

Abstract
The B30.2/SPRY domain is present in ∼700 eukaryotic (∼150 human) proteins, including medically important proteins such as TRIM5α and Pyrin. Nonetheless, the functional role of this modular domain remained unclear. Here, we report the crystal structure of an SPRY-SOCS box family protein GUSTAVUS in complex with Elongins B and C, revealing a highly distorted two-layered β-sandwich core structure of its B30.2/SPRY domain. Ensuing studies identified one end of the β-sandwich as the surface interacting with an RNA helicase VASA with a 40 nM dissociation constant. The sequence variation in TRIM5α responsible for HIV-1 restriction and most of the mutations in Pyrin causing familial Mediterranean fever map on this surface, implicating the corresponding region in many B30.2/SPRY domains as the ligand-binding site. The amino acids lining the binding surface are highly variable among the B30.2/SPRY domains, suggesting that these domains are protein-interacting modules, which recognize a specific individual partner protein rather than a consensus sequence motif.
Keywords: B30.2/SPRY, GUSTAVUS, Pyrin, Structure, TRIM5alpha
Introduction
The B30.2 domain was first identified as a protein domain encoded by an exon (named B30-2) in the human class I major histocompatibility complex region (chromosome 6p21.3) (Vernet et al, 1993), whereas the SPRY domain was first identified in a Dictyostelium discoidueum kinase splA and mammalian Ca2+-release channels ryanodine receptors (Ponting et al, 1997). The two domains share sequence similarity such that a SPRY domain can be identified in virtually all the known B30.2 domain-containing proteins in the sequence-based domain classification databases Pfam (Bateman et al, 2004) and SMART (Schultz et al, 1998). Notably, the SPRY domains are shorter at the N-teminus than the B30.2 domains and appear as a subdomain of the latter. Out of 642 SPRY domain-containing proteins classified in the SMART database, 516 possess a PRY domain (50–60 residues) right next to the N-terminus of the SPRY domain. The functional relationship between the PRY and SPRY domains has been unknown. The B30.2/SPRY (for B30.2 and/or SPRY) domain is present in a large number of proteins with diverse individual functions in different biological processes (Henry et al, 1998; Rhodes et al, 2005). The functional significance of the B30.2/SPRY domains is clear from numerous biochemical or genetic studies. The following is the description of such studies on four different proteins: TRIM5α, Pyrin, MID1, and GUSTAVUS.
TRIM5α is a potent retroviral restriction factor that blocks retroviruses, such as human immunodeficiency virus type 1 (HIV-1) or HIV-2 at the early postentry phase in a species-specific manner via a poorly understood mechanism, which involves viral capsid (Stremlau et al, 2004; Nisole et al, 2005). The protein belongs to the tripartite motif (TRIM) family of proteins, which contain a RING domain, B-box zinc finger(s), and a coiled-coil domain (Reddy et al, 1992). TRIM5α contains a B30.2/SPRY domain C-terminal to the coiled-coil domain. TRIM5α has been suggested to be an E3 ubiquitin ligase that results in the ubiquitination of the capsid protein of retroviruses, thereby adversely affecting capsid disassembly or targeting the protein for proteosomal degradation (Stremlau et al, 2004; Nisole et al, 2005). Recently, the specificity in the retroviral restriction was shown to arise from species-specific amino-acid changes and/or significant length polymorphism in the B30.2/SPRY domain of the TRIM5α proteins (Nisole et al, 2005; Sawyer et al, 2005; Song et al, 2005; Yap et al, 2005).
Missense mutations in Pyrin/Marenostrin/MEFV are the cause of familial Mediterranean fever (FMF), which is characterized by recurrent episodes of fever and serosal inflammation (Consortium, 1997; Bakkaloglu, 2003). The biological function of Pyrin is not clear, but a probable role of Pyrin may be the homeostatic control of inflammation (Consortium, 1997). Human Pyrin contains an N-terminal Pyrin domain, a B-box, a coiled-coil, and a C-terminal B30.2/SPRY domain (Henry et al, 1998). Most FMF-causing mutations are located in the SPRY domain and are thought to produce Pyrin protein with diminished or altered function (Consortium, 1997; Schaner et al, 2001).
A partial loss of mutations in the SPRY domain of an E3 ubiquitin ligase MID1/Midline 1/Midin/TRIM18 is responsible for Opitz syndrome (OS), characterized by multiple congenital abnormalities of midline structures due to defects in the embryonic development at the midline. MID1 also belongs to the TRIM family and shares the same modular organization as TRIM5α. How the defect caused by the mutations results in the OS disease is unknown. Most of the OS-causing mutations are clustered at the C-terminus of the MID1 gene corresponding to the coiled-coil region and the B30.2/SPRY domain, while the rest of the mutations are spread over almost all parts of the protein (De Falco et al, 2003; Schweiger and Schneider, 2003).
GUSTAVUS/CG2944-PF belongs to the SPRY domain-containing SOCS box protein family (Hilton et al, 1998). It is composed of an N-terminal short stretch of ∼40 residues, a central B30.2/SPRY domain, and a C-terminal SOCS box. The SOCS box is an ∼50 amino-acid stretch, first identified in the suppressor of cytokines signaling (SOCS) protein family (Starr et al, 1997). The SOCS box contains a consensus BC box that binds to the heterodimer of Elongins B and C (Kamura et al, 1998). The interaction could couple a target protein to ubiquitination–proteosomal degradation (Pintard et al, 2004). Independently of Elongins B and C, GUSTAVUS interacts with an RNA helicase VASA, a key protein in establishing a translational activity required for accumulation of several germ-line proteins at a specialized cytoplasm of developing Drosophila oocytes, called the pole plasm (Carrera et al, 2000). Through the interaction, GUSTAVUS plays an essential role in the localization of VASA at the posterior pole of the oocyte (Styhler et al, 2002). A genetic mutation in the gus locus or a deletion of the GUSTAVUS-interacting segment of VASA prevented the posterior localization of VASA and caused female sterility. The function of GUSTAVUS in the germ cell lineage should be conserved throughout metazoan evolution, as extremely similar mammalian orthologues are found, including human SPRY domain-containing SOCS box (SSB) proteins, SSB-1 and -4, which exhibit ∼69% sequence identity with GUSTAVUS.
A few binding analyses suggest that the B30.2/SPRY domain is a protein-interacting module. Deletion of the B30.2/SPRY domain in RanBPM, Ro52, and TRIM11 abrogated the interaction of the proteins with their binding partner proteins (Rhodes et al, 2002; Wang et al, 2002; Niikura et al, 2003). The B30.2/SPRY domain of GNIP/TRIM7 was sufficient for the interaction with glycogenin in yeast two-hybrid assay (Zhai et al, 2004). Moreover, in vitro binding was observed between xanthine dehydrogenase and the glutathione S-transferase-fused cytoplasmic domain of butyrophilin (BTN1A1), a transmembrane protein composed of two extracellular immunoglobulin-like domains and a cytoplasmic B30.2 domain (Ishii et al, 1995). However, it is not clear whether these domains may recognize a consensus sequence or not. In addition, although the sequence homology among the B30.2/SPRY domains suggests the presence of a ligand- or substrate-binding site commonly located on their structures, relevant information is totally lacking. Furthermore, the relationship between the B30.2, SPRY, and PRY domains remains confusing. Herein, combined with biochemical and biophysical studies, the structure of a B30.2/SPRY domain of GUSTAVUS provides answers to these questions. Mapping of sequence variations or disease-associated mutations identified for TRIM5α, Pyrin, and MID1 onto the presented canonical structure provides insights into the functional role of the B30.2/SPRY domains.
Results
Structure determination
The reported structure is for a truncated version of GUSTAVUS in a ternary complex with Elongins B and C at 1.8 Å resolution. Initially, we cloned six different B30.2/SPRY-containing proteins: mouse SSB-1, -2, -3, -4, human TRIM5α, and Drosophila GUSTAVUS. Of these, only full-length GUSTAVUS was expressed as a soluble protein and yielded microcrystals. After an intensive but unrewarding effort to improve the crystals, several GUSTAVUS constructs were designed, expressed, and subjected to crystallization. One of the constructs was devoid of 28 residues at both N- and C-terminus, but contained the central B30.2/SPRY domain and the C-terminal BC box. The truncation of the C-terminal region was based on the observation that it was easily cleaved off during the purification and crystallization of full-length GUSTAVUS. With this construct, we explored a possibility that the complex of Elongins B and C binds to the BC box of GUSTAVUS and provides favorable crystal packing interactions. Indeed, well-diffracting crystals were obtained with truncated GUSTAVUS containing residues 29–253 (hereafter referred to as GUS) in complex with mouse Elongins B and C (hereafter referred to as ElonginBC), and as we wished, the ElonginBC molecules provided the GUS molecules with crystal packing interactions. The choice of mouse ElonginBC instead of the Drosophila counterpart was based on the observation that Elongin C, which constitutes the BC box-binding surface, exhibits 90% amino-acid sequence identity between the two species. The structure of the ternary complex was solved with single-wavelength anomalous dispersion (SAD) phasing by using a crystal of selenomethionine-substituted proteins.
Overall structure
GUS interacts with ElonginBC through the BC box, and in this ternary complex, the three proteins are linearly arranged (Figure 1A). The crystal packing interactions between symmetry-related molecules do not indicate an oligomerization of the ternary complex, which is consistent with the molecular weight of the complex (50 kDa) estimated by gel filtration (data not shown). The B30.2/SPRY domain adopts a highly distorted, compact β-sandwich fold with two additional short α-helices at the N-terminus (Figure 1A). Both the N- and C-terminal ends of the B30.2/SPRY domain are close to each other. This structural feature and the monomeric nature suggest that the B30.2/SPRY domains in general are suitable for serving as a modular domain in multidomain proteins. The BC box of the C-terminal SOCS box forms the third α-helix and interacts with Elongin C of the ElonginBC complex (Figure 1A). Given the structure of ElonginBC in complex with the von Hippel Lindau protein (pVHL) reported previously (Stebbins et al, 1999), a detailed description of the interactions between the BC box of GUS and ElonginBC is refrained here. However, a distinction in the intermolecular interactions is notable as described in Supplementary Figure 1.
Figure 1.
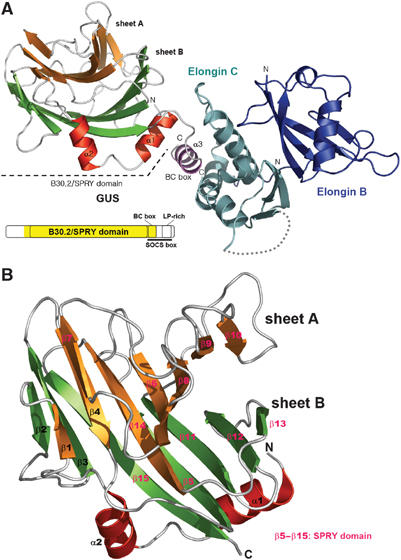
Structure of the GUS:ElonginBC complex. (A) Ribbon drawing of the GUS:ElonginBC complex. The α-helices of GUS are in either red or magenta (for BC box), sheet A in orange, and sheet B in green. The domain organization of GUSTAVUS is shown. The region in yellow on the diagram indicates the fragment of the protein used for the structure determination. (B) Ribbon drawing of the B30.2/SPRY domain of GUS. The secondary structural elements are sequentially labeled and colored as in (A). The SPRY domain in GUS is indicated by color-coding the secondary structural elements.
Structure of B30.2/SPRY domain and its structural homologs
The β-sandwich of the B30.2/SPRY domain consists of two layers of β-sheets (Figure 1B): sheet A composed of eight strands (β1, β4, β6, β7, β8, β9, β10, and β14) and sheet B composed of seven strands (β2, β3, β5, β11, β12, β13, and β15). All the β-strands are in antiparallel arrangement. Loops of varying length connecting the β-strands extend out from both ends of the β-sandwich. Helix α1 sits atop a part of the saddle-like platform formed by β5, β11, β12, and β15. The interactions between α1 and the β-sheet are extensive and exclusively hydrophobic (Supplementary Figure 2), indicating that the helix is structurally important probably for stabilizing the curvature of the highly distorted β-sandwich. The short two-turn helix α2 interacts with a part of one edge of the β-sandwich. The interaction between the two involves many water molecules and is predominantly hydrophilic, indicating that the presence of this helix may not be absolutely required for the β-sandwich scaffolding. A database search using the program Dali (Holm and Sander, 1995) revealed that the B30.2/SPRY domain of GUS is quite similar to eight proteins in the three-dimensional structure and folding topology (Z-score>6.5). They are the carbohydrate recognition domain of human galectin-3, a hypothetical protein yesu and β-1,4-xylosidase of Bacillus subtilis, a domain of human thrombospondin-1, a glycosyl hydrolase of Clostridium perfringens, sialidase of Trypanosoma rangeli, alginate lyase of Corynebacterium, and a domain in congerin I of Conger myriaster. Despite the significant structural homology, none of these proteins shows sequence similarity to any known B30.2/SPRY domains. The SCOP database (Murzin et al, 1995) classifies six of these structural homologs under the concanavalin A-like lectins/glucanases fold, while the remaining two have not been classified in the database yet. The ligand-binding sites have been identified for the domains of gallectin-3 (Seetharaman et al, 1998) and congerin I (Shirai et al, 1999). They are commonly located on a β-sheet, which corresponds to sheet A of GUS. However, the corresponding groove is fully occupied by loop β7–β8 in the GUS structure, and thus the surface on sheet A is unlikely to be responsible for a potential intermolecular interaction of GUS.
Comparison with other B30.2 and PRY-SPRY domains
A sequence analysis of GUS by SMART (Schultz et al, 1998) identifies an SPRY domain (residues 99–234), but not a PRY domain, while InterProScan (Zdobnov and Apweiler, 2001) identifies the B30.2 domain, which spans most of the SPRY domain and 60 residues preceding this domain. The preceding residues, corresponding to the PRY domain of tandem PRY-SPRY domains, constitute the secondary structural elements α1, α2, β1, β2, β3, and β4. Since GUS is a one-domain structure, we asked whether an SPRY domain and the preceding ∼60 residues in other proteins also constitute a single domain structure. Figure 2A shows a multiple sequence alignment and secondary structure prediction of phylogentically dispersed and relatively well-studied SPRY domain-containing proteins. As expected, they exhibited poor overall sequence homology, which greatly diminishes in the N-teminal ∼60 residues. Nevertheless, each of these proteins contains 12–13 predicted β-strands, whose locations on the primary sequences are markedly similar across the alignment. According to the prediction, the N-terminal ∼60 amino-acid segments, some of which are classified as the PRY domain, commonly contain at least one α-helix and three β-strands corresponding to the structurally important helix α1 and to β2 through β4 of the GUS structure. Therefore, the N-terminal segment is likely to constitute a single-domain structure together with the SPRY domain in each protein. Probably, the genes coding for the PRY-SPRY and B30.2 domains have derived from the same ancestral gene, and the first part of the gene encoding the N-terminal ∼60 residues has diversified much further than the rest of the gene through evolution.
Figure 2.
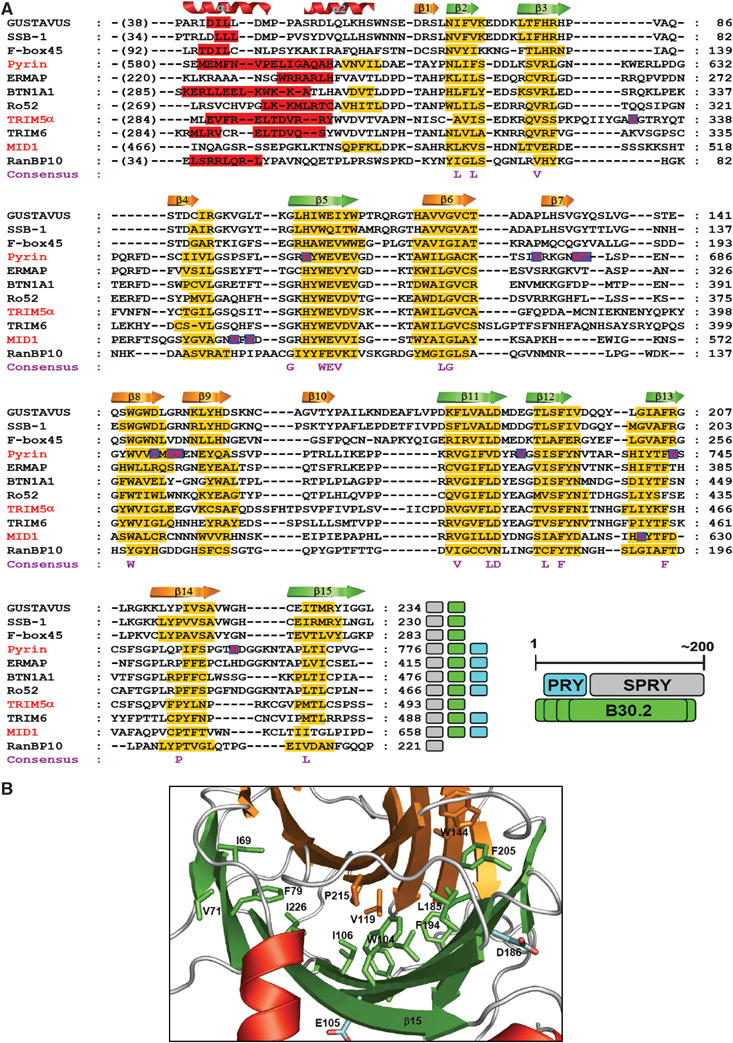
Multiple sequence alignment and consensus residues. (A) Sequence alignment and secondary structure prediction of B30.2/SPRY domains. In total, 20 sequences of SPRY domains and ∼60 preceding amino acids were aligned (Supplementary Figure 3), and 11 of these are shown. The sequences were first aligned using ClustalX (Thompson et al, 1997) for a global alignment, followed by manual adjustments based on the consensus secondary structure prediction using NPSA (Deleage et al, 1997) and Jpred (Cuff et al, 1998). The secondary structures predicted by Jpred are indicated by red bars for α-helices and yellow bars for β-strands. The crystallographically determined secondary structural elements of GUS are shown at the top of the alignment. The letters at the bottom of the alignment indicate the similarity consensus residues conserved in the 20 sequences. The red letters in blue box indicate the positions of the mutations causing FMF (in pyrin), OS (in MID1), or the single amino-acid substitution in human TRIM5α that confers the ability to restrict HIV-1. The accession numbers for the sequences are listed in the Materials and methods section. The colored boxes at the end of each sequence indicate the domains identified in the databases: gray, SPRY domain; green, B30.2 domain; cyan, PRY domain. The relationship between the three domains on the primary structure is schematically drawn. Varying length of the B30.2 domains is indicated by multiple lines. (B) Location of the consensus residues. The consensus residues (shown in A) are represented as sticks on the ribbon drawing of the B30.2/SPRY domain of GUS and most of them are labeled. The hydrophobic residues on sheets A and B are in orange and green, respectively, while the hydrophilic residues are in cyan.
Consensus residues are all buried
The consensus residues among the 20 aligned sequences (Figure 2A, Supplementary Figure 3) are mostly hydrophobic amino acids and exclusively located on the secondary structural elements. The structure of GUS shows that all the hydrophobic consensus residues are buried completely in between two β-sheets of GUS (Figure 2B). Likewise, all the polar consensus residues are nearly buried and involved in hydrophilic as well as hydrophobic interactions with nearby residues (data not shown). Therefore, the consensus residues are important for maintaining the structural scaffold, strongly suggesting that the tertiary structures of all the B30.2/SPRY domains are similar. The absence of any conserved residues on the protein surface distinguishes the B30.2/SPRY domain from other modular domains, such as SH2 and SH3 domains, which have highly conserved or invariant residues exposed on the ligand-binding surface and interact with a specific sequence motif (Waksman et al, 1992; Musacchio et al, 1994).
Mapping species-specific sequence variation and length polymorphism in TRIM5α
The B30.2/SPRY domain of the primate TRIM5α proteins is the sole region where the species-specific variations responsible for retroviral restriction are found (Figure 3A). A 13 amino-acid stretch in this domain is enriched with positively selected residues and/or is the place for the length polymorphism critical for retroviral restriction (Sawyer et al, 2005; Song et al, 2005). For example, a single amino-acid substitution of Arg332 on this segment of human TRIM5α with proline, the corresponding amino acid in rhesus monkey TRIM5α, resulted in a human protein that restricted HIV-1 nearly as efficiently as the rhesus monkey protein (Nisole et al, 2005; Stremlau et al, 2005; Yap et al, 2005). The polypeptide segment corresponds to β3–β4 loop of GUS composed of only three amino acids, but is predicted to be a long loop composed of 19 residues in TRIM5α (Figures 2A and 3B). β3–β4 loop, together with four other loops extending out from one end of the β-sheet, constitutes one face of the molecule. This surface (Figure 3B, designated as surface A) structurally corresponds to the complementary-determining region (CDR) of heterodimeric immunoglobulins (Davies and Cohen, 1996) or the phosphopeptide-binding region of Forkhead-associated (FHA) domains (Durocher et al, 2000). In addition to the 13 amino-acid stretch, two other regions, in which individual residue variations as well as significant length polymorphism occur, contribute partially to antiretroviral specificity of the primate TRIM5α proteins (Song et al, 2005). These two regions correspond to the loops between β6 and β8 and between β9 and β11 of GUS that form a part of surface A (Figure 3B, colored in pink), strongly suggesting that the length polymorphism can be easily accommodated without perturbing the putative β-sandwich structures of the TRIM5α proteins and that these putatively exposed loops, together with β3–β4 loop, are well suited for serving as the interface for antiretroviral activity.
Figure 3.
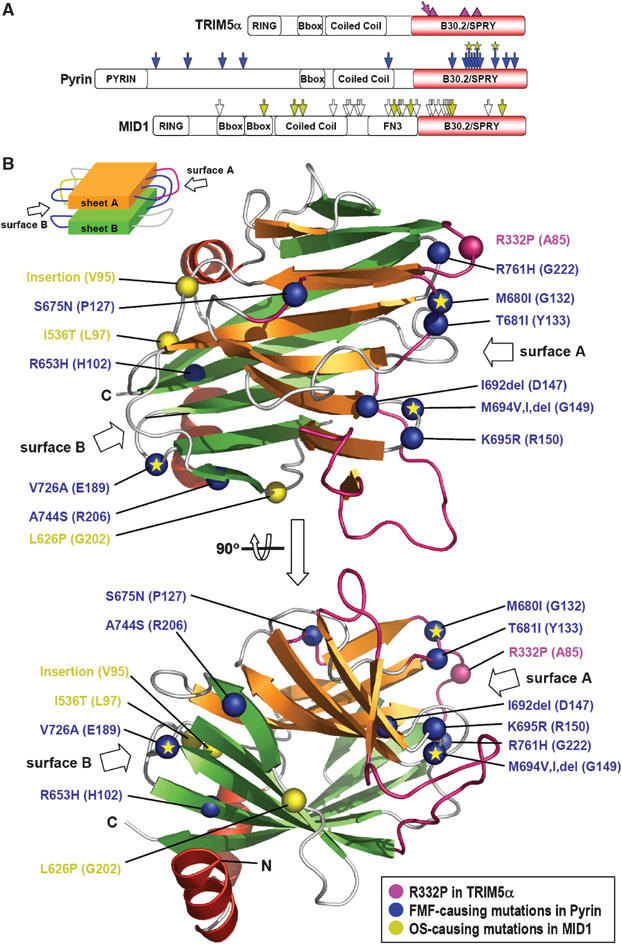
Sequence variations or mutations in TRIM5α, Pyrin, and MID1. (A) Location on the primary structures. The diagrams depict the primary structures of the three proteins and the domains they possess. The locations of the sequence variations or disease-causing mutations are marked on the diagrams for TRIM5α: pink triangles, locations of the significant sequence variation and length polymorphism in primate TRIM5α proteins; pink arrow, the substitution of R332P in human TRIM5α that confers the ability to restrict HIV-1, for Pyrin: blue arrows, the FMF-causing point mutations including three mutational hot spots marked with an asterisk, and for MID1: white arrows, the OS-causing frame shift or nonsense mutations; yellow arrows, point mutations, insertion, or deletion of amino acids. (B) Location of the corresponding residues of GUS on the tertiary structure. The sequence variations or mutations in the three proteins are mapped on the structure of the B30.2/SPRY domain of GUS. The mutation sites are indicated by large Cα atom spheres and labels shown in the same color of the arrows in (A). The residues of GUS corresponding to the mutation points are in the parentheses. The loop regions in pink correspond to the locations of the length polymorphism in the primate TRIM5α proteins. ‘Insertion' stands for the eight amino-acid insertional mutation in MID1, and ‘del' stands for deletion of a residue in Pyrin. A schematic drawing of the β-sandwich structure of GUS is shown to aid the recognition of surface A and surface B.
Mapping mutations in Pyrin responsible for FMF
All the known mutations in the B30.2/SPRY domain in human Pyrin were mapped on the structure of GUS (Figure 3B). They are the amino-acid changes at the positions of Arg653, Ser675, Met680, Thr681, Ile692, Met694, Lys695, Val726, Ala744, or Arg761 (Schaner et al, 2001). Of these, Met680, Met694, and Val726 are most commonly mutated in FMF patients (Bakkaloglu, 2003). Remarkably, six out of 10 residues including the mutational hot spots of M680 and M694 are concentrated on surface A. Except for one residue (Ser675), which is located on a loop above sheet A, the three other residues are clustered on the opposite surface (Figure 3B, designated as surface B). Both surfaces A and B are composed exclusively of loops in the GUS structure, strongly suggesting that the phenotypic changes caused by the mutations do not arise from a defect in the structural integrity of the proteins, but most likely from an alteration or a failure in the interaction with their binding partner.
Mapping mutations in MID1 responsible for OS
Two OS-causing point mutations and one insertional mutation on the B30.2/SPRY were identified (Figure 3A). These are the point mutations of I536T and L626P and an eight amino-acid insertion next to Val534 (De Falco et al, 2003; Schweiger and Schneider, 2003). Remarkably, the residues of GUS corresponding to the three mutation points are all located on surface B and exposed to the bulk solvent (Figure 3B). The observation suggests that surface B may serve as a ligand-binding site in a subset of B30.2/SPRY domains including that of MID1.
The B30.2/SPRY domain of GUS binds VASA tightly
VASA is known to interact with GUS in the yeast two-hybrid system (Styhler et al, 2002). Consistently, a (His)6-tag pull-down experiment showed that full-length VASA binds GUS tightly. A gel filtration analysis indicated a 1:1 stoichiometry of binding between GUS and VASA, both as a monomer (data not shown). Next, we sought to determine the minimal region of VASA that is sufficient to bind GUS. Five different deletion mutants of VASA were constructed, and their binding to GUS was tested by the (His)6-tag pull-down experiment (Figure 4A). The strong binding affinity for GUS was retained in a 30 amino-acid stretch of VASA (residues 174–203). This is consistent with the yeast-two hybrid experiment showing that a deletion of the identical region of VASA abolished the interaction between the two proteins (Styhler et al, 2002). A 20 amino-acid stretch of VASA (residues 174–193) exhibited a slightly diminished interaction with GUS compared with other mutants (data not shown). In a quantitative analysis of the binding interaction by isothermal titration calorimetry (ITC), the full-length VASA and a synthetic peptide identical to the 30 amino-acid stretch of VASA interacted with GUS with nearly the same and significantly high affinity (apparent dissociation constants (KDs) of approximately 40 nM) (Figure 4B). GUS in complex with ElonginBC interacted with the VASA peptide with a KD of 48 nM, indicating that the BC box is not involved in the binding of VASA and the bound ElonginBC molecules do not affect the interaction between GUS and the VASA peptide. Although we did not measure the binding affinity of full-length GUSTAVUS for VASA, the pronounced affinity of GUS for VASA suggests that the B30.2/SPRY domain of GUS is primarily, if not solely, responsible for the interaction with VASA.
Figure 4.

Identification of the GUS-binding region of VASA. (A) (His)6-tag pull-down experiment. Each of the indicated (His)6-tagged MBP-fused VASA deletion mutants and GUS without a tag were mixed and incubated with Ni-NTA agarose resin (QIAGEN). The resin was vigorously washed and subjected to denaturing gel electrophoresis. (B) ITC analysis of the interaction between GUS and VASA. The ITC experiments were carried out by titrating 5 μl of 0.15 mM of GUS into the solution of full-length VASA (5 μM) or titrating 5 μl of 0.2 mM of the 30 amino-acid VASA peptide into the solution of GUS (9 μM) or GUS in complex with ElonginBC (9 μM). KD values were deduced from curve fittings of the integrated heat per mol of added ligand and presented in the table. The titration curves obtained for the formation of the indicated complexes are shown in the insets.
Ligand-binding surface of GUS
To define the VASA-binding surface on the B30.2/SPRY domain of GUS, we tried to crystallize GUS or GUS:ElonginBC in complex with the VASA peptide. However, both the samples were refractory to crystallization, and, subsequently, we resorted to a mutational study. Given the FMF-causing mutations in Pyrin and the species-specific amino-acid variations in TRIM5α densely concentrated on surface A, we presumed that this region is the common ligand-binding surface of many SPRY domains, including that of GUS. We changed Tyr133, Gly149, Arg150, or Trp221 on surface A non-homologously into Ala, Tyr, Trp, or Leu, respectively (Figure 5A). It should be noted that Tyr133, Gly149, and Arg150 correspond to Thr681, Met694, and Lys695 of Pyrin, whose individual mutation is the cause of FMF. All the mutants were purified in complex with ElonginBC and the effect of these non-homologous substitutions on the binding affinity of GUS for the VASA peptide was estimated by ITC. The GUS (W221L) mutant exhibited a significant but measurable decrease in the binding affinity (KD of 4.5 μM) compared with the wild-type GUS (Figure 5B). The binding affinity of the other three mutants (Y133A, G149Y, and R150W) for the VASA peptide was too low to produce a meaningful titration curve required for the determination of the KD values by this method. To confirm this result, each of the mutants in complex with ElonginBC was mixed with the VASA peptide, dialyzed, and analyzed by denaturing gel electrophoresis. The bound VASA peptide was observed on the gel with the wild-type GUS, but not with the mutant proteins (Figure 5C). Consistently, each of the GUS mutants in complex with ElonginBC did not exhibit noticeable binding to full-length VASA (Figure 5D). For a comparison and as a control, we generated two GUS mutants containing a non-homologous substitution, W189A or R206S. Trp189 and Arg206 correspond to the two of the three mutation points of Pyrin mapped on surface B (Figure 5A). The two mutants exhibited virtually the same binding affinity for the VASA peptide as wild-type GUS in the calorimetric analysis (Figure 5B). Consistent results were obtained in the electrophoretic analyses, in which the GUS mutants exhibited the same band patterns on the denaturing and native gels as the wild-type protein (Figure 5C and D). Combined together, the data suggest that surface A, but not surface B, is the VASA-binding surface of the B30.2/SPRY domain of GUS. All the mutant proteins described above displayed unaltered biophysical properties as analyzed by native gel electrophoresis, gel-filtration, and circular dichroism spectroscopy (data not shown), assuring that the altered affinity of the mutants for the VASA peptide or protein is not attributable to a change in the structural integrity of the resulting proteins.
Figure 5.

Identification of the VASA-binding surface of GUS. (A) Locations of the site-directed mutagenesis on the B30.2/SPRY domain of GUS. The green dotted circles and labels on surface A indicate the mutations that decreased the binding affinity for the VASA peptide. The dotted circles in magenta indicate the mutations on surface B, which did not affect the binding affinity. The residues of GUS corresponding to the sequence variation and the disease-related mutations in TRIM5α, Pyrin, and MID1 are highlighted by the indicated colors. (B) ITC analysis of the interaction between GUS mutants and the VASA peptide. The titration curves and curve fittings are shown for two indicated GUS mutants and the KD values determined for the three GUS mutants are tabulated. (C) Denaturing gel elelctrophoretic analysis of the interaction between GUS mutants and the VASA peptide. Wild-type GUS and the mutants in complex with ElonginBC (each at 10 μM) were mixed with VASA peptide (30 μM) and incubated for 2 h. The mixtures were dialyzed for one day at 4°C, and each sample in the dialysis bags was subjected to denaturing gel electrophoresis. The mutants containing the amino-acid substitution on surface A or surface B are labeled with green and magenta letters, respectively. (D) Native gel electrophoretic analysis of the interaction between GUS mutants and full-length VASA. Wild-type and mutant GUS proteins in complex with ElonginBC (each at 10 μM) were mixed with VASA (5 μM) and incubated for 4 h. The analysis of the reaction mixtures by gel electrophoresis is shown. The amino-acid substitutions in the mutant proteins are color-coded as in C. While VASA mixed with wild-type GUS migrates to a distinct location, signifying a complex formation between the two, neither reduction of the band intensity nor appearance of a new band is observable for VASA mixed with the indicated GUS mutants.
Discussion
Domain boundaries, protein fold, and ligand-binding of B30.2/SPRY domains
The structure of the B30.2/SPRY domain of GUS can be described as a variation of the β-sandwich structure with a complex topology, which is most homologous to the concanavalin A-like lectins/glucanases folding scaffold. The low sequence similarity between the B30.2/SPRY domains, especially at the N-terminal ∼60 residues, obscures the boundaries of the domains. As a consequence, a new domain name PRY was coined for a group of similar sequence segments N-terminal to the SPRY domains. The diversified N-terminal sequences are likely to be an integral part of a single domain structure, according to the presented structural information and the sequence comparison and the secondary structure prediction of the B30.2/SPRY domains (Supplmentary Figure 3).
The monomeric state of GUS in solution and the 1:1 stoichiometry of binding between GUS and VASA suggest that the B30.2/SPRY domains generally do not induce oligomerization of the proteins containing this domain, and interact with a cognate ligand in a monomeric state. However, combined with an oligomerization domain, some B30.2/SPRY domains may interact with homooligomeric ligands. Consistently, TRIM5α forms a homotrimer owing to its coiled-coil domain, and the B30.2/SPRY domain of the protein in a homotrimeric state was suggested to interact with pseudo-threefold symmetrical structures on retroviral capsid (Mische et al, 2005).
Surface A structurally coincides with the CDR region of immunoglobulins, the phosphopeptide-binding surface of the FHA domains, and the carbohydrate-binding region of legume lectins (Sharma and Surolia, 1997). An important distinction of the B30.2/SPRY domains from the FHA domains and legume lectins is the absence of any conserved residues at the protein-binding surface. This feature is shared by the CDR region of immunoglobulins and readily indicates that the B30.2/SPRY domains have a varying individual specificity for binding a specific partner protein or different ligand rather than a consensus sequence or a specific motif. Besides surface A, surface B may also serve as the ligand-binding site in some of B30.2/SPRY domain-containing proteins, since the three OS-causing and three FMF-causing mutations map on this surface (Figure 3B). A relevant and open question is whether the B30.2/SPRY domain may be a module with an adaptor function to mediate interactions of two different proteins by using both surface A and surface B.
B30.2/SPRY domains as a protein-interacting module
The SMART database identifies 149 SPRY domain-containing human proteins. They can be roughly classified into four groups: the RING domain-containing proteins (∼33% of the total) including the TRIM family members (∼19%), the butyrophilin family proteins (∼15%), the TRIM family-like proteins lacking the RING domain and butyrophilin family-like proteins lacking the extracellular domain (∼12%), and other proteins (∼40%) such as SSB protein family members. The B30.2/SPRY domain in these proteins is likely to function through protein–protein interaction, in the light of several biochemical evidences of the direct interaction of this domain with a protein. These include the VASA-binding activity of GUS shown in this study, the interaction of the B30.2/SPRY domain-containing cytoplasmic domain of butyrophilin with xanthine dehydrogenase (Ishii et al, 1995), and that between the B30.2/SPRY domain of TRIM7 and glycogenin (Zhai et al, 2004). In contrast, there has been no report implicating the B30.2/SPRY domain as a module having an enzyme activity or interacting with a small molecule. In this context, the critical role of B30.2/SPRY domain of the TRIM5α proteins may lie in specific recognition of the differences in viral capsid proteins by surface A of this domain. Likewise, Surface A and/or B of Pyrin and MID1 would recognize(s) an unknown partner protein to exert the function of the proteins.
Concluding remarks
The presented work revealed the structure, the domain boundaries, and the target protein-binding surface of the B30.2/SPRY domain of GUS, which provide an insight into how the B30.2/SPRY domains may function through protein–protein interaction. At least majority of the B30.2/SPRY domains, if not all, would be protein-interacting modules that bind a partner protein with the target specificity determined by the amino-acid sequence variation on the ligand-binding surface. These ubiquitously present domains are reminiscent of the variable domains of immunoglobulins, in that both the proteins adopt a β-sandwich fold as a core structure and display a highly variable ligand-binding surface(s). This structural property of the B30.2/SPRY domain, the same fold but different specificity for a target protein, appears to be utilized in a vast number of functionally unrelated proteins. The presented structure and conclusions drawn from this study should serve as a canonical model and framework for studying numerous B30.2/SPRY domain-containing proteins.
Materials and methods
Protein expression and purification of GUS:ElonginBC
The Drosophila GUS gene coding for residues 29–253 was amplified by polymerase chain reaction (PCR) and ligated into the pPosKJ vector (Kwon et al, 2005). Each of the coding sequences for mouse Elongin B (full length) and Elongin C (residues 17–112) was first cloned into pET30a (Novagen). From these vectors, a two-promoter expression vector was constructed in which the two genes were under the control of T7 promoters. The two vectors were transformed in Escherichia coli BL21 (DE3) RIG strain (Novagen). The proteins were expressed at 15°C overnight and purified using a Ni-NTA column (QIAGEN) and a Hitrap Q anion exchange column (Amersham Biosciences). The N-terminal (His)6-tagged Vitreoscilla hemoglobin attached to GUS was removed by TEV protease and the reaction mixture was loaded onto an Ni-NTA column from which the protein complex was eluted unbound. The complex was further purified with a Hitrap Q column and a Superdex 200 gel filtration column (Amersham Biosciences). The fraction containing the complex was concentrated to 30 mg/ml in 20 mM HEPES buffer (pH 7.5) containing 2 mM β-mercaptoethanol (β-ME) and used for crystallization. The selenomethionine-substituted GUS:ElonginBC complex was produced in E. coli B834 (DE3) methionine auxotroph (Novagen) and purified as described above.
Construction and purification of GUS mutants
A standard PCR protocol was used to generate GUS mutant genes. Each of the GUS mutants was coexpressed with ElonginBC and purified as the ternary complex according to the procedure used for the purification of wild-type GUS in complex with ElonginBC. The final samples were dialyzed against a buffer solution composed of 20 mM sodium phosphate (pH 7.5), 150 mM sodium chloride, and 2 mM β-ME for ITC analysis.
Construction and purification of VASA and deletion mutants
Drosophila VASA gene was ligated into the pProEX vector (Invitrogen) and expressed in E. coli BL21 (DE3) RIG strain (Novagen). The expressed protein was purified using a Ni-NTA and Hitrap Q column, and then ATP was added in the protein solution to the final concentration 50 μM. The N-terminal (His)6-tag was removed by TEV protease and the reaction mixture was loaded onto an Ni-NTA column from which the VASA protein was eluted unbound. The protein was further purified on a Hitrap Q column and dialyzed in the buffer composed of 20 mM sodium phosphate (pH 7.5), 150 mM sodium chloride, and 2 mM β-ME. The coding sequences for five fragments of VASA (residue 174–661, 174–458, 130–203, 174–203, 174–193) were ligated into the modified pMAL-c2 vector (pMAL-c2: New England Biolabs) to produce the deletion mutants as N-terminal (His)6-tagged MBP-fused proteins. Each mutant protein was purified using a Ni-NTA column and a Hitrap Q column.
Crystallization and structure determination
Crystals of the GUS:ElonginBC ternary complex were obtained by the hanging-drop vapor-diffusion method at 20°C by mixing and equilibrating 1 μl of each of the protein solution and a precipitant solution containing 18% (w/v) PEG 2000, 0.2 M ammonium tartrate, and 0.1 M sodium potassium phosphate. The crystals belonged to the space group P212121, and contained one heterotrimer in the asymmetric unit. A SAD data set was collected with a crystal of the selenomethionine-substituted complex on the beamline 6B at the Pohang Accelerator Laboratory, Korea. All diffraction data were processed using the programs DENZO and SCALEPACK (Otwinowski and Minor, 1997). Subsequent calculations were carried out using the CNS program suite (Brunger et al, 1998). Six selenium sites in the asymmetric unit were located and used for phase determination at 2.8 Å and phases were subsequently improved by solvent flipping as implemented in CNS. The quality of the electron density map was excellent and guided unambiguous model building of the GUS molecule. The position of the ElonginBC complex was determined by the molecular replacement method using the previously reported structure of pVHL:ElonginBC (Stebbins et al, 1999). The model building and refinement were performed with the programs O (Jones et al, 1991) using 1.8 Å native data collected on the beamline 4 A at the Pohang Accelerator Laboratory, Korea. The final model does not include the following residues: GUS, residues 29–34, 252–253; Elongin B, 99–118; Elongin C, 50–57. These residues constitute flexible loops whose electron densities were unobserved or very weak. Crystallographic data statistics are summarized in Supplementary Table I.
Isothermal titration calorimetry
All measurements were carried out at 26°C on a MicroCalorimetry System (Microcal). GUS or the protein in complex with ElonginBC and GUS mutants in complex with ElonginBC were dialyzed against a buffer solution containing 20 mM sodium phosphate (pH 7.5) and 150 mM sodium chloride. The chemically synthesized 30 amino-acid VASA peptide (Peptron) or full-length VASA were dissolved in the same buffer. The samples were degassed for 20 min and centrifuged to remove any residuals prior to the measurements. Dilution enthalpies were determined in separate experiments (titrant into buffer) and subtracted from the enthalpies of the binding between the protein and the titrant. Data were analyzed using the Origin software (OriginLab Corp.).
CD spectroscopy
Data were collected on a JASCO J-715. CD spectra were recorded with protein samples (2.5–10 mM) in 20 mM phosphate buffer (pH 7.5) at 25°C over the range of 200–250 nm in a nitrogen atmosphere. Each spectrum is the accumulation of three scans corrected by subtracting signals from the buffer control.
Accession codes
The PDB identification codes for the proteins cited in the text are the carbohydrate recognition domain of human galectin-3 (1A3K), yesu (1OQ1), β-1,4-xylosidase (1YIF), a domain of human thrombospondin-1 (1UX6), glycosyl hydrolase (1UPS), sialidase (1MZ5), alginate lyase (1UAI), and a domain of congerin I (1C1L). The accession numbers in the sequence databases are GUSTAVUS (NP_724402.1), SSB-1 (NP_079382.2), SOCS-1 (NP_003736.1), WSB-1 (NP_056441.6), ASB-1 (NP_057198.1), RAR (AAA17031.1), MUF1(NP_006360.3), HIV-1 VIF (NP_057198.1), Elongin A (NP_003189.1) and pVHL (AAB64200.1), F-box45 (XP_117294.3), Pyrin (NP_000234.1), ERMAP (CAH72715.1), BTN1A1 (NP_001723.1), Ro52 (AAH10861.1), TRIM5α (NP_149023.1), TRIM6 (NP_477514.1), MID1 (NP_150632.1), and RanBP10 (NP_065901.1).
Coordinates
The coordinates of the GUS:ElonginBC structure have been deposited in the Protein Data Bank (accession code 2FNJ).
Supplementary Material
Supplementary Information
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Table 1
Acknowledgments
We greatly appreciate Dr P Lasko for providing the full-length VASA gene. The study made use of beamlines 6B and 4A at Pohang Accelerator Laboratory, Pohang, Korea, and was supported by Creative Research Initiatives of the Korean Ministry of Science & Technology. J-SW was supported by the Brain Korea 21 Project.
Competing interests statement The authors declare that they have no competing financial interests.
References
- Bakkaloglu A (2003) Familial Mediterranean fever. Pediatr Nephrol 18: 853–859 [DOI] [PubMed] [Google Scholar]
- Bateman A, Coin L, Durbin R, Finn RD, Hollich V, Griffiths-Jones S, Khanna A, Marshall M, Moxon S, Sonnhammer EL, Studholme DJ, Yeats C, Eddy SR (2004) The Pfam protein families database. Nucleic Acids Res 32: D138–D141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL (1998) Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr 54: 905–921 [DOI] [PubMed] [Google Scholar]
- Carrera P, Johnstone O, Nakamura A, Casanova J, Jackle H, Lasko P (2000) VASA mediates translation through interaction with a Drosophila yIF2 homolog. Mol Cell 5: 181–187 [DOI] [PubMed] [Google Scholar]
- Cuff JA, Clamp ME, Siddiqui AS, Finlay M, Barton GJ (1998) JPred: a consensus secondary structure prediction server. Bioinformatics 14: 892–893 [DOI] [PubMed] [Google Scholar]
- Davies DR, Cohen GH (1996) Interactions of protein antigens with antibodies. Proc Natl Acad Sci USA 93: 7–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Falco F, Cainarca S, Andolfi G, Ferrentino R, Berti C, Rodriguez Criado G, Rittinger O, Dennis N, Odent S, Rastogi A, Liebelt J, Chitayat D, Winter R, Jawanda H, Ballabio A, Franco B, Meroni G (2003) X-linked Opitz syndrome: novel mutations in the MID1 gene and redefinition of the clinical spectrum. Am J Med Genet A 120: 222–228 [DOI] [PubMed] [Google Scholar]
- Deleage G, Blanchet C, Geourjon C (1997) Protein structure prediction. Implications for the biologist. Biochimie 79: 681–686 [DOI] [PubMed] [Google Scholar]
- Durocher D, Taylor IA, Sarbassova D, Haire LF, Westcott SL, Jackson SP, Smerdon SJ, Yaffe MB (2000) The molecular basis of FHA domain:phosphopeptide binding specificity and implications for phospho-dependent signaling mechanisms. Mol Cell 6: 1169–1182 [DOI] [PubMed] [Google Scholar]
- Henry J, Mather IH, McDermott MF, Pontarotti P (1998) B30.2-like domain proteins: update and new insights into a rapidly expanding family of proteins. Mol Biol Evol 15: 1696–1705 [DOI] [PubMed] [Google Scholar]
- Hilton DJ, Richardson RT, Alexander WS, Viney EM, Willson TA, Sprigg NS, Starr R, Nicholson SE, Metcalf D, Nicola NA (1998) Twenty proteins containing a C-terminal SOCS box form five structural classes. Proc Natl Acad Sci USA 95: 114–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm L, Sander C (1995) Dali: a network tool for protein structure comparison. Trends Biochem Sci 20: 478–480 [DOI] [PubMed] [Google Scholar]
- Ishii T, Aoki N, Noda A, Adachi T, Nakamura R, Matsuda T (1995) Carboxy-terminal cytoplasmic domain of mouse butyrophilin specifically associates with a 150-kDa protein of mammary epithelial cells and milk fat globule membrane. Biochim Biophys Acta 1245: 285–292 [DOI] [PubMed] [Google Scholar]
- Jones TA, Zou JY, Cowan SW, Kjeldgaard M (1991) Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A 47: 110–119 [DOI] [PubMed] [Google Scholar]
- Kamura T, Sato S, Haque D, Liu L, Kaelin WG Jr, Conaway RC, Conaway JW (1998) The Elongin BC complex interacts with the conserved SOCS-box motif present in members of the SOCS, ras, WD-40 repeat, and ankyrin repeat families. Genes Dev 12: 3872–3881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon SY, Choi YJ, Kang TH, Lee KH, Cha SS, Kim GH, Lee HS, Kim KT, Kim KJ (2005) Highly efficient protein expression and purification using bacterial hemoglobin fusion vector. Plasmid 53: 274–282 [DOI] [PubMed] [Google Scholar]
- Mische CC, Javanbakht H, Song B, Diaz-Griffero F, Stremlau M, Strack B, Si Z, Sodroski J (2005) Retroviral restriction factor TRIM5alpha is a trimer. J Virol 79: 14446–14450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murzin AG, Brenner SE, Hubbard T, Chothia C (1995) SCOP: a structural classification of proteins database for the investigation of sequences and structures. J Mol Biol 247: 536–540 [DOI] [PubMed] [Google Scholar]
- Musacchio A, Saraste M, Wilmanns M (1994) High-resolution crystal structures of tyrosine kinase SH3 domains complexed with proline-rich peptides. Nat Struct Biol 1: 546–551 [DOI] [PubMed] [Google Scholar]
- Niikura T, Hashimoto Y, Tajima H, Ishizaka M, Yamagishi Y, Kawasumi M, Nawa M, Terashita K, Aiso S, Nishimoto I (2003) A tripartite motif protein TRIM11 binds and destabilizes Humanin, a neuroprotective peptide against Alzheimer's disease-relevant insults. Eur J Neurosci 17: 1150–1158 [DOI] [PubMed] [Google Scholar]
- Nisole S, Stoye JP, Saib A (2005) TRIM family proteins: retroviral restriction and antiviral defence. Nat Rev Microbiol 3: 799–808 [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276: 307–326 [DOI] [PubMed] [Google Scholar]
- Pintard L, Willems A, Peter M (2004) Cullin-based ubiquitin ligases: Cul3-BTB complexes join the family. EMBO J 23: 1681–1687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponting C, Schultz J, Bork P (1997) SPRY domains in ryanodine receptors (Ca(2+)-release channels). Trends Biochem Sci 22: 193–194 [DOI] [PubMed] [Google Scholar]
- Reddy BA, Etkin LD, Freemont PS (1992) A novel zinc finger coiled-coil domain in a family of nuclear proteins. Trends Biochem Sci 17: 344–345 [DOI] [PubMed] [Google Scholar]
- Rhodes DA, de Bono B, Trowsdale J (2005) Relationship between SPRY and B30.2 protein domains. Evolution of a component of immune defence? Immunology 116: 411–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes DA, Ihrke G, Reinicke AT, Malcherek G, Towey M, Isenberg DA, Trowsdale J (2002) The 52 000 MW Ro/SS-A autoantigen in Sjogren's syndrome/systemic lupus erythematosus (Ro52) is an interferon-gamma inducible tripartite motif protein associated with membrane proximal structures. Immunology 106: 246–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawyer SL, Wu LI, Emerman M, Malik HS (2005) Positive selection of primate TRIM5alpha identifies a critical species-specific retroviral restriction domain. Proc Natl Acad Sci USA 102: 2832–2837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaner P, Richards N, Wadhwa A, Aksentijevich I, Kastner D, Tucker P, Gumucio D (2001) Episodic evolution of pyrin in primates: human mutations recapitulate ancestral amino acid states. Nat Genet 27: 318–321 [DOI] [PubMed] [Google Scholar]
- Schultz J, Milpetz F, Bork P, Ponting CP (1998) SMART, a simple modular architecture research tool: identification of signaling domains. Proc Natl Acad Sci USA 95: 5857–5864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweiger S, Schneider R (2003) The MID1/PP2A complex: a key to the pathogenesis of Opitz BBB/G syndrome. Bioessays 25: 356–366 [DOI] [PubMed] [Google Scholar]
- Seetharaman J, Kanigsberg A, Slaaby R, Leffler H, Barondes SH, Rini JM (1998) X-ray crystal structure of the human galectin-3 carbohydrate recognition domain at 2.1-Å resolution. J Biol Chem 273: 13047–13052 [DOI] [PubMed] [Google Scholar]
- Sharma V, Surolia A (1997) Analyses of carbohydrate recognition by legume lectins: size of the combining site loops and their primary specificity. J Mol Biol 267: 433–445 [DOI] [PubMed] [Google Scholar]
- Shirai T, Mitsuyama C, Niwa Y, Matsui Y, Hotta H, Yamane T, Kamiya H, Ishii C, Ogawa T, Muramoto K (1999) High-resolution structure of the conger eel galectin, congerin I, in lactose-liganded and ligand-free forms: emergence of a new structure class by accelerated evolution. Struct Fold Des 7: 1223–1233 [DOI] [PubMed] [Google Scholar]
- Song B, Javanbakht H, Perron M, Park do H, Stremlau M, Sodroski J (2005) Retrovirus restriction by TRIM5alpha variants from old world and new world primates. J Virol 79: 3930–3937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starr R, Willson TA, Viney EM, Murray LJ, Rayner JR, Jenkins BJ, Gonda TJ, Alexander WS, Metcalf D, Nicola NA, Hilton DJ (1997) A family of cytokine-inducible inhibitors of signalling. Nature 387: 917–921 [DOI] [PubMed] [Google Scholar]
- Stebbins CE, Kaelin WG Jr, Pavletich NP (1999) Structure of the VHL–ElonginC–ElonginB complex: implications for VHL tumor suppressor function. Science 284: 455–461 [DOI] [PubMed] [Google Scholar]
- Stremlau M, Owens CM, Perron MJ, Kiessling M, Autissier P, Sodroski J (2004) The cytoplasmic body component TRIM5alpha restricts HIV-1 infection in Old World monkeys. Nature 427: 848–853 [DOI] [PubMed] [Google Scholar]
- Stremlau M, Perron M, Welikala S, Sodroski J (2005) Species-specific variation in the B30.2(SPRY) domain of TRIM5alpha determines the potency of human immunodeficiency virus restriction. J Virol 79: 3139–3145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Styhler S, Nakamura A, Lasko P (2002) VASA localization requires the SPRY-domain and SOCS-box containing protein, GUSTAVUS. Dev Cell 3: 865–876 [DOI] [PubMed] [Google Scholar]
- The International FMF Consortium (1997) Ancient missense mutations in a new member of the RoRet gene family are likely to cause familial Mediterranean fever. Cell 90: 797–807 [DOI] [PubMed] [Google Scholar]
- Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25: 4876–4882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernet C, Boretto J, Mattei MG, Takahashi M, Jack LJ, Mather IH, Rouquier S, Pontarotti P (1993) Evolutionary study of multigenic families mapping close to the human MHC class I region. J Mol Evol 37: 600–612 [DOI] [PubMed] [Google Scholar]
- Waksman G, Kominos D, Robertson SC, Pant N, Baltimore D, Birge RB, Cowburn D, Hanafusa H, Mayer BJ, Overduin M, Resh MD, Rios CB, Silverman L, Kuriyan J (1992) Crystal structure of the phosphotyrosine recognition domain SH2 of v-src complexed with tyrosine-phosphorylated peptides. Nature 358: 646–653 [DOI] [PubMed] [Google Scholar]
- Wang D, Li Z, Messing EM, Wu G (2002) Activation of Ras/Erk pathway by a novel MET-interacting protein RanBPM. J Biol Chem 277: 36216–36222 [DOI] [PubMed] [Google Scholar]
- Yap MW, Nisole S, Stoye JP (2005) A single amino acid change in the SPRY domain of human Trim5alpha leads to HIV-1 restriction. Curr Biol 15: 73–78 [DOI] [PubMed] [Google Scholar]
- Zdobnov EM, Apweiler R (2001) InterProScan—an integration platform for the signature-recognition methods in InterPro. Bioinformatics 17: 847–848 [DOI] [PubMed] [Google Scholar]
- Zhai L, Dietrich A, Skurat AV, Roach PJ (2004) Structure-function analysis of GNIP, the glycogenin-interacting protein. Arch Biochem Biophys 421: 236–242 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Table 1