG3-CT-1999-00573) and the Karolinska Institute. MM-C has been the recipient of a postdoctoral fellowship from the Ministerio de Educación y Ciencia, Spain.
References
- Alvarez E, Zhou W, Witta SE, Freed CR (2005) Characterization of the Bex gene family in humans, mice, and rats. Gene 357: 18–28 [DOI] [PubMed] [Google Scholar]
- Benedetti M, Levi A, Chao MV (1993) Differential expression of nerve growth factor receptors leads to altered binding affinity and neurotrophin responsiveness. Proc Natl Acad Sci USA 90: 7859–7863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilderback TR, Grigsby RJ, Dobrowsky RT (1997) Association of p75(NTR) with caveolin and localization of neurotrophin-induced sphingomyelin hydrolysis to caveolae. J Biol Chem 272: 10922–10927 [DOI] [PubMed] [Google Scholar]
- Bronfman FC, Fainzilber M (2004) Multi-tasking by the p75 neurotrophin receptor: sorting things out? EMBO Rep 5: 867–871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronfman FC, Tcherpakov M, Jovin TM, Fainzilber M (2003) Ligand-induced internalization of the p75 neurotrophin receptor: a slow route to the signaling endosome. J Neurosci 23: 3209–3220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AL, Kay GF (1999) Bex1, a gene with increased expression in parthenogenetic embryos, is a member of a novel gene family on the mouse X chromosome. Hum Mol Genet 8: 611–619 [DOI] [PubMed] [Google Scholar]
- Carter BD, Kaltschmidt C, Kaltschmidt B, Offenhäuser N, Böhm-Matthaei R, Baeuerle PA, Barde Y-A (1996) Selective activation of NFκ-B by nerve growth factor through the neurotrophin receptor p75. Science 272: 542–545 [DOI] [PubMed] [Google Scholar]
- Casaccia-Bonnefil P, Carter BD, Dobrowsky RT, Chao MV (1996) Death of oligodendrocytes mediated by the interaction of nerve growth factor with its receptor p75. Nature 383: 716–719 [DOI] [PubMed] [Google Scholar]
- Casademunt E, Carter BD, Benzel I, Frade JM, Dechant G, Barde YA (1999) The zinc finger protein NRIF interacts with the neurotrophin receptor p75(NTR) and participates in programmed cell death. EMBO J 18: 6050–6061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chittka A, Arevalo JC, Rodriguez-Guzman M, Perez P, Chao MV, Sendtner M (2004) The p75NTR-interacting protein SC1 inhibits cell cycle progression by transcriptional repression of cyclin E. J Cell Biol 164: 985–996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chittka A, Chao MV (1999) Identification of a zinc finger protein whose subcellular distribution is regulated by serum and nerve growth factor. Proc Natl Acad Sci USA 96: 10705–10710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotrina ML, Gonzalez-Hoyuela M, Barbas JA, Rodriguez-Tebar A (2000) Programmed cell death in the developing somites is promoted by nerve growth factor via its p75(NTR) receptor. Dev Biol 228: 326–336 [DOI] [PubMed] [Google Scholar]
- DeFreitas MF, McQuillen PS, Shatz CJ (2001) A novel p75NTR signaling pathway promotes survival, not death, of immunopurified neocortical subplate neurons. J Neurosci 21: 5121–5129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrowsky R, Werner M, Castellino A, Chao M, Hannun Y (1994) Activation of the sphingomyelin cycle through the low-affinity neurotrophin receptor. Science 265: 1596–1599 [DOI] [PubMed] [Google Scholar]
- Ernfors P, Hallböök F, Ebendal T, Shooter E, Radeke MJ, Misko TP, Persson H (1988) Developmental and regional expression of ß-nerve growth factor receptor mRNA in the chick and rat. Neuron 1: 983–996 [DOI] [PubMed] [Google Scholar]
- Foehr ED, Lin X, O'Mahony A, Geleziunas R, Bradshaw RA, Greene WC (2000) NF-kappa B signaling promotes both cell survival and neurite process formation in nerve growth factor-stimulated PC12 cells. J Neurosci 20: 7556–7563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frade JM (2005) Nuclear translocation of the p75 neurotrophin receptor cytoplasmic domain in response to neurotrophin binding. J Neurosci 25: 1407–1411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman WJ (2000) Neurotrophins induce death of hippocampal neurons via the p75 receptor. J Neurosci 20: 6340–6346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentry JJ, Barker PA, Carter BD (2004) The p75 neurotrophin receptor: multiple interactors and numerous functions. Prog Brain Res 146: 25–39 [DOI] [PubMed] [Google Scholar]
- Greene LA, Tischler AS (1976) Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc Natl Acad Sci USA 73: 2424–2428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamanoue M, Middleton G, Wyatt S, Jaffrey E, Hay RT, Davies AM (1999) p75-mediated NF-kB activation enhances the survival response of developing sensory neurons to NGF. Mol Cell Neurosci 14: 28–40 [DOI] [PubMed] [Google Scholar]
- Harrington AW, Kim JY, Yoon SO (2002) Activation of Rac GTPase by p75 is necessary for c-jun N-terminal kinase-mediated apoptosis. J Neurosci 22: 156–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosomi S, Yamashita T, Aoki M, Tohyama M (2003) The p75 receptor is required for BDNF-induced differentiation of neural precursor cells. Biochem Biophys Res Commun 301: 1011–1015 [DOI] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF (2003) TRK receptors: roles in neuronal signal transduction. Annu Rev Biochem 27: 27. [DOI] [PubMed] [Google Scholar]
- Huber LJ, Chao MV (1995) Mesenchymal and neuronal cell expression of the p75 neurotrophin receptor gene occur by different mechanisms. Dev Biol 167: 227–238 [DOI] [PubMed] [Google Scholar]
- Irie S, Hachiya T, Rabizadeh S, Maruyama W, Mukai J, Li Y, Reed JC, Bredesen DE, Sato TA (1999) Functional interaction of Fas-associated phosphatase-1 (FAP-1) with p75(NTR) and their effect on NF-kappaB activation. FEBS Lett 460: 191–198 [DOI] [PubMed] [Google Scholar]
- Jeanmougin F, Thompson JD, Gouy M, Higgins DG, Gibson TJ (1998) Multiple sequence alignment with Clustal X. Trends Biochem Sci 23: 403–405 [DOI] [PubMed] [Google Scholar]
- Jung KM, Tan S, Landman N, Petrova K, Murray S, Lewis R, Kim PK, Kim DS, Ryu SH, Chao MV, Kim TW (2003) Regulated intramembrane proteolysis of the p75 neurotrophin receptor modulates its association with the TrkA receptor. J Biol Chem 11: 11. [DOI] [PubMed] [Google Scholar]
- Kanning KC, Hudson M, Amieux PS, Wiley JC, Bothwell M, Schecterson LC (2003) Proteolytic processing of the p75 neurotrophin receptor and two homologs generates C-terminal fragments with signaling capability. J Neurosci 23: 5425–5436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan DR, Miller FD (2000) Neurotrophin signal transduction in the nervous system. Curr Opin Neurobiol 10: 381–391 [DOI] [PubMed] [Google Scholar]
- Kendall SE, Goldhawk DE, Kubu C, Barker PA, Verdi JM (2002) Expression analysis of a novel p75(NTR) signaling protein, which regulates cell cycle progression and apoptosis. Mech Dev 117: 187–200 [DOI] [PubMed] [Google Scholar]
- Kendall SE, Ryczko MC, Mehan M, Verdi JM (2003) Characterization of NADE, NRIF and SC-1 gene expression during mouse neurogenesis. Brain Res Dev Brain Res 144: 151–158 [DOI] [PubMed] [Google Scholar]
- Khursigara G, Bertin J, Yano H, Moffett H, DiStefano PS, Chao MV (2001) A prosurvival function for the p75 receptor death domain mediated via the caspase recruitment domain receptor-interacting protein 2. J Neurosci 21: 5854–5863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khursigara G, Orlinick JR, Chao MV (1999) Association of the p75 neurotrophin receptor with TRAF6. J Biol Chem 274: 2597–2600 [DOI] [PubMed] [Google Scholar]
- Koo JH, Saraswati M, Margolis FL (2005) Immunolocalization of Bex protein in the mouse brain and olfactory system. J Comp Neurol 487: 1–14 [DOI] [PubMed] [Google Scholar]
- Lee KF, Li E, Huber LJ, Landis SC, Sharpe AH, Chao MV, Jaenisch R (1992) Targeted mutation of the gene encoding the low affinity NGF receptor p75 leads to deficits in the peripheral sensory nervous system. Cell 69: 737–749 [DOI] [PubMed] [Google Scholar]
- Liepinsh E, Ilag LL, Otting G, Ibanez CF (1997) NMR structure of the death domain of the p75 neurotrophin receptor. EMBO J 16: 4999–5005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Sanchez N, Frade JM (2002) Control of the cell cycle by neurotrophins: lessons from the p75 neurotrophin receptor. Histol Histopathol 17: 1227–1237 [DOI] [PubMed] [Google Scholar]
- Mamidipudi V, Li X, Wooten MW (2002) Identification of interleukin 1 receptor-associated kinase as a conserved component in the p75-neurotrophin receptor activation of nuclear factor-kappa B. J Biol Chem 277: 28010–28018 [DOI] [PubMed] [Google Scholar]
- Mukai J, Hachiya T, Shoji-Hoshino S, Kimura MT, Nadano D, Suvanto P, Hanaoka T, Li Y, Irie S, Greene LA, Sato TA (2000) NADE, a p75NTR-associated cell death executor, is involved in signal transduction mediated by the common neurotrophin receptor p75NTR. J Biol Chem 275: 17566–17570 [DOI] [PubMed] [Google Scholar]
- Naumann T, Casademunt E, Hollerbach E, Hofmann J, Dechant G, Frotscher M, Barde YA (2002) Complete deletion of the neurotrophin receptor p75NTR leads to long-lasting increases in the number of basal forebrain cholinergic neurons. J Neurosci 22: 2409–2418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabizadeh S, Bitler CM, Butcher LL, Bredesen DE (1994) Expression of the low-affinity nerve growth factor receptor enhances beta-amyloid peptide toxicity. Proc Natl Acad Sci USA 91: 10703–10706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux PP, Barker PA (2002) Neurotrophin signaling through the p75 neurotrophin receptor. Prog Neurobiol 67: 203–233 [DOI] [PubMed] [Google Scholar]
- Rudkin BB, Lazarovici P, Levi BZ, Abe Y, Fujita K, Guroff G (1989) Cell cycle-specific action of nerve growth factor in PC12 cells: differentiation without proliferation. EMBO J 8: 3319–3325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salehi AH, Roux PP, Kubu CJ, Zeindler C, Bhakar A, Tannis LL, Verdi JM, Barker PA (2000) NRAGE, a novel MAGE protein, interacts with the p75 neurotrophin receptor and facilitates nerve growth factor-dependent apoptosis. Neuron 27: 279–288 [DOI] [PubMed] [Google Scholar]
- Scott R, Eketjäll S, Aineskog H, Ibáñez CF (2005) Distinct turnover of alternatively-spliced isoforms of the RET kinase receptor mediated by differential recruitment of the Cbl ubiquitin ligase. J Biol Chem 280: 13442–13449 [DOI] [PubMed] [Google Scholar]
- Sole C, Dolcet X, Segura MF, Gutierrez H, Diaz-Meco MT, Gozzelino R, Sanchis D, Bayascas JR, Gallego C, Moscat J, Davies AM, Comella JX (2004) The death receptor antagonist FAIM promotes neurite outgrowth by a mechanism that depends on ERK and NF-kapp B signaling. J Cell Biol 167: 479–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tcherpakov M, Bronfman FC, Conticello SG, Vaskovsky A, Levy Z, Niinobe M, Yoshikawa K, Arenas E, Fainzilber M (2002) The p75 neurotrophin receptor interacts with multiple MAGE proteins. J Biol Chem 277: 49101–49104 [DOI] [PubMed] [Google Scholar]
- Teng KK, Hempstead BL (2004) Neurotrophins and their receptors: signaling trios in complex biological systems. Cell Mol Life Sci 61: 35–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urdiales JL, Becker E, Andrieu M, Thomas A, Jullien J, van Grunsven LA, Menut S, Evan GI, Martin-Zanca D, Rudkin BB (1998) Cell cycle phase-specific surface expression of nerve growth factor receptors TrkA and p75(NTR). J Neurosci 18: 6767–6775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Grunsven LA, Thomas A, Urdiales JL, Machenaud S, Choler P, Durand I, Rudkin BB (1996) Nerve growth factor-induced accumulation of PC12 cells expressing cyclin D1: evidence for a G1 phase block. Oncogene 12: 855–862 [PubMed] [Google Scholar]
- von Schack D, Casademunt E, Schweigreiter R, Meyer M, Bibel M, Dechant G (2001) Complete ablation of the neurotrophin receptor p75(NTR) causes defects both in the nervous and the vascular system. Nat Neurosci 4: 977–978 [DOI] [PubMed] [Google Scholar]
- Wood JN (1995) Regulation of NF-kappa B activity in rat dorsal root ganglia and PC12 cells by tumour necrosis factor and nerve growth factor. Neurosci Lett 192: 41–44 [DOI] [PubMed] [Google Scholar]
- Wooten MW, Seibenhener ML, Mamidipudi V, Diaz-Meco MT, Barker PA, Moscat J (2001) The atypical protein kinase C-interacting protein p62 is a scaffold for NF-kappaB activation by nerve growth factor. J Biol Chem 276: 7709–7712 [DOI] [PubMed] [Google Scholar]
- Yamashita T, Tucker KL, Barde YA (1999) Neurotrophin binding to the p75 receptor modulates Rho activity and axonal outgrowth. Neuron 24: 585–593 [DOI] [PubMed] [Google Scholar]
- Ye X, Mehlen P, Rabizadeh S, VanArsdale T, Zhang H, Shin H, Wang JJ, Leo E, Zapata J, Hauser CA, Reed JC, Bredesen DE (1999) TRAF family proteins interact with the common neurotrophin receptor and modulate apoptosis induction. J Biol Chem 274: 30202–30208 [DOI] [PubMed] [Google Scholar]
- Yoon SO, Casaccia-Bonnefil P, Carter B, Chao MV (1998) Competitive signaling between TrkA and p75 nerve growth factor receptors determines cell survival. J Neurosci 18: 3273–3281 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information