

Abstract
Through its involvement in inflammation, opsonization, and cytolysis, the complement protects against infectious agents. Although most of the complement proteins are synthesized in the central nervous system (CNS), the role of the complement system in the normal or ischemic CNS remains unclear. Here we demonstrate for the first time that neural progenitor cells and immature neurons express receptors for complement fragments C3a and C5a (C3a receptor (C3aR) and C5a receptor). Mice that are deficient in complement factor C3 (C3−/−) lack C3a and are unable to generate C5a through proteolytic cleavage of C5 by C5-convertase. Intriguingly, basal neurogenesis is decreased both in C3−/− mice and in mice lacking C3aR or mice treated with a C3aR antagonist. The C3−/− mice had impaired ischemia-induced neurogenesis both in the subventricular zone, the main source of neural progenitor cells in adult brain, and in the ischemic region, despite normal proliferative response and larger infarct volumes. Thus, in the adult mammalian CNS, complement activation products promote both basal and ischemia-induced neurogenesis.
Keywords: C3, cerebral ischemia, complement system, mice, neurogenesis
Introduction
The complement system, a component of the humoral immune system, is involved in inflammation, opsonization, and cytolysis. Many cell types produce or express complement proteins. In the central nervous system (CNS), astrocytes, microglia, and nerve cells are, together, capable of synthesizing or expressing most of the complement proteins (Spiegel et al, 1998; Thomas et al, 2000; D'Ambrosio et al, 2001). However, the role of the complement system in the normal CNS remains unclear. Since the principal manifestation of complement deficiency in the CNS is increased susceptibility to meningococcal meningitis, it has been suggested that these proteins mainly provide defense against infectious agents (Spiegel et al, 1998).
C3a and C5a, generated upon complement activation through proteolytic cleavage of the third complement component (C3) and fifth complement component (C5), respectively, are small polypeptides with potent anaphylatoxic properties. C3a and C5a exert their functions at picomolar to nanomolar concentrations by binding to specific receptors, C3a receptor (C3aR) and C5a receptor (C5aR), respectively, that are members of the rhodopsin family of seven-transmembrane G-protein-coupled receptors (Ember et al, 1998). C3a and C5a are both chemoattractant molecules, but whereas C5a has broad proinflammatory effects, the effects of C3a are more selective and rather anti-inflammatory (Ember et al, 1998). Both C3aR and C5aR are expressed in the normal uninjured CNS, predominantly by hippocampal and cortical neurons (Davoust et al, 1999; O'Barr et al, 2001). In vitro studies have shown that C5a is mitogenic for undifferentiated human neuroblastoma cells, whereas it is neuroprotective for terminally differentiated cells (O'Barr et al, 2001). Oligodendrocyte progenitors express C5aR in vitro, but C5aR mRNA expression is downregulated upon differentiation into mature oligodendrocytes (Nataf et al, 2001).
The complement system has recently been proposed to participate in tissue regeneration. In amphibians, C3 expression was detected in regenerating limbs but not in developing limbs (Del Rio-Tsonis et al, 1998). The C3 and C5 proteins are expressed in regenerating newt limb and lens (Kimura et al, 2003). The newt ortholog of CD59, a membrane regulator of the terminal complement pathway, has been implicated in blastema positional identity during adult limb regeneration (Morais da Silva et al, 2002). In a mammalian system, C3a and C5a are critical for hepatocyte proliferation and liver regeneration (Mastellos et al, 2001; Strey et al, 2003; Daveau et al, 2004). Finally, C3a promotes homing (Reca et al, 2003), chemotaxis (Honczarenko et al, 2005), and retention of hematopoietic stem and progenitor cells in the bone marrow (Ratajczak et al, 2004), although the issue of C3aR expression on these cells and its involvement in these phenomena remains controversial.
We investigated whether neural stem cells express receptors for complement-derived anaphylatoxins and whether complement activation products might play a role in adult mammalian neurogenesis. We show that neural stem cells and neural progenitor cells express C3aR and C5aR, and that the complement system positively regulates basal adult neurogenesis as well as the number of newly born neurons after cerebral ischemia.
Results
C3aR and C5aR are expressed on neural stem cells and neural progenitors
C3aR has been shown to be expressed by hematopoietic stem cells and to play a role in homing to bone marrow (Reca et al, 2003). Oligodendrocyte progenitors express C5aR in vitro (Nataf et al, 2001). To investigate whether complement-derived anaphylatoxin receptors are present on neural stem cells, we stained clonally derived neural stem cells from adult rat hippocampus with antibodies against C3aR and C5aR. Both receptors appeared to be localized in the cell membrane and were homogeneously distributed (Figure 1A and C). As demonstrated by nestin positivity, the cells retained their stem cell phenotype (Figure 1A and C). The specificity of the anti-C3aR and anti-C5aR antibodies, respectively, was confirmed by the absence of immunostaining on brain sections from mice deficient in C3aR (Kildsgaard et al, 2000) and C5aR (Hopken et al, 1996), respectively (Figure 2A and B).
Figure 1.

Expression of C3aR and C5aR on cultured neural stem cells. Immunostaining of clonally derived neural stem cells from adult rat hippocampus for C3aR (A) and C5aR (C) shows homogeneous distribution of both receptors in the cell membrane. Nestin immunostaining confirmed that the cells retain their stem cell phenotype. The cells were infected with retrovirus to express green fluorescent protein (GFP). Negative control immunostaining, in which the primary antibodies against C3aR, C5aR, and nestin were omitted (B, D). Scale bar equals 20 μm.
Figure 2.
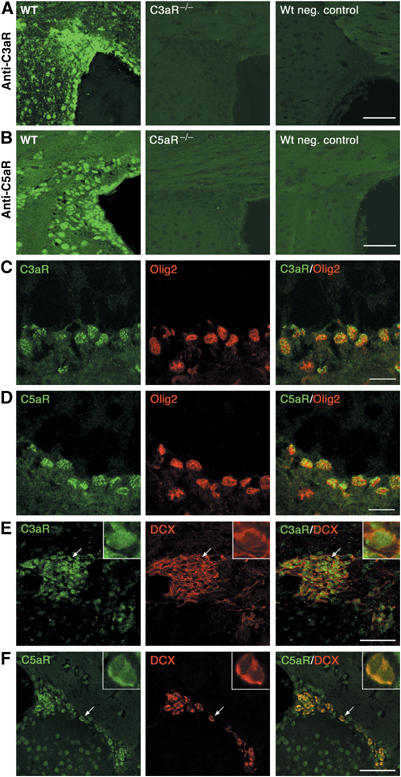
Expression of C3aR and C5aR on transit-amplifying precursors and migrating neuroblasts in vivo. Immunostaining of paraffin-embedded brain sections from adult mice with antibodies against C3aR (A) and C5aR (B). Both receptors are expressed by cells in the SVZ and rostral migratory stream of WT mice. The specificity of the anti-C3aR and anti-C5aR antibodies, respectively, was confirmed by the absence of immunostaining on brain sections from C3aR−/− and C5aR−/−mice. In the negative control immunostainings of WT brain sections, the primary antibodies were omitted. Olig2-positive transit-amplifying precursor cells in the SVZ are colabeled by anibodies against C3aR (C) and C5aR (D). Dcxpos migrating neuroblasts in the rostral migratory stream express C3aR (E) and C5aR (F). Arrows indicate double-labeled cells in the insert. Scale bars equal 50 μm (A, B) 10 μm (C, D) and 60 μm (E, F).
Oligodendrocyte transcription factor 2 (Olig2) is a marker of fast-dividing transit-amplifying precursor cells in the subventricular zone (SVZ) (Hack et al, 2004, 2005). Using brain sections from wild-type (WT) C57BL/6 mice, we showed that these cells express C3aR and C5aR (Figure 2C and D). Colabeling with doublecortin (Dcx), a marker of migrating neuroblasts, which are normally present in the SVZ and rostral migratory stream of adult brain (Nacher et al, 2001), showed that, in both regions, all Dcx-positive (Dcxpos) cells were also positive for C5aR and many were positive for C3aR (Figure 2E and F). Immunostaining for NeuN, a marker of differentiated neurons, showed that over 95% of NeuNpos cells were also positive for the C3aR and C5aR, whereas we did not find any colocalization of C3aR or C5aR with glial fibrillary acidic protein (GFAP), which marks adult neural stem cells as well as mature astrocytes (Doetsch et al, 1999) (data not shown). Thus, neural stem cells in vitro as well as murine neural progenitor cells, migrating neuroblasts, and mature neurons in vivo express C3aR and C5aR.
Reduced basal neurogenesis in the absence of signaling through C3aR
To assess whether signaling through the C3aR might play a role in basal neurogenesis, we injected WT C57BL/6 mice with a nonpeptide antagonist of the C3aR, SB290157 (Ames et al, 2001) for 10 days. Control as well as C3−/− mice were injected with vehicle. For the first 7 days, the mice also received bromodeoxyuridine (BrdU). Proliferating neural stem cells (GFAPposBrdUpos), transit-amplifying cells (Olig2posBrdUpos), migrating neuroblasts (DcxposBrdUpos), newly formed neurons (NeuNposBrdUpos), and BrdUpos cells were counted in the two principal sites of adult neurogenesis, SVZ and the dentate gyrus subgranular zone (SGZ) of the hippocampus, in the dentate gyrus granule cell layer (GCL) as well as in the olfactory bulb (OB), the final destination for the neuroblasts originating in SVZ under basal conditions (Gage, 2000; Alvarez-Buylla and Garcia-Verdugo, 2002; Doetsch, 2003).
The number of DcxposBrdUpos cells in OB and SGZ was reduced by 33–34% in both the C3−/− mice (P<0.0005) and the mice that received the C3aR antagonist compared to control mice (P<0.0005) (Figure 3). The quantification of NeuNposBrdUpos cells in OB and SGZ/GCL showed a 22–25% reduction in the number of these cells in both the C3−/− mice (P<0.05 and <0.005, respectively) and the C3aR antagonist-treated mice (P<0.005) (Figure 4). The number of BrdUpos, GFAPposBrdUpos, and Olig2posBrdUpos cells in any of the regions was not affected by the C3 deficiency or C3aR antagonist treatment (data not shown).
Figure 3.
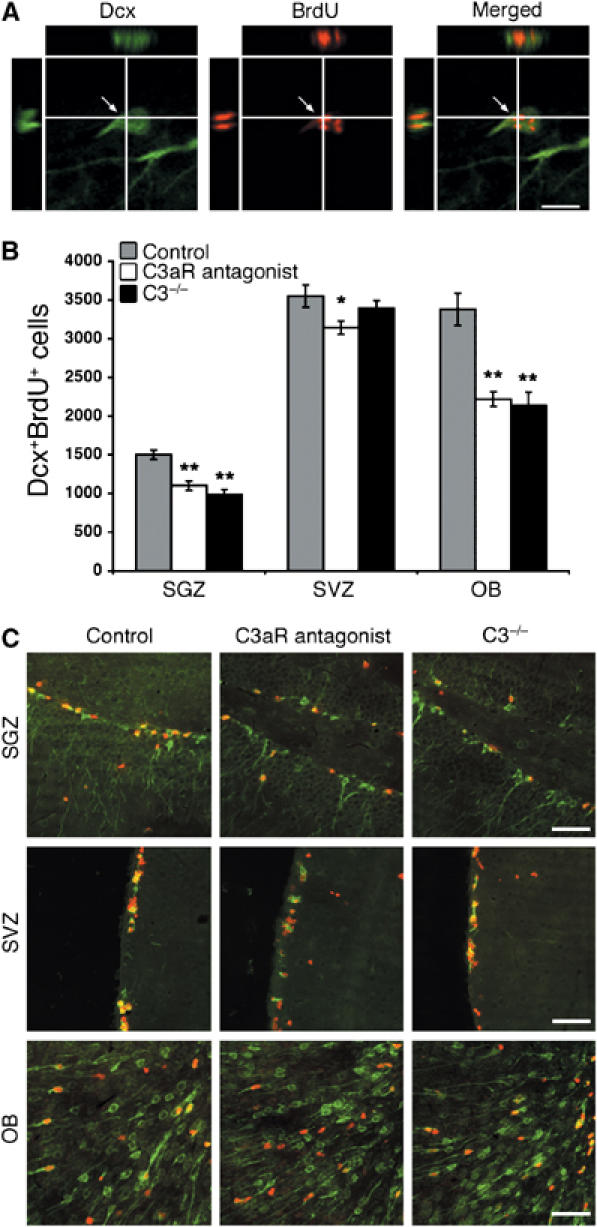
Proliferating migrating neuroblast in the dentate gyrus SGZ visualized by confocal microscopy in an orthogonal projection composed of 14 optical z-planes, 0.5 μm thick (A). The number of DcxposBrdUpos cells in SGZ, SVZ, and OB in control mice (n=12), C3−/− mice (n=6), and mice treated with C3aR antagonist (n=10) (B). Representative low-power images of DcxposBrdUpos cells in SGZ, SVZ, and OB in the three groups of mice; Dcx, green, BrdU, red (C). Values are the number of cells per region. *P<0.05, **P<0.0005. Scale bars equal 10 μm (A) and 200 μm (C).
Figure 4.
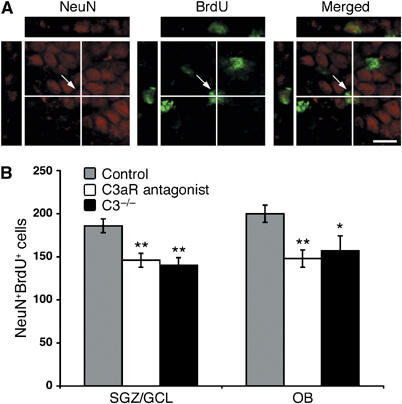
A newly formed neuron in the dentate gyrus GCL visualized by confocal microscopy in an orthogonal projection composed of 14 optical z-planes, 0.5 μm thick (A). The number of NeuNposBrdUpos cells in the SGZ/GCL and OB in control mice (n=12), C3−/− mice (n=6) and mice treated with C3aR antagonist (n=10) (B). Values are the number of cells per region. *P<0.05, **P<0.005. Scale bar equals 10 μm.
To further confirm the role of C3aR in neurogenesis, we injected C3aR−/− and WT C57BL/6 mice with BrdU for 7 days and assessed the number of BrdUpos, DcxposBrdUpos, and NeuNposBrdUpos cells in SVZ, OB, and SGZ/GCL 14 days later. In the C3aR−/− mice, the number of DcxposBrdUpos cells in OB and SGZ was 22 and 28% lower, respectively (P<0.05) (Figure 5A). Similarly, the C3aR−/− mice showed a 22–28% reduction in the number of NeuNposBrdUposcells in OB and SGZ/GCL (P<0.05), (Figure 5B). There was no difference in the number of BrdUpos cells between the groups (data not shown).
Figure 5.
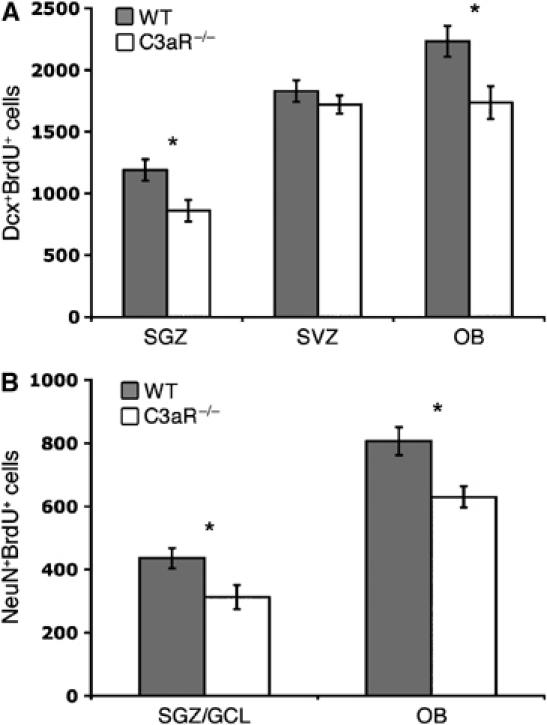
Basal neurogenesis is reduced in C3aR−/− mice. Compared to control (WT) mice (n=6), the C3aR−/− mice (n=6) have lower number of newly formed migrating neuroblasts in the SGZ and OB (A) and lower number of newly formed neurons in SGZ/the dentate gyrus GCL and OB (B). Values are the number of cells per region. *P<0.05. The higher number of NeuNposBrdUpos cells in control mice in this figure compared to Figure 4 is likely due to a difference in survival time after the last BrdU injection (14 versus 3 days).
Taken together, these findings indicate that signaling through the C3aR positively regulates basal neurogenesis, presumably by stimulating differentiation of neural progenitors. Since the newly formed cells have to migrate over a considerable distance from SVZ to their final destination in OB, the observed findings in OB can be the result of a combined defect in cell migration and differentiation in the absence of C3aR signaling.
Reduced ischemia-induced neurogenesis in SVZ in C3−/− mice
C3−/− mice lack C3a and are unable to generate C5a through proteolytic cleavage of C5 by C5-convertase (Pekna et al, 1998). To determine if complement activation plays a role in ischemia-triggered neurogenesis, we subjected C3−/− mice and WT controls to middle cerebral artery (MCA) occlusion (MCAO), a model of cerebral ischemia that leads to an ischemic infarct in the cerebral cortex and striatum. After ischemia, neural progenitor cells were reported to proliferate in the SVZ ipsilateral to the insult, migrate into the damaged area, and differentiate into neurons (Arvidsson et al, 2002; Parent et al, 2002; Jin et al, 2003). Therefore, we compared the numbers of Dcxpos migrating neuroblasts in the SVZ in the two groups of mice 7 days after ischemia. We found that the WT mice had 46% more Dcxpos cells in the ipsilateral than in the contralateral SVZ (P<0.005) compared to increase by 26% in the C3−/− mice (P=0.064).
Moreover, the C3−/− mice had 24% fewer Dcxpos cells in the ipsilateral SVZ than WT mice (P<0.05); both groups had a similar number of these cells in the contralateral SVZ (Figure 6A).
Figure 6.
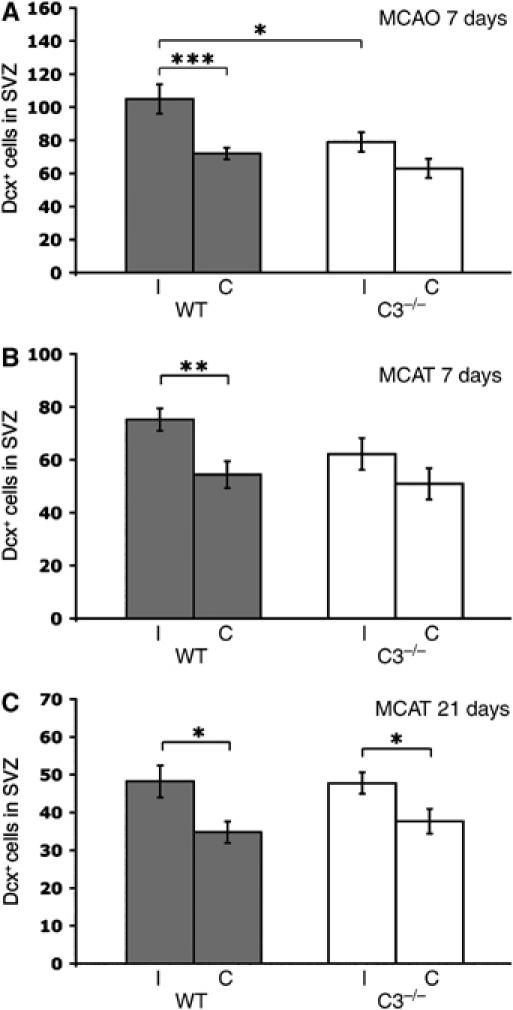
Number of Dcxpos migrating neuroblasts in the SVZ in the ipsilateral (I) and contralateral (C) hemispheres of WT (n=7, 8, 8) and C3−/− (n=9, 7, 11) mice. The mice were subjected to focal cerebral ischemia by MCAO or MCAT. Dcxpos cells in the SVZ were counted in three sections per mouse 7 days after MCAO (A) and MCAT (B) and 21 days after MCAT (C). Values are the number of cells per section. *P<0.05, **P<0.01, ***P<0.005.
To confirm the findings from the MCAO study and to be able to assess ischemia-induced neurogenesis at a later time point, we used a MCA transection (MCAT) model in which the infarct volume is approximately 20% of that after MCAO (Welsh et al, 1987; Fotheringham et al, 2000). At both 7 and 21 days after MCAT, the WT mice had 38% more Dcxpos cells in the ipsilateral than in the contralateral SVZ (P<0.01 and <0.05, respectively). Interestingly, the C3−/− mice showed a less pronounced ischemia-induced increase in the number of Dcxpos cells with only 22 and 27%, respectively, more Dcxpos cells in the ipsilateral compared to the contralateral SVZ (P=0.2 and <0.05, respectively), (Figure 6B and C). These data suggest that the complement acts as a positive regulator of the ischemia-induced neural progenitor differentiation in SVZ. Alternatively, complement activation products may stimulate neural progenitor proliferation after ischemia. To distinguish between these two possibilities, we evaluated cell proliferation in SVZ using in vivo BrdU labeling. Before and for 7 days after MCAT, mice were injected with BrdU. In both groups, BrdUpos cells were more numerous in the ipsilateral than in the contralateral SVZ at 7 days (92.8±4.0 versus 77.9±4.1, P<0.05 for C3−/− mice and 90.0±3.9 versus 70.7±4.4, P<0.005 for WT mice) and 21 days after MCAT (14.5±1.0 versus 10.6±0.8, P<0.01 and 16.3±1.5 versus 10.9±1.1, P<0.05). Thus, complement stimulates neural progenitor differentiation in SVZ after ischemia.
Reduced ischemia-induced neurogenesis in the penumbra and infarct area in C3−/− mice
To determine whether complement affects the number of neural progenitor cells in the penumbra and infarct area, we stained the sections for nestin, a marker of neural progenitor cells that is also expressed by reactive astrocytes (Lendahl et al, 1990), and glial fibrillary protein (GFAP), a marker of reactive astrocytes. Compared with controls, the C3−/− mice had 50% fewer nestinpos GFAPneg neural progenitor cells in the infarct area (3.6±0.97 versus 8.3±1.76 cells/section, P<0.05) and in the penumbra (15.9±2.02 versus 28.4±2.22 cells/section, P<0.005) 7 days after MCAO and 30% fewer nestinpos GFAPneg cells in the penumbra (11.3±0.2 versus 16.1±0.5 cells/10 mm2, P<0.00001) 7 days after MCAT (Figure 7). Furthermore, the fraction of GFAPneg cells among the nestinpos cells was reduced by 29% in the C3−/− mice (5.4±0.1 versus 7.6±0.2%, P<0.00001). No nestinpos cells were detected in the penumbra of either group of mice at 21 days. It is not clear whether these cells are locally derived or originate in the neurogenic SVZ, as there is evidence to support both alternatives (Reynolds and Weiss, 1992; Palmer et al, 1999; Kondo and Raff, 2000; Gould et al, 2001). Regardless of their origin, the response of neural progenitors in the penumbra is positively regulated by complement-derived stimuli.
Figure 7.

Neural progenitor cells in the infarct area and penumbra of WT (n=7, 8, 8) and C3−/− (n=9, 7, 11) mice after focal cerebral ischemia. Immunostaining for nestin and GFAP was used to visualize nestinpos GFAPneg neural progenitor cells (A). These cells were counted in 2–3 sections/mouse in the infarct area and penumbra 7 days after MCAO and in the penumbra 7 days after MCAT (B). Values are the number of cells per section (MCAO) and the number of cells/10 mm2 (MCAT). Representative low-power images of the penumbra in the two groups of mice; nestin, red, GFAP, green. Arrows indicate nestinpos GFAPneg cells. Broken line depicts the infarct border. i, infarct (C). *P<0.05, ***P<0.005, *****P<0.00001. Scale bars equal 15 μm (A) and 200 μm (C).
To address whether the reduction in the number of neural progenitor cells and migrating neuroblasts might lead to reduced formation of new neurons, we used combined immunostaining for NeuN and BrdU. Intriguingly, the C3−/− mice had fewer newly formed neurons (NeuNposBrdUpos cells) in the penumbra both 7 days (7.5±0.4 versus 10.0±0.5 cells/10 mm2, P<0.001) and 21 days after MCAT (9.7±0.6 versus 13.3±0.9 cells/10 mm2, P<0.005) (Figure 8). Also, the fraction of NeuNpos cells among the BrdUpos cells was reduced in the C3−/− mice (3.4±0.2 versus 4.76±0.3%, P<0.005 and 4.9±0.4 versus 6.5±0.4%, P<0.05).
Figure 8.
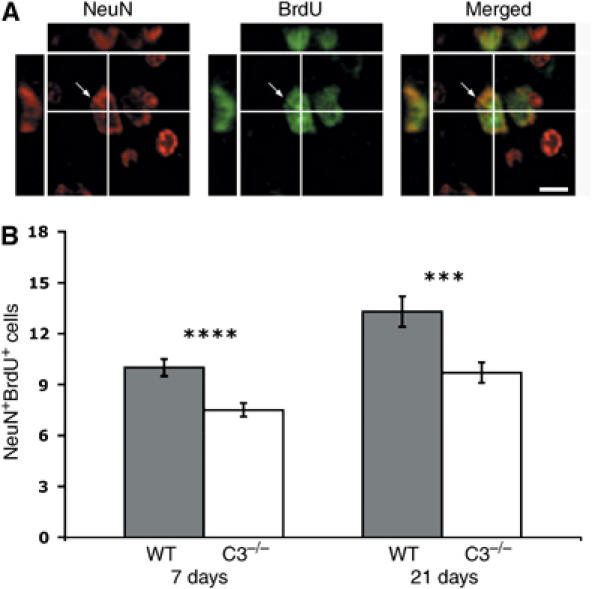
Ischemia-induced generation of new neurons. A newly formed NeuNposBrdUpos neuron in the penumbra visualized by confocal microscopy in an orthogonal projection composed of 14 optical z-planes, 0.5 μm thick (A). Number of NeuNposBrdUpos cells in the penumbra of WT (n=10, 11) and C3−/− (n=9, 11) mice 7 and 21 days after MCAT (B). The number of NeuNposBrdUpos cells/10 mm2 was counted in the penumbra on 2–3 sections/mouse. ***P<0.005, ****P<0.001. Scale bar equals 15 μm.
Postmitotic neurons can incorporate BrdU through stress-induced cell cycle activation before undergoing delayed neuronal death by apoptosis (Sanes and Okun, 1972; Osuga et al, 2000; Katchanov et al, 2001; Liu and Greene, 2001; Verdaguer et al, 2002). To determine whether the NeuNposBrdUpos cells in the penumbra are undergoing ischemia-induced apoptosis, we performed triple-label immunostaining with antibodies against BrdU, NeuN, and activated caspase-3, the latter being expressed in association with delayed ischemic neuronal death (Namura et al, 1998). No activated caspase-3pos cells were detected in the infarct area or within the penumbra 21 days after MCAT; however, 7 days after MCAT, caspase-3 immunoreactivity was confined to the infarct and NeuN to the penumbra (Figure 9). No NeuNposBrdUpos cells were also positive for activated caspase-3, which proves that the NeuNposBrdUpos cells were not undergoing caspase-3-dependent apoptosis and supports the argument that they are neurons newly formed after the ischemic injury. Thus, even in the ischemic region, complement activation-derived signals positively regulate the number of both neural progenitor cells and newly formed neurons.
Figure 9.

The NeuNposBrdUpos cells are not undergoing ischemia-induced apoptosis. Immunostaining for BrdU, NeuN and caspase-3 on day 7 (A) and day 21 (B) after MCAT. Broken line depicts the infarct border. p, penumbra; i, infarct. Higher magnification images of boxed areas in (A) showing penumbra (P) and infarct (I). Scale bar equals 120 μm (A, B) and 60 μm (P, I).
No activated caspase-3pos cells were detected in the SVZ in either of the groups (not shown). It is therefore unlikely that increased cell death accounts for the observed difference in the number of Dcxpos cells or that the complement-derived signals affect the survival of neural progenitor cells.
Impaired ischemia-induced neurogenesis in C3−/− mice is not the consequence of reduced infarct size
Earlier studies using a transient cerebral ischemia model have shown that inhibition of complement activation reduced infarction volume (Akita et al, 2003; De Simoni et al, 2003). To determine whether the reduced ischemia-induced neurogenic response in the C3−/− mice is the consequence of reduced production or release of neurogenic factors due to reduced ischemic damage, we evaluated the brain tissue volume lost to infarction. We found that the cerebral infarct volume was 24% larger in C3−/− mice than in the WT mice (16.3±0.31 versus 13.1±1.07 mm3, P<0.01) 7 days after MCAO. At 7 days after MCAT the infarct volume did not significantly differ between the groups, although there was a tendency toward larger volume in the C3−/− mice (2.5±0.5 versus 1.6±0.3 mm3, P=0.14). However, 21 days after MCAT, the C3−/− mice had lost twice as much tissue as controls (2.3±0.2 versus 1.1±0.2 mm3, P<0.0005). The neuronal density, as assessed by the quantification of NeuNpos cells, in the penumbra 7 days after ischemia did not differ between the C3−/− and control mice (1393±78.1 and 1330±92.3 cells/10 mm2) (Figure 10).
Figure 10.
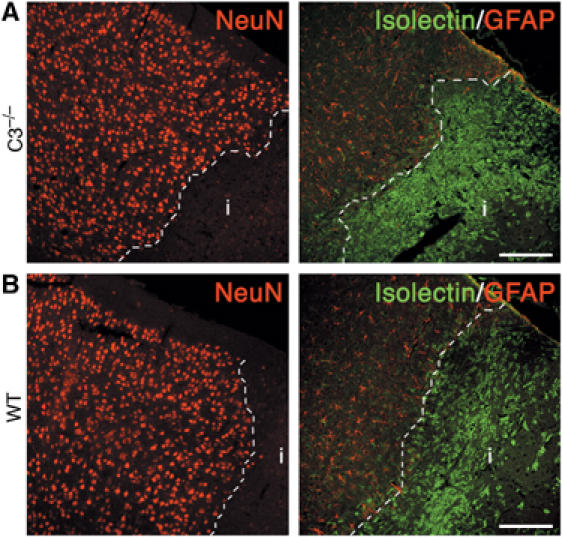
Representative low-power images of the density of neurons (NeuNpos), reactive astrocytes (GFAPpos), and microglia (isolectinpos) at the infarct border in C3−/− (A) and WT (B) mice 7 days after cerebral ischemia. The sections were stained with antibodies against NeuN and GFAP, and with isolectin. Broken line depicts the infarct border. i, infarct. Scale bars equal 150 μm.
We also assessed the activation of astrocytes and recruitment of microglia. Immunostaining for GFAP 7 days after ischemia showed a massive presence of highly GFAPpos astrocytes at the infarct border in both C3−/− and WT mice. Similarly, isolectin staining that identifies microglia and endothelial cells demonstrated a rim of activated microglial cells around the ischemic lesion and revealed migration of these cells into the infarct area in both groups of mice. The width of the GFAPpos band was comparable in C3−/− and WT mice (456±62.3 and 440±25.1 μm), as was the density of the GFAPpos cells (824±28.0 and 729±77.0 cells/10 mm2) (Figure 10). Similarly, there was no difference in width of the isolectinpos band (367±18.8 and 319±20.3 μm) or the density of isolectinpos cells (142±28.0 and 173±26.4 cells/10 mm2) between the groups (Figure 10).
The data imply that impaired neurogenesis observed in the C3−/− mice cannot be explained by reduced availability of complement-independent neurogenic stimuli due to smaller ischemic damage, and that the lack of C3 is not associated with impaired activation of astrocytes or microglia.
Discussion
It has been demonstrated that essentially all of the activation components, regulatory molecules, and receptors of the complement system are produced by astrocytes, microglia, and neurons (Spiegel et al, 1998; Thomas et al, 2000; D'Ambrosio et al, 2001). Although neurons are the predominant cell type expressing C3aR and C5aR in the CNS (Stahel et al, 1997; Davoust et al, 1999; O'Barr et al, 2001), the effects of C3a and C5a on these cells have been largely speculative, ranging from chemotaxis of neuroblasts to the production of adhesion molecules or neurotrophins (Nataf et al, 1999). The expression of C3aR and C5aR on cultured neural stem cells, on transit-amplifying precursors, and migrating neuroblasts in vivo, and reduced basal neurogenesis in the absence of C3aR signaling in mice shown in this study establish a novel role of complement-derived anaphylatoxins in normal CNS. Although the mechanism of C3a and C5a generation in CNS during development and under basal conditions is currently unknown, our results suggest that these peptides may have a function in mammalian cerebrogenesis and basal adult neurogenesis. Loss-of-function mutant mouse strains for C3aR (Kildsgaard et al, 2000) and C5aR (Hopken et al, 1996) may prove useful in elucidating the roles of C3a and C5a in CNS development.
There is experimental evidence that complement activation contributes to inflammation and tissue destruction in CNS disease and ischemia. Complement-deficient mice are protected from demyelinization in an animal model of multiple sclerosis (Nataf et al, 2000) and inhibition of complement activation by C1 inhibitor reduced brain infarction volume (Akita et al, 2003; De Simoni et al, 2003). Our data provide experimental support for a recent notion that products of inflammation may not simply play an adverse role but also collaborate to guide somatic stem cell behavior during perturbation of an organ (Imitola et al, 2004). Our findings suggest that complement has a role in CNS regeneration. It is conceivable that the mechanism involves signaling through C3aR, since the C3−/− mice lack C3a, the only known ligand for C3aR. Our data do not prove or rule out that signaling through C5aR has a regulatory effect on adult neurogenesis. C5aR signaling may be partially functional in the absence of C3, as C5a can be generated directly from C5 by activated phagocytic cells (Huber-Lang et al, 2002). A recent study demonstrated that inhibition of complement activation aggravated amyloid plaque formation and neurodegeneration in an experimental model of Alzheimer's disease (Wyss-Coray et al, 2002), an observation consistent with the involvement of intracerebral complement in brain tissue repair. The notion that complement promotes brain tissue repair after CNS injury is further corroborated by recent reports that implicate C3aR and C5aR in the regeneration of other organs (Del Rio-Tsonis et al, 1998; Mastellos et al, 2001; Reca et al, 2003; Strey et al, 2003; Daveau et al, 2004).
Remarkably, the impaired neurogenesis in the C3−/− mice was associated with increased amounts of brain tissue lost to infarction. Thus, the impaired neurogenesis in the C3−/− mice cannot be explained by reduced production of complement-independent neurogenic stimuli due to lesser tissue damage. It is likely that, besides impaired neurogenic response, the lack of other functions of the complement activation-derived fragments contributed to the increase in infarction volume in the C3−/− mice. For example, C3a and C5a were shown to be neuroprotective (Murkherjee and Passinetti, 2001; O'Barr et al, 2001; van Beek et al, 2001) and exposure to C3a induced de novo expression of nerve growth factor, a molecule involved in neuronal growth and survival, in microglial cells in vitro (Heese et al, 1998). Our data provide experimental in vivo support for the pivotal role that complement activation products and other inflammation-associated mechanisms play in neuroprotection and CNS repair.
Notably, ischemia-induced recruitment of microglia and reactive gliosis were not affected in the C3−/− mice. As a major function of C5a is chemotaxis (Ember et al, 1998) and C5a can be generated in the absence of C3 (Huber-Lang et al, 2002), it is plausible that C5a mediates recruitment of microglia and astrocyte activation in response to brain ischemia in these mice. Alternatively, complement-derived signals are not involved in mediating these phenomena, or other inflammatory mediators sufficiently compensate for these functions of C5a and/or C3a in their absence.
Whereas deficiency of C3 led to increased infarction volume in our model of permanent focal cerebral ischemia, others reported smaller infarction volume 48 h after reperfusion in transient cerebral ischemia models when complement activation was inhibited by C1 inhibitor (Akita et al, 2003; De Simoni et al, 2003). These seemingly controversial results are likely due to the dual role complement plays in brain ischemia. Complement activation in the brain appears to be a double-edged sword in that it can exacerbate tissue damage or promote neuronal survival and tissue remodeling depending on the pathophysiological context (van Beek et al, 2003). In addition, the balance between the beneficial and detrimental effects of complement activation may change over time. The differences in outcome between other studies (Akita et al, 2003; De Simoni et al, 2003) and ours can conceivably be explained not only by the difference in ischemia model but also by the different time of evaluation of infarction volume. The aspect of time therefore seems to be critical for the design and interpretation of future studies of the role of complement activation in cerebral ischemia.
In conclusion, the data presented here implicate complement as a positive regulator of basal and ischemia-induced neurogenesis in mice. Future studies to confirm a direct effect of C3a- and C5a-dependent signaling on neural stem cells and immature neurons under basal conditions and after ischemia are warranted. A better understanding of the dual role of complement in cerebral ischemia and possibly other CNS pathologies may help us to design more effective therapeutic strategies by orchestrating the judicial use of complement-inhibitory agents that neutralize the adverse aspects of complement activation while enhancing those that are neuroprotective and facilitate repair.
Materials and methods
Mice
C3−/−mice (Pekna et al, 1998) were backcrossed onto the C57BL/6 genetic background (Charles River, Uppsala, Sweden) for nine (MCAO) or 13 generations (MCAT). Heterozygous mice were then intercrossed to generate homozygous C3−/− mice. C3aR−/− mice (Kildsgaard et al, 2000) were backcrossed onto the C57BL/6 genetic background (Jackson Laboratories, Bar Harbour, Maine, USA) for 10 generations. Heterozygous mice were then intercrossed to generate homozygous C3aR−/− mice. Gender- and age-matched WT C57BL/6 mice of the same substrain as the mutant mice served as controls. For the C3aR antagonist study, male C57BL/6 mice (Charles River) were used.
Surgical procedures
At 9–11 weeks of age, control (n=7) and C3−/− (n=9) mice were anesthetized with 2.5% isoflurane (Forene, Abbott, Solna, Sweden) in oxygen, 0.5 l/min, during surgery. The body temperature was maintained at 34.5–36.5°C. The MCA was occluded with an intraluminal filament as described (Hara et al, 1996). The reduction in blood flow was monitored with a laser Doppler probe (Moor Instruments, Devon, UK) mounted on the skull above the MCA. Mice in which blood flow was not reduced by more than 70% were excluded from the study. Left MCAT was performed as described (Welsh et al, 1987; Fotheringham et al, 2000) with slight modifications. Mice were anesthetized and body temperature was maintained at 37°C. Under the operating microscope, the left MCA was exposed, occluded at two points by bipolar coagulation, and transected to ensure permanent disruption. Mice were injected with BrdU (Sigma-Aldrich, St Louis, MO, USA) in PBS, 200 mg/kg, i.p. twice daily for 7 days starting just before surgery. On day 7 or 21 after surgery, the 19–20-week old control (n=11 and 10, respectively) and C3−/− (n=12 and 12, respectively) mice were deeply anesthetized and perfused with 0.1 M phosphate buffer, followed by 4% paraformaldehyde in 0.1 M phosphate buffer.
C3aR antagonist and BrdU injections
Male mice (n=10), 8-week old, were injected i.p. C3aR antagonist (Ames et al, 2001) (Calbiochem, San Diego, CA, USA; 500 μg/mouse) diluted in PBS and DMSO (1.16% v/v) twice daily for 10 days. Control (n=12) and C3−/− (n=6) mice were given PBS and DMSO (1.16% v/v). During the first 7 days, all mice received BrdU (Sigma-Aldrich; 200 mg/kg). On day 10 after the first injection, the mice were deeply anesthetized and perfused as above. Male C3aR−/− (n=6) and control (n=6) mice, 12-week old, were injected with BrdU as above and killed on day 21 after the first injection.
Histology and morphometric evaluation of infarct volume
The brains were embedded in paraffin, cut into 8-μm sections, and stained with hematoxylin and erytrosin. Infarct size was assessed morphometrically with the Easy Image program (Bergström Instruments, Stockholm, Sweden). Infarct volumes were determined by planimetry of serial sections. For the preparation of frozen brain sections, adult C57BL/6 mice were perfused with sterile PBS. The brains were snap frozen in optimal cutting temperature (OCT) embedding medium and cut into 10-μm sections.
Immunohistochemistry
For immunohistochemical evaluation, the sections were deparaffinized, permeabilized in 0.01 M citric acid (pH 6.0), heated twice for 5 min in a microwave oven, and blocked with 1% BSA and 0.05% Triton-X-100 in PBS. For colabeling of Olig2 with C3aR and C5aR, acetone-fixed frozen sections were used. Negative control was performed by omission of primary antibody, unless stated otherwise. For cell counting the sections were selected as follows, unless stated otherwise: bregma 0.62–0.45 mm for the penumbra and infarct area, and 0.14 to −0.03 mm for SVZ.
Neural stem cells and transit-amplifying precursor cells in SVZ were visualized with rabbit anti-GFAP antibody (DAKO; 1:100) and rabbit-anti Olig2 (Arnett et al, 2004; 1:10 000), respectively, followed by Alexa488-conjugated anti-rabbit Ig (Molecular Probes; 1:500). Migrating neural progenitor cells were detected with goat anti-Dcx antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA; 1:50), followed by Alexa488-conjugated donkey anti-goat Ig (Molecular Probes, Eugene, OR, USA; 1:100). In the MCAO and MCAT experiments, Dcxpos cells in the SVZ were counted in both hemispheres on three sections per mouse. To label proliferating cells, sections were further stained with mouse anti-BrdU antibody. The BrdUpos, GFAPposBrdUpos, Olig2posBrdUpos, and DcxposBrdUpos cells were counted in one hemisphere on 4–12 sections 160 μm apart/region in the SVZ (bregma 0.14–1.1 mm), the granular layer of OB (bregma 3.2–4.28 mm), and SGZ (bregma −1.34 to −3.64 mm).
Neural progenitor cells in the penumbra were stained with mouse anti-nestin monoclonal antibody (BD Biosciences, Erembodegem, Belgium; 1:100), followed by TRITC-conjugated anti-mouse Ig (DAKO A/S, Glostrup, Denmark; 1:30). Reactive astrocytes were stained with rabbit anti-GFAP antibody (DAKO; 1:100), followed by Alexa488-conjugated anti-rabbit Ig (Molecular Probes; 1:500). Nestinpos GFAPneg nonendothelial cells (neural progenitors) in the penumbra and infarct area were counted on 2–3 sections per mouse. Nestinpos GFAPneg endothelial cells were excluded based on their morphological appearance (branching, lumen-forming structures).
Newly formed neurons were stained with biotinylated anti-NeuN monoclonal antibody (Chemicon, Temecula, CA, USA; 1:100), followed by Cy3-conjugated streptavidin (Sigma-Aldrich; 1:100) or Alexa633-conjugated streptavidin (Molecular Probes; 1:500) in combination with FITC-conjugated rat anti-BrdU antibody (Accurate Chemical, Westbury, NY, USA; 1:75). NeuNpos and NeuNposBrdUpos cells were counted in the penumbra on 2–3 sections per mouse. BrdUpos cells were also counted in the SVZ. In the C3aR antagonist study, the sections were further stained with mouse anti-BrdU (DAKO; 1:100) and Alexa568-conjugated goat anti-mouse Ig (Molecular Probes; 1:500). The BrdUpos and NeuNposBrdUpos cells were counted in one hemisphere on 4–12 sections 160 μm apart/region in the granular layer of OB (bregma 3.2–4.28 mm) and SGZ/GCL (bregma −1.34 to −3.64 mm).
Cells undergoing ischemia-induced apoptosis were stained with rabbit polyclonal antibody against cleaved (17–20 kDa) caspase-3 (BD Pharmingen, San Diego, CA, USA; 1:100), followed by Alexa568-conjugated goat anti-rabbit Ig (Molecular Probes; 1:500).
C5aR was stained with rabbit anti-mouse C5aR antibody (Morgan et al, 1993; van Beek et al, 2000; O'Barr et al, 2001) (a kind gift from Dr J Ember, BD Pharmingen; 1:100) or rat anti-mouse C5aR clone 10/92 (Soruri et al, 2003; 10 μg/ml), followed by Alexa568-conjugated goat anti-rabbit Ig (Molecular Probes; 1:500) and Alexa568-conjugated goat anti-rat Ig (Molecular Probes; 1:500), respectively. C3aR was detected with chicken anti-mouse C3aR antibody (Accurate Chemical; 1:50), followed by biotinylated anti-chicken Ig (Molecular Probes; 1:250) and Cy3-conjugated streptavidin (Sigma-Aldrich; 1:100) or rat anti-mouse C3aR clone 1G4 (Zwirner et al., in preparation; 10 μg/ml), followed by Alexa568-conjugated goat anti-rat Ig (Molecular Probes; 1:500).
Reactive astrocytes were stained with anti-GFAP antibody (DAKO; 1:100) and inflammatory cells by biotinylated lectin (Sigma-Aldrich; 1:10), followed by TRITC-conjugated swine anti-rabbit Ig and FITC-conjugated streptavidin (both from DAKO). Quantification was performed by counting GFAPpos and isolectinpos cells and by measuring the width of the band of positive cells at the infarct border on 2 sections/mouse.
All evaluations were performed in a blinded manner. The cells in the SVZ, OB, penumbra, and infarction area were counted by epifluorescence microscopy on an Eclipse 80i microscope (Nikon, Tokyo, Japan). The cells in SGZ and GCL were counted by confocal microscopy using Radiance 2000 and Laser Sharp 2000 software (Bio-Rad, Hertfordshire, UK).
Neural stem cell culture in vitro
Neural stem cells derived from adult hippocampus (clone HCNA 94/GFPH, passage 15) were cultured as described (Palmer et al, 1997; Takahashi et al, 1999). Before immunostaining, the cells were fixed in methanol at −20°C for 5 min and then washed in PBS, which was also used as a dilution buffer. After blocking with 5% normal goat serum (DAKO), the cells were stained with Abs against C3aR, C5aR, and nestin. The secondary Abs (all from Molecular Probes) were Alexa633 goat anti-rabbit Ig (1:250), biotinylated anti-chicken Ig (1:100), Alexa633 streptavidin (1:100), and Alexa568 goat anti-mouse Ig (1:500). Immunofluorescence was analyzed on an Eclipse 80i microscope (Nikon).
Data analysis
Data are expressed as mean±s.e.m. The two-tailed t-test was used for statistical analysis. Differences were regarded as significant at P<0.05.
Acknowledgments
We thank K Larsson for valuable help; F Gage for the HCNA 94/GFPH cells and valuable comments on this article; D Rowitch and C Stiles for the anti-Olig2 antibody; and S Dickson for the frozen brain sections. This work was supported by grants from the Swedish Research Council (projects 13470 and 11548), the Swedish Cancer Foundation (project 3622), King Gustaf V's 80 Years Foundation, Swedish Stroke Foundation (to MiP and to MaP), Heart-Lung Foundation, the Swedish Society for Medicine, Swedish Society for Medical Research, Göteborg Medical Society, W and M Lundgren Foundation, and Volvo Assar Gabrielsson Foundation. Some of the confocal microscope images were taken at the Center for Cellular Imaging at the Sahlgrenska Academy.
References
- Akita N, Nakase H, Kaido T, Kanemoto Y, Sakaki T (2003) Protective effect of C1 esterase inhibitor on reperfusion injury in the rat middle cerebral artery occlusion model. Neurosurgery 52: 395–400 [DOI] [PubMed] [Google Scholar]
- Alvarez-Buylla A, Garcia-Verdugo JM (2002) Neurogenesis in adult subventricular zone. J Neurosci 22: 629–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ames RS, Lee D, Foley JJ, Jurewicz AJ, Tornetta MA, Bautsch W, Settmacher B, Klos A, Erhard KF, Cousins RD, Sulpizio AC, Hieble JP, McCafferty G, Ward KW, Adams JL, Bondinell WE, Underwood DC, Osborn RR, Badger AM, Sarau HM (2001) Identification of a selective nonpeptide antagonist of the anaphylatoxin C3a receptor that demonstrates antiinflammatory activity in animal models. J Immunol 166: 6341–6348 [DOI] [PubMed] [Google Scholar]
- Arnett HA, Fancy SP, Alberta JA, Zhao C, Plant SR, Kaing S, Raine CS, Rowitch DH, Franklin RJ, Stiles CD (2004) bHLH transcription factor Olig1 is required to repair demyelinated lesions in the CNS. Science 306: 2111–2115 [DOI] [PubMed] [Google Scholar]
- Arvidsson A, Collin T, Kirik D, Kokaia Z, Lindvall O (2002) Neuronal replacement from endogenous precursors in the adult brain after stroke. Nat Med 8: 963–970 [DOI] [PubMed] [Google Scholar]
- D'Ambrosio AL, Pinsky DJ, Connolly ES (2001) The role of the complement cascade in ischemia/reperfusion injury: implications for neuroprotection. Mol Med 7: 367–382 [PMC free article] [PubMed] [Google Scholar]
- Daveau M, Benard M, Scotte M, Schouft M-T, Hiron M, Francois A, Salier J-P, Fontaine M (2004) Expression of a functional C5a receptor in regenerating hepatocytes and its involvement in a proliferative signalling pathway in rat. J Immunol 173: 3418–3424 [DOI] [PubMed] [Google Scholar]
- Davoust N, Jones J, Stahel PF, Ames RS, Barnum SR (1999) Receptor for the C3a anaphylatoxin is expressed by neurons and glial cells. Glia 26: 201–211 [DOI] [PubMed] [Google Scholar]
- De Simoni MG, Storini C, Barba M, Catapano L, Arabia AM, Rossi E, Bergamaschini L (2003) Neuroprotection by complement (C1) inhibitor in mouse transient brain ischemia. J Cereb Blood Flow Metab 23: 232–239 [DOI] [PubMed] [Google Scholar]
- Del Rio-Tsonis K, Tsonis PA, Zarkadis IK, Tsagas AG, Lambris JD (1998) Expression of the third component of complement, C3, in regenerating limb blastema cells of urodeles. J Immunol 161: 6819–6824 [PubMed] [Google Scholar]
- Doetsch F (2003) The glial identity of neural stem cells. Nat Neurosci 6: 1127–1134 [DOI] [PubMed] [Google Scholar]
- Doetsch F, Caille I, Lim DA, Garcia-Verdugo JM, Alvarez-Buylla A (1999) Subventricular zone astrocytes are neural stem cells in the adult mammalian brain. Cell 97: 703–716 [DOI] [PubMed] [Google Scholar]
- Ember JA, Jagels MA, Hugli T (1998) Characterization of complement anaphylatoxins and biological responses. In The Human Complement System in Health and Disease, Volanakis JE, Frank MM (eds), pp 241–284. New York: Marcel-Dekker [Google Scholar]
- Fotheringham AP, Davies CA, Davies I (2000) Oedema and glial cell involvement in the aged mouse brain after permanent focal ischemia. Neuropathol Appl Neurobiol 26: 412–423 [DOI] [PubMed] [Google Scholar]
- Gage FH (2000) Mammalian neural stem cells. Science 287: 1433–1438 [DOI] [PubMed] [Google Scholar]
- Gould E, Vail N, Wagers M, Gross CG (2001) Adult-generated hippocampal and neocortical neurons in macaques have a transient existence. Proc Natl Acad Sci USA 98: 10910–10917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hack MA, Saghatelyan A, de Chevigny A, Pfeifer A, Ashery-Padan R, Lledo P-M, Götz M (2005) Neuronal fate determinants of adult olfactory bulb neurogenesis. Nat Neurosci 8: 865–872 [DOI] [PubMed] [Google Scholar]
- Hack MA, Sugimori M, Lundberg C, Nakafuku M, Götz M (2004) Regionalization and fate specification in neurospheres: the role of Olig2 and Pax6. Mol Cell Neurosci 25: 664–678 [DOI] [PubMed] [Google Scholar]
- Hara H, Huang PL, Panahian N, Fishman MC, Moskowitz MA (1996) Reduced brain edema and infarction volume in mice lacking the neuronal isoform of nitric oxide synthase after transient MCA occlusion. J Cereb Blood Flow Metab 4: 605–611 [DOI] [PubMed] [Google Scholar]
- Heese K, Hock C, Otten U (1998) Inflammatory signals induce neurotropin expression in human microglial cells. J Neurochem 70: 699–707 [DOI] [PubMed] [Google Scholar]
- Honczarenko M, Ratajczak MZ, Nicholson-Weller A, Silberstein LE (2005) Complement C3a enhances CXCL12 (SDF-1)-mediated chemotaxis of bone marrow hematopoietic cells independently of C3a receptor. J Immunol 175: 3698–3706 [DOI] [PubMed] [Google Scholar]
- Hopken UE, Lu B, Gerard NP, Gerard C (1996) The C5a chemoattractant receptor mediates mucosal defense to infection. Nature 383: 86–89 [DOI] [PubMed] [Google Scholar]
- Huber-Lang M, Younkin EM, Sarma JV, Riedermann N, McGuire SR, Lu KT, Kunkel R, Younger JG, Zetoune FS, Ward PA (2002) Generation of C5a by phagocytic cells. Am J Pathol 161: 1849–1859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imitola J, Raddassi K, In Park K, Mueller F-J, Nieto M, Teng YD, Frenkel D, Li J, DSidman RL, Walsh CA, Snyder EY, Khoury SJ (2004) Directed migration of neural stem cells to sites of CNS injury by the stromal cell-derived factor 1 alpha/CXC chemokine receptor 4 pathway. Proc Natl Acad Sci USA 101: 18117–18122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin K, Sun Y, Xie L, Peel A, Ou Mao X, Batteur S, Greenberg DA (2003) Directed migration of neuronal precursors into the ischemic cerebral cortex and striatum. Mol Cell Neurosci 24: 171–189 [DOI] [PubMed] [Google Scholar]
- Katchanov J, Harms C, Gertz K, Hauck L, Waeber C, Hirt L, Priller J, von Harsdorf R, Bruck W, Hörtnagl H, Dirnagl U, Bhide PG, Endres M (2001) Mild cerebral ischemia induces loss of cyclin-dependent kinase inhibitors and activation of cell cycle machinery before delayed neuronal cell death. J Neurosci 21: 5045–5053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kildsgaard J, Hollmann TJ, Matthews KW, Bian K, Murad F, Wetsel RA (2000) Targeted disruption of the C3a receptor gene demonstrates a novel protective anti-inflammatory role for C3a in endotoxin shock. J Immunol 165: 5406–5409 [DOI] [PubMed] [Google Scholar]
- Kimura Y, Madhavan M, Call MK, Santiago W, Tsonis PA, Lambris JD, Del Rio-Tsonis K (2003) Expression of complement 3 and complement 5 in newt limb and lens regeneration. J Immunol 170: 2331–2339 [DOI] [PubMed] [Google Scholar]
- Kondo T, Raff M (2000) Oligodendrocyte precursor cells reprogrammed to become multipotential CNS stem cells. Science 289: 1754–1757 [DOI] [PubMed] [Google Scholar]
- Lendahl U, Zimmermann LB, McKay RD (1990) CNS stem cells express a new class of intermediate filament protein. Cell 60: 585–595 [DOI] [PubMed] [Google Scholar]
- Liu DX, Greene LA (2001) Neuronal apoptosis at the G1/S cell cycle checkpoint. Cell Tissue Res 305: 217–228 [DOI] [PubMed] [Google Scholar]
- Mastellos D, Papadimitriou JC, Franchini S, Tsonis PA, Lambris JD (2001) A novel role of complement: mice deficient in the fifth component of complement (C5) exhibit impaired liver regeneration. J Immunol 166: 2479–2486 [DOI] [PubMed] [Google Scholar]
- Morais da Silva S, Gates PB, Brockers JP (2002) The newt ortholog of CD59 is implicated in proximodistal identity during amphibian limb regeneration. Dev Cell 3: 547–555 [DOI] [PubMed] [Google Scholar]
- Morgan EL, Ember JA, Sanderson SD, Scolz W, Buchner R, Ye RD, Hugli TE (1993) Anti-C5a receptor antibodies. Characterization of neutralizing antibodies specific for a peptide, C3aR9-29, derived from the predicted amino-terminal sequence of the human C3a receptor. J Immunol 151: 377–388 [PubMed] [Google Scholar]
- Murkherjee P, Passinetti GM (2001) Complement anaphylatoxin C5a neuroprotects through mitogen-activated protein kinase-dependent inhibition of caspase 3. J Neurochem 77: 43–49 [DOI] [PubMed] [Google Scholar]
- Nacher J, Crespo C, McEwen BS (2001) Doublecortin expression in the adult rat telencephalon. Eur J Neurosci 14: 629–644 [DOI] [PubMed] [Google Scholar]
- Namura S, Zhu J, Fink K, Endres M, Srinivasan A, Tomaselli KJ, Yuan J, Moskowitz MA (1998) Activation and cleavage of caspase-3 in apoptosis induced by experimental cerebral ischemia. J Neurosci 18: 3659–3668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nataf S, Carroll SL, Wetsel RA, Szalai AJ, Barnum SR (2000) Attenuation of experimental autoimmune demyelinization in complement-deficient mice. J Immunol 165: 5867–5873 [DOI] [PubMed] [Google Scholar]
- Nataf S, Levison SW, Barnum SR (2001) Expression of the anaphylatoxin C5a receptor in the oligodendrocyte lineage. Brain Res 894: 321–326 [DOI] [PubMed] [Google Scholar]
- Nataf S, Stahel PF, Davoust N, Barnum SR (1999) Complement anaphylatoxin receptors on neurons: new tricks for old receptors? Trends Neurosci 22: 397–402 [DOI] [PubMed] [Google Scholar]
- O'Barr SA, Caguioa J, Gruol D, Perkins G, Ember JA, Hugli T, Cooper NR (2001) Neuronal expression of a functional receptor for the C5a complement activation fragment. J Immunol 166: 4154–4162 [DOI] [PubMed] [Google Scholar]
- Osuga H, Osuga S, Wang F, Fetni R, Hogan MJ, Slack RS, Hakim AM, Ikeda J-E, Park DS (2000) Cyclin-dependent kinases as a therapeutic target for stroke. Proc Natl Acad Sci USA 97: 10254–10259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer TD, Markakis EA, Willhoitte AR, Safar F, Gage FH (1999) Fibroblast growth factor-2 activates a latent neurogenic program in neural stem cells from diverse regions of the adult CNS. J Neurosci 19: 8487–8497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer TD, Takahashi J, Gage FH (1997) The adult rat hippocampus contains primordial neural stem cells. Mol Cell Neurosci 6: 389–404 [DOI] [PubMed] [Google Scholar]
- Parent JM, Vexler ZS, Gong C, Derugin N, Ferriero DM (2002) Rat forebrain neurogenesis and striatal neuron replacement after focal stroke. Ann Neurol 52: 802–813 [DOI] [PubMed] [Google Scholar]
- Pekna M, Hietala MA, Rosklint T, Betsholtz C, Pekny M (1998) Targeted disruption of the murine gene coding for the third complement component (C3). Scand J Immunol 47: 25–29 [DOI] [PubMed] [Google Scholar]
- Ratajczak J, Reca R, Kucia M, Majka M, Allendorf DJ, Baran JT, Janowska-Wieczorek A, Wetsel RA, Ross GD, Ratajczak MZ (2004) Mobilization studies in mice deficient in either C3 or C3a receptor (C3aR) reveal a novel role for complement in retention of hematopoietic stem/progenitor cells in bone marrow. Blood 103: 2071–2078 [DOI] [PubMed] [Google Scholar]
- Reca R, Mastellos D, Majka M, Marquez L, Ratajczak J, Franchini S, Glodek A, Honczarenko M, Spruce LA, Janowska-Wieczorek A, Lambris JD, Ratajczak MZ (2003) Functional receptor for C3a anaphylatoxin is expressed by normal hematopoietic stem/progenitor cells, and C3a enhances their homing related responses to SDF-1. Blood 101: 3784–3793 [DOI] [PubMed] [Google Scholar]
- Reynolds BA, Weiss S (1992) Generation of neurons and astrocytes from isolated cells from the adult mammalian central nervous system. Science 255: 1707–1710 [DOI] [PubMed] [Google Scholar]
- Sanes JR, Okun LM (1972) Induction of DNA synthesis in cultured neurons by ultraviolet light or methyl methane sulfonate. J Cell Biol 53: 587–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soruri A, Kim S, Kiafard Z, Zwirner J (2003) Characterization of C5aR expression on murine myeloid and lymphoid cells by the use of a novel monoclonal antibody. Immunol Lett 88: 47–52 [DOI] [PubMed] [Google Scholar]
- Spiegel K, Emmerling MR, Barnum SR (1998) Strategies for inhibition of complement activation in the treatment of neurodegenerative diseases. In Neuroinflammation: Mechanisms and Management, Wood PL (ed), pp 129–176. Totowa: Humana Press Inc [Google Scholar]
- Stahel PF, Kossmann T, Morganti-Kossmann MC, Hans VHJ, Barnum SR (1997) Experimental diffuse axonal injury induces enhanced neuronal C5a receptor mRNA expression in rats. Mol Brain Res 50: 205–212 [DOI] [PubMed] [Google Scholar]
- Strey CW, Markiewski M, Mastellos D, Tudoran R, Spruce LA, Greenbaum LE, Lambris JD (2003) The proinflammatory mediators C3a and C5a are essential for liver regeneration. J Exp Med 198: 913–923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi J, Palmer TD, Gage FH (1999) Retinoic acid and neurotrophins collaborate to regulate neurogenesis in adult-derived neural stem cell cultures. J Neurobiol 38: 65–81 [PubMed] [Google Scholar]
- Thomas A, Gasque P, Vaudry H, Gonzalez B, Fontaine M (2000) Expression of a complete and functional complement system by human neuronal cells in vitro. Int Immunol 12: 1015–1023 [DOI] [PubMed] [Google Scholar]
- van Beek J, Bernaudin M, Petit E, Gasque P, Nouvelot A, MacKenzie ET, Fontaine M (2000) Expression of receptors for complement anaphylatoxins C3a and C5a following permanent focal cerebral ischemia in the mouse. Exp Neurol 161: 373–382 [DOI] [PubMed] [Google Scholar]
- van Beek J, Elward K, Gasque P (2003) Activation of complement in the central nervous system. Roles in neurodegeneration and neuroprotection. Ann NY Acad Sci 992: 56–71 [DOI] [PubMed] [Google Scholar]
- van Beek J, Nicole O, Ali C, Ischenko A, MacKenzie ET, Buisson A, Fontaine M (2001) Complement anaphylatoxin C3a is selectively protective against NMDA-induced neuronal cell death. Neuroreport 12: 289–293 [DOI] [PubMed] [Google Scholar]
- Verdaguer E, Garcia-Jorda E, Canudas AM, Dominiguez E, Jimenez A, Pubill D, Escubedo E, Caramasa Merce Pallas J, Camins A (2002) Kainic acid-induced apoptosis in cerebral granule neurons: an attempt at cell cycle-re-entry. Neuroreport 13: 413–416 [DOI] [PubMed] [Google Scholar]
- Welsh FA, Sakamoto T, McKee AE, Sims RE (1987) Effect of lactacidosis on pyridine nucleotide stability during ischemia in mouse brain. J Neurochem 49: 846–851 [DOI] [PubMed] [Google Scholar]
- Wyss-Coray T, Yan F, Lin AH, Lambris JD, Alexander JJ, Quigg RJ, Masliah E (2002) Prominent neurodegeneration and increased plaque formation in complement-inhibited Alzheimer's mice. Proc Natl Acad Sci USA 99: 10837–10842 [DOI] [PMC free article] [PubMed] [Google Scholar]