

Abstract
The MDM2 homolog MDMX is an important regulator of p53 during mouse embryonic development. DNA damage promotes MDMX phosphorylation, nuclear translocation, and degradation by MDM2. Here we show that MDMX copurifies with 14-3-3, and DNA damage stimulates MDMX binding to 14-3-3. Chk2-mediated phosphorylation of MDMX on S367 is important for stimulating 14-3-3 binding, MDMX nuclear import by a cryptic nuclear import signal, and degradation by MDM2. Mutation of MDMX S367 inhibits ubiquitination and degradation by MDM2, and prevents MDMX nuclear import. Expression of 14-3-3 stimulates the degradation of phosphorylated MDMX. Chk2 and 14-3-3 cooperatively stimulate MDMX ubiquitination and overcome the inhibition of p53 by MDMX. These results suggest that MDMX–14-3-3 interaction plays a role in p53 response to DNA damage by regulating MDMX localization and stability.
Keywords: Chk2, MDM2, MDMX, p53, 14-3-3
Introduction
The p53 tumor suppressor is critical for maintenance of genomic stability and protection against malignant transformation. P53 is regulated by multiple signaling pathways and can respond to a wide range of stress conditions, allowing it to act as a tumor suppressor in many cell types (Harris and Levine, 2005). P53 turnover is regulated by MDM2, which binds p53 and functions as an ubiquitin E3 ligase to promote p53 degradation by the proteasomes (Zhang and Xiong, 2001). Stress signals such as DNA damage induce p53 accumulation by phosphorylation (Prives and Hall, 1999). Mitogenic signals activate p53 by induction of the p14ARF tumor suppressor, which inhibits the ability of MDM2 to ubiquitinate p53 (Zhang and Xiong, 2001).
Mammalian cells also express an MDM2 homolog called MDMX (Shvarts et al, 1996). MDMX shares strong homology to MDM2 at the amino-acid sequence level, and can bind to p53 and inhibit its transcriptional function. Knockout of MDM2 in mice results in embryonic lethality due to hyperactivation of p53 (Montes de Oca Luna et al, 1995). Several studies showed that MDMX-null mice also die in utero in a p53-dependent fashion, which can be rescued by crossing into the p53-null background (Parant et al, 2001; Finch et al, 2002; Migliorini et al, 2002b). Therefore, MDMX is also a critical regulator of p53.
Although MDMX alone does not promote p53 ubiquitination or degradation (Stad et al, 2001), recent evidence showed that MDMX cooperates with MDM2 to promote p53 degradation (Gu et al, 2002). Human tumor cell lines with wild-type p53 often overexpress MDMX (Ramos et al, 2001), suggesting that MDMX may contribute to p53 inactivation during tumorigenesis. Therefore, regulation of MDMX expression level may be an important mechanism of p53 activation during stress response. Several reports showed that MDMX can be ubiquitinated and degraded by MDM2 (de Graaf et al, 2003; Kawai et al, 2003; Pan and Chen, 2003). MDMX and MDM2 form heterodimers through the C-terminal RING domains and the MDM2 RING domain alone is sufficient to degrade MDMX (Sharp et al, 1999; Tanimura et al, 1999). DNA damage induced by ionizing irradiation also promotes MDMX nuclear translocation and degradation by the proteasomes (Li et al, 2002; Pan and Chen, 2003).
The 14-3-3 family are scaffold proteins that regulate many cellular functions by interacting with other proteins, often binding to phosphorylated serine or threonine residues (Fu et al, 2000; Tzivion et al, 2001). There are seven 14-3-3 isoforms in humans. Most of the isoforms are expressed in all tissues, although 14-3-3σ is restricted to epithelial cells (Hermeking, 2003). Although highly homologous in sequence and structure, different 14-3-3 isoforms have distinct biological functions, as demonstrated by the phenotypes of 14-3-3ɛ and ζ disruption in Drosophila (Su et al, 2001). There is also extensive evidence that 14-3-3σ plays significant roles in regulating cell differentiation and cancer development (Hermeking, 2003).
Several lines of evidence suggest that 14-3-3 proteins are involved in regulating p53. Involvement of 14-3-3 in the p53 pathway was first revealed by the ability of p53 to activate expression of 14-3-3σ at the transcriptional level, which can lead to G2/M cell cycle arrest (Hermeking et al, 1997). Furthermore, 14-3-3σ, γ, ɛ, and τ isoforms can bind to p53 C-terminus and activate p53 transcriptional activity (Stavridi et al, 2001; Yang et al, 2003). P53–14-3-3 interaction requires phosphorylation of S378, and is stimulated by dephosphorylation of the adjacent S376 after DNA damage through an ATM-dependent mechanism (Waterman et al, 1998). Mutation of S376 compromises the ability of p53 to induce p21 expression and G1 arrest after ionizing irradiation (Stavridi et al, 2001). 14-3-3σ interaction with p53 also inhibits p53 ubiquitination and degradation by MDM2 (Yang et al, 2003), possibly contributing to stabilization of p53 after DNA damage.
In this report, we present evidence that MDMX interacts with multiple 14-3-3 isoforms. MDMX–14-3-3 binding is stimulated by DNA damage. This is associated with increased phosphorylation of S367, which is a residue required for interaction with 14-3-3. S367 is also required for efficient ubiquitination of MDMX by MDM2, and 14-3-3 stimulates degradation of MDMX phosphorylated on S367. Chk2 kinase-mediated phosphorylation of S367 is required for MDMX nuclear import and efficient degradation after DNA damage. These results suggest that 14-3-3 proteins participate in regulating p53 response to DNA damage in part by interacting with MDMX.
Results
MDMX interacts with 14-3-3
In order to determine whether MDMX interacts with novel cellular proteins, HeLa cells were stably transfected with FLAG-tagged MDMX. When FLAG-MDMX was purified by immunoprecipitation, two proteins reproducibly copurified with MDMX (Figure 1A). Peptide sequencing by mass spectrometry determined the 34 kDa protein as casein kinase 1 alpha, and the 24 kDa protein as 14-3-3τ isoform. In a separate report, we showed that CK1α interaction with the central region of MDMX promotes phosphorylation of S289 and cooperates with MDMX to inactivate p53 (Chen et al, 2005b). MDMX binding to 14-3-3 appeared to be independent of CK1α and is the focus of this study.
Figure 1.
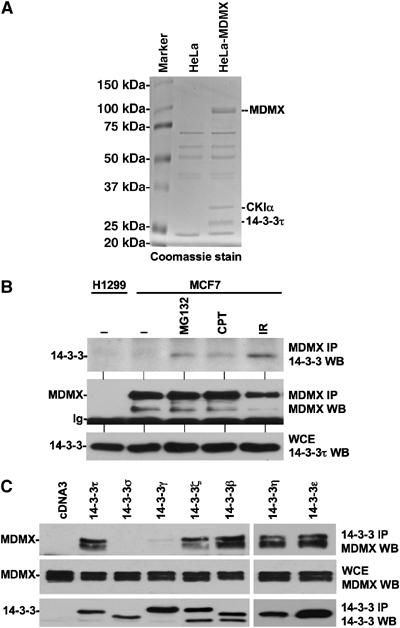
MDMX forms a complex with 14-3-3. (A) Copurification of 14-3-3 with MDMX. HeLa cells stably transfected with FLAG-MDMX were immunoprecipitated using anti-FLAG M2 antibody, the purified MDMX and associated proteins were detected by Coomassie stain. The two marked bands were identified as CK1α and 14-3-3τ by mass spectrometric peptide sequencing. (B) Interaction of endogenous 14-3-3 and MDMX. MCF7 cells were treated with 30 μM MG132 for 4 h, 0.5 μM camptothecin for 16 h, or irradiated with 10 Gy γ irradiation for 4 h. Cell lysates were immunoprecipitated with MDMX antibody 8C6, followed by anti-14-3-3τ Western blot. (C) Interaction of MDMX with 14-3-3 isoforms. H1299 cells transiently cotransfected with MDMX and epitope-tagged 14-3-3 plasmids were immunoprecipitated using anti-FLAG (for τ, σ, γ), anti-myc (for ζ, β), and anti-His6 (for ɛ, η) antibodies.
To confirm that endogenous MDMX interacts with 14-3-3, MCF7 cells were immunoprecipitated with MDMX monoclonal antibody and probed for 14-3-3 using a 14-3-3τ specific antibody. Endogenous MDMX interaction with 14-3-3 was nearly below our detection limit in the absence of stress. However, DNA-damaging treatments with camptothecin or ionizing irradiation significantly stimulated MDMX–14-3-3τ binding (Figure 1B). Furthermore, treatment with the proteasome inhibitor MG132 also increased MDMX–14-3-3 binding, suggesting that a population of unstable MDMX may preferentially bind to 14-3-3. DNA damage stimulation of MDMX–14-3-3 binding has also been reported in a recent publication (Okamoto et al, 2005).
One of the dominant 14-3-3 isoforms expressed in HeLa is 14-3-3τ (Nomura et al, 2003). This may be the reason that 14-3-3τ was found in the MDMX complex from this cell line. To determine whether MDMX also interacts with other 14-3-3 isoforms, MDMX was cotransfected with the entire 14-3-3 panel (τ, σ, β, γ, ζ, ɛ, η). The epitope tagged 14-3-3 was immunoprecipitated and the co-precipitated MDMX was detected by Western blot. The results showed absence of binding to 14-3-3σ, weak binding to γ, and strong binding to τ, β, ζ, ɛ, and η (Figure 1C). This result suggested that MDMX interacts with multiple 14-3-3 isoforms. As such, siRNA knockdown of 14-3-3τ alone did not have observable effect on MDMX and p53 response to DNA damage (data not shown).
Identification of 14-3-3 binding site on MDMX
A major function of 14-3-3 is to bind to proteins with phosphorylated serine and threonine residues. The increased MDMX binding to 14-3-3 after DNA-damaging treatment suggested that MDMX phosphorylation is involved in regulating 14-3-3 binding. We recently found that DNA damage induces the appearance of MDMX with reduced mobility on SDS–PAGE. Inhibition of proteasome using MG132 can further preserve this labile population. Mass spectrometric analysis identified several phosphorylation sites on MDMX after DNA damage by γ irradiation, including S342, S367, S391, and S403 (Chen et al, 2005a) (Figure 2A). A recent report by Pereg et al (2005) also identified phosphorylation on S342, S367, and S403, and showed that S403 is a target for ATM after DNA damage.
Figure 2.
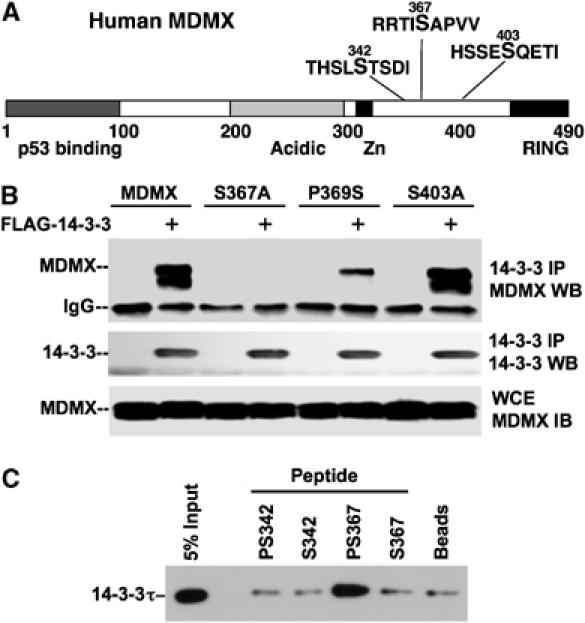
Identification of 14-3-3 binding site on MDMX. (A) Location of phosphorylation sites on MDMX. Phosphorylation of S342, S367, and S403 was detected by mass spectrometry and verified by phosphorylation-specific antibody analyses in previous studies. (B) MDMX mutant binding to 14-3-3. H1299 cells were transiently cotransfected with MDMX mutant and FLAG-14-3-3τ plasmids for 48 h, followed by anti-FLAG IP and MDMX Western blot. (C) 14-3-3 binding by phosphorylated S367 peptide. MDMX peptides with or without phosphorylation on S342 and S367 were chemically crosslinked to agarose beads. The beads were incubated with whole-cell extract, washed, and bound 14-3-3τ detected by Western blot.
Of particular interest, S367 is present in a sequence context favorable for interaction with 14-3-3 after phosphorylation (xSxPx) (Yaffe et al, 1997). We tested this possibility by generating S367A and P369S mutations to target the phosphorylation site and the adjacent proline residue important for providing the optimal sequence context. Mutants of adjacent phosphorylated sites S342A and S403A were also tested. The MDMX mutants were coexpressed with FLAG-14-3-3τ in H1299 cells and analyzed by anti-FLAG IP followed by MDMX Western blot. The results showed that MDMX binding to 14-3-3 was abrogated by the S367A mutation and significantly reduced by P369S mutation (Figure 2B). Furthermore, S342A and S403A mutations had no effect on MDMX–14-3-3 binding (Figure 2B and data not shown). These results suggested that S367 is the major binding site for 14-3-3.
To confirm that phosphorylated S367 is directly involved in binding to 14-3-3, peptides with phosphorylated or unphosphorylated S367 were conjugated to agarose beads. Whole-cell extract was incubated with peptide-loaded beads and the amount of 14-3-3τ captured was determined by Western blot. The result showed that peptide with phosphorylated S367 specifically captured 14-3-3τ from the extract. Unphosphorylated S367 peptide, phosphorylated or unphosphorylated S342 peptides showed no binding above background noise (Figure 2C). These results further demonstrated that phosphorylated S367 is the 14-3-3 binding site on MDMX.
S367 is a major phosphorylation site in vivo
If S367 plays a significant role in the regulation of MDMX after DNA damage, we would expect that this site should be strongly phosphorylated. Therefore, we determined the relative modification levels of several MDMX phosphorylation sites in vivo by metabolic labeling and two-dimensional (2D) phosphopeptide analysis. 293T cells were transiently transfected with MDMX mutants with serine to alanine mutations at each phosphorylation site and labeled with 32P-orthophosphate. The labeled MDMX was immunoprecipitated, gel purified, digested with Asp-N endoproteinase, and the peptides were analyzed by 2D peptide mapping. The labeling of wild-type and MDMX point mutant enabled us to assign some of the phosphopeptide spots on the 2D peptide map to particular serine residues (Figure 3). The results showed that the most intensely labeled MDMX peptide was eliminated by the S367A mutation, suggesting that S367 was the major phosphorylation site on MDMX under the labeling condition (Figure 3). Although phosphorylation of S403 was detectable by mass spectrometry and phosphorylation-specific antibody, we were not able to definitively assign this site to a particular spot on the 2D peptide map of Asp-N digestion (data not shown). This may be due to weak signal or poor solubility of the phosphopeptide containing S403.
Figure 3.
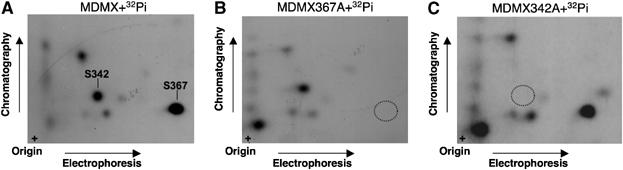
S367 is a major MDMX phosphorylation site in vivo. 293T cells were transiently transfected with MDMX mutants with serine-to-alanine mutations at the indicated phosphorylation site and labeled with 32P-orthophosphate. The labeled MDMX was immunoprecipitated, digested with Asp-N endoproteinase, and analyzed by 2D peptide mapping. (A) Wild-type MDMX. (B, C) MDMX point mutants.
We also tested whether metabolic labeling and 2D peptide mapping can be used to detect γ irradiation-stimulated phosphorylation of MDMX. The results indicated that treatment of cells with 10 Gy γ irradiation immediately before the 4 h labeling with 0.2 mCi/ml 32P-orthophosphate did not have an effect on the labeling level of S367 relative to other spots (data not shown), suggesting that the amount of irradiation received from the 32P-orthophosphate incubation alone was sufficient to induce high-level modification of S367.
Chk2 phosphorylation of S367 is important for stimulating 14-3-3 binding
The sequence context of S367 (RRTIS367APVV) fits the consensus substrate motif LxRxxS/T for Chk2 kinase, as identified by oriented peptide libraries in vitro (O'Neill et al, 2002). In order to study the regulation of MDMX by phosphorylation, we recently generated phosphorylation-specific antibody against S367. Using this antibody, we found that S367 was phosphorylated at a low basal level in undamaged cells, but significantly phosphorylated after DNA damage by γ irradiation. Furthermore, in vitro kinase reaction showed that purified Chk2 kinase preferentially phosphorylates S367 of recombinant MDMX (Chen et al, 2005a). As expected, DNA damage by γ irradiation or UV induced S367 phosphorylation in wild-type HCT116 cells, but not in HCT116-Chk2−/− cells (Figure 4A) (Jallepalli et al, 2003). Therefore, Chk2 kinase is important for both ionizing irradiation and UV-induced phosphorylation of S367. Phosphorylation of MDMX on at least some of the other sites (possibly on S403 by ATM) was still evident after γ irradiation in Chk2−/− cells, generating a visible shift in MDMX mobility as in wild-type HCT116 cells (Figure 4A).
Figure 4.
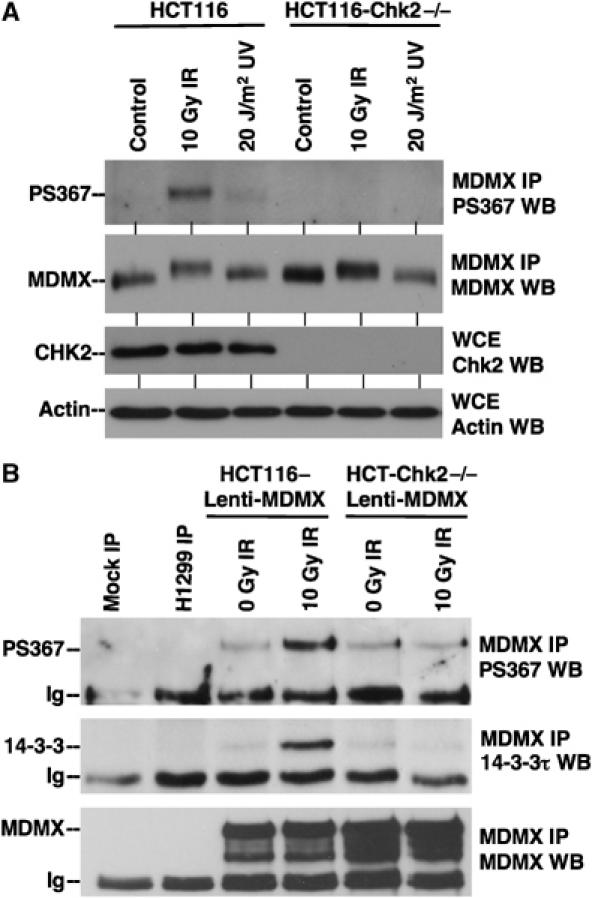
Chk2 is required for DNA damage-induced phosphorylation of S367 and binding to 14-3-3. (A) Wild-type and Chk2-null HCT116 cells were treated with 10 Gy γ irradiation or 20 J/m2 for 4 h in the presence of 30 μM MG132 to preserve phosphorylated MDMX. MDMX was immunoprecipitated with 8C6 and analyzed using anti-PS367 antibody by Western blot. The membrane was reprobed with 8C6 to confirm the level of total MDMX. (B) HCT116 cells were stably infected with MDMX lentivirus to facilitate detection of MDMX binding to endogenous 14-3-3. Cells were treated with 10 Gy γ irradiation for 4 h in the presence of 30 μM MG132. MDMX was immunoprecipitated and analyzed using anti-PS367 antibody (top panel). H1299 cells with very low MDMX level were used as negative control. MDMX immunoprecipitate was analyzed for the co-precipitation of 14-3-3τ.
To test the role of Chk2 in DNA damage-induced MDMX–14-3-3 binding, HCT116-Chk−/− cells were infected with lentivirus expressing MDMX to obtain a level necessary for detection of endogenous 14-3-3 binding. The cells were treated with 10 Gy irradiation in the presence of MG132 (to prevent degradation of phosphorylated MDMX); MDMX was immunoprecipitated and analyzed for the co-precipitation of 14-3-3. The results showed that loss of Chk2 expression completely abrogated the stimulation of 14-3-3 binding by DNA damage (Figure 4B). These results showed that Chk2 phosphorylation of S367 is required for stimulation of MDMX–14-3-3 binding after DNA damage.
Chk2 phosphorylation of MDMX is required for nuclear import and degradation
Previous studies revealed that transfected MDMX showed predominantly cytoplasmic distribution and can be induced to accumulate in the nucleus by binding to MDM2, or through an MDM2-independent mechanism after DNA damage (Stad et al, 2001; Li et al, 2002; Migliorini et al, 2002a). The increased MDMX–14-3-3 binding after DNA damage suggested that Chk2-regulated 14-3-3 binding plays a role in MDMX nuclear translocation. To test this hypothesis, we generated a stable U2OS cell line overexpressing the S367A mutant. Immunofluorescence staining showed that after γ irradiation, wild-type MDMX underwent nuclear translocation as reported previously, whereas the S367A mutant showed significant defect in nuclear translocation (Figure 5A). However, both wild-type MDMX and S367A were targeted into the nucleus when cotransfected with MDM2 (data not shown), indicating that S367A mutation does not affect MDM2-mediated nuclear import. Therefore, phosphorylation of S367 is required for the DNA damage-induced, MDM2-independent nuclear import, possibly mediated by 14-3-3 binding.
Figure 5.
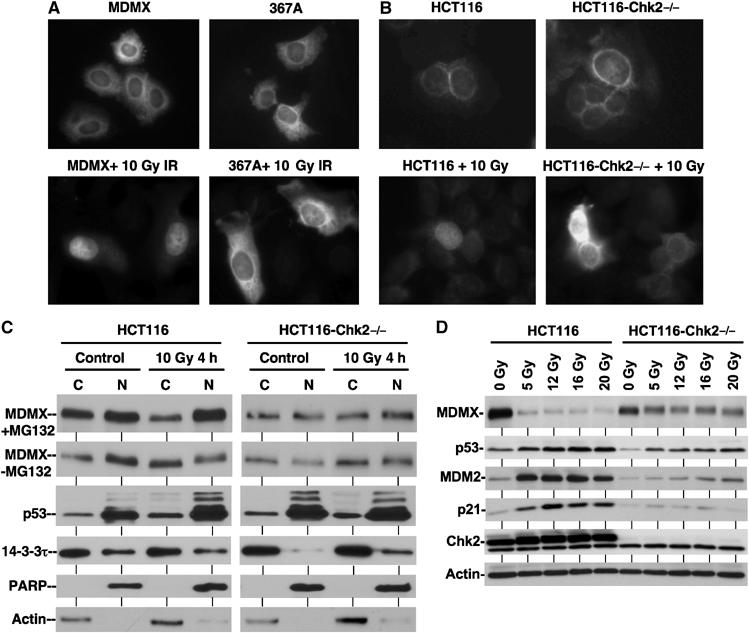
Chk2 phosphorylation of MDMX is required for nuclear import and degradation. (A) S367A mutant does not undergo nuclear translocation after DNA damage. U2OS cells stably transfected with wild-type MDMX or S367A mutant were treated with 0.5 μM CPT for 18 h and stained using 8C6. (B) DNA damage does not induce MDMX nuclear import in Chk2-null cells. HCT116 wild-type and Chk2-null cells stably infected with lentivirus expressing MDMX were treated with 10 Gy irradiation for 18 h and stained with 8C6 antibody. (C) Analysis of endogenous MDMX distribution by subcellular fractionation. HCT116 cells were treated with 10 Gy γ irradiation in the presence or absence of 30 μM MG132 for 4 h. Nuclear and cytoplasmic fractions were analyzed by Western blot. To facilitate comparison of ratio, loading of IR-treated C/N pair was empirically increased to obtain MDMX signal similar to the untreated pair. (D) Chk2-null cells were partially defective for MDMX degradation. HCT116 cells were treated with indicated doses of γ irradiation for 4 h and analyzed by Western blot for different markers.
The ability of S367A mutation to block MDMX nuclear translocation suggested that phosphorylation by Chk2 is required for DNA damage-regulated MDMX nuclear import. To test this prediction, wild-type and HCT116-Chk2−/− cells were stably infected with lentivirus vector expressing MDMX and treated with γ irradiation. Immunofluorescence staining of MDMX revealed that DNA damage induced MDMX nuclear import in wild-type HCT116 cells, but not in HCT116-Chk2−/− cells (Figure 5B). Therefore, Chk2 phosphorylation of MDMX on S367 is important for its nuclear translocation after DNA damage.
To further test the role of Chk2 in MDMX nuclear import, we examined the localization of endogenous MDMX by nuclear/cytoplasmic fractionation. The results showed that MDMX in wild-type HCT116 cells was distributed in both the cytoplasm and nucleus, with moderate preference for the nucleus (Figure 5C). Nuclear distribution became prominent 4 h after γ irradiation when MG132 was also added to prevent degradation of phosphorylated MDMX. Without MG132 treatment, γ irradiation actually reduced the amount of nuclear MDMX due to degradation (Figure 5C). The nuclear MDMX in wild-type HCT116 cells showed reduced gel mobility after irradiation, suggesting that the phosphorylated form was preferentially transferred to the nucleus (Figure 5C, second panel). In contrast, MDMX was almost equally distributed between the cytoplasm and nucleus in Chk2-null cells, and this pattern was not significantly altered by γ irradiation and MG132 cotreatment (Figure 5C). For comparison, p53 in the same experiment was predominantly localized in the nucleus irrespective of the treatments (Figure 5C). Additional time-course experiments showed that in the absence of MG132, MDMX level in both compartments of wild-type HCT116 cells decreased rapidly after irradiation. However, in the presence of MG132, cytoplasmic MDMX decreased, whereas nuclear MDMX remained the same or increased after irradiation (Supplementary Figure S1). These results showed that nuclear translocation and degradation of MDMX after DNA damage require Chk2 activity. Inhibition of the proteasome caused accumulation of the phosphorylated MDMX in the nucleus.
Direct comparison of total MDMX levels in HCT116 with and without Chk2 also showed that loss of Chk2 prevented efficient degradation of MDMX after γ irradiation. This was associated with moderately reduced stabilization of p53 and weaker induction of p21 expression (Figure 5D). These results suggested that Chk2 is critical for efficient degradation of MDMX and full activation of p53 after DNA damage, possibly in part through regulation of MDMX–14-3-3 interaction and promoting MDMX nuclear import.
S367 phosphorylated MDMX preferentially localizes to the nucleus
The correlation between Chk2-mediated S367 phosphorylation and MDMX nuclear import suggested that phosphorylated MDMX should preferentially accumulate in the nucleus. To test this prediction, HeLa cells stably expressing high levels of MDMX were stained with MDMX 8C6 antibody and PS367 antibody. As expected, cytoplasmic MDMX prior to DNA damage was not stained by the PS367 antibody. After γ irradiation or treatment with camptothecin, the majority of MDMX accumulated in the nucleus and displayed a strong nuclear staining by the PS367 antibody (Figure 6A). Transient transfection of MDMX into U2OS cells resulted in a subpopulation of cells showing diffused cytoplasmic and nuclear distribution. Double IF staining of these cells also showed that PS367 antibody preferentially stained the nucleus (Figure 6B). These results are consistent with the cell fractionation experiment showing that nuclear MDMX has a slower electrophoretic mobility after DNA damage (Figure 5C).
Figure 6.
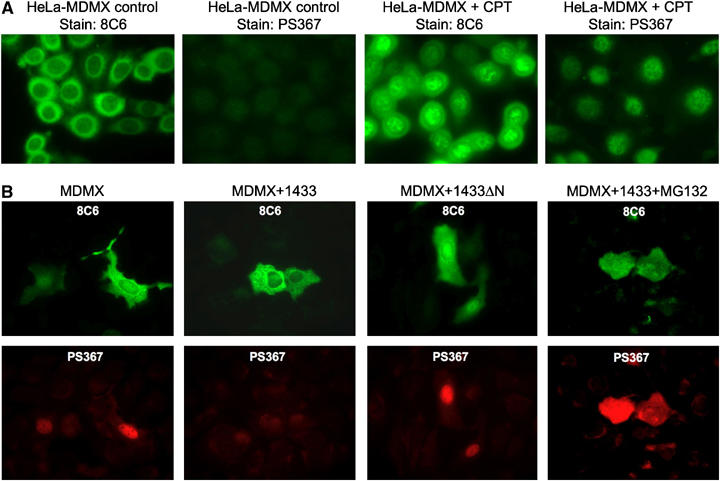
S367 phosphorylated MDMX preferentially localizes to the nucleus. (A) HeLa cells stably transfected with MDMX were treated with 0.5 μM CPT for 18 h and stained using 8C6 or PS367 antibody. (B) 14-3-3 expression promotes elimination of phosphorylated MDMX from the nucleus. U2OS cells were transiently cotransfected with MDMX and 14-3-3τ plasmids for 24 h and treated with 30 μM MG132 for 4 h. Cells were double stained using 8C6 for total MDMX (green) and PS367 antibody for phosphorylated MDMX (red).
Phosphorylated MDMX is rapidly degraded after DNA damage, whereas mutation of S367 increases MDMX resistance to ubiquitination and degradation by MDM2 (Chen et al, 2005a). These observations, together with the results in Figure 5C, suggested that 14-3-3 binding helps to promote degradation of nuclear MDMX. To test this hypothesis, MDMX was transiently cotransfected with 14-3-3 into U2OS cells, which express relatively high levels of endogenous MDM2. Staining of MDMX by 8C6 antibody showed that 14-3-3 overexpression promoted the elimination of nuclear MDMX, resulting in more prominent cytoplasmic distribution (Figure 6B). One interpretation of this result is that 14-3-3 overexpression promotes MDMX nuclear export. However, PS367 staining also showed a loss of phosphorylated MDMX after 14-3-3 cotransfection, suggesting that 14-3-3 promotes degradation of phosphorylated MDMX. Furthermore, when MDMX and 14-3-3 cotransfected cells were treated with MG132, accumulation of phosphorylated MDMX was observed (Figure 6B). The diffused localization of phosphorylated MDMX after MG132 treatment may be due to overexpression, which was not seen in the fractionation of endogenous MDMX (Figure 5C). Overall, the results favor the interpretation that 14-3-3 binding promotes MDMX nuclear import and degradation in the nucleus.
MDMX contains a cryptic nuclear import signal that mediates Chk2-regulated nuclear import
To determine how phosphorylation and 14-3-3 binding promote MDMX nuclear import, we tested whether MDMX contains a cryptic nuclear import signal (NLS) that may be activated by 14-3-3 binding. When GFP was fused to the N-terminus of full-length MDMX, the fusion protein became constitutively cytoplasmic even after DNA damage (data not shown), indicating an interference of nuclear import by the GFP moiety. However, GFP-MDMX300–490 was constitutively nuclear localized (Figure 7A), suggesting the exposure of a cryptic NLS in this region. Inspection of MDMX sequence suggested that the only candidate is a stretch of basic residues within the RING domain (465RRLKK), which is similar to the cryptic nucleolar targeting signal on MDM2 (465KKLKK) (Lohrum et al, 2000). A K468E mutation introduced into GFP-MDMX300–490 resulted in diffused localization (Figure 7A), demonstrating that it was required for nuclear localization. Furthermore, full-length MDMX with K468E mutation failed to undergo nuclear translocation after DNA damage (Figure 7B), despite normal levels of S367 phosphorylation and 14-3-3 binding (data not shown). This mutation had no effect on MDM2-mediated nuclear import of MDMX in cotransfection assays (data not shown), suggesting that it is specifically required for the MDM2-independent nuclear import.
Figure 7.
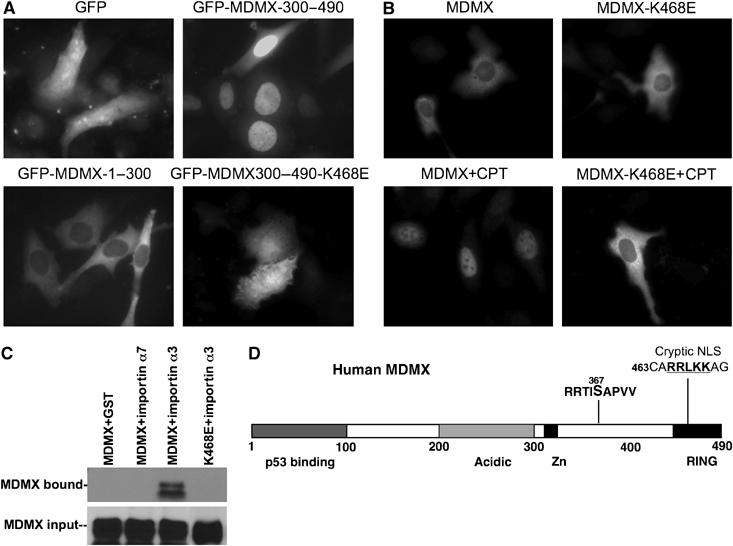
MDMX nuclear import after DNA damage requires a cryptic NLS. (A) GFP–MDMX fusions were stably transfected into U2OS cells and photographed for localization using fluorescence from the GFP tag. (B) Full-length MDMX with the K468E mutation was transiently transfected into U2OS cells, treated for 18 h with 0.5 μM CPT, and stained using 8C6 antibody. (C) GST-importin-loaded beads were incubated with lysate of HCT116 cells transiently transfected with wild-type or mutant MDMX. Bound MDMX were detected by Western blot. (D) Diagram of MDMX indicating the location of the cryptic NLS.
Classic NLS-mediated nuclear import requires binding of the NLS to a family of cytoplasmic receptors (importin α) (Weis, 2003). When GST-importin α1, 3, 5, 6, and 7 fusion proteins were incubated with cell lysate containing MDMX, importin α3 showed significant binding to MDMX (Liu et al, 2005), which was abrogated by the K468E mutation (Figure 7C). This result was consistent with a role of the 465RRLKK sequence in mediating MDMX nuclear import. However, GST-importin α3 pulldown efficiency was not affected by Chk2 phosphorylation of S367 (data not shown), suggesting that the in vitro binding assay does not recapitulate all aspects of the import process. These results showed that MDMX contains a cryptic NLS sequence that mediates Chk2 and 14-3-3-regulated nuclear import after DNA damage.
14-3-3 cooperates with Chk2 to promote MDMX ubiquitination
To further investigate the role of the 14-3-3 binding site on regulation of MDMX ubiquitination, the effect of Chk2 on wild-type MDMX and the S367 mutant was examined. HCT116-Chk2−/− cells were transiently transfected with MDMX, MDM2, His6-ubiquitin, and Chk2 plasmids. MDMX ubiquitination was determined by Ni-NTA purification of ubiquitinated proteins, followed by MDMX Western blot. The results showed that expression of wild-type Chk2, but not the kinase-deficient Chk2-A347 mutant (Chehab et al, 2000), strongly stimulated the poly-ubiquitination and degradation of MDMX by MDM2 (Figure 8A). In contrast, ubiquitination and degradation of the MDMX-S367A mutant was not stimulated by Chk2 (Figure 8A). Therefore, phosphorylation of S367 is important for efficient ubiquitination of MDMX and is necessary for the regulation by Chk2.
Figure 8.
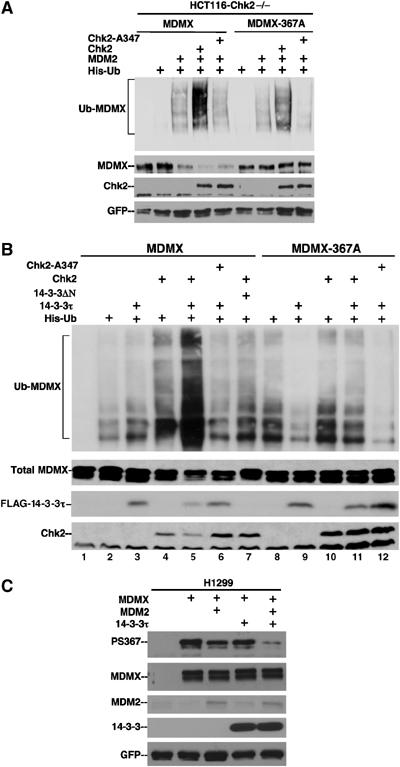
Chk2 promotes ubiquitination and degradation of MDMX through modification of the 14-3-3 binding site. (A) HCT116-Chk2−/− cells were transiently transfected with MDMX, MDM2, Chk2, and His6-ubiquitin. MDMX ubiquitination was detected by Ni-NTA purification, followed by MDMX Western blot. Whole-cell extract was analyzed for MDMX level by Western blot. Chk2-A347 is a kinase-deficient mutant. (B) HCT116-Chk2−/− cells were transfected with His6-ubiquitin, MDMX, 14-3-3τ, and Chk2 plasmids. MDMX ubiquitination was detected by Ni-NTA purification, followed by MDMX Western blot. 14-3-3ΔN is a 30–245 fragment of 14-3-3τ lacking the dimerization domain. (C) 14-3-3 stimulates degradation of PS367 MDMX. H1299 cells were transiently transfected with MDMX, MDM2, and 14-3-3τ for 48 h. MDMX was immunoprecipitated with 8C6 and probed with PS367 antibody for the phosphorylated form, or with 8C6 for total MDMX level.
To determine whether 14-3-3 and Chk2-mediated phosphorylation cooperate to stimulate the ubiquitination of MDMX, HCT116-Chk2−/− cells were transfected with His6-ubiquitin, MDMX, 14-3-3, and Chk2 expression plasmids. Under dose-limiting conditions where 14-3-3 and Chk2 alone had negligible effects on MDMX ubiquitination by endogenous MDM2 (50–100 ng plasmid for a 6 cm plate), coexpression of 14-3-3 and Chk2 significantly enhanced MDMX ubiquitination and degradation (Figure 8B, lane 5 versus 2 and 3). Furthermore, mutation of S367 abrogated the cooperative effect of 14-3-3 and Chk2 (Figure 8B, lane 11 versus 9 and 10). Therefore, Chk2-mediated phosphorylation cooperates with 14-3-3 to stimulate MDMX ubiquitination and degradation.
To further test whether 14-3-3 stimulates degradation of MDMX phosphorylated on S367, MDMX and MDM2 were cotransfected with 14-3-3 in H1299 cells. The transfection procedure alone induced significant phosphorylation of MDMX on S367 without DNA-damaging treatments. The levels of total MDMX level and PS367 MDMX level were determined by IP–Western blot. The result showed that when MDM2 level was suboptimal for significant degradation of total MDMX, 14-3-3 cotransfection selectively stimulated the degradation of MDMX phosphorylated on S367 (Figure 8C). Under conditions of MDM2 overexpression, the levels of total MDMX and phosphorylated MDMX were degraded at a similar rate (data not shown), possibly because phosphorylation on S367 was no longer a rate-limiting step. These results showed that 14-3-3 stimulates MDM2 degradation of MDMX phosphorylated at S367.
To further test the effect of Chk2 and 14-3-3 on the ability of MDMX to inhibit p53 transcriptional activity, HCT116-Chk2−/− cells were transfected with the p53-responsive BP100-luciferase reporter (Freedman et al, 1997). Transfected BP100-luc alone was activated by endogenous p53 and produced baseline readout of p53 activity. Cotransfection of MDMX reduced p53 activity by ∼2-fold (Figure 9A), which was typical for MDMX and weaker than the effect of MDM2 in such assay. Coexpression of 14-3-3 and Chk2 partially overcome the inhibition of p53 activity by wild-type MDMX, consistent with the ability of this combination in promoting MDMX degradation. This effect required active Chk2 and was not seen using kinase-deficient Chk2–347A mutant (Figure 9A). Importantly, the MDMX 367A mutant was not regulated by 14-3-3 and Chk2 (Figure 9B), confirming a requirement for phosphorylation and 14-3-3 binding. These results demonstrated that Chk2 phosphorylation of MDMX on S367 and recruitment of 14-3-3 cooperates to abrogate its inhibitory effect on p53.
Figure 9.
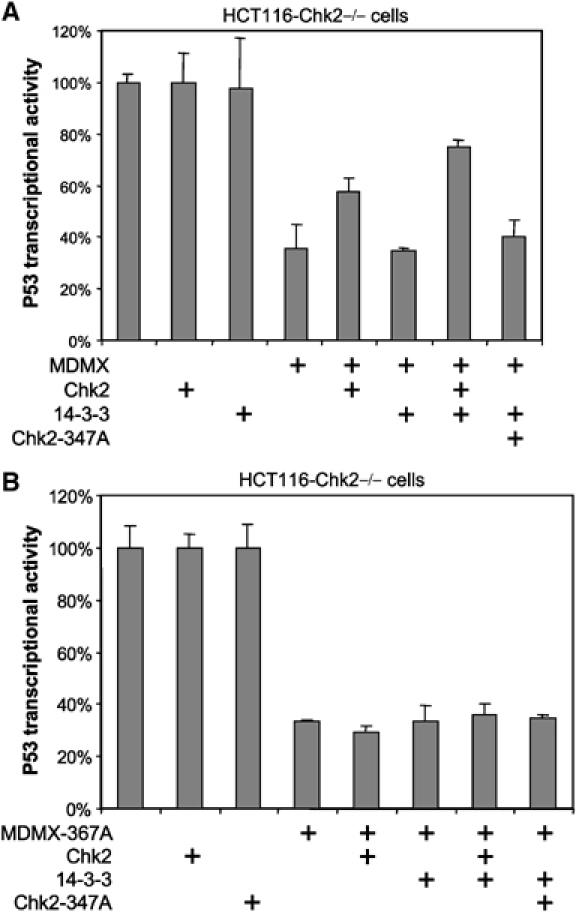
Chk2 and 14-3-3 cooperate to neutralize MDMX inhibition of p53. (A) HCT116-Chk2−/− cells were transfected with p53-responsive BP100-luc reporter to detect endogenous p53 transcriptional activity. The effects of wild-type MDMX, 14-3-3 and Chk2 coexpression on p53 activity were detected by luciferase assay and normalized to CMV-lacZ transfection efficiency control. (B) A control experiment identical to (A), except that the MDMX 367A mutant was used.
Discussion
The results described above showed that MDMX specifically interacts with 14-3-3. S367 is the major 14-3-3 binding site on MDMX and is significantly phosphorylated after DNA damage in a Chk2-dependent fashion, resulting in increased 14-3-3 binding. S367 phosphorylation and 14-3-3 binding stimulate degradation of MDMX by MDM2. Furthermore, phosphorylation of S367 is required for MDMX nuclear import after DNA damage, possibly by activating a cryptic NLS in the RING domain. These results suggest that 14-3-3 proteins regulate MDMX localization and degradation in response to DNA damage, and this effect may contribute to the efficient activation of p53.
Recent findings showed that, after DNA damage, ATM is critical for inducing phosphorylation of MDMX on multiple sites. ATM directly phosphorylates S403, and is also required for phosphorylation of S367 and S342 by activating Chk2. S367 is also the most heavily phosphorylated MDMX residue after ionizing irradiation, and S367 mutation has the most significant impact on MDMX ubiquitination and degradation by MDM2. The recruitment of 14-3-3 by phosphorylated S367 suggests that 14-3-3 is an important regulator of MDMX. It is worth noting that 14-3-3 was initially copurified with MDMX from HeLa cells in the absence of DNA damage, suggesting a basal level of S367 phosphorylation. Chk1 is a likely candidate in this process because it has housekeeping functions and is active in unperturbed cells (Bartek and Lukas, 2003). However, ionizing irradiation and UV did not stimulate S367 phosphorylation in Chk2-null cells, suggesting that Chk2 is critical for the DNA damage response.
Our data showed that phosphorylation of S367 by Chk2 is important for nuclear translocation of MDMX after DNA damage. Mutation of S367 or loss of Chk2 function prevents MDMX nuclear import induced by DNA damage. MDMX has multiple mechanisms for nuclear import. Interaction with MDM2 can target MDMX into the nucleus using the NLS on MDM2. As expected, this effect does not require MDMX S367 phosphorylation or the MDMX cryptic NLS. A second mechanism requires phosphorylation of S367 and possibly involves 14-3-3 recruitment. 14-3-3 binding to phosphorylated S367 may induce conformational change of the RING domain and activate the cryptic NLS. Phosphorylated MDMX also binds to MDM2 with higher affinity (Chen et al, 2005a), consistent with conformational change in the RING domain. Therefore, MDMX belongs to a rare group of proteins that are targeted to the nucleus by 14-3-3 binding.
How 14-3-3 binding stimulates MDMX degradation remains to be further investigated. Nuclear translocation should facilitate interaction with MDM2 in the nucleus, resulting in ubiquitination and degradation of MDMX. Conformational change induced by phosphorylation and 14-3-3 binding may increase affinity to MDM2, or increase the ability of MDMX RING domain to activate MDM2 E3 function after forming the heterodimer. A recent report showed that the de-ubiquitinating enzyme HAUSP binds to MDMX and regulates MDMX stability by de-ubiquitination (Meulmeester et al, 2005). Interaction between MDMX and HAUSP was reduced after DNA damage and is thought to contribute to MDMX destabilization. It is possible that 14-3-3 binding displaces HAUSP and contributes to increased MDMX ubiquitination.
Efficient activation of p53 after DNA damage is likely to be achieved by phosphorylation of multiple targets, including p53, MDM2, and MDMX. The results described in this report add another level of complexity to p53 signaling. Further elucidation of the physiological functions of S367 phosphorylation will require a knock-in or gene replacement approach. It will also be important to determine whether other types of stress also target MDMX or p53 by regulating MDMX–14-3-3 interaction.
Materials and methods
Cell lines and plasmids
H1299, U2OS, MCF-7, and 293T cells were maintained in DMEM medium with 10% fetal bovine serum. HCT116 and HCT116-Chk2−/− cells were kindly provided by Dr Bert Vogelstein and maintained in McCoy 5A medium with 10% fetal bovine serum. Human MDMX cDNA was kindly provided by Dr Donna George (Sharp et al, 1999). FLAG-tagged Chk2 wild-type and A347 mutant were provided by Dr Thanos Halazonetis (Chehab et al, 2000). Expression plasmid for FLAG-14-3-3τ was generated by PCR amplification of 14-3-3τ cDNA from a HeLa cDNA library and subcloning into pcDNA3. Expression plasmids for 14-3-3β, ɛ and η were provided by Dr Haian Fu. 14-3-3γ was provided by Dr Hua Lu, 14-3-3σ was provided by Dr Mong-Hong Lee, and 14-3-3ζ was provided by Dr Anthony J Muslin. GST-importin α constructs were provided by Dr Nancy Reich. For affinity purification and mass spectrometric analysis, a FLAG epitope tag was added to the C-terminus of myc-MDMX by PCR to create double-tagged myc-MDMX-Flag. All MDM2 and MDMX constructs used in this study were based on human cDNA clones.
Affinity purification of MDMX-associated protein
HeLa-S cells stably transfected with FLAG-tagged MDMX were grown as a suspension culture. Cells from 500 ml culture (∼2 × 108 cells) were lysed in 10 ml lysis buffer (50 mM Tris–HCl (pH 8.0), 5 mM EDTA, 150 mM NaCl, 0.5% NP40, 1 mM PMSF), centrifuged for 5 min at 10 000 g, and the insoluble debris were discarded. The lysate was precleared with 100 μl bed volume of protein A Sepharose beads for 30 min, and then incubated with 50 μl bed volume of M2-agarose bead (Sigma) for 4 h at 4°C. The beads were washed extensively with lysis buffer, and MDMX and its associated proteins were eluted with 70 μl of 20 mM Tris (pH 8.0), 2% SDS, and 200 μg/ml FLAG epitope peptide (Sigma) for 15 min. The eluted proteins were fractionated on SDS–PAGE and stained with Coomassie Blue. Proteins copurified with MDMX were excised from the gel and subjected to protease digestion and peptide sequencing by mass spectrometry at the Harvard Microchemistry Laboratory.
Protein analysis
To detect proteins by Western blot, cells were lysed in lysis buffer (50 mM Tris–HCl (pH 8.0), 5 mM EDTA, 150 mM NaCl, 0.5% NP40, 1 mM PMSF, 200 nM okadaic acid), centrifuged for 5 min at 10 000 g, and the insoluble debris were discarded. Cell lysate (10–50 μg protein) was fractionated by SDS–PAGE using a gradient gel and transferred to Immobilon P filters (Millipore). The filter was blocked for 1 h with phosphate-buffered saline (PBS) containing 5% non-fat dry milk and 0.1% Tween-20. The following monoclonal antibodies were used: 3G9 for MDM2 (Chen et al, 1993); DO-1 (Pharmingen) for p53 Western blot; 8C6 monoclonal or a rabbit polyclonal serum for MDMX Western blot and IP (Li et al, 2002), PS367 antibody for phosphorylated MDMX (Chen et al, 2005a). The filter was developed using ECL-plus reagent (Amersham).
Immunoprecipitation assay
Cells were lysed in lysis buffer (50 mM Tris–HCl (pH 8.0), 5 mM EDTA, 150 mM NaCl, 0.5% NP40, 1 mM PMSF), centrifuged for 5 min at 10 000 g, and the insoluble debris were discarded. Cell lysate (500–1000 μg protein) was immunoprecipitated using 100 μl 8C6 hybridoma supernatant against MDMX and protein G agarose beads for 4 h at 4°C. The beads were washed extensively with lysis buffer, boiled in SDS sample buffer, fractionated by SDS–PAGE, and analyzed by anti-14-3-3τ Western blot using an isoform-specific rabbit polyclonal antibody (Santa Cruz Biotechnology).
Capture of 14-3-3 using MDMX phosphopeptides
MDMX peptides (30 μg) S342 (HSLSTSDIT), PS342 (HSL(pS)TSDIT), S367 (RTISAPVVR), and PS367 (RTI(pS)APVVR) were crosslinked to 30 μl of CarboxyLink beads (Pierce) according to the instructions from the manufacturer. The beads (15 μl) were incubated with 200 μg lysate of MCF7 cells stably transfected with FLAG-14-3-3τ (expressed at a level similar to endogenous 14-3-3τ) for 3 h at 4°C, washed with lysis buffer, and analyzed by anti-FLAG Western blot.
In vivo ubiquitination assay
HCT116-Chk2−/− cells in 6 cm plates were transfected with combinations of 0.5 μg His6-ubiquitin expression plasmid, 1 μg MDMX, 0.5 μg MDM2, and 1 μg Chk2 expression plasmids using Lipofectamine Plus reagents (Life Technologies). For detection of 14-3-3 and Chk2 cooperation, 0.5 μg His6-ubiquitin, 1 μg MDMX, 0.1 μg Chk2, and 0.05 μg 14-3-3τ plasmids were cotransfected. At 24 h after transfection, cells were lysed in buffer A (6 M guanidinium–HCl, 0.1 M Na2HPO4/NaH2PO4, 0.01 M Tris–HCl (pH 8.0), 5 mM imidazole, 10 mM β-mercaptoethanol) and incubated with Ni2+-NTA beads (Qiagen) for 4 h at room temperature. The beads were washed with buffer A, B (8 M urea, 0.1 M Na2PO4/NaH2PO4, 0.01 M Tris–HCl (pH 8.0), 10 mM β-mercaptoethanol), C (8 M urea, 0.1 M Na2PO4/NaH2PO4, 0.01 M Tris–HCl (pH 6.3), 10 mM β-mercaptoethanol), and bound proteins were eluted with buffer D (200 mM imidazole, 0.15 M Tris–HCl (pH 6.7), 30% glycerol, 0.72 M β-mercaptoethanol, 5% SDS). The eluted proteins were analyzed by Western blot for the presence of conjugated MDMX using 8C6 antibody.
Luciferase assay
HCT116-Chk2−/− cells were plated in 24-well plates (50 000/well) for 24 h and transfected with a mixture containing 20 ng p53-responsive BP100-luciferase, 40 ng MDMX, 5 ng CMV-lacZ, 2 ng 14-3-3τ, and 5 ng Chk2 plasmids. Transfection was achieved using Lipofectamine PLUS reagents (Invitrogen) and cells were analyzed for luciferase and β galactosidase expression after 24 h. The ratio of luciferase/β galactosidase activity was used as an indicator of p53 transcriptional activity.
Immunofluorescence staining
Cells cultured on chamber slides were transfected with indicated combinations of MDMX and 14-3-3τ expression plasmids using Lipofectamine PLUS reagents. At 24 h after transfection, cells were fixed with acetone-methanol (1:1) for 3 min at room temperature, blocked with PBS+10% normal goat serum (NGS) for 20 min, and incubated with a mixture of anti-MDMX 8C6 hybridoma supernatant and rabbit-anti-PS367 antibody in PBS+10% NGS for 2 h. The slides were washed with PBS+0.1% Triton X-100, incubated with rhodamine-goat-anti-rabbit IgG and FITC-goat-anti-mouse IgG in PBS+10% NGS for 1 h, washed with PBS+0.1% Triton X-100 and mounted.
Nuclear cytoplasmic fractionation
Approximately 1 × 107 cells were pelleted and suspended in 500 μl of 1 × Hypotonic buffer (40 mM HEPES (pH 7.9), 2 mM EDTA, 2 mM EGTA, 20 mM NaF, 1 mM DTT, 1 mM PMSF, 1 mM Na3VO4) containing protease inhibitor cocktail (Sigma). After incubation on ice for 2 min, cells were homogenized with 10 strokes in a Dounce homogenizer. Samples were centrifuged at 4°C at 1000 g for 5 min. Supernatant was collected (Cytosolic fraction) and the pellet was washed with nuclear wash buffer (50 mM NaCl, 10 mM HEPES (pH 8.0), 25% glycerol, 0.1 mM EDTA, 1 mM NaF, 20 mM Na3VO4) and centrifuged briefly at 4°C. The pellet was then lysed on a rotating wheel at 4°C for 20 min in 100 μl of high-salt buffer (840 mM KCl, 40 mM HEPES, 2 mM EDTA, 2 mM EGTA, 40% glycerol, 1 mM DTT) containing protease inhibitors and phosphatase inhibitors. After centrifugation at 10 000 g for 20 min, supernatant was collected (nuclear fraction). Protein concentrations in the cytoplasmic and nuclear fractions were determined, and identical amounts of protein were subjected to Western blot analyses.
In vivo phospholabeling and phosphopeptide analysis
To detect MDMX phosphorylation in vivo, 293T cells in 10 cm plates were transiently transfected with 10 μg MDMX expression plasmids using the calcium phosphate precipitation method. At 40 h after transfection, cells were washed with DMEM without phosphate and incubated with 32P-orthophosphate (0.2 mCi/ml) in DMEM without phosphate for 4 h. Cell lysate was immunoprecipitated with 8C6 and analyzed by SDS–PAGE and autoradiography. Nylon membrane containing radiolabeled MDMX bands were excised and incubated with 50 ng endoproteinase Asp-N (Sigma) for 16 h in 50 mM ammonium bicarbonate at 37°C. MDMX peptides were oxidized with performic acid and resolved by electrophoresis on a thin-layer cellulose plate for 30 min at 1.0 kV in formic acid/glacial acetic acid/water (1:3.1:35.9; pH 1.9) using the HTLE-7002 apparatus. This was followed by chromatography in the second dimension in n-butyl alcohol/pyridine/glacial acetic acid/water (5:3.3:1:4) for 16 h. The phosphopeptides were visualized by autoradiography.
Supplementary Material
Supplemental Data
Acknowledgments
We thank the Moffitt Molecular Biology Core for DNA sequence analyses, Dr William Lane for protein identification by mass spectrometry, Dr Haian Fu for 14-3-3 clones, Dr Nancy Reich for GST-importin α constructs, and Dr Hua Lu for stimulating discussions. This work was supported by grants from the American Cancer Society and the NIH to J Chen. C LeBron is supported by a supplement from the NIH.
References
- Bartek J, Lukas J (2003) Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell 3: 421–429 [DOI] [PubMed] [Google Scholar]
- Chehab NH, Malikzay A, Appel M, Halazonetis TD (2000) Chk2/hCds1 functions as a DNA damage checkpoint in G(1) by stabilizing p53. Genes Dev 14: 278–288 [PMC free article] [PubMed] [Google Scholar]
- Chen J, Marechal V, Levine AJ (1993) Mapping of the p53 and mdm-2 interaction domains. Mol Cell Biol 13: 4107–4114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Gilkes DM, Pan Y, Lane WS, Chen J (2005a) ATM and Chk2-dependent phosphorylation of MDMX contribute to p53 activation after DNA damage. EMBO J 24: 3411–3422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Li C, Pan Y, Chen J (2005b) Regulation of p53–MDMX interaction by casein kinase 1 alpha. Mol Cell Biol 25: 6509–6520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Graaf P, Little NA, Ramos YF, Meulmeester E, Letteboer SJ, Jochemsen AG (2003) Hdmx protein stability is regulated by the ubiquitin ligase activity of Mdm2. J Biol Chem 278: 38315–38324 [DOI] [PubMed] [Google Scholar]
- Finch RA, Donoviel DB, Potter D, Shi M, Fan A, Freed DD, Wang CY, Zambrowicz BP, Ramirez-Solis R, Sands AT, Zhang N (2002) mdmx is a negative regulator of p53 activity in vivo. Cancer Res 62: 3221–3225 [PubMed] [Google Scholar]
- Freedman DA, Epstein CB, Roth JC, Levine AJ (1997) A genetic approach to mapping the p53 binding site in the MDM2 protein. Mol Med 3: 248–259 [PMC free article] [PubMed] [Google Scholar]
- Fu H, Subramanian RR, Masters SC (2000) 14-3-3 proteins: structure, function, and regulation. Annu Rev Pharmacol Toxicol 40: 617–647 [DOI] [PubMed] [Google Scholar]
- Gu J, Kawai H, Nie L, Kitao H, Wiederschain D, Jochemsen AG, Parant J, Lozano G, Yuan ZM (2002) Mutual dependence of MDM2 and MDMX in their functional inactivation of p53. J Biol Chem 277: 19251–19254 [DOI] [PubMed] [Google Scholar]
- Harris SL, Levine AJ (2005) The p53 pathway: positive and negative feedback loops. Oncogene 24: 2899–2908 [DOI] [PubMed] [Google Scholar]
- Hermeking H (2003) The 14-3-3 cancer connection. Nat Rev Cancer 3: 931–943 [DOI] [PubMed] [Google Scholar]
- Hermeking H, Lengauer C, Polyak K, He TC, Zhang L, Thiagalingam S, Kinzler KW, Vogelstein B (1997) 14-3-3 sigma is a p53-regulated inhibitor of G2/M progression. Mol Cell 1: 3–11 [DOI] [PubMed] [Google Scholar]
- Jallepalli PV, Lengauer C, Vogelstein B, Bunz F (2003) The Chk2 tumor suppressor is not required for p53 responses in human cancer cells. J Biol Chem 278: 20475–20479 [DOI] [PubMed] [Google Scholar]
- Kawai H, Wiederschain D, Kitao H, Stuart J, Tsai KK, Yuan ZM (2003) DNA damage-induced MDMX degradation is mediated by MDM2. J Biol Chem 278: 45946–45953 [DOI] [PubMed] [Google Scholar]
- Li C, Chen L, Chen J (2002) DNA damage induces MDMX nuclear translocation by p53-dependent and -independent mechanisms. Mol Cell Biol 22: 7562–7571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, McBride KM, Reich NC (2005) STAT3 nuclear import is independent of tyrosine phosphorylation and mediated by importin-alpha3. Proc Natl Acad Sci USA 102: 8150–8155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohrum MA, Ashcroft M, Kubbutat MH, Vousden KH (2000) Identification of a cryptic nucleolar-localization signal in MDM2. Nat Cell Biol 2: 179–181 [DOI] [PubMed] [Google Scholar]
- Meulmeester E, Maurice MM, Boutell C, Teunisse AF, Ovaa H, Abraham TE, Dirks RW, Jochemsen AG (2005) Loss of HAUSP-mediated deubiquitination contributes to DNA damage-induced destabilization of Hdmx and Hdm2. Mol Cell 18: 565–576 [DOI] [PubMed] [Google Scholar]
- Migliorini D, Danovi D, Colombo E, Carbone R, Pelicci PG, Marine JC (2002a) Hdmx recruitment into the nucleus by Hdm2 is essential for its ability to regulate p53 stability and transactivation. J Biol Chem 277: 7318–7323 [DOI] [PubMed] [Google Scholar]
- Migliorini D, Lazzerini Denchi E, Danovi D, Jochemsen A, Capillo M, Gobbi A, Helin K, Pelicci PG, Marine JC (2002b) Mdm4 (Mdmx) regulates p53-induced growth arrest and neuronal cell death during early embryonic mouse development. Mol Cell Biol 22: 5527–5538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montes de Oca Luna R, Wagner DS, Lozano G (1995) Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature 378: 203–206 [DOI] [PubMed] [Google Scholar]
- Nomura M, Shimizu S, Sugiyama T, Narita M, Ito T, Matsuda H, Tsujimoto Y (2003) 14-3-3 Interacts directly with and negatively regulates pro-apoptotic Bax. J Biol Chem 278: 2058–2065 [DOI] [PubMed] [Google Scholar]
- Okamoto K, Kashima K, Pereg Y, Ishida M, Yamazaki S, Nota A, Teunisse A, Migliorini D, Kitabayashi I, Marine JC, Prives C, Shiloh Y, Jochemsen AG, Taya Y (2005) DNA damage-induced phosphorylation of MdmX at serine 367 activates p53 by targeting MdmX for Mdm2-dependent degradation. Mol Cell Biol 25: 9608–9620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Neill T, Giarratani L, Chen P, Iyer L, Lee CH, Bobiak M, Kanai F, Zhou BB, Chung JH, Rathbun GA (2002) Determination of substrate motifs for human Chk1 and hCds1/Chk2 by the oriented peptide library approach. J Biol Chem 277: 16102–16115 [DOI] [PubMed] [Google Scholar]
- Pan Y, Chen J (2003) MDM2 promotes ubiquitination and degradation of MDMX. Mol Cell Biol 23: 5113–5121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parant J, Chavez-Reyes A, Little NA, Yan W, Reinke V, Jochemsen AG, Lozano G (2001) Rescue of embryonic lethality in Mdm4-null mice by loss of Trp53 suggests a nonoverlapping pathway with MDM2 to regulate p53. Nat Genet 29: 92–95 [DOI] [PubMed] [Google Scholar]
- Pereg Y, Shkedy D, de Graaf P, Meulmeester E, Edelson-Averbukh M, Salek M, Biton S, Teunisse AF, Lehmann WD, Jochemsen AG, Shiloh Y (2005) Phosphorylation of Hdmx mediates its Hdm2- and ATM-dependent degradation in response to DNA damage. Proc Natl Acad Sci USA 102: 5056–5061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prives C, Hall PA (1999) The p53 pathway. J Pathol 187: 112–126 [DOI] [PubMed] [Google Scholar]
- Ramos YF, Stad R, Attema J, Peltenburg LT, van der Eb AJ, Jochemsen AG (2001) Aberrant expression of HDMX proteins in tumor cells correlates with wild-type p53. Cancer Res 61: 1839–1842 [PubMed] [Google Scholar]
- Sharp DA, Kratowicz SA, Sank MJ, George DL (1999) Stabilization of the MDM2 oncoprotein by interaction with the structurally related MDMX protein. J Biol Chem 274: 38189–38196 [DOI] [PubMed] [Google Scholar]
- Shvarts A, Steegenga WT, Riteco N, van Laar T, Dekker P, Bazuine M, van Ham RC, van der Houven van Oordt W, Hateboer G, van der Eb AJ, Jochemsen AG (1996) MDMX: a novel p53-binding protein with some functional properties of MDM2. EMBO J 15: 5349–5357 [PMC free article] [PubMed] [Google Scholar]
- Stad R, Little NA, Xirodimas DP, Frenk R, van der Eb AJ, Lane DP, Saville MK, Jochemsen AG (2001) Mdmx stabilizes p53 and Mdm2 via two distinct mechanisms. EMBO Rep 2: 1029–1034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavridi ES, Chehab NH, Malikzay A, Halazonetis TD (2001) Substitutions that compromise the ionizing radiation-induced association of p53 with 14-3-3 proteins also compromise the ability of p53 to induce cell cycle arrest. Cancer Res 61: 7030–7033 [PubMed] [Google Scholar]
- Su TT, Parry DH, Donahoe B, Chien CT, O'Farrell PH, Purdy A (2001) Cell cycle roles for two 14-3-3 proteins during Drosophila development. J Cell Sci 114: 3445–3454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanimura S, Ohtsuka S, Mitsui K, Shirouzu K, Yoshimura A, Ohtsubo M (1999) MDM2 interacts with MDMX through their RING finger domains. FEBS Lett 447: 5–9 [DOI] [PubMed] [Google Scholar]
- Tzivion G, Shen YH, Zhu J (2001) 14-3-3 proteins; bringing new definitions to scaffolding. Oncogene 20: 6331–6338 [DOI] [PubMed] [Google Scholar]
- Waterman MJ, Stavridi ES, Waterman JL, Halazonetis TD (1998) ATM-dependent activation of p53 involves dephosphorylation and association with 14-3-3 proteins. Nat Genet 19: 175–178 [DOI] [PubMed] [Google Scholar]
- Weis K (2003) Regulating access to the genome: nucleocytoplasmic transport throughout the cell cycle. Cell 112: 441–451 [DOI] [PubMed] [Google Scholar]
- Yaffe MB, Rittinger K, Volinia S, Caron PR, Aitken A, Leffers H, Gamblin SJ, Smerdon SJ, Cantley LC (1997) The structural basis for 14-3-3:phosphopeptide binding specificity. Cell 91: 961–971 [DOI] [PubMed] [Google Scholar]
- Yang HY, Wen YY, Chen CH, Lozano G, Lee MH (2003) 14-3-3 sigma positively regulates p53 and suppresses tumor growth. Mol Cell Biol 23: 7096–7107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Xiong Y (2001) Control of p53 ubiquitination and nuclear export by MDM2 and ARF. Cell Growth Differ 12: 175–186 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Data