

Abstract
It has been shown that MDMX inhibits the activity of the tumor suppressor p53 by primarily cooperating with the p53 feedback regulator MDM2. Here, our study shows that this inhibition can be overcome by 14-3-3γ and Chk1. 14-3-3γ was identified as an MDMX-associated protein via an immuno-affinity purification-coupled mass spectrometry. Consistently, 14-3-3γ directly interacted with MDMX in vitro, and this interaction was stimulated by MDMX phosphorylation in vitro and in cells. Interestingly, in response to UV irradiation, the wild-type, but not the kinase-dead mutant, Chk1 phosphorylated MDMX at serine 367, enhanced the 14-3-3γ-MDMX binding and the cytoplasmic retaining of MDMX. The Chk1 specific inhibitor UCN-01 repressed all of these effects. Moreover, overexpression of 14-3-3γ, but not its mutant K50E, which did not bind to MDMX, suppressed MDMX-enhanced p53 ubiquitination, leading to p53 stabilization and activation. Finally, ablation of 14-3-3γ by siRNA reduced UV-induced p53 level and G1 arrest. Thus, these results demonstrate 14-3-3γ and Chk1 as two novel regulators of MDMX in response to UV irradiation.
Keywords: 14-3-3γ, Chk1, MDMX, p53, UV response
Introduction
In response to stresses, the tumor suppressor p53 protein is stabilized and activated to induce cell growth arrest and/or apoptosis, consequently protecting normal cells from undergoing neoplasmic transformation (Vogelstein et al, 2000). These functions of p53 primarily attribute to its nuclear transcriptional activity to induce the expression of a broad panel of genes. Some of these genes encode proteins, such as p21 or 14-3-3σ, responsible for stopping the cell growth (el-Deiry et al, 1993; Harper et al, 1993; Dulic et al, 1994; Hermeking et al, 1997; Bunz et al, 1998), and others encode proteins, such as Bax or PIGs, important for apoptosis (Miyashita and Reed, 1995; Venot et al, 1998). Also, p53 directly travels to the mitochondria, triggering apoptosis (Mihara et al, 2003; Chipuk et al, 2004; Leu et al, 2004). To control these cytotoxic effects of p53, cells have developed a negative feedback regulatory loop to monitor its level. The key component of this loop is MDM2 (also called HDM2 in human) (Barak et al, 1993; Fakharzadeh et al, 1993; Wu et al, 1993). MDM2 mediates p53 proteasomal degradation (Haupt et al, 1997; Kubbutat et al, 1997) through its E3 ubiquitin ligase activity (Honda et al, 1997). To warrant the efficient execution of this feedback regulation, the cells also recruit an assistant for MDM2.
This assistant protein is MDMX (also called MDM4, or HDMX for human) (Shvarts et al, 1996). MDMX resembles MDM2 at the N-terminal p53-binding and the C-terminal ring-finger domains (Sharp et al, 1999). Like MDM2, MDMX binds to p53 and inhibits its functions (Jackson and Berberich, 2000; Rallapalli et al, 2003). Similar to the mdm2–p53 double-knockout (KO) phenotype (Jones et al, 1995; Montes de Oca Luna et al, 1995), deleting the p53 gene also rescues the lethal phenotype of mdmx null mice (Parant et al, 2001; Migliorini et al, 2002b), suggesting that MDMX is an essential MDM2 partner. But, unlike MDM2, MDMX transcript is not regulated by p53 (Shvarts et al, 1996) and MDMX by itself is unable to ubiquitinate p53 (Jackson and Berberich, 2000; Stad et al, 2000). Also, MDMX resides mostly in the cytoplasm (Rallapalli et al, 1999), but can be recruited to the nucleus by MDM2 (Gu et al, 2002; Migliorini et al, 2002a), which is induced by γ irradiation (Li et al, 2002). MDMX is thought to enhance MDM2-mediated p53 ubiquitination and degradation (Ghosh et al, 2003), which are prevented by siRNAs against MDMX (Gu et al, 2002; Linares et al, 2003). Hence, to activate p53, stress signals must turn on cellular mechanisms that surmount the negative control by MDM2 and MDMX.
Indeed, the MDM2–p53 loop is highly tuned through post-translational regulations in response to stresses (Appella and Anderson, 2001). For instance, UV activates the ATR kinase, which phosphorylates p53 at serine 15 or MDM2 at serine 407 (Tibbetts et al, 1999), or Chk1 that in turn phosphorylates p53 at serine 15 and serine 20 (Shieh et al, 2000), resulting in p53 activation. Also, 14-3-3σ was shown to activate p53 by binding to and protecting it from MDM2 attack (Yang et al, 2003). 14-3-3σ is one of the seven 28–33 kDa 14-3-3 proteins, which mostly localize in the cytoplasm. These acidic proteins are ubiquitously expressed and play multiple roles in cellular signaling, trafficking, apoptosis, cell cycle, and stress response. They prefer binding to serine/threonine-phosphoryated proteins at the consensus-binding motifs RSXpS/T/XP (type 1) or RXXXpS/TXP (type 2) (Muslin et al, 1996; Yaffe et al, 1997), where X stands for any amino acids. Thus, those post-translational regulations ultimately activate p53 by blocking the MDM2 feedback loop. Although recent studies have begun to link post-translational regulations with MDMX (Elias et al, 2005; Meulmeester et al, 2005; Pereg et al, 2005), less is known about how stress signals may activate p53 by influencing MDMX function and why MDMX mostly resides in the cytoplasm.
To address these issues, we generated a stable Flag-MDMX expression cell line using human embryonic kidney (HEK) epithelial 293 cells. Using this cell line for immuno-affinity purification followed by mass spectrometry, we identified 14-3-3γ as an MDMX-associated protein from the cytoplasmic extracts. Further analyses verified the association of this γ form with MDMX in cells. This binding is enhanced by Chk1-mediated phosphorylation of MDMX at serine 367 in response to UV, resulting in p53 activation.
Results
MDMX associates with 14-3-3γ in cells
To identify cellular proteins that may regulate MDMX function, we carried out an immuno-affinity purification using anti-Flag antibody-conjugated beads and cytoplasmic extracts prepared from Flag-MDMX-expressed HEK 293 cells. Proteins eluted from the beads with Flag peptides were analyzed by SDS–PAGE and colloidal blue staining. Three major distinct bands between the 64 and the 26 kDa markers were specifically pulled down from the Flag-MDMX-expressed 293 cell extracts, but not from the mock-transfected extracts (Figure 1A). Mass spectrometric analysis of these bands revealed that the largest band above the 50 kDa marker was an MDMX fragment, and that several peptide sequences derived from the two bands above the 26 kDa marker matched the β, ɛ, γ, σ, τ, and ζ isoforms of the 14-3-3 family. This result suggests that MDMX might bind to 14-3-3s. Sequence analysis of MDMX revealed a potential 14-3-3-binding domain RRTISAP between amino acids 363 and 369 (Figure 1B). This motif in MDMX is evolutionarily conserved, but does not exist in the same region of MDM2 (Figure 1B).
Figure 1.
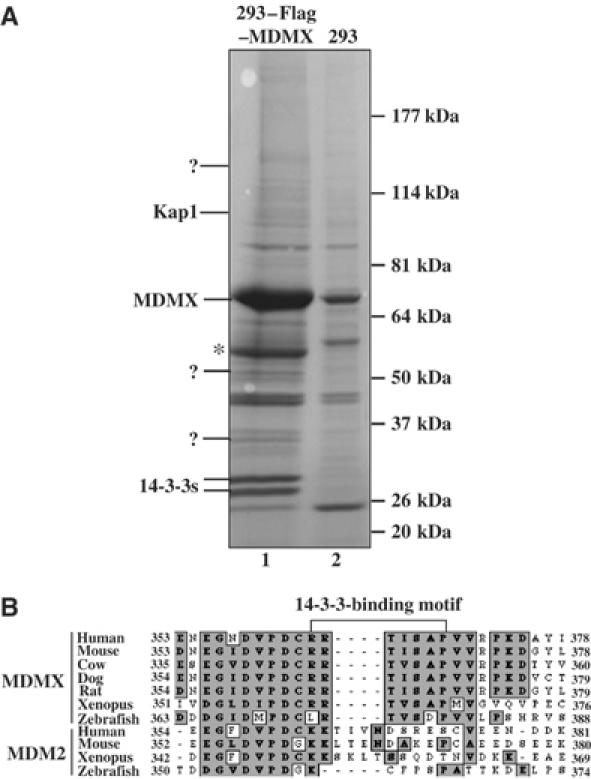
Identification of 14-3-3s as Flag-MDMX-associated proteins by immunoaffinity purification. (A) Colloidal Blue staining of proteins eluted from Flag antibody beads loaded with either empty vector expressing 293 cytoplasmic extracts (lane 2) or the cytoplasmic extracts from the 293–Flag–MDMX cell line (lane 1). MDMX-associated polypeptides were subjected to mass spectrometry. MDMX and 14-3-3 bands are indicated. The question mark denotes unidentified polypeptides. *Indicates the short MDMX fragment. Peptide sequences of 14-3-3 proteins from mass spectrometry are: β, TAFDEAIAELDTLNEESYK; ɛ, AAFDDAIAELDTLSEESYK; γ, AYSEAHEISK; θ/τ, QTIDNSQGAYQEAFDISK; ζ, KGIVDQSQQAYQEAFEISK. ? denotes unknown peptides. (B) MDMX or MDM2 peptide sequences from different species were aligned using the MacVector software. A putative 14-3-3-binding motif as indicated was identified in MDMX, but not in the same region of MDM2. Consensus sequences were highlighted in gray.
To determine which 14-3-3 isoform may bind to MDMX, we conducted transient transfection assays using 293 and H1299 cells with the mammalian expression vectors encoding each of these 14-3-3 isoforms and MDMX followed by immunoprecipitation (IP)–Western blot (WB). Representative results using 293 cells are shown in Figure 2A and B. Interestingly, MDMX bound to 14-3-3γ more efficiently than to the σ, τ, ɛ, β, or ζ isoforms, as tested using IP with anti-Flag (Figure 2A) and anti-GFP (Figure 2B) (see Supplementary Figure S1 for 14-3-3ɛ; data for β and ζ isoforms are not shown). Consistently, endogenous MDMX and 14-3-3γ were coimmunoprecipitated with the anti-14-3-3γ or anti-MDMX, but not anti-His, antibody from 293 cells (Figure 2C). By contrast, endogenous 14-3-3ɛ did not efficiently bind to endogenous MDMX (Figure 2C, right panel), neither did 14-3-3σ (data not shown). Although 14-3-3ɛ was pulled down through affinity purification (Figure 1A), this result might be due to the large quantity of proteins used in the purification. These results suggest that MDMX prefers binding to 14-3-3γ in cells. Thus, we focused on examining the role of 14-3-3γ in regulating MDMX function in this study.
Figure 2.
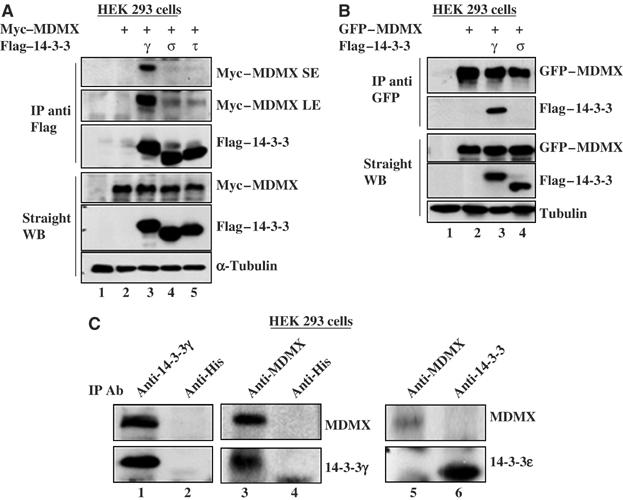
14-3-3γ interacts with MDMX in cells. (A) HEK 293 cells (panel A) or H1299 cells (data not shown) were transfected with 3 μg of c-myc–MDMX along or together with 3 μg of Flag-14-3-3γ, σ or τ vector. Cell lysates (300 μg) were immunoprecipitated with the anti-Flag antibody followed by WB, and expression of exogenous proteins was assessed by WB using 50 μg per lane, as indicated. SE: short exposure. LE: long exposure. (B) MDMX interacts with 14-3-3γ in cells. 293 cells were transfected with 3 μg GFP-MDMX vector along or together with 3 μg Flag-14-3-3γ, or σ vector. Proteins were detected by IP-WB as indicated. Similar assays were carried out using the vector encoding β, ɛ, or ζ (data not shown). (C) Endogenous 14-3-3γ and MDMX interact with each other in 293 cells. Whole-cell lysates (500 μg protein/sample) were immunoprecipitated with the monoclonal anti-14-3-3γ, anti-MDMX, anti-14-3-3ɛ, or anti-4xHis (control, lanes 2 and 4) antibody followed by WB using antibodies as indicated on the right.
14-3-3γ directly interacts with MDMX in vitro with a high affinity to MDMX phosphopeptides
To determine whether MDMX binds to 14-3-3γ directly, we performed an in vitro protein–protein interaction assay using bacterially expressed and purified GST-14-3-3γ and his-MDMX (Figure 3A). Indeed, MDMX bound to GST-14-3-3γ (lane 2), but not GST alone (lane 1). This binding was reduced by a 15-mer peptide that contains the serine-phosphorylated 14-3-3-binding consensus sequence RSASpEP, but not by its nonphosphorylated counterpart, in a dose-dependent manner (Figure 3A). The same result was obtained when a serine-phosphorylated 15-mer peptide containing the sequence 363RRTISpAP369 derived from MDMX was used under the same experimental setting (Figure 3B). The interaction between 14-3-3γ and MDMX was reduced >90% when fourfold (molar ratio) of the MDMX phosphopeptide over MDMX was used (lane 3), suggesting that 14-3-3γ displays a higher affinity to the phosphorylated MDMX peptide. But, at the same concentrations, the nonphosphorylated MDMX peptide had no apparent effect (lane 7). These results suggest that 14-3-3γ binds to MDMX in vitro with a high affinity to the serine-phosphorylated RRTISpAP peptide of MDMX.
Figure 3.
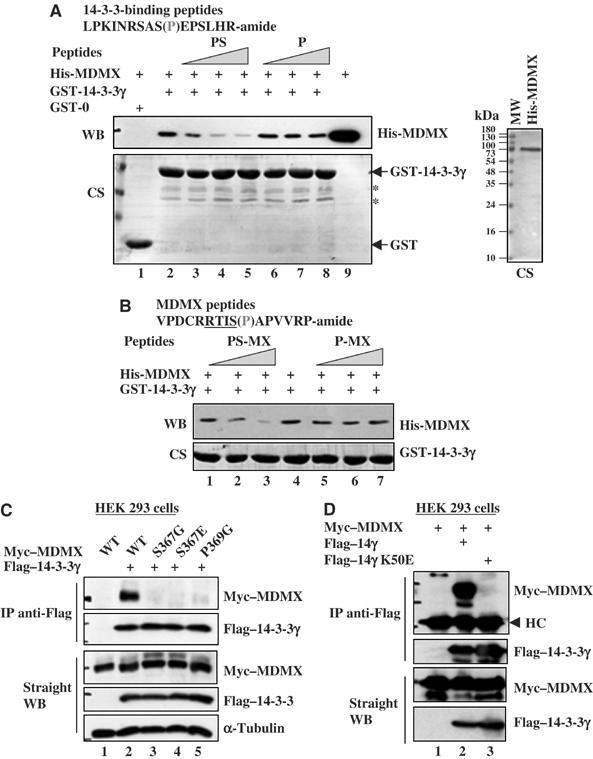
The 14-3-3-binding motif of MDMX and the target-binding pocket of 14-3-3γ are essential for the MDMX–14-3-3γ interaction. (A) Recombinant human MDMX interacts with GST-14-3-3γ in vitro, which is competed by phosphorylated 14-3-3-binding peptides. For peptide competition assays, phosphopeptides (PS) and nonphosphopeptide (P) were used. Purified His-MDMX (2 μg proteins) is shown in the right panel. CS stands for Coomassie staining. *Indicates the minor proteins pulled down. (B) The MDMX phosphopeptide competes with His-MDMX for 14-3-3γ binding in vitro. The assay was conducted as that in panel A (PS-MX, serine 367 phosphorylated MDMX peptide; P-MX, non-phosphorylated MDMX peptide). (C) Point mutations at the 14-3-3-binding region of MDMX eliminate the association of MDMX with 14-3-3γ. 293 cells were transfected with 3 μg plasmid encoding MDMX (wild type)-myc or myc-tagged point mutations of MDMX along or together with 3 μg plasmid encoding Flag-tagged 14-3-3γ. Cell lysates were used for IP–WB and direct WB as that for Figure 2. (D) Point mutation at the protein-binding domain of 14-3-3γ abolishes the 14-3-3γ–MDMX interaction. Myc–MDMX (3 μg) vector alone (lane 1) or together with 3 μg of Flag-tagged 14-3-3γ (lane 2) or Flag-14-3-3γ K50E (lane 3) was used for transfection. Cell lysates were used for IP–WB and direct WB.
The RRTISpAP motif and serine 367 of MDMX are crucial for binding to 14-3-3γ
To determine if the RRTISpAP motif and serine 367 in MDMX are important for 14-3-3γ binding, we replaced serine (S) 367 with either glycine (G) or glutamic acid (E), and proline (P) 369 with glycine (G) in the c-myc–MDMX vector. Using these mutants as well as MDMX and 14-3-3γ, we conducted a set of transient transfection experiments followed by IP with anti-Flag antibody and WB. Mutation of either S367 or P369 in MDMX completely abolished the interaction between MDMX and 14-3-3γ (Figure 3C). The negative result with the S367E mutant was expected, as 14-3-3s prefer contacting with the phospho group at the target site to the side chain of acidic residues (Zhang et al, 1997; Ku et al, 1998). Hence, this result suggests that the RRTISpAP motif and serine 367 are essential for the binding of MDMX to 14-3-3γ.
Next, we wanted to determine if the target-binding domain of 14-3-3γ is also critical for MDMX binding, using a 14-3-3γ mutant with substitution of lysine (K) 50 by glutamic acid (E). This K50E mutant has been shown to lose the ability to bind to its target proteins (Zhang et al, 1997). In an experiment similar to that shown in Figure 3C, except the K50E mutant, we found that this mutant failed to bind MDMX in cells (Figure 3D). These results indicate that 14-3-3γ utilizes its target-binding domain at the K50 position to contact with the 363RRTISpAP369 motif of MDMX in cells.
UV stimulates the 14-3-3γ–MDMX interaction in the presence of Chk1
Next we wanted to determine if the 14-3-3γ–MDMX interaction is affected by stresses. To do so, we irradiated the transfected 293 cells with 30 J/m2 UV-C or 7 Gy of γ irradiation, and harvested them for IP-WB using antibodies, as shown in Figure 4A. Interestingly, the 14-3-3γ-MDMX binding was enhanced by UV-C (lane 3), but reduced by γ irradiation (lane 4). Consistently, UV-C also led to the cytoplasmic accumulation of endogenous MDMX, and 14-3-3γ existed primarily in the cytoplasm (Figure 4B and C and also see Supplementary Figure S2). By contrast, γ irradiation resulted in nuclear accumulation of MDMX (Figure 4B and C), as expected (Gu et al, 2002; Li et al, 2002; Migliorini et al, 2002a). This result, with the study showing enhancement of MDMX degradation by γ irradiation (Chen et al, 2005), could explain why γ irradiation reduced the interaction of 14-3-3γ with MDMX (Figure 4A). We could not merge the images of MDMX and 14-3-3γ because we only found the monoclonal antibodies against each of them suitable for immunofluorescent staining. These results suggest that UV may activate a kinase that phosphorylates MDMX at serine 367 and enhances its binding to 14-3-3γ.
Figure 4.
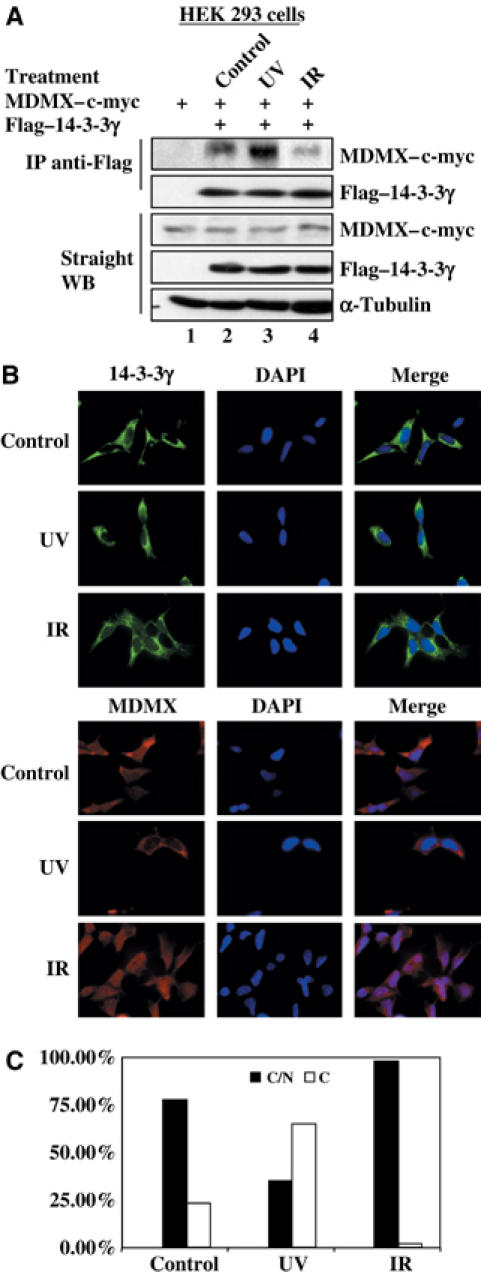
UV irradiation induces MDMX–14-3-3γ association and MDMX nuclear export. (A) UVC enhances the interaction of MDMX with 14-3-3γ in cells. HEK 293 cells were transfected with 3 μg of myc–MDMX plasmid alone or together with 3 μg of Flag-tagged 14-3-3γ plasmid. Transfected cells were treated with 30 J/m2 UVC or 7 Gy ionizing irradiation (IR) at 3 h before harvest. Cell lysates were used for IP with the anti-Flag antibody followed by WB with anti-myc and anti-Flag antibodies, as well as for WB using antibodies as indicated on the right. (B) Subcellular localization of MDMX and 14-3-3γ after irradiation. After irradiation as described above, HEK 293 cells were fixed for immunofluorescent staining with the anti-MDMX antibody 8C6 or the anti-14-3-3γ antibody Ab2 and DAPI staining as indicated. (C) Percentage of cells that displays subcellular distributions of MDMX was plotted. In all, 300 cells of each sample were counted and classified by MDMX subcellular localization. C/N indicates cytosol and nucleus, and C denotes cytosol. We rarely identified a single cell displaying nuclear MDMX only.
Analysis of the RRTISpAP sequence of MDMX revealed serine 367 as a potential target for Chk1, which is UV-activated (Liu et al, 2000) and prefers targeting the consensus motif (RXXS) (O'Neill et al, 2002). Next we tested whether Chk1 enhances the MDMX–14-3-3γ interaction by performing an experiment similar to that in Figure 4A, except that Chk1 and p38MAPK were included. As shown in Figure 5A, overexpression of Chk1, but not p38MAPK, in UV-irradiated 293 cells enhanced the MDMX–14-3-3γ interaction, suggesting that Chk1, but not p38MAPK, may phosphorylate MDMX. To confirm this result, we also introduced the Chk1 kinase-dead mutant into 293 cells followed by UV irradiation. UV irradiation enhanced the 14-3-3γ–MDMX interaction in the presence of Chk1, but not its KD mutant (Figure 5B). Without UV irradiation, ectopic Chk1 did not affect this interaction (data not shown). These results suggest that Chk1 might be the kinase responsible for the UV-enhanced interaction of MDMX with 14-3-3γ.
Figure 5.
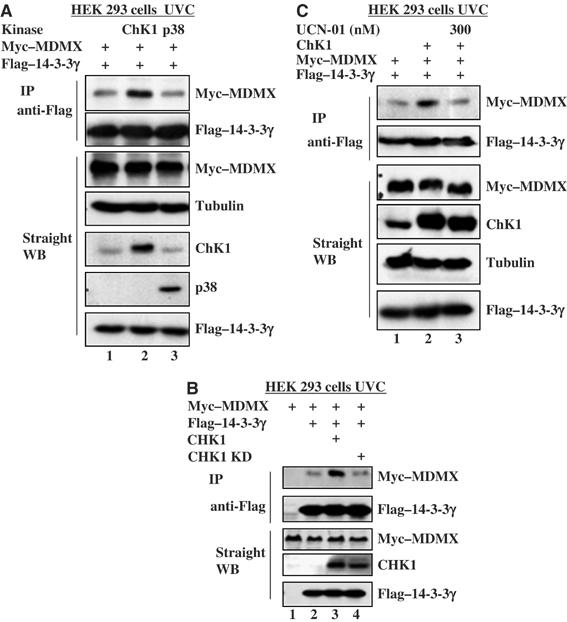
Chk1 enhances MDMX–14-3-3γ association after UV irradiation. (A) Chk1, but not p38 MAP kinase, enhances MDMX–14-3-3γ association after UVC irradiation. HEK 293 cells were transfected with 2 μg of myc–MDMX and 2 μg of Flag-tagged 14-3-3γ or together with 2 μg of Chk1 or p38 MAP kinase vector. Transfected cells were treated with 30 J/m2 UVC 3 h before harvesting. Cell lysates (300 μg) were used for IP-WB or WB as that for Figure 2, using antibodies as indicated on the right. (B) Wild-type, but not kinase-dead, Chk1 enhances the interaction between MDMX and 14-3-3γ after UV irradiation. HEK 293 cells were transfected with 2 μg of myc–MDMX vector and 2 μg of Flag-14-3-3γ vector with or without 2 μg plasmid encoding Chk1 wild-type or kinase-dead form (Chk1 KD). One set of transfected cells was treated with 30 J/m2 UVC 3 h before harvest. IP–WB and straight WB were carried out using whole-cell lysates as described in Figure 2. (C) UNC-01 represses Chk1-enhanced MDMX–14-3-3γ interaction. An assay similar to that in panel B was conducted, except that 300 nM UCN-01 (lane 3) or DMSO (other lanes) was added into the media right after UV irradiation.
To verify this likelihood, we employed a Chk1 inhibitor called UCN-01 in the same set of experiments. UCN-01 is a potent kinase inhibitor highly specific for Chk1 (100-fold higher than for Chk2) (Busby et al, 2000; Graves et al, 2000). We wanted to determine whether UNC-01 inhibits UV- and Chk1-induced MDMX–14-3-3γ interaction in cells. First, our in vitro kinase assay showed that UNC-01 effectively inhibited Chk1 activity on p53. In all, 300 nM of the inhibitor led to 80% reduction of p53 phosphorylation (Supplementary Figure S4a). Next, 300 nM of UCN-01 was used for in vivo protein–protein interaction assays using IP–WB. Again, UV irradiation enhanced the MDMX–14-3-3γ interaction in the presence of Chk1, but its inhibitor UCN-01 markedly reduced this interaction (Figure 5C). Taken together, these results demonstrate that Chk1 is the kinase responsible for the UV-induced MDMX–14-3-3γ interaction in cells.
Chk1 phosphorylates serine 367 of MDMX in vitro and in cells
To test if Chk1 phosphorylates MDMX, we conducted an IP-kinase assay using the Flag-Chk1-containing 293 cell lysates as the kinase resource, and using his-MDMX, its mutants S367G and S367E as substrates. This immunoprecipitated Flag-Chk1 phosphorylated MDMX, but not its two mutants in vitro (Figure 6A). Furthermore, we purified GST-Chk1 and GST-Chk1 KD from baculovirus-infected insect cells through GSH-agarose beads. This purified Chk1 was active, as Chk1, but not its kinase-dead mutant, was able to phosphorylate p53 as well as GST-MDMX in vitro (Supplementary Figure S3A and B). Hence, Chk1 can indeed phosphorylate MDMX at serine 367 in vitro.
Figure 6.
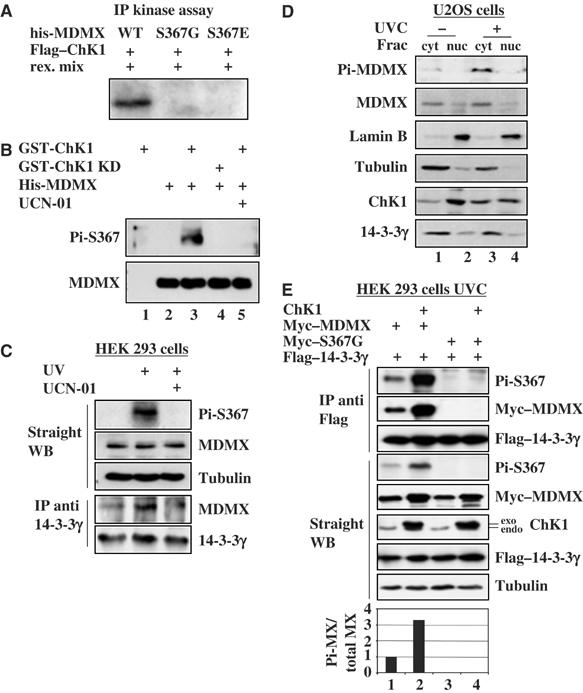
Chk1 phosphorylates MDMX in vitro and in cells. (A) MDMX is phosphorylated by Flag-Chk1 in vitro. His-MDMX wild type (500 ng), His-MDMX S367G or His-MDMX S367E was incubated with Flag-Chk1 beads prepared from overexpressed 293 cell lysates as described in Materials and methods. Phosphorylated proteins were detected by autoradiography. (B) The anti-serine 367 phosphorylation antibody detects MDMX phosphorylation by Chk1 in vitro. Purified His-MDMX (300 ng) was incubated with 500 ng GST-Chk1 or GST-Chk1 KD. Total or phosphorylated MDMX was detected by WB with the anti-MDMX or anti-Pi-S367 antibody. (C) UVC induces endogenous MDMX phosphorylation and its association with 14-3-3γ. 293 cells were irradiated with 30 J/m2 UVC 3 h before harvest (lanes 2 and 3). UCN-01 (300 nM) was added into media right after UV treatment (lane 3). In all, 300 μg of cell lysates were used for IP with the anti-14-3-3γ antibody and 50 μg was used for WB using antibodies as indicated on the right. (D) UV-induced MDMX serine 367 phosphorylation occurs in the cytoplasm. Treated (30 J/m2 UVC for 3 h) or nontreated U2OS cells were harvested and subjected to subcellular fractionation. Each fraction (45 μg) was resolved by SDS–PAGE followed by WB as indicated on the left. Lamin B and tubulin were used as the nuclear and cytosolic markers, respectively. Cyt, cytosolic fraction; nuc, nuclear fraction. (E) UVC induces Chk1 phosphorylation of MDMX and the MDMX–14-3-3γ association. 293 cells were transfected with 2 μg of Flag-14-3-3γ and 2 μg of myc–MDMX or S367G myc–MDMX vector with or without 2 μg of Chk1 vector. Transfected cells were treated with 30 J/m2 UVC 3 h before harvest. Cell lysates were for IP with the monoclonal anti-Flag antibody followed by WB, or for WB using antibodies as indicated on the right. The ratio of S367 phosphorylated MDMX to total MDMX in straight WB was plotted on the graph.
To determine whether Chk1 also phosphorylates serine 367 of MDMX in cells, we developed a polyclonal antibody against the serine 367 phosphorylated peptide (Figure 3B). This antibody was highly specific to this phosphorylated peptide, as 1:4000 dilution of this antibody clearly detected 1 nmol of the phosphopeptides, but not even 10 nmol of non-phosphopeptides (Supplementary Figure S3C). We purified this antibody through the phosphopeptide conjugated column. Using the purified antibody, we found that it detected the phosphorylated species of MDMX by Chk1, but not by its KD mutant, in vitro (Figure 6B). Again, UCN-01 completely inhibited Chk1-mediated MDMX phosphorylation (lane 5). Remarkably, this serine 367 antibody detected UV-induced endogenous MDMX phosphorylation that was inhibited by UCN-01 (Figure 6C). UV also induced the interaction between endogenous MDMX and 14-3-3γ, which was inhibited by UCN-01 as well (Figure 6C, lower panels). Consistent with the result in Figure 4, phosphorylated MDMX as well as total MDMX appeared to accumulate in the cytoplasm after UV irradiation (Figure 6D). Chk1 level also increased in the cytoplasm after UV irradiation in comparison with nonirradiated cells (Figure 6D), suggesting that Chk1 might phosphorylate MDMX in the cytoplasm. The anti-phosphoserine 367 antibody also detected Chk1-mediated phosphorylation of ectopic MDMX, but not of its S367G mutant, in UV-irradiated 293 cells (Figure 6E). 14-3-3γ bound to Chk1-phosphorylated MDMX (Figure 6E). The residual signal of MDMX in the absence of ectopic Chk1 detected by this serine 367 antibody (lane 1) might be due to MDMX phosphorylation by UV-activated endogenous Chk1. These results demonstrate that UV activates Chk1, which in turn phosphorylates MDMX at serine 367, consequently enhancing the interaction of 14-3-3γ with MDMX in cells.
14-3-3γ induces p53 and its activity
To determine the functional consequence of the MDMX–14-3-3γ interaction, human osteosarcoma U2OS (p53-proficient) or Saos-2 (p53-deficient) cells were transfected with the Flag-14-3-3γ or myc–MDMX- plasmid alone or together and harvested 36 h after transfection for WB. In line with the results in Figures 1, 2, 3, 4, 5 and 6, ectopic 14-3-3γ induced the level of endogenous p53 as well as p21 in U2OS cells (Figure 7A). As expected (Finch et al, 2002; Linares et al, 2003), overexpression of MDMX reduced the level of p53 and p21 (Figure 7A). Strikingly, 14-3-3γ rescued this reduction (lane 4), suggesting that 14-3-3γ can activate p53 by repressing MDMX function. This p21 induction by 14-3-3γ was not evident in Saos-2 cells (Figure 7B), suggesting that the induction seen in U2OS cells is p53-dependent. Consistently, 14-3-3γ also rescued the repression of p53-dependent transcriptional activity by MDMX as measured in the lucifierase reporter assay (Figure 7C). This rescue was not observed when 14-3-3γ K50E, which failed to bind MDMX (Figures 2 and 3), was used (Figure 7C). Consequently, overexpression of 14-3-3γ, but not its K50E mutant, led to cell growth arrest at G1 phase in U2OS (Figure 7D and E), but not in Saos-2 cells (Figure 7F). Remarkably, ablation of 14-3-3γ by its siRNA, but not scramble siRNA, in U2OS cells reduced UV-induced p53 level and G1 arrest (Figure 8A and B). Taken together, these results indicate that 14-3-3γ plays a key role in UV-induced p53 activation and G1 arrest by binding to Chk1-phosphorylated MDMX.
Figure 7.
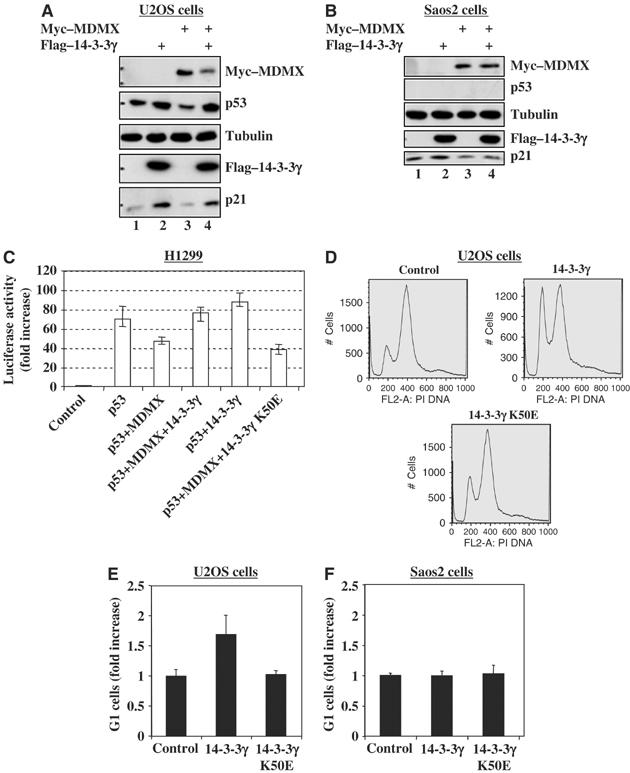
14-3-3γ stimulates p53 activity and G1 arrest. (A, B) Ecotopic expression of 14-3-3γ induces p21 dependently on p53. U2OS (panel A) or Saos 2 (panel B) cells were transfected with a combination of c-myc–MDMX and Flag-14-3-3γ vectors as indicated. Cells lyastes (20 μg) were used for WB with antibodies as indicated on the right. (C) 14-3-3γ induces p53 transactivation activity by suppressing MDMX function. H1299 cells were transiently transfected with plasmids as indicated on the bottom. Luciferase activity is presented in the fold of increase compared to control transfection. The error bars indicate standard deviations from three independent experiments. (D–F) 14-3-3γ induces G1 cell arrest in U2OS, but not Saos2, cells. U2OS or Saos2 cells were transfected with plasmids as indicated, for FACS analysis. 105 cells were counted in each sample. G1 cells from U2OS (D–E) or Saos2 (F) were presented in the fold of increase compared to a control. The error bars indicate standard deviations from three independent experiments.
Figure 8.
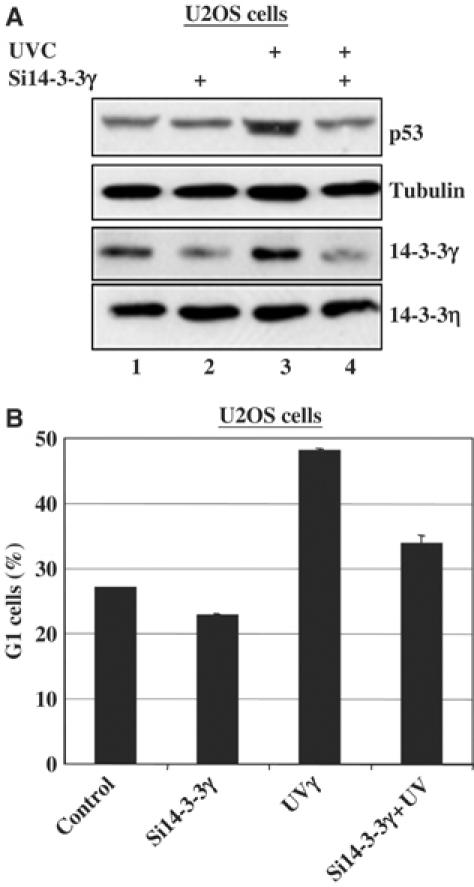
Ablation of 14-3-3γ by siRNA suppresses UV-induced p53 level and G1 arrest. (A) U2OS cells transfected with scrambled or 14-3-3γ siRNAs were irradiated with or without 30 J/m2 UVC for 3 h before harvest. Cell lyastes (15 μg) were used for WB with antibodies as indicated on the right. (B) U2OS cells were treated in the same way as that for panel A, except that the cells were harvested for FACS analysis. In all, 50 000 cells were counted in each sample. Percentages of G1 cells are plotted on the graph and the error bars indicate standard deviations from three independent experiments.
14-3-3γ represses MDMX-enhanced p53 ubiquitination
It has been shown that MDMX enhances MDM2-mediated p53 ubiquitination and degradation, though it alone does not ubiquitinate p53 (Linares et al, 2003). To determine whether the induction of p53 by 14-3-3γ is due to the inhibition of MDMX-enhanced p53 ubiquitination by this protein, we performed an in vivo ubiquitination assay. H1299 cells (p53-deficient but MDM2-proficient) were transfected with the plasmids encoding 14-3-3γ, K50E, MDMX, p53 and His-ubiquitin in different combinations as shown in Figure 8A. Cells were harvested for WB and an in vivo ubiquitination assay as described in Materials and methods. As expected (Linares et al, 2003), ectopic MDMX in H1299 enhanced p53 ubiquitination (Figure 8A). Ectopic 14-3-3γ, but not its K50E mutant, inhibited this MDMX-enhanced p53 ubiquitination (Figure 8A). Of note, 14-3-3γ, but not its mutant, also slightly decreased p53 ubiquitination in the absence of the exogenous MDMX (lanes 5 and 7). This result, consistent with the above results, indicates that 14-3-3γ alleviates MDMX-stimulated p53 ubiquitination by binding to MDMX, as 14-3-3γ did not bind to MDM2 or p53 in the absence of MDMX in cells (Figure 9B), and nor to MDM2 in vitro (Figure 9C).
Figure 9.
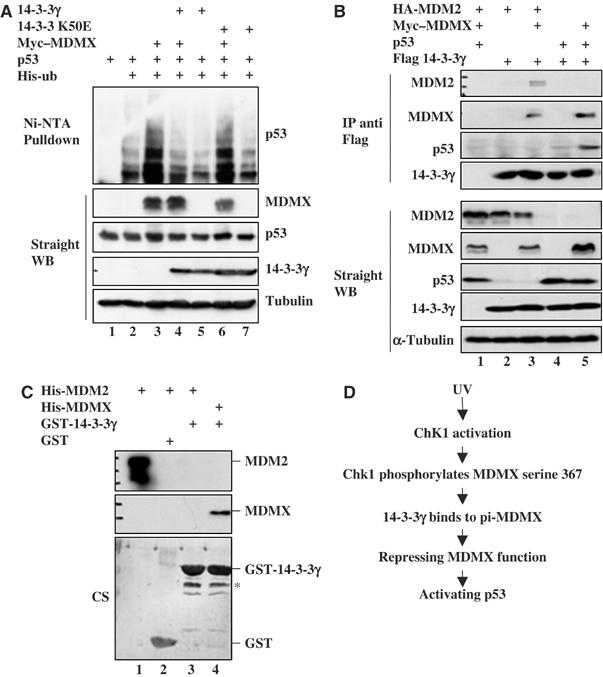
14-3-3γ inhibits p53 ubiquitination by binding to MDMX, but not MDM2 or p53. (A) 14-3-3γ inhibits p53 ubiquitination in cells. H1299 cells were transfected with His6-ubiquitin (2 μg), p53 (0.8 μg), Falg-14-3-3γ (1.6 μg) or c-myc–MDMX (1.6 μg) expression plasmids as indicated on the top. Ubiquitinated p53 proteins were detected by the anti-p53 (DO-1) antibody. Protein levels were detected as indicated on the right. (B) 14-3-3γ does not bind to MDM2 or p53 in cells. H1299 cells were transfected with plasmids as indicated on the top. Cells lysates (300 μg) were immunoprecipitated with the monoclonal anti-Flag antibody followed by WB. In all, 50 μg of lysates were used for WB with antibodies, as indicated on the left. (C) MDM2 does not bind to 14-3-3γ in vitro. GST fusion protein pulldown assay were carried out as that in Figure 3A. His-MDMX (500 ng) or His MDM2 (500 ng) was used for the binding assay. Bound proteins were detected by WB using the anti-MDM2 (2A10) or anti-MDMX antibody (8C6). GST-0 and GST-14-3-3γ levels were determined by Coomassie staining (CS) of the membrane after WB. ‘*' indicates the minor proteins. (D) A schematic model for p53 activation by14-3-3γ after UV irradiation.
Discussion
The oncoprotein MDMX plays an essential role in the MDM2–p53 feedback loop to further consolidate the inhibitory regulation of p53 by MDM2 (Marine and Jochemsen, 2005). Interrupting this loop is crucial for activating p53 and preventing cell transformation (Vousden, 2002). Our study unveils 14-3-3γ and Chk1 as two novel MDMX regulators to block this loop.
Our study demonstrates a direct and phosphorylation-stimulated interaction between 14-3-3γ and MDMX in response to UV. Previously, 14-3-3σ was shown to be induced by p53 to mediate p53-dependent G2 arrest (Hermeking et al, 1997) and in turn to activate p53 by binding to it (Yang et al, 2003). Unlike 14-3-3σ, 14-3-3γ did not bind to p53 (Figure 9), instead strongly bound to MDMX in cells (Figures 2, 3, 4 5 and 6). Consistently, there exists a 14-3-3-binding motif 363RRTISpAP369 in MDMX, but not in the same region of MDM2 (Figure 1B). Mutagenesis analyses of S367 or P369 in MDMX and of K50 in 14-3-3γ demonstrate the importance of these motifs in their interaction as well as functional regulation (Figures 3, 4, 5, 6, 7, 8 and 9). Although MDM2 displays two tentative 14-3-3-binding sites in different regions as detected using the ‘Scansite' program (Obenauer et al, 2003), our results demonstrate that 14-3-3γ does not bind to MDM2 in vitro and in cells (Figure 9).
One remaining puzzle is whether 14-3-3γ requires homo- or hetero-dimerization to bind to MDMX. In contrast to 14-3-3σ, which prefers forming homodimers in cells (Benzinger et al, 2005; Wilker et al, 2005), 14-3-3γ may form a heterodimer with other isoforms in order to associate with MDMX, as more than two isoforms were copurified with Flag-MDMX from cells (Figure 1A). Also, a report published during the revision of our article has shown that exogenous MDMX also associates with the β, ɛ and τ isoforms in addition to 14-3-3γ (Okamoto et al, 2005). By contrast, we found that MDMX binds to the γ isoform more efficiently than to other isoforms (Figure 2 and Supplementary Figure S1; data not shown). This discrepancy might be due to different cells or washing conditions used by the two laboratories. Or, it suggests that 14-3-3γ might possess a higher affinity to MDMX than others. The latter speculation is supported by our observation that endogenous 14-3-3γ, but not 14-3-3ɛ, bound to MDMX without apparent stresses (Figure 2C). Also, ablation of 14-3-3γ by siRNA alleviated UV-induced p53 activation and G1 arrest (Figure 8), suggesting that 14-3-3γ plays a role in UV-induced p53 activation (Figures 4, 5, 6, 7 and 8), at least at the early response to UV irradiation (Supplementary Figure S2).
It has been shown that UV activates several kinases, including Chk1 that activates p53 (Appella and Anderson, 2001). Interestingly, Chk1, but not its kinase-dead mutant, also stimulated the 14-3-3γ–MDMX interaction in response to UV (Figure 5B). This stimulation was specific for Chk1, as p38MAPK did not affect the 14-3-3γ–MDMX binding in response to UV (Figure 5A), and the Chk1-specific inhibitor UCN-01 effectively inhibited this stimulation (Figures 5C and 6). Also Chk1 phosphorylated MDMX, but not the MDMX S367G or S367E mutant, in vitro and in cells (Figure 6). These results demonstrate that Chk1 is a bona fide kinase for serine 367 and enhances the MDMX–14-3-3γ binding in response to UV. But it remains likely that the Chk1–14-3-3γ–MDMX pathway may respond to different stresses and that serine 367 may be phosphorylated by other kinases, such as Chk2 (Chen et al, 2005), as other DNA-damaging agents were recently shown to induce the interaction of MDMX with 14-3-3β (Okamoto et al, 2005).
The functional consequence of the interaction between 14-3-3γ and MDMX is p53 activation and G1 arrest (Figure 7). One mechanism underlying this action is the inhibition of MDMX-enhanced p53 ubiquitination by 14-3-3γ (Figure 9A). This inhibition might not be due to the competition for protein binding, as MDM2 and p53 were pulled down with Flag-14-3-3γ in the presence of MDMX by the anti-Flag antibody (Figure 9B). It is likely that 14-3-3γ might interfere with the ubiquitin conjugation process by associating with MDMX as a stereo block. An alternative mechanism would be the nuclear exclusion of MDMX by associating with cytoplasmic 14-3-3γ in response to UV (Figures 1, 4 and 6). By contrast, γ irradiation may reduce this interaction by enhancing the nuclear import (Figure 4) and MDMX degradation by MDM2 (Chen et al, 2005). This is not the case after UV (Figures 4 and 6) as it decreases MDM2 level (Zeng et al, 2000). It still remains unclear if the 14-3-3γ–MDMX binding affects MDMX stability.
In sum, our study suggests a model for the action of 14-3-3γ working with Chk1 on the MDM2–MDMX–p53 pathway in response to UV irradiation (Figure 9D). This model raises some important questions. For instance, in which cellular compartment does Chk1 phosphorylate MDMX? Though our result suggests that it may occur in the cytoplasm (Figure 6D), this notion needs to be verified. Also, is 14-3-3γ mutated in human cancers? Addressing these and other tempting questions as aforementioned would shed light on our better understanding of the role of 14-3-3γ in the MDMX–MDM2–p53 pathway.
Materials and methods
Cell culture
HEK epithelial 293 cells, human lung non-small-cell carcinoma H1299 cells, human p53-proficient osteosarcoma U2OS cells, and human p53-null osteosarcoma Saos-2 cells were cultured as described previously (Jin et al, 2002; Zeng et al, 2002).
Buffers
See Supplementary data for details.
Antibodies and plasmids
Monoclonal anti-Flag, anti-Flag-M2 agarose and anti-α-tubulin antibodies were purchased from Sigma. Polyclonal anti-GFP antibody, monoclonal anti-14-3-3ɛ antibody (8C3), polyclonal anti-14-3-3γ antibody (C-16), monoclonal anti-Chk1 antibody, polyclonal anti-p53 antibody (FL393) and monoclonal anti-p53 antibody (DO-1) were purchased from Santa Cruz Biotechnology. Monoclonal anti-MDM2 antibodies 4B11 and 2A10 were described previously (Zeng et al, 1999). Monoclonal anti-p21waf1/cip1 antibody (Ab11), monoclonal anti-14-3-3σ antibody (Ab1) and monoclonal anti-14-3-3γ antibody (Ab2) were purchased from Neomarker Biotech. Monoclonal anti-myc tag (9E10) antibody was purchased from Upstate. Monoclonal anti 4xHis antibody was purchased from Qiagen. pCDNA3-Ha-MDM2 and pCMV-p53 plasmids were described previously (Jin et al, 2002). To generate human 14-3-3γ expression construct pcDNA3-2Flag-14-3-3γ, the full-length 14-3-3γ cDNA was amplified by reverse transcriptase PCR (RT–PCR) from HeLa cell mRNA with primers 5′-CGCGGATCCATGGTGGACCGCGAGCAACTG-3′ and 5′-CCGGAATTCTTAAT-TGTTGCCTTCGCCGC-3′. The PCR product was subcloned into the pcDNA3-2Flag vector. The mutant K50E vector was generated from pcDNA3-2Flag-14-3-3γ by mutating K50–E50. The human MDMX construct was a gift from Aart Jochemsen (Leiden University, The Netherlands). The pcDNA3–Flag–14-3-3σ was a gift from Haian Fu (Emory University). 14-3-3ɛ and β cDNAs were cloned by RT–PCR and inserted into the pcDNA3–Myc vector. The pcDNA3–myc–14-3-3ζ was described previously (Luo et al, 1995). The monoclonal anti-MDMX antibody (8C6), the Flag–14-3-3τ and pcDNA3–c-myc–MDMX- plasmids were kind gifts from Jiandong Chen (H Lee Moffitt Comprehensive Cancer Center, Tampa, FL). Point mutation vectors MDMX–S367G–myc, myc–MDMX–S367E, and myc–MDMX–P367G were generated from pCDNA3–myc–MDMX vector by replacing S367 to G or E, and P369 to G, respectively. Mammalian expression vectors encoding Chk1 or Chk1 KD were obtained from Mathew Thayer (Oregon Health & Science University, Portland, OR). GST-Chk1 and GST-Chk1 KD viruses were obtained from Carol Prives (Columbia University, New York, NY). p38 MAP kinase vector was described previously (Keller et al, 1999).
Transient transfection and WB analyses
HEK 293, H1299, U2OS or Saos2 cells (70% confluence) were transfected with combinations of pcDNA3–Flag–14-3-3γ, pcDNA3–myc–MDMX or mutants (see figure legends for the amount of plasmids used) with TransFectin lipid reagent (Bio-Rad). At 48 h post-transfection, cells were harvested and lysed in lysis buffer. Clarified whole-cell lysates (50 μg protein) were loaded directly onto an SDS gel for WB with antibodies as indicated in each figure.
Establishment of Flag-MDMX expression cell lines
HEK 293 cells were transfected with pcDNA3-Flag-MDMX or pcDNA3 vector. Transfected cells expressing Flag-MDMX were selected in the presence of neomycin (0.5 mg/ml) and screened by WB with the anti-Flag antibody.
Affinity purification of human MDMX-associated protein complexes
Approximately 109 HEK 293 cells were used for the preparation of nuclear extract and cytoplasm fraction (S100) by a previously described method (Dignam et al, 1983). The anti-Flag M2 agarose beads were washed with phosphate-buffered saline (PBS) and suspended in PBS as 50% slurry. In all, 50 mg of S100 fractions from either 293–Flag–MDMX cells or empty vector expressing 293 cells were incubated with 0.2 ml of anti-Flag-M2 beads at 4°C for 4 h. The beads were washed four times in lysis buffer. The bead-bound proteins were eluted in 0.2 ml of lysis buffer containing 0.4 mg of synthetic Flag peptides/ml. Eluted proteins were loaded onto an SDS–5–17% gradient polyacrylamide gel for Colloidal Blue staining after electrophoresis. Specific bands from 293–Flag–MDMX fractions were excised and subjected to mass spectrometry.
In vivo ubiquitination assay
In vivo ubiquitination assay was conducted as described previously (Jin et al, 2003) (see Supplementary data for details).
Immunofluorescent staining and fluorescent microscopic analysis
HEK 293 cells were irradiated with 30 J/m2 UVC or 7 Gy of γ ray. At 3 h post-treatment, cells were fixed for immunofluorescent staining with the anti-MDMX antibody 8C6 or monoclonal anti-14-3-3γ antibody Ab2 followed by the Alexa Fluor 546 (red) goat anti-mouse and the Alexa Fluor 488 (green) goat anti-mouse antibodies (Molecular Probes, OR), respectively. DNA was stained with DAPI. Stained cells were analyzed under the Zeiss Axiovert 200 M fluorescent microscope (Zeiss, Germany).
GST fusion protein–protein association assay
The fusion proteins were expressed in E. coli and purified on a glutathione-Sepharose 12B column. Protein–protein association assays were conducted as reported using fusion protein-containing beads (Zeng et al, 1999). See Supplementary data for details.
Luciferase assays
H1299 cells were transfected with the pCMV-β-galactoside reporter plasmid (0.1 μg) and a luciferase reporter plasmid (0.1 μg) driven by two copies of the p53RE motif derived from the MDM2 promoter (Wu et al, 1993), together with a combination of different plasmids (total plasmid DNA 1 μg/well) as indicated in the figure legend with TransFectin (Bio-Rad). At 48 h post-transfection, cells were harvested for luciferase assays as described previously (Zeng et al, 2001, 2002). Luciferase activity was normalized by a factor of β-galactosidase activity in the same assay.
Chk1 kinase assays
Chk1 kinase assays were conducted as described previously (Zhao and Piwnica-Worms, 2001). See Supplementary data for IP-kinase and cold kinase assays.
SiRNA transfection
SiRNA specific to human 14-3-3γ (sc-29582) was purchased from Santa Cruz Biotechnology, Inc. and transfected into U2OS cells using oligofectAMINE reagent (Invitrogen) as described (Jin et al, 2003). One set of transfected cells was treated with 30 J/m2 UVC 3 h before harvest. Cells were harvested 48 h post-transfection and lysed in lysis buffer for SDS–PAGE and WB. The other set of transfected and UV-irradiated cells were harvested for FACS analysis.
Subcellular fractionation
See Supplementary data for details.
UCN-01 treatment
UCN-01 was generously provided by Sally Hausman at NIH/NCI. UCN-01 was dissolved in DMSO and titrated from 100 to 500 nM for inhibiting p53 phosphorylation by GST-Chk1. Based on the titration result, 300 nM of UCN-01 or DMSO as a control was used in either in vitro assay or cultured cells.
FACS analysis
U2OS or Saos2 cells were transfected with plasmids encoding Flag-14-3-3γ, Flag-14-3-3γ K50E, or empty vector. Transfected cells were treated with 150 ng/ml nocodazole for 16 h before harvesting. At 40 h post-transfection, cells were harvested and re-suspended in 100 μl of PBS, and transferred to a polystyrene tube for FACS analysis as detailed in Supplementary data.
Supplementary Material
Supplementary Materials
Acknowledgments
We want to thank Drs Jiandong Chen, Carol Prives, Sally Hausman, Aart G Jochemsen, Haian Fu, and Matt Thayer for providing plasmids and antibodies. This work is supported by NCI grants CA93614, CA095441 and CA 079721 to HL.
References
- Appella E, Anderson CW (2001) Post-translational modifications and activation of p53 by genotoxic stresses. Eur J Biochem 268: 2764–2772 [DOI] [PubMed] [Google Scholar]
- Barak Y, Juven T, Haffner R, Oren M (1993) mdm2 expression is induced by wild type p53 activity. EMBO J 12: 461–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benzinger A, Popowicz GM, Joy JK, Majumdar S, Holak TA, Hermeking H (2005) The crystal structure of the non-liganded 14-3-3 sigma protein: insights into determinants of isoform specific ligand binding and dimerization. Cell Res 15: 219–227 [DOI] [PubMed] [Google Scholar]
- Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, Sedivy JM, Kinzler KW, Vogelstein B (1998) Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 282: 1497–1501 [DOI] [PubMed] [Google Scholar]
- Busby EC, Leistritz DF, Abraham RT, Karnitz LM, Sarkaria JN (2000) The radiosensitizing agent 7-hydroxystaurosporine (UCN-01) inhibits the DNA damage checkpoint kinase hChk1. Cancer Res 60: 2108–2112 [PubMed] [Google Scholar]
- Chen L, Gilkes DM, Pan Y, Lane WS, Chen J (2005) ATM and Chk2-dependent phosphorylation of MDMX contribute to p53 activation after DNA damage. EMBO J 24: 3411–3422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chipuk JE, Kuwana T, Bouchier-Hayes L, Droin NM, Newmeyer DD, Schuler M, Green DR (2004) Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science 303: 1010–1014 [DOI] [PubMed] [Google Scholar]
- Dignam JD, Lebovitz RM, Roeder RG (1983) Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res 11: 1475–1489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulic V, Kaufmann WK, Wilson SJ, Tlsty TD, Lees E, Harper JW, Elledge SJ, Reed SI (1994) p53-dependent inhibition of cyclin-dependent kinase activities in human fibroblasts during radiation-induced G1 arrest. Cell 76: 1013–1023 [DOI] [PubMed] [Google Scholar]
- el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, Vogelstein B (1993) WAF1, a potential mediator of p53 tumor suppression. Cell 75: 817–825 [DOI] [PubMed] [Google Scholar]
- Elias B, Laine A, Ronai Z (2005) Phosphorylation of MdmX by CDK2/Cdc2(p34) is required for nuclear export of Mdm2. Oncogene 24: 2574–2579 [DOI] [PubMed] [Google Scholar]
- Fakharzadeh SS, Rosenblum-Vos L, Murphy M, Hoffman EK, George DL (1993) Structure and organization of amplified DNA on double minutes containing the mdm2 oncogene. Genomics 15: 283–290 [DOI] [PubMed] [Google Scholar]
- Finch RA, Donoviel DB, Potter D, Shi M, Fan A, Freed DD, Wang CY, Zambrowicz BP, Ramirez-Solis R, Sands AT, Zhang N (2002) mdmx is a negative regulator of p53 activity in vivo. Cancer Res 62: 3221–3225 [PubMed] [Google Scholar]
- Ghosh M, Huang K, Berberich SJ (2003) Overexpression of Mdm2 and MdmX fusion proteins alters p53 mediated transactivation, ubiquitination, and degradation. Biochemistry 42: 2291–2299 [DOI] [PubMed] [Google Scholar]
- Graves PR, Yu L, Schwarz JK, Gales J, Sausville EA, O'Connor PM, Piwnica-Worms H (2000) The Chk1 protein kinase and the Cdc25C regulatory pathways are targets of the anticancer agent UCN-01. J Biol Chem 275: 5600–5605 [DOI] [PubMed] [Google Scholar]
- Gu J, Kawai H, Nie L, Kitao H, Wiederschain D, Jochemsen AG, Parant J, Lozano G, Yuan ZM (2002) Mutual dependence of MDM2 and MDMX in their functional inactivation of p53. J Biol Chem 277: 19251–19254 [DOI] [PubMed] [Google Scholar]
- Harper JW, Adami GR, Wei N, Keyomarsi K, Elledge SJ (1993) The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell 75: 805–816 [DOI] [PubMed] [Google Scholar]
- Haupt Y, Maya R, Kazaz A, Oren M (1997) Mdm2 promotes the rapid degradation of p53. Nature 387: 296–299 [DOI] [PubMed] [Google Scholar]
- Hermeking H, Lengauer C, Polyak K, He TC, Zhang L, Thiagalingam S, Kinzler KW, Vogelstein B (1997) 14-3-3 sigma is a p53-regulated inhibitor of G2/M progression. Mol Cell 1: 3–11 [DOI] [PubMed] [Google Scholar]
- Honda R, Tanaka H, Yasuda H (1997) Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett 420: 25–27 [DOI] [PubMed] [Google Scholar]
- Jackson MW, Berberich SJ (2000) MdmX protects p53 from Mdm2-mediated degradation. Mol Cell Biol 20: 1001–1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y, Lee H, Zeng SX, Dai MS, Lu H (2003) MDM2 promotes p21waf1/cip1 proteasomal turnover independently of ubiquitylation. EMBO J 22: 6365–6377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y, Zeng SX, Dai MS, Yang XJ, Lu H (2002) MDM2 inhibits PCAF (p300/CREB-binding protein-associated factor)-mediated p53 acetylation. J Biol Chem 277: 30838–30843 [DOI] [PubMed] [Google Scholar]
- Jones SN, Roe AE, Donehower LA, Bradley A (1995) Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature 378: 206–208 [DOI] [PubMed] [Google Scholar]
- Keller D, Zeng X, Li X, Kapoor M, Iordanov MS, Taya Y, Lozano G, Magun B, Lu H (1999) The p38MAPK inhibitor SB203580 alleviates ultraviolet-induced phosphorylation at serine 389 but not serine 15 and activation of p53. Biochem Biophys Res Commun 261: 464–471 [DOI] [PubMed] [Google Scholar]
- Ku NO, Liao J, Omary MB (1998) Phosphorylation of human keratin 18 serine 33 regulates binding to 14-3-3 proteins. EMBO J 17: 1892–1906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubbutat MH, Jones SN, Vousden KH (1997) Regulation of p53 stability by Mdm2. Nature 387: 299–303 [DOI] [PubMed] [Google Scholar]
- Leu JI, Dumont P, Hafey M, Murphy ME, George DL (2004) Mitochondrial p53 activates Bak and causes disruption of a Bak–Mcl1 complex. Nat Cell Biol 6: 443–450 [DOI] [PubMed] [Google Scholar]
- Li C, Chen L, Chen J (2002) DNA damage induces MDMX nuclear translocation by p53-dependent and -independent mechanisms. Mol Cell Biol 22: 7562–7571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linares LK, Hengstermann A, Ciechanover A, Muller S, Scheffner M (2003) HdmX stimulates Hdm2-mediated ubiquitination and degradation of p53. Proc Natl Acad Sci USA 100: 12009–12014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Guntuku S, Cui XS, Matsuoka S, Cortez D, Tamai K, Luo G, Carattini-Rivera S, DeMayo F, Bradley A, Donehower LA, Elledge SJ (2000) Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev 14: 1448–1459 [PMC free article] [PubMed] [Google Scholar]
- Luo ZJ, Zhang XF, Rapp U, Avruch J (1995) Identification of the 14.3.3 zeta domains important for self-association and Raf binding. J Biol Chem 270: 23681–23687 [DOI] [PubMed] [Google Scholar]
- Marine JC, Jochemsen AG (2005) Mdmx as an essential regulator of p53 activity. Biochem Biophys Res Commun 331: 750–760 [DOI] [PubMed] [Google Scholar]
- Meulmeester E, Maurice MM, Boutell C, Teunisse AF, Ovaa H, Abraham TE, Dirks RW, Jochemsen AG (2005) Loss of HAUSP-mediated deubiquitination contributes to DNA damage-induced destabilization of Hdmx and Hdm2. Mol Cell 18: 565–576 [DOI] [PubMed] [Google Scholar]
- Migliorini D, Danovi D, Colombo E, Carbone R, Pelicci PG, Marine JC (2002a) Hdmx recruitment into the nucleus by Hdm2 is essential for its ability to regulate p53 stability and transactivation. J Biol Chem 277: 7318–7323 [DOI] [PubMed] [Google Scholar]
- Migliorini D, Denchi EL, Danovi D, Jochemsen A, Capillo M, Gobbi A, Helin K, Pelicci PG, Marine JC (2002b) Mdm4 (Mdmx) regulates p53-induced growth arrest and neuronal cell death during early embryonic mouse development. Mol Cell Biol 22: 5527–5538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihara M, Erster S, Zaika A, Petrenko O, Chittenden T, Pancoska P, Moll UM (2003) p53 has a direct apoptogenic role at the mitochondria. Mol Cell 11: 577–590 [DOI] [PubMed] [Google Scholar]
- Miyashita T, Reed JC (1995) Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell 80: 293–299 [DOI] [PubMed] [Google Scholar]
- Montes de Oca Luna R, Wagner DS, Lozano G (1995) Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature 378: 203–206 [DOI] [PubMed] [Google Scholar]
- Muslin AJ, Tanner JW, Allen PM, Shaw AS (1996) Interaction of 14-3-3 with signaling proteins is mediated by the recognition of phosphoserine. Cell 84: 889–897 [DOI] [PubMed] [Google Scholar]
- O'Neill T, Giarratani L, Chen P, Iyer L, Lee CH, Bobiak M, Kanai F, Zhou BB, Chung JH, Rathbun GA (2002) Determination of substrate motifs for human Chk1 and hCds1/Chk2 by the oriented peptide library approach. J Biol Chem 277: 16102–16115 [DOI] [PubMed] [Google Scholar]
- Obenauer JC, Cantley LC, Yaffe MB (2003) Scansite 2.0: proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res 31: 3635–3641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto K, Kashima K, Pereg Y, Ishida M, Yamazaki S, Nota A, Teunisse A, Migliorini D, Kitabayashi I, Marine JC, Prives C, Shiloh Y, Jochemsen AG, Taya Y (2005) DNA damage-induced phosphorylation of MdmX at serine 367 activates p53 by targeting MdmX for Mdm2-dependent degradation. Mol Cell Biol 25: 9608–9620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parant J, Chavez-Reyes A, Little NA, Yan W, Reinke V, Jochemsen AG, Lozano G (2001) Rescue of embryonic lethality in Mdm4-null mice by loss of Trp53 suggests a nonoverlapping pathway with MDM2 to regulate p53. Nat Genet 29: 92–95 [DOI] [PubMed] [Google Scholar]
- Pereg Y, Shkedy D, de Graaf P, Meulmeester E, Edelson-Averbukh M, Salek M, Biton S, Teunisse AF, Lehmann WD, Jochemsen AG, Shiloh Y (2005) Phosphorylation of Hdmx mediates its Hdm2- and ATM-dependent degradation in response to DNA damage. Proc Natl Acad Sci USA 102: 5056–5061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rallapalli R, Strachan G, Cho B, Mercer WE, Hall DJ (1999) A novel MDMX transcript expressed in a variety of transformed cell lines encodes a truncated protein with potent p53 repressive activity. J Biol Chem 274: 8299–8308 [DOI] [PubMed] [Google Scholar]
- Rallapalli R, Strachan G, Tuan RS, Hall DJ (2003) Identification of a domain within MDMX-S that is responsible for its high affinity interaction with p53 and high-level expression in mammalian cells. J Cell Biochem 89: 563–575 [DOI] [PubMed] [Google Scholar]
- Sharp DA, Kratowicz SA, Sank MJ, George DL (1999) Stabilization of the MDM2 oncoprotein by interaction with the structurally related MDMX protein. J Biol Chem 274: 38189–38196 [DOI] [PubMed] [Google Scholar]
- Shieh SY, Ahn J, Tamai K, Taya Y, Prives C (2000) The human homologs of checkpoint kinases Chk1 and Cds1 (Chk2) phosphorylate p53 at multiple DNA damage-inducible sites. Genes Dev 14: 289–300 [PMC free article] [PubMed] [Google Scholar]
- Shvarts A, Steegenga WT, Riteco N, van Laar T, Dekker P, Bazuine M, van Ham RC, van der Houven van Oordt W, Hateboer G, van der Eb AJ, Jochemsen AG (1996) MDMX: a novel p53-binding protein with some functional properties of MDM2. EMBO J 15: 5349–5357 [PMC free article] [PubMed] [Google Scholar]
- Stad R, Ramos YF, Little N, Grivell S, Attema J, van Der Eb AJ, Jochemsen AG (2000) Hdmx stabilizes Mdm2 and p53. J Biol Chem 275: 28039–28044 [DOI] [PubMed] [Google Scholar]
- Tibbetts RS, Brumbaugh KM, Williams JM, Sarkaria JN, Cliby WA, Shieh SY, Taya Y, Prives C, Abraham RT (1999) A role for ATR in the DNA damage-induced phosphorylation of p53. Genes Dev 13: 152–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venot C, Maratrat M, Dureuil C, Conseiller E, Bracco L, Debussche L (1998) The requirement for the p53 proline-rich functional domain for mediation of apoptosis is correlated with specific PIG3 gene transactivation and with transcriptional repression. EMBO J 17: 4668–4679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelstein B, Lane D, Levine AJ (2000) Surfing the p53 network. Nature 408: 307–310 [DOI] [PubMed] [Google Scholar]
- Vousden KH (2002) Activation of the p53 tumor suppressor protein. Biochim Biophys Acta 1602: 47–59 [DOI] [PubMed] [Google Scholar]
- Wilker EW, Grant RA, Artim SC, Yaffe MB (2005) A structural basis for 14-3-3sigma functional specificity. J Biol Chem 280: 18891–18898 [DOI] [PubMed] [Google Scholar]
- Wu X, Bayle JH, Olson D, Levine AJ (1993) The p53-mdm-2 autoregulatory feedback loop. Genes Dev 7: 1126–1132 [DOI] [PubMed] [Google Scholar]
- Yaffe MB, Rittinger K, Volinia S, Caron PR, Aitken A, Leffers H, Gamblin SJ, Smerdon SJ, Cantley LC (1997) The structural basis for 14-3-3:phosphopeptide binding specificity. Cell 91: 961–971 [DOI] [PubMed] [Google Scholar]
- Yang HY, Wen YY, Chen CH, Lozano G, Lee MH (2003) 14-3-3 sigma positively regulates p53 and suppresses tumor growth. Mol Cell Biol 23: 7096–7107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng SX, Dai MS, Keller DM, Lu H (2002) SSRP1 functions as a co-activator of the transcriptional activator p63. EMBO J 21: 5487–5497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng X, Chen L, Jost CA, Maya R, Keller D, Wang X, Kaelin WG Jr, Oren M, Chen J, Lu H (1999) MDM2 suppresses p73 function without promoting p73 degradation. Mol Cell Biol 19: 3257–3266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng X, Keller D, Wu L, Lu H (2000) UV but not gamma irradiation accelerates p53-induced apoptosis of teratocarcinoma cells by repressing MDM2 transcription. Cancer Res 60: 6184–6188 [PubMed] [Google Scholar]
- Zeng X, Lee H, Zhang Q, Lu H (2001) p300 does not require its acetylase activity to stimulate p73 function. J Biol Chem 276: 48–52 [DOI] [PubMed] [Google Scholar]
- Zhang L, Wang H, Liu D, Liddington R, Fu H (1997) Raf-1 kinase and exoenzyme S interact with 14-3-3zeta through a common site involving lysine 49. J Biol Chem 272: 13717–13724 [DOI] [PubMed] [Google Scholar]
- Zhao H, Piwnica-Worms H (2001) ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol Cell Biol 21: 4129–4139 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Materials