

Abstract
The spindle assembly checkpoint (SAC) monitors chromosome attachment to spindle microtubules. SAC proteins operate at kinetochores, scaffolds mediating chromosome-microtubule attachment. The ubiquitous SAC constituents Mad1 and Mad2 are recruited to kinetochores in prometaphase. Mad2 sequesters Cdc20 to prevent its ability to mediate anaphase onset. Its function is counteracted by p31comet (formerly CMT2). Upon binding Cdc20, Mad2 changes its conformation from O-Mad2 (Open) to C-Mad2 (Closed). A Mad1-bound C-Mad2 template, to which O-Mad2 binds prior to being converted into Cdc20-bound C-Mad2, assists this process. A molecular understanding of this prion-like property of Mad2 is missing. We characterized the molecular determinants of the O-Mad2:C-Mad2 conformational dimer and derived a rationalization of the binding interface in terms of symmetric and asymmetric components. Mutation of individual interface residues abrogates the SAC in Saccharomyces cerevisiae. NMR chemical shift perturbations indicate that O-Mad2 undergoes a major conformational rearrangement upon binding C-Mad2, suggesting that dimerization facilitates the structural conversion of O-Mad2 required to bind Cdc20. We also show that the negative effects of p31comet on the SAC are based on its competition with O-Mad2 for C-Mad2 binding.
Keywords: Cdc20, centromere, kinetochore, mitosis, mitotic arrest deficient, spindle assembly checkpoint
Introduction
In mitosis, the spindle assembly checkpoint (SAC) monitors bipolar attachment of sister chromatids to microtubules emanating from opposite spindle poles (Musacchio and Hardwick, 2002; Bharadwaj and Yu, 2004). Microtubule capture takes place at kinetochores, specialized proteinaceous structures assembling at the centromere (Cleveland et al, 2003). The SAC might sense both the lack of microtubule attachment at kinetochores and the lack of tension between sister centromeres caused by incorrect attachments (Pinsky and Biggins, 2005). Under these conditions, the SAC generates a diffusible wait-anaphase signal that inhibits cell cycle progression. The target of the SAC is the E3 ubiquitin ligase anaphase promoting complex or cyclosome (APC/C), which links ubiquitin to Cyclin B and Securin, causing their proteasome-mediated degradation and promoting anaphase (Musacchio and Hardwick, 2002; Bharadwaj and Yu, 2004).
SAC components include the members of the mitotic arrest deficient (MAD) and budding uninhibited by benzimidazole (BUB) families (Hoyt et al, 1991; Li and Murray, 1991), which are ubiquitous in eukaryotes (Musacchio and Hardwick, 2002; Bharadwaj and Yu, 2004). All SAC proteins are recruited to kinetochores in prometaphase, where they monitor the attachment of spindle microtubules. Mad2 is a critical SAC component that binds and sequesters Cdc20, an essential accessory subunit of the APC/C. Mutations in the Mad2-binding site of Cdc20 abrogate the SAC (Fang et al, 1998; Hwang et al, 1998; Kallio et al, 1998; Kim et al, 1998; Wassmann and Benezra, 1998). BubR1, another SAC component, also binds Cdc20 directly causing its inhibition (Tang et al, 2001; Fang, 2002). Mad2 and BubR1, together with the BubR1 associated subunit Bub3, may form a quaternary complex with Cdc20 named MCC, for mitotic checkpoint complex (Hardwick et al, 2000; Fraschini et al, 2001; Sudakin et al, 2001; Chen, 2002; Morrow et al, 2005; Poddar et al, 2005). The precise composition and function of the MCC remain elusive (Chan et al, 2005).
Another SAC component, the coiled-coil protein Mad1, is required in vivo to form the Mad2:Cdc20 complex (Hwang et al, 1998; Chung and Chen, 2002). Mad1 forms a tight 2:2 complex with Mad2, the Mad1:Mad2 core complex (Sironi et al, 2001, 2002). This is loaded onto kinetochores in prometaphase by a kinetochore-binding domain located in the N-terminal coiled-coil region of Mad1 (Chen et al, 1998, 1999; Chung and Chen, 2002; Sironi et al, 2002). A 15-residue Mad2-binding motif in the C-terminal half of each Mad1 protomer creates the tight Mad1:Mad2 tetramer (Luo et al, 2002, 2004; Sironi et al, 2002).
Mad2 adopts two conformations (Luo et al, 2000, 2002, 2004; Sironi et al, 2002), O-Mad2 and C-Mad2 (for Open and Closed, alternatively known as N1 and N2, for Native conformation 1 and 2). These conformations differ in the C-terminal 50 residues of Mad2, the ‘safety belt' (Sironi et al, 2002). Mad2 adopts the C-Mad2 conformation when bound to Mad1. Conversely, cytosolic Mad2 folds as O-Mad2 (Luo et al, 2004; De Antoni et al, 2005b). The O-Mad2 and C-Mad2 conformers of Mad2 form a ‘conformational dimer' (Luo et al, 2004; De Antoni et al, 2005a, 2005b; Hagan and Sorger, 2005; Hardwick, 2005; Nasmyth, 2005). Conformational dimerization is essential to recruit O-Mad2 to Mad1-bound C-Mad2 at the kinetochore (Hagan and Sorger, 2005; Hardwick, 2005; Nasmyth, 2005; De Antoni et al, 2005a). O-Mad2 mutants that bind Mad1 but cannot sustain the interaction with C-Mad2 are not recruited to kinetochores. Conversely, O-Mad2 mutants that are unable to bind Mad1, but retain the ability to bind C-Mad2 are recruited normally to kinetochores (De Antoni et al, 2005a). Thus, the primary function of Mad1 is to localize C-Mad2 at the kinetochore. Because the Mad1:C-Mad2 complex is extremely stable and does not release Mad2 (De Antoni et al, 2005a), the Mad2 monomers eventually reaching Cdc20 appear to be retrieved from a pool of O-Mad2 that cycles at the kinetochore thanks to its interaction with Mad1-bound C-Mad2 (Figure 1A). This provides a molecular explanation to fluorescence recovery after photobleaching (FRAP) experiments showing that Mad1 and 50% of Mad2 (C-Mad2 bound to Mad1) are stably bound to kinetochores during checkpoint activation, while a remaining 50% of Mad2 (O-Mad2) turns over at the kinetochore with short halftimes (Shah et al, 2004).
Figure 1.
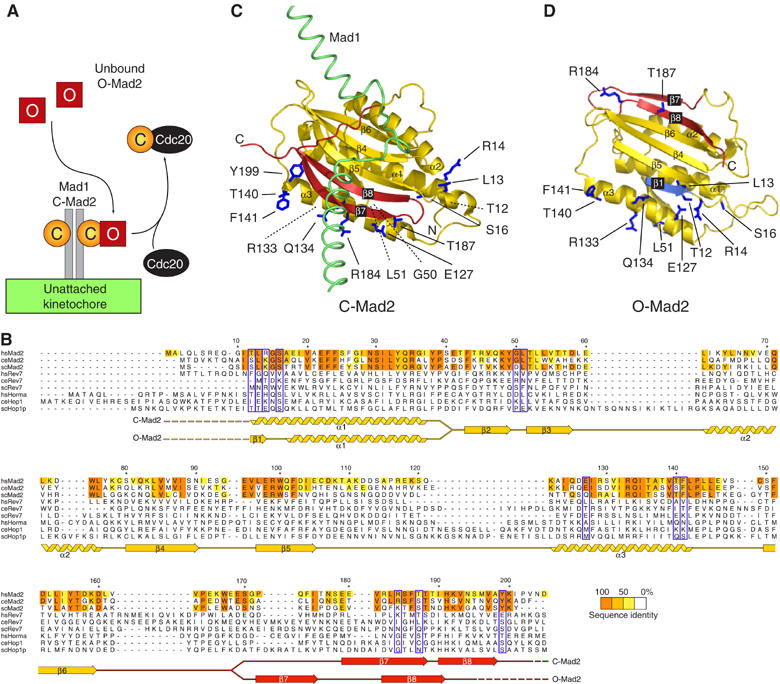
Design of mutants impairing the O:C-Mad2 dimerization. (A) The ‘Mad2-template' model. O-Mad2 (red square) binds C-Mad2 (yellow circle) tightly associated with Mad1. This causes kinetochore recruitment of O-Mad2 (De Antoni et al, 2005a). O-Mad2 is quickly released and binds Cdc20. The model implies conformational dimerization of O-Mad2 and C-Mad2. (B) Sequence alignment of HORMA domains with secondary structure of HsMad2 in O-Mad2 and C-Mad2. We aligned 31 sequences of Mad2, Rev7 and Hop1 orthologues (Supplementary Figure 1). Mad2 conservation was evaluated on a larger alignment of 14 Mad2 sequences. Surface residues conserved in Mad2—but not in Rev7 and Hop1—were selected for mutation (blue boxes). (C) C-Mad2 bound to the Mad2-binding site of Mad1 (green, PDB ID 1GO4). The ‘safety belt' is colored red. Parts of the structure that are invariant in O-Mad2 and C-Mad2 are colored yellow. Side chains of mutated residues are dark blue. Dashed lines mark residues whose side chain is hidden from the current view. (D) Same representation as above for O-Mad2 (PDB ID 1DUJ). The first β-strand β1 (residues 1–15) is shown in light blue.
Mad2 adopts the C-Mad2 conformation also when bound to Cdc20. This is explained by the fact that the Mad2-binding motifs of Mad1 and Cdc20 conform to a similar consensus sequence (Luo et al, 2002; Sironi et al, 2002). Thus, the mechanism of Mad2 activation towards Cdc20 binding implies that a C-Mad2 conformer stably bound to Mad1 binds an O-Mad2 conformer from the cytosol to promote its conversion into C-Mad2 bound to Cdc20 (Figure 1A). This hypothesis has been named ‘Mad2 template' hypothesis because it envisions a C-Mad2-mediated transformation of O-Mad2 into Cdc20-bound C-Mad2 (De Antoni et al, 2005a).
Like O-Mad2, p31comet (previously CMT2) selectively binds the C-Mad2 conformer and is unable to form a stable complex with O-Mad2 (Xia et al, 2004). Interestingly, p31comet is a strong SAC inhibitor (Habu et al, 2002; Xia et al, 2004). At present, the mechanism by which the binding of p31comet to C-Mad2 inhibits the SAC is unclear. Because p31comet and O-Mad2 are comparable in terms of their ability to bind selectively to C-Mad2, these proteins might be expected to compete for C-Mad2 binding to control the state of activation of the SAC.
We have extended our characterization of the mechanism of conformational dimerization of Mad2 and provide here an account of our results. The new evidence can be interpreted straightforwardly on the basis of the ‘Mad2 template' model.
Results
Relationship of Mad2 with other members of the HORMA domain family
Two critical properties of Mad2 are important for the SAC: (1) Mad2 adopts O-Mad2 and C-Mad2 conformations and (2) O-Mad2 and C-Mad2 form conformational dimers. These properties, originally identified in human (Hs) Mad2, are conserved in Saccharomyces cerevisiae, suggesting that the binding interface of the O:C Mad2 dimer is conserved in evolution (LN, GR, A De Antoni, S Pasqualato, SP and AM, in preparation). The HsMad2R133A (Arg133 to Ala) and HsMad2Q134A (Gln134 to Ala) point mutants are unable to form tight Mad2 conformational dimers (Sironi et al, 2001, 2002; De Antoni et al, 2005a, 2005b). Arg133 and Gln134 are conserved in all Mad2 orthologues (Figure 1B). The equivalent residues (Arg126 and Gln127) in budding yeast are essential for the SAC (LN, GR, A De Antoni, S Pasqualato, SP and AM, in preparation).
The Mad2 fold is known as HORMA domain and is shared by Rev7, a regulatory subunit of DNA polymerase ζ, and by the meiosis-specific recombination protein Hop1 (Aravind and Koonin, 1998). Residues that are important for the fold of the HORMA domain are invariant in Mad2, Rev7 and Hop1 (Figure 1B. Supplementary Figure 1 reports the full alignment). Arg35 and Glu98 (HsMad2 numbering), which form a buried salt bridge, are completely conserved. It is unknown whether the ability of Mad2 of adopting two conformations and its conformational dimerization apply to Rev7 and Hop1. Arg133 and Gln134, which mediate the O-Mad2:C-Mad2 interaction, are only conserved within the Mad2 branch of the HORMA domain. Thus, even if Rev7 and Hop1 adopted open and closed conformers like Mad2, these may be unable to form conformational dimers.
To identify additional residues at the O-Mad2:C-Mad2 interface, we restricted our analysis to residues that, similarly to Arg133 and Gln134, are exclusively conserved in the Mad2 subfamily. We further limited the search to solvent-exposed residues. Besides Arg133, Gln134 and a few other residues whose alanine mutants have been characterized (Sironi et al, 2001), we identified a new set of 12 residues that to a varying degree are specifically conserved in the Mad2 subfamily and solvent-exposed. These include Thr12, Leu13, Arg14, Ser16, Gly50, Leu51, Glu127, Thr140, Phe141, Arg184, Thr187 and Tyr199 (blue-boxed in Figure 1B and displayed in Figure 1C). Being located distantly from the Mad1- and Cdc20-binding site of Mad2 (Luo et al, 2002; Sironi et al, 2002), none of these residues is expected to be important for Mad2 binding to Mad1 or Cdc20.
New Mad2 mutants impaired in conformational dimerization
The on-rate for binding of human O-Mad2 to Cdc20 in vitro is slow (∼400/Ms; Martin Vink, MM, LM and AM, unpublished data, 2006). However, the interaction can be visualized using a GST pull-down assay (De Antoni et al, 2005a) in which C-Mad2 is created by a 1–2 h incubation of wild-type Mad2 (Mad2wt) with GST-Cdc20111−138 (which contains the Mad2-binding region of Cdc20) immobilized onto glutathione–sepharose (GSH) beads (Figure 2A). As O-Mad2, we resort to using Mad2ΔC, a Mad2 deletion mutant lacking the last 10 C-terminal residues. Mad2ΔC is locked as O-Mad2 and is unable to bind Cdc20 stably (Sironi et al, 2001, 2002; De Antoni et al, 2005a, 2005b). Consistently, while Mad2ΔC was unable to bind GST-Cdc20111−138 (Figure 2B, lane 4), it remained bound to GSH beads containing immobilized GST-Cdc20111−138 if the beads were preincubated with Mad2wt to create C-Mad2 (Figure 2B, lane 5). Mad2ΔC can be practically regarded as a wild-type form of O-Mad2 for what concerns its structural stability and its ability to bind C-Mad2 (De Antoni et al, 2005a). Its use is particularly useful because Mad2ΔC cannot stably convert into C-Mad2, providing a source of pure O-Mad2. We adopted this assay to study the effects on the O-Mad2:C-Mad2 interaction of alanine mutants of the 12 residues listed above. Ser and Thr residues within this list were additionally mutated into glutamic acid. All mutants expressed in a soluble form in bacteria, bound Cdc20 normally (see below), and did not show evident signs of aggregation. A subset of mutants was analyzed by size-exclusion chromatography (SEC) and found to elute as expected for a Mad2 monomer (Supplementary Figure 2). Based on our previous characterization of the stability of Mad2R133A and Mad2ΔC (De Antoni et al, 2005a), we believe that the stability of the Mad2 mutants analyzed here is very similar to that of Mad2wt.
Figure 2.
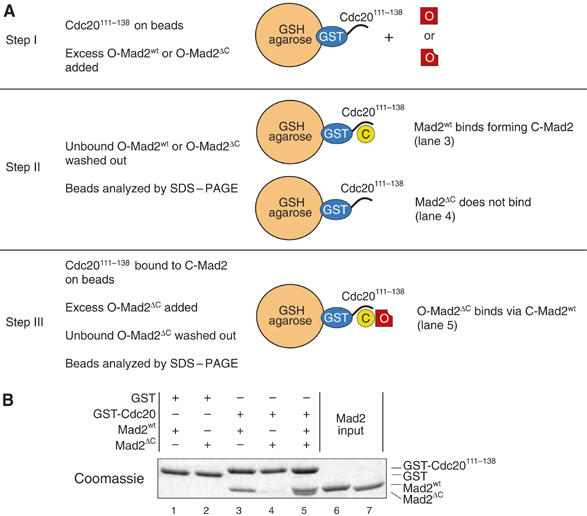
Visualization of the O-Mad2:C-Mad2 interaction. (A) Rationale of the solid-phase binding assay revealing the interaction of O-Mad2 with C-Mad2. (B) GST-Cdc20111−138 bound to GSH beads was incubated with Mad2wt, Mad2ΔC or both sequentially. Beads were washed and bound proteins analyzed by SDS–PAGE and Coomassie staining. Mad2ΔC cannot close on Cdc20111−138, while Mad2wt closes on the Cdc20 peptides and recruits Mad2ΔC onto the beads. The gel is a representative example of four independent experiments.
Because O-Mad2 and C-Mad2 form a conformational dimer, while neither O-Mad2 nor C-Mad2 dimerize without the other conformer (De Antoni et al, 2005b), there is a logical requirement for the two surfaces involved in the conformational dimerization of O-Mad2 and C-Mad2 to be different. Thus, the effects of point mutations need to be tested both in the frame of the C-Mad2 conformer for binding to wild-type O-Mad2 and of the O-Mad2 conformer for binding to wild-type C-Mad2. We first asked if O-Mad2ΔC was able to bind mutant C-Mad2 conformers. Similar amounts of wild-type or mutant Mad2 were incubated with GST-Cdc20111−138 on GSH beads. Unbound Mad2 was washed away, and purified Mad2ΔC was added. After washing away Mad2ΔC in excess, proteins on beads were revealed by SDS–PAGE (Figure 3).
Figure 3.
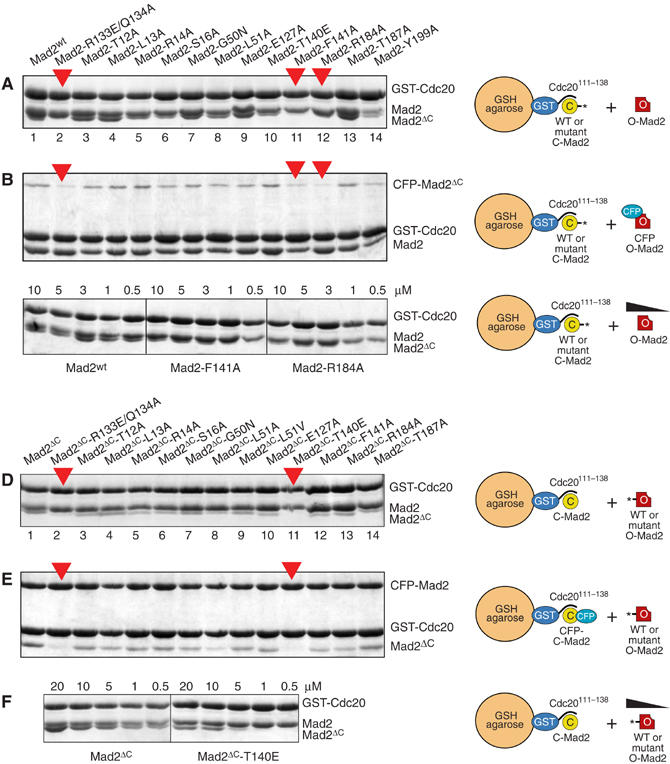
Mapping of residues involved in O-Mad2:C-Mad2 dimerization. (A) GST pull-downs (see Figure 2). Mad2wt and mutants were incubated for 1 h at 25°C with 1 μM GST-Cdc20111−138 preadsorbed on GSH beads to form C-Mad2. Excess Mad2 was washed away and equal amounts of O-Mad2ΔC were added. After 1 h, beads were washed twice and boiled in sample buffer. Bound species were separated by SDS–PAGE and Coomassie-stained. Red arrows indicate C-Mad2 mutants impaired in Mad2ΔC binding. (B) As in (A), but using CFP-Mad2ΔC instead of Mad2ΔC to obtain better separation from Mad2wt. (C) C-Mad2wt, C-Mad2F141A or C-Mad2R184A prebound to GST-Cdc20 were incubated with decreasing concentrations of O-Mad2ΔC. Binding of O-Mad2ΔC to Mad2wt was detected at concentrations as low as 0.5 μM. No binding of O-Mad2ΔC to C-Mad2F141A or C-Mad2R184A was observed at concentrations below 3 μM. (D) Equal amounts of Mad2wt were incubated with GST-Cdc20111−138 on GSH beads to create C-Mad2. O-Mad2ΔC and mutant variants were then added. (E) As in (D), but using CFP-Mad2wt as C-Mad2. The O-Mad2ΔC−T140E mutant deficient in binding to C-Mad2 is indicated with a red arrow. (F) C-Mad2wt prebound to GST-Cdc20 was incubated with decreasing concentrations of O-Mad2ΔC or O-Mad2ΔC−T140E. Binding of O-Mad2ΔC to C-Mad2wt was detected at concentrations as low as 0.5 μM. No binding of Mad2ΔC−T140E to C-Mad2wt was observed at concentrations below 5 μM. All gels are representative examples of at least three independent experiments.
O-Mad2ΔC bound normally to C-Mad2wt (Figure 3A, lane 1). Conversely, O-Mad2ΔC failed to bind C-Mad2R133E/Q134A (lane 2), a double point mutant that binds Cdc20 with the same affinity of Mad2wt but whose C-Mad2 conformer is unable to bind O-Mad2 (De Antoni et al, 2005a). All mutant proteins (lanes 3–14) interacted normally with GST-Cdc20111−138. O-Mad2ΔC bound all mutants, except for C-Mad2F141A and C-Mad2R184A. To increase the separation of the C-Mad2 variants and Mad2ΔC by SDS–PAGE, we repeated the GST pull-downs using CFP-Mad2ΔC (whose molecular weight is ∼47 kDa) as a replacement for O-Mad2. This confirmed that C-Mad2F141A and C-Mad2R184A are impaired in binding O-Mad2 (Figure 3B).
To gain a semiquantitative estimate of the effects of the mutations, we carried out a titration experiment in which decreasing concentrations of pure Mad2ΔC (from 10 to 0.5 μM) were incubated with pure C-Mad2wt, C-Mad2F141A and C-Mad2R184A already bound to GST-Cdc20111−138 (1 μM). O-Mad2ΔC bound C-Mad2wt at all concentrations, including the minimal concentration of 0.5 μM. Conversely, O-Mad2ΔC failed to bind C-Mad2F141A at concentrations below 3 μM, while binding to C-Mad2R184A was significantly reduced already at 10 μM (Figure 3C). Mutations that interfere with the O-Mad2:C-Mad2 interaction cause the monomerization of pure HsMad2 (Sironi et al, 2001; De Antoni et al, 2005b). Consistently, Mad2F141A and Mad2R184A eluted as expected for monomeric species from an SEC column (Supplementary Figure 2; the Mad2T140E mutant described below was not tested).
To test the effects of mutations on the O-Mad2 side, the same mutations were introduced in O-Mad2ΔC (except for Y199, which maps to the segment deleted in O-Mad2ΔC) and their binding to C-Mad2wt was tested. C-Mad2wt or CFP-C-Mad2wt bound to GST-Cdc20111−138 on GSH beads (∼1 μM) were incubated with O-Mad2ΔC or its mutant variants (Figure 3D and E). Unbound O-Mad2ΔC was washed out and the species on beads were resolved by SDS–PAGE. Replacement of Thr140 with Glu produced a severe perturbation of binding (substitution of Thr140 with Ala produced a less-penetrant phenotype—not shown). The L51A mutation also perturbed binding, but to a lesser extent relative to T140E. A titration experiment indicated that the T140E mutation causes a ∼10-fold reduction in binding affinity (Figure 3F). At present, there is no evidence that T140 is phosphorylated in vivo (Wassmann et al, 2003), and the enhanced effects of mutating this residue to Glu relative to Ala might be due to perturbations of the charge distribution at the O-Mad2:C-Mad2 interface.
The C-Mad2 interactor p31comet prevents O:C-Mad2 dimer association
The Mad2-binding protein p31comet has been implicated in SAC silencing (Habu et al, 2002; Xia et al, 2004), but a plausible molecular explanation of its role is missing. It was argued that p31comet facilitates the disassembly of Cdc20 from Mad2 in mid-mitosis, freeing Cdc20 (Habu et al, 2002). Subsequently, it was found that p31comet is incorporated into a complex with Mad2, Cdc20 and the APC/C (Xia et al, 2004). This interaction is explained by the ability of p31comet to bind selectively to the C-Mad2 conformer (Xia et al, 2004).
Because the ‘Mad2 template' model predicts that C-Mad2 is required to activate O-Mad2 for binding Cdc20, p31comet might downregulate the SAC by interfering with the ability of O-Mad2 to bind C-Mad2. Although it is clear that p31comet binds C-Mad2 (Xia et al, 2004), it is unknown whether this interaction is competitive with the binding of O-Mad2 to C-Mad2. To test this, we adapted the GST pull-down assay in Figure 2A for a competition experiment in which the ability of O-Mad2ΔC to bind GST-Cdc20111−138:C-Mad2 was tested in the presence of increasing amounts of p31comet. Both O-Mad2ΔC and p31comet bound to C-Mad2 on GSH beads when incubated individually (Figure 4A, lanes 2 and 6, respectively). At the concentrations tested, p31comet effectively competed with O-Mad2ΔC for binding C-Mad2. At roughly equimolar p31comet and O-Mad2ΔC concentrations, p31comet remained bound to C-Mad2. Thus, p31comet is a high-affinity ligand of C-Mad2 whose binding prevents the dimerization of C-Mad2 with O-Mad2.
Figure 4.
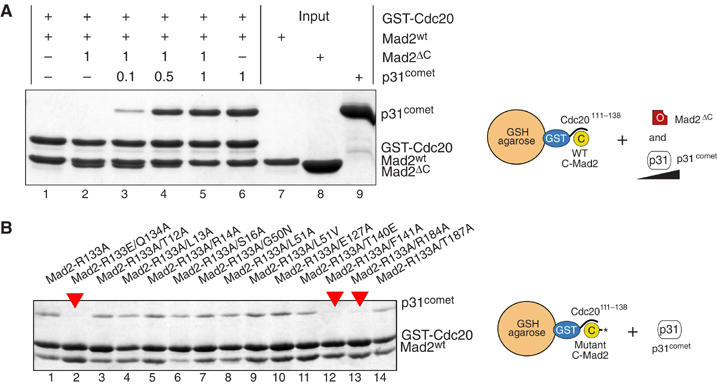
p31comet is a competitive inhibitor of O-Mad2:C-Mad2 assembly. (A) Mad2ΔC and p31comet compete for C-Mad2wt bound to GST-Cdc20. Mad2ΔC (3 μM) and p31comet (0.3–3 μM) can separately bind to ∼ 1 μM Cdc20:C-Mad2wt on beads (lanes 2 and 6, respectively). Relative stoichiometries of Mad2ΔC and p31comet are indicated. When mixed together (lane 5), Mad2ΔC and p31comet are unable to enter a single complex. Already at equimolar concentrations, p31comet prevents O-Mad2ΔC from binding C-Mad2 (lane 5). (B) GST-Cdc20 pull-down assay with double C-Mad2 point mutants additionally harboring the R133A mutation. Experiments were repeated three times with identical results.
To map more finely the p31comet-binding site on C-Mad2, we performed a GST pull-down experiment similar to the one used to map the O:C-Mad2 interaction surfaces. p31comet (at concentrations from 0.03 to 1.0 μM) was incubated with C-Mad2wt and C-Mad2 mutants bound to GST-Cdc20111−138. None of the C-Mad2 point mutants, including Mad2R133A or Mad2Q134A, was defective in binding p31comet in this assay (data not shown). This was somewhat unexpected, because if p31comet binds C-Mad2 competitively with O-Mad2 (Figure 4A), and Arg133 and Gln134 are located on the C-Mad2 surface required to bind O-Mad2, these residues would also be predicted to be part of the C-Mad2 interface required for binding p31comet. The interaction of p31comet with C-Mad2 is stronger than that of O-Mad2 (KD ∼25 nM and ∼1 μM, respectively; Martin Vink, MM, LM and AM, unpublished data, 2006), single point mutations such as those tested above might be insufficient to disrupt the tight p31comet:C-Mad2 complex. We reasoned that the combination of multiple mutations might exacerbate their effects and decided to test the mutations in the frame of Mad2R133A. This revealed that Mad2R133A−Q134A, Mad2R133A−F141A and Mad2R133A−R184A (red arrows in Figure 4B) are impaired in p31comet binding. Q134A, F141A and R184A are the same mutations impairing the binding of C-Mad2 to O-Mad, strongly suggesting that C-Mad2 binds O-Mad2 and p31comet with significantly overlapping surfaces.
Validation of mutants in S. cerevisiae
R133 and Q134 of HsMad2 are conserved in the Mad2 subfamily (Figure 1A). Recently, we have shown that the interaction of O-Mad2 with C-Mad2 is completely conserved in S. cerevisiae (Sc), and that alanine mutants of ScMad2R126A and ScMad2Q127A (equivalent to R133 and Q134 of HsMad2) are unable to rescue the checkpoint deficiency of a mad2Δ strain (LN, GR, A De Antoni, S Pasqualato, SP and AM, in preparation). Thr140, Phe141 and Arg184 are, respectively, equivalent to Thr133, Phe134 and Lys179 of ScMad2 (Figure 5A). To assess if these residues are required for Mad2 function, we assayed the ability of ScMad2wt, ScMad2T133A, ScMad2F134A and ScMad2K179A to restore the SAC deficiency of a mad2Δ strain of S. cerevisiae. Cells arrested in G1 with α-factor were released in the cell cycle in the presence of the spindle-depolymerizing drug nocodazole to activate the SAC. To assess SAC proficiency, we monitored (1) the ability to arrest cells in mitosis and to prevent re-replication, (2) lack of rebudding and (3) retention of sister chromatid cohesion (Figure 5). When exposed to nocodazole, wild-type cells completed DNA replication at about 60 min after release from the G1 block and arrested as budded cells with a 2C DNA content (Figure 5B). These cells did not rebud or separate sister chromatids (Figure 5C), indicative of an active SAC. Conversely, mad2 cells were unable to arrest, lost sister chromatid cohesion, rebudded and re-replicated their DNA, indicative of a disrupted SAC.
Figure 5.
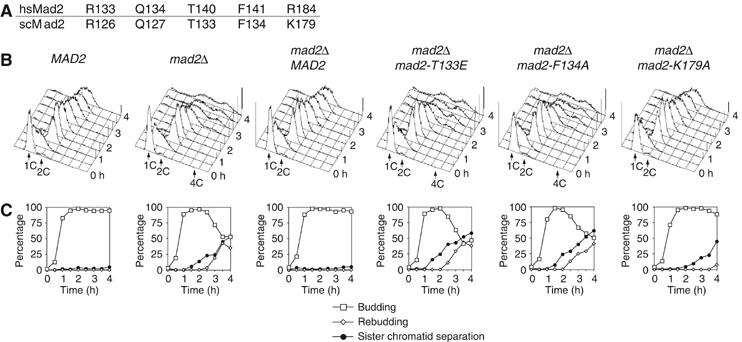
mad2T133A, mad2F134A and mad2K179A do not complement the deletion of the MAD2 gene in S. cerevisiae. (A) Numbering of equivalent human (Hs) and yeast (Sc) Mad2 residues discussed in the text. (B, C) Strains with the indicated genotypes were grown to log phase, arrested in G1 by α factor and released in fresh medium containing nocodazole. At the indicated times, cell samples were withdrawn for (B) FACS analysis of DNA contents, and (C) to score the percentage of budded and rebudded cells, as well as the percentage of sister chromatid separation.
To test the ability of different MAD2 alleles to complement the loss of the SAC, we integrated wild-type and mutant MAD2 alleles at the LEU2 locus of the mad2Δ strain. ScMad2wt, ScMad2T133A, ScMad2F134A and ScMad2K179A were all expressed at levels similar to those of endogenous Mad2 (not shown). Expression of ScMad2wt in the mad2Δ strain restored the SAC (Figure 5B and C). Expression of ScMad2T133A and ScMad2F134A, however, failed to complement the lack of MAD2. Cells expressing these proteins underwent sister chromatid separation, rebudding and re-replication with very similar timings relative to the bare mad2Δ strain. While being defective, Mad2K179A may retain residual functionality, as revealed by the fact that loss of sister chromatid cohesion and rebudding were delayed relative to the other two mutants.
Symmetry and asymmetry at the O-Mad2:C-Mad2 interface
Because R133E and Q134A affect the binding of mutant C-Mad2 to wild-type O-Mad2, as well as the binding of mutant O-Mad2 to wild-type C-Mad2 (De Antoni et al, 2005a, 2005b), R133 and Q134 must be part of the O-Mad2 interface required to bind C-Mad2, and of the C-Mad2 interface required to bind O-Mad2. Mutation of T140, on the other hand, affects the ability of O-Mad2 to bind C-Mad2 (Figure 3D). R133, Q134 and T140 reside on the helix α3 of O-Mad2 (Figure 6A). On the other hand, mutation of Phe141 and Arg184 affects the binding of mutant C-Mad2 to wild-type O-Mad2. Thus, Arg133, Gln134, Phe141 and Arg184 are likely to be located on the region of C-Mad2 that binds O-Mad2 (Figure 6B). Also, these residues cluster on a well-defined surface of C-Mad2 spanning the α3-helix and the β7 strand. Of note, Arg184 is located in the C-terminal segment of Mad2 that undergoes the structural rearrangement upon binding to Mad1 or Cdc20. Arg184 is close to Arg133 and Gln134 in C-Mad2, but is distant from these residues in O-Mad2 (Figure 6A). Thus, the mutational analysis suggests the presence at the O-Mad2:C-Mad2 interface of (1) ‘symmetric' elements, including R133 and Q134, which are involved both on O-Mad2 and C-Mad2; (2) ‘asymmetric' elements, such as Arg184 in C-Mad2, which is required exclusively on the C-Mad2 conformer. The asymmetry of the interaction surface is crucial to explain why only dimers of different conformers can form (De Antoni et al, 2005a, 2005b) and (3) ‘semisymmetric' elements, such as Thr140 of O-Mad2 and Phe141 of C-Mad2. We define this class as ‘semisymmetric' rather than ‘asymmetric' because although the C- and O-Mad2 conformers use these residues differentially (see above), they are very close in space. These mutations might have a different weight on the ability of the O-Mad2 and C-Mad2 conformers to bind their cognate Mad2 conformer, an effect that our relatively crude binding assay might fail to detect. The residues, however, further implicate the α3-helix as a critical element of the interface.
Figure 6.
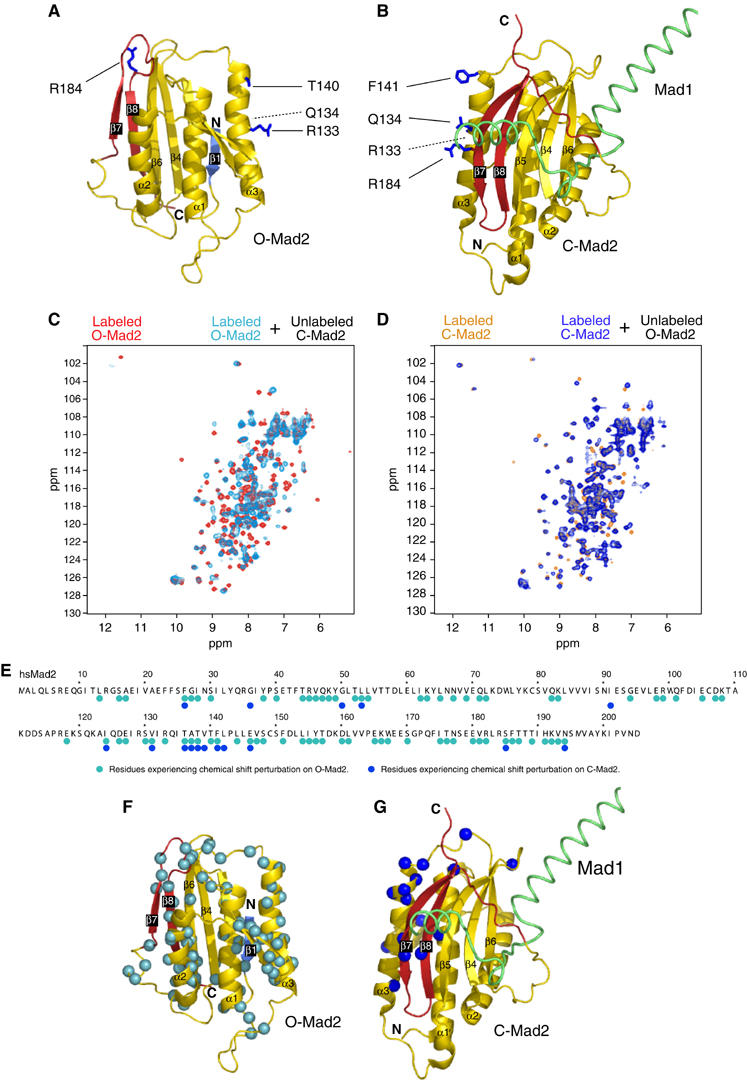
NMR analysis of O-Mad2:C-Mad2 dimer. (A) Ribbon model of O-Mad2. Secondary structure is color-coded as in Figure 1. O-Mad2 residues required to bind C-Mad2 are in blue and ball-and-sticks. (B) C-Mad2 analyzed as in (A). (C) NMR chemical shift perturbation. The spectrum of O-Mad2ΔC (red) was superimposed with that of 1H,15N-labeled O-Mad2ΔC bound to equimolar unlabeled C-Mad2wt:Cdc20 (cyan). (D) The spectrum of C-Mad2wt (orange) was superimposed with that of 1H,15N-labeled C-Mad2wt bound to equimolar unlabeled O-Mad2ΔC (dark blue). (E) Summary of chemical shift perturbations monitored in 2D 1H,15N-HSQC experiments reported exclusively for residues that could be unambiguously assigned in the free and bound state. (F) The Cα atoms of residues in O-Mad2 whose amide's chemical shifts had changed are displayed as cyan spheres. (G) As in panel (F) for C-Mad2, with chemical shift perturbations indicated by dark blue spheres.
NMR spectroscopy analysis of the C-Mad2:O-Mad2 interaction
We characterized the structural features of the O-Mad2:C-Mad2 conformational dimer using NMR spectroscopy. The binding interface was mapped based on chemical shift perturbations between the free O-Mad2 and C-Mad2 proteins and the O-Mad2:C-Mad2 dimer. The chemical shifts were measured with two-dimensional (2D) 1H,15N correlation experiments on samples comprising 1H,15N-labeled O-Mad2 and unlabeled C-Mad2, or vice versa on samples containing unlabeled O-Mad2 and 1H,15N-labeled C-Mad2 (Figure 6C and D).
In the NMR spectra of C-Mad2, only a few residues are affected upon complex formation with O-Mad2. The binding surface identified by these chemical shift perturbations includes residues in helix α3, consistent with the mutational analysis. Furthermore, amides in strands β7, β8 and β5 in the central β-sheet as well as exposed residues in helix α1 and in the β2–β3 hairpin are affected (Figure 6). The well-defined and localized chemical shift changes suggest that C-Mad2 binds to O-Mad2 as a rigid body without large conformational rearrangements. This is consistent with the idea that C-Mad2 acts as a platform for the binding of O-Mad2 and does not modify its conformation in this process, compatibly with its function as a template for the modification of O-Mad2 into C-Mad2.
In contrast, chemical shift changes in the NMR spectra of O-Mad2 upon binding to C-Mad2 are widespread and involve almost all secondary structure elements except the β4 and β5 strands (Figure 6). The extensive chemical shift perturbations presumably result not only from direct contacts at the interface with C-Mad2 but may indicate that O-Mad2 undergoes a further conformational rearrangement upon complex formation. The Mad2-template model predicts that O-Mad2 changes its conformation into C-Mad2 as a direct or indirect consequence of its interaction with C-Mad2. While our present NMR analysis precludes a more detailed assessment of the conformational change impinging on O-Mad2, it suggests that O-Mad2 might adopt an intermediate conformation upon binding C-Mad2, possibly defining a structural intermediate in the conversion to C-Mad2.
A model for the conversion of O-Mad2 into C-Mad2
Only the initial (O-Mad2) and final (C-Mad2) stages of the Mad2 transition are known in detail (Luo et al, 2000, 2002; Sironi et al, 2002). Our NMR analysis, however, implies that O-Mad2 might significantly modify its conformation upon binding to C-Mad2. Thus, C-Mad2 might mould an intermediate conformation on O-Mad2 to accelerate the complex structural transformation of O-Mad2 to C-Mad2. Our NMR analysis does not allow a detailed description of the structural change underlying the Mad2 structural transition. It is clear from Figure 1C and D, however, that the formation of C-Mad2 implies that the C-terminal ‘safety belt' (strands β7–β8) replaces the N-terminal β1 strand of O-Mad2, which is paired to β5. In C-Mad2, β5 now pairs with β8, while the residues corresponding to the β1 strand of O-Mad2 are found as an extension at the N-terminus of α1. We refer to this segment of C-Mad2 as β1* to emphasize the fact that it constituted the β1 strand in O-Mad2. The transformation of β1* is nontrivial. The β1 strand and the α1-helix are located on opposite faces of O-Mad2 (Figure 1C and D). Thus, the β1* segment literally has to cross the Mad2 core to occupy its final location at the N-terminus of α1 in C-Mad2. We speculate that this transformation of Mad2 might be facilitated if the α3-helix, which is positioned like a gate on the path of β1*, were temporarily displaced (Figure 7A). Thus, the function of the C-Mad2 template might be that of docking the α3-helix of O-Mad2 to allow its temporary displacement from the Mad2 core and the passage of β1* through the core. This hypothesis is compatible with the observation that the β5–α3 loop, whose sequence is generally poorly conserved in the HORMA domain alignment (Figure 1B), is however always rather long (15 residues or longer). This extended loop might be required to allow the temporary displacement of α3 (Figure 7A).
Figure 7.
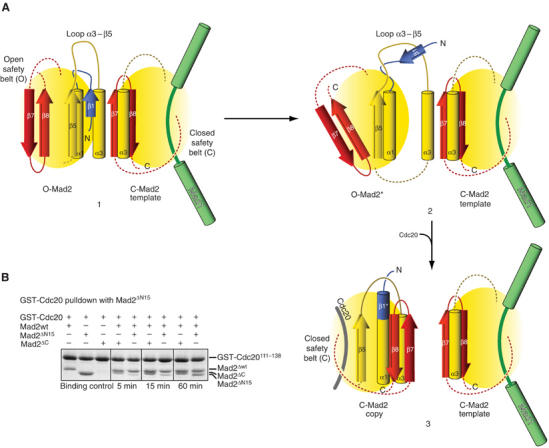
Model of O-Mad2:C-Mad2 interaction. (A) A cartoon summarizing the possible role of Mad1-bound C-Mad2 in the conversion of O-Mad2 into Cdc20-bound C-Mad2. Only the critical secondary structure elements are displayed. Panel 1 shows that the α3-helix of O-Mad2 docks on the α3-helix and β8 strand of C-Mad2. While the α3-helix remains docked onto C-Mad2, a displacement of the O-Mad2 core allows the passage of β1. The displacement is made possible by the long and flexible α3–β5 loop. After displacing β1, the C-terminal tail can move into its final position upon capturing Cdc20. (B) Mad2ΔN15, Mad2ΔC and Mad2ΔN15 were tested for association with C-Mad2wt prebound to GST-Cdc20111−138. Mad2ΔC binds readily to C-Mad2, while Mad2ΔN15 is a slow binder. The experiment was repeated at least three times with basically identical results.
If the β1–β5 pairing counteracts the relocation of the C-terminal safety belt, the removal of β1 might be predicted to favor the repositioning of the β7–β8 safety belt in the position observed in C-Mad2. We thus created Mad2ΔN15, a Mad2 deletion mutant lacking 15 residues from the N-terminus and having therefore the whole β1 strand deleted. We have shown previously that the dimerization of Mad2 is limited to opposite conformers (De Antoni et al, 2005b). To assess the conformation of Mad2ΔN15, we tested its ability to bind C-Mad2. For this, we prebound Mad2wt to GST-Cdc20111−138 to create C-Mad2 as in Figure 2. We then compared the ability of Mad2ΔN15 to bind C-Mad2 with that of Mad2ΔC (Figure 7B). Mad2ΔC bound readily to GST111−138:C-Mad2, and achieved maximal binding already after a 5′ incubation. Mad2ΔN15, on the other hand, required a significantly longer time to bind C-Mad2 so that maximal binding was only observed after 60 min of incubation. This suggests that Mad2ΔN15 prefers the C-Mad2 conformation while being still capable of converting slowly into O-Mad2. To corroborate the idea that Mad2ΔN15 exists predominantly as C-Mad2, we tested its ability to bind O-Mad2ΔC. Confirming our prediction, Mad2ΔN15 and O-Mad2ΔC, which individually eluted as monomers from an SEC column, formed a dimer when mixed stoichiometrically (Supplementary Figure 3. The legend to Supplementary Figure 2 explains the rationale of this experiment).
Discussion
The ‘Mad2 template' hypothesis (Figure 1A) predicts that the SAC starts with the encounter of two separate pools of Mad2 consisting of (1) C-Mad2 stably bound to Mad1 and (2) cytosolic O-Mad2 destined to turn into C-Mad2 upon binding to Cdc20 (De Antoni et al, 2005a). The biochemical mechanism of the ‘Mad2 template' model is conserved in S. cerevisiae. Mad2 mutants that interfere with the conformational dimerization of Mad2, such as ScMad2R126A and ScMad2Q127A, are unable to restore the SAC in a mad2Δ strain (LN, GR, A De Antoni, S Pasqualato, SP and AM, in preparation). Here, we significantly extend these observations by showing that ScMad2T133A, ScMad2F134A and ScMad2K179A are also unable to sustain the SAC in a mad2Δ strain. Together with R126A and Q127A, this makes a total of five residues whose mutation impairs the conformational dimerization of Mad2 and concomitantly causes an SAC deficiency in S. cerevisiae. All five residues are almost completely conserved in the Mad2 family, indicating that the mechanism affected by the mutation is conserved. These observations provide very strong genetic support to the ‘Mad2 template' model.
The mechanism by which the interaction of C-Mad2 and O-Mad2 is regulated will require extensive investigation. We take a step in this direction by reporting that p31comet is a competitive inhibitor of the recruitment of O-Mad2 onto C-Mad2. Previously, it has been shown that p31comet negatively regulates the SAC and is a specific ligand of C-Mad2 (Habu et al, 2002; Xia et al, 2004). However, the observation that p31comet does not compete with the interaction of Mad2 with Cdc20 prevented an immediate understanding of the biochemical bases of its function. Our discovery that p31comet and O-Mad2 bind C-Mad2 competitively can be easily integrated in the ‘Mad2 template' model by assuming that p31comet acts as a screen to separate the O-Mad2 and C-Mad2 pools, limiting the number of O-Mad2 molecules that can be transferred to Cdc20 using a C-Mad2 template (Supplementary Figure 4).
We speculate that the Mad1:C-Mad2 complex is recruited to the kinetochore together with p31comet (Supplementary Figure 4), is activated there after weakening of the interaction of p31comet with C-Mad2, and is finally released from the kinetochore by a microtubule-dependent dynein-dependent transport system (Howell et al, 2001; Shannon et al, 2002). Our observations in vitro indicate that unperturbed p31comet is a significantly tighter C-Mad2 ligand than O-Mad2 (Martin Vink, MM, LM and AM, unpublished data, 2006). It is possible that the relative affinities of the two C-Mad2 interactors are modulated to ensure the accurate timing of checkpoint activation and inactivation. Further studies will test this prediction and address the details of the regulation of the p31comet:C-Mad2 complex. The model predicts that checkpoint regulation might be rather different in different organisms, because p31comet homologues have not yet been identified in S. cerevisiae. Furthermore, dynein has not been identified in the nucleus in S. cerevisiae, and is therefore unlikely to play a role in checkpoint inactivation in this organism.
Understanding the physical significance of the O-Mad2:C-Mad2 interaction, that is, the manner by which it promotes the binding of O-Mad2 to Cdc20, has become crucial. The interaction is limited to opposite Mad2 conformers (De Antoni et al, 2005a, 2005b), suggesting that the binding surfaces on the interacting partners are at least partly asymmetric. We report that the surface of C-Mad2 involved in binding O-Mad2 contains the Mad2 C-terminal tail, the safety belt whose conformation marks the differences between the two Mad2 conformers. Conversely, this part of the structure is not required for the binding of O-Mad2 to C-Mad2. Other elements of the interaction surfaces of O-Mad2 and C-Mad2, on the other hand, are conserved on both conformers, in particular the patch encompassing the helices α3 of the two protomers. Owing to the asymmetric nature of the interaction surface, however, these ‘symmetrical' patches might be at least in principle involved in a completely nonsymmetrical interaction.
So far, the O-Mad2:C-Mad2 dimer has escaped structural analysis and we have been unable to obtain crystals of this complex. The highly dynamic nature of the interaction, combined with our NMR analysis, which indicates that O-Mad2 undergoes a conformation change upon binding to C-Mad2, provides a simple explanation for these difficulties. To gain an insight into the structural rearrangement of O-Mad2 upon binding to C-Mad2, strategies will have to be developed to stabilize the complex. From a structural perspective, we note that the conversion of O-Mad2 into C-Mad2 requires the fracture of the exposed β-sheet of Mad2 along the β1–β5 strands (Figure 7). In C-Mad2, β7 substitutes β1, while the latter is transformed into a helical extension at the N-terminus of α1. This very complex rearrangement may be rate limiting for the ability of Mad2 to bind Cdc20, and we have indeed shown that the on-rate for the binding of O-Mad2 to GST-Cdc20111−138 is very slow (Martin Vink and Andrea Musacchio, unpublished data, 2006).
Based on these considerations, we suspect that the interaction of O-Mad2 with C-Mad2 accelerates the rate of conversion of O-Mad2 into C-Mad2 bound to Cdc20. This view depicts C-Mad2 as a catalyst for the transformation of O-Mad2 into C-Mad2, and the catalytic nature of the O-Mad2:C-Mad2 interaction might be interpreted as a stabilization of the complex transition state of the conversion of O-Mad2 into C-Mad2. Thus, the biological significance of the ‘Mad2 template' model may coincide with a catalytic function of C-Mad2 on O-Mad2. We suspect that studies aiming to evaluate the correctness of this prediction should make use of real-time sensors of the conversion of O-Mad2 to C-Mad2. These have not been described yet and their creation may be nontrivial.
Materials and methods
Plasmids and proteins
Human Mad2wt (1–205), Mad2ΔC (1–195) and Mad2ΔN15 (16–205) were expressed from pET43 (Novagen) as N-terminal hexa-histidine tag fusions. Point mutations were introduced with QuickChange (Stratagene) and PCR primers indicated in Supplementary Table I. All constructs were sequenced. pGEX-Cdc20111−138 has been described (Sironi et al, 2001). p31comet was expressed from pET21a with a C-terminal hexa-histidine tag. All Mad2 forms were expressed in BL21-pLysS(DE3). For large-scale purification, Mad2 mutants were isolated on NTA beads (Qiagen) and further purified on a gel filtration S75 column (Amersham). p31comet was expressed in BL21-Rosetta and purified analogously.
GST pull-down, titration and competition assays
For GST-Cdc20111−138 pull-down experiments in Figures 3A and 4A, Mad2 or Mad2ΔC mutants in bacterial lysates were quantified by SDS–PAGE. To test MadΔC mutants, 1 μM GST-Cdc20 on beads was incubated with 2 μM Mad2wt. The beads were washed and Mad2ΔC or mutants were added at 2 μM. Reactions were protracted for 2 h at room temperature (TR), after which beads were washed and bound proteins analyzed by SDS–PAGE. The supernatant of the incubation was used as control for the relative concentrations of the Mad2ΔC mutants used in the experiment (not shown). To test full-length Mad2 mutants, equal amounts of each mutant were incubated with GST-Cdc20111−138 on GSH beads. Beads were washed before addition of Mad2ΔC. Reagents were mixed, incubated for 2 h at 20°C and proteins on beads were separated on SDS–PAGE. The same procedure was adopted for the GST pull-down experiments performed with C-Mad2R133A double mutants and p31comet. To monitor relative affinities of Mad2 mutants, the GST pull-down assay described above was carried out with increasing concentrations of purified Mad2ΔC either wild-type or point-mutated. To evaluate competition between Mad2ΔC and p31comet, reaction mix consisting of 1 μM GST-Cdc20-bound Mad2wt, stoichiometric amounts of Mad2ΔC and increasing concentrations of p31comet were assembled. After 2 h at 20°C, the beads were washed and bound species were resolved by SDS–PAGE.
NMR spectroscopy
Hs Mad2wt and Mad2ΔC were expressed as N-terminal His6 fusion from a pET43 vector in Escherichia coli BL21(DE3). Isotopically labeled (90% 2H, 13C and/or 15N) Mad2 was prepared by growing bacteria in minimal medium supplemented with [U-13C]glucose and/or 15NH4Cl in D2O. The Mad2 proteins were isolated on Ni-NTA agarose (Qiagen) and subsequently purified by gel filtration through a Superdex 75 (16/60) column (Pharmacia) in 50 mM sodium phosphate and 300 mM NaCl at pH 6.8. Samples were used at concentrations of 0.5–1.0 mM. NMR spectra were acquired at 37°C on a Bruker DRX600 spectrometer equipped with a cryogenic probe. The backbone chemical shifts of O-Mad2 and C-Mad2 were based on BMRB entries 4775 (Luo et al, 2000) and 5299 (Luo et al, 2002), respectively. Chemical shift assignments were confirmed and extended using triple resonance experiments (Sattler et al, 1999) applied to two samples, comprising either 2H,13C-,15N-labeled full-length Mad2 bound to unlabeled Mad1 peptide and unlabeled Mad2ΔC or 2H,13C,15N-labeled Mad2ΔC bound to unlabeled full-length Mad2/Mad1 peptide. The assignments for the Mad2 molecules in the heterodimeric complexes were obtained comparing TROSY-HNCA experiments on 2H,13C,15N-labeled samples. Chemical shift perturbations (Δδ=[(Δδ1H)2+(1/5 Δδ15N)2], in parts per million) were monitored in 2D 1H,15N-HSQC experiments for residues that could be unambiguously assigned in the free and bound state. The chemical shift perturbations indicated in Figure 6F and G by spheres refer exclusively to these residues.
Yeast strains, media and reagents
Standard genetic techniques were used to manipulate yeast strains (Sherman, 2002). All yeast strains were derivatives of, or were backcrossed at least three times with, W303 (ade2-1, trp1-1, leu2-3, 112, his3-11, 15, ura3, ssd1). Supplementary Table II reports a full list of strains used in this study and their genotype. Cells were grown in YEP medium (1% yeast extract, 2% bactopeptone, 50 mg/l adenine) supplemented with 2% glucose (YEPD). The α factor was used at 2 μg/ml, nocodazole at 15 μg/ml. All strains were normally grown at 25°C. An ScMAD2 HindIII/BglII fragment, containing the whole coding region plus ∼400 bp of upstream and ∼280 bp of downstream sequence, was cloned in HindIII/BamHI of Yiplac128. The resulting pSP42 plasmid was integrated at the LEU2 locus by EcoRV digestion. Integrations were checked by Southern analysis. Mutant alleles were generated using Quikchange (Stratagene).
Flow cytometry and analysis of sister chromatid separation
Flow cytometric DNA quantitation was determined on a Becton-Dickinson FACScan as described (Epstein and Cross, 1992). Sister chromatid separation was followed on ethanol-fixed cells by visualizing tetracycline-repressor-GFP fusion proteins bound to tandem repeats of tet operators integrated at about 35 kb away from the centromere of chromosome V (Michaelis et al, 1997).
Supplementary Material
Supplementary Figures
Acknowledgments
We thank Anna De Antoni, K Nasmyth, W Zachariae, K Hardwick for providing reagents. MS thanks the Deutsche Forschungsgemeinschaft and the Center for Biomagnetic Resonance in Frankfurt (Germany) for high-field NMR measurement time. LN received supports from the European School of Molecular Medicine (SEMM). SP is grateful to the Associazione Italiana per la Ricerca sul Cancro (AIRC) for funding. AM is grateful to AIRC, Human Frontier Science Program, EU FP6 programs 3D-Repertoire and Mitocheck, Fondazione Telethon, Fondo FIRB and Fondazione Cariplo for funding.
References
- Aravind L, Koonin EV (1998) The HORMA domain: a common structural denominator in mitotic checkpoints, chromosome synapsis and DNA repair. Trends Biochem Sci 23: 284–286 [DOI] [PubMed] [Google Scholar]
- Bharadwaj R, Yu H (2004) The spindle checkpoint, aneuploidy, and cancer. Oncogene 23: 2016–2027 [DOI] [PubMed] [Google Scholar]
- Chan GK, Liu ST, Yen TJ (2005) Kinetochore structure and function. Trends Cell Biol 15: 589–598 [DOI] [PubMed] [Google Scholar]
- Chen R-H (2002) BubR1 is essential for kinetochore localization of other spindle checkpoint proteins and its phosphorylation requires Mad1. J Cell Biol 158: 487–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen RH, Brady DM, Smith D, Murray AW, Hardwick KG (1999) The spindle checkpoint of budding yeast depends on a tight complex between the Mad1 and Mad2 proteins. Mol Biol Cell 10: 2607–2618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen RH, Shevchenko A, Mann M, Murray AW (1998) Spindle checkpoint protein Xmad1 recruits Xmad2 to unattached kinetochores. J Cell Biol 143: 283–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung E, Chen R-H (2002) Spindle checkpoint requires Mad1-bound and Mad1-free Mad2. Mol Biol Cell 13: 1501–1511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleveland DW, Mao Y, Sullivan KF (2003) Centromeres and kinetochores: from epigenetics to mitotic checkpoint signaling. Cell 112: 407–421 [DOI] [PubMed] [Google Scholar]
- De Antoni A, Pearson CG, Cimini D, Canman JC, Sala V, Nezi L, Mapelli M, Sironi L, Faretta M, Salmon ED, Musacchio A (2005a) The mad1/mad2 complex as a template for mad2 activation in the spindle assembly checkpoint. Curr Biol 15: 214–225 [DOI] [PubMed] [Google Scholar]
- De Antoni A, Sala V, Musacchio A (2005b) Explaining the oligomerization properties of the spindle assembly checkpoint protein Mad2. Philos Trans R Soc Lond B Biol Sci 360: 637–647, discussion 447–638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epstein CB, Cross FR (1992) CLB5: a novel B cyclin from budding yeast with a role in S phase. Genes Dev 6: 1695–1706 [DOI] [PubMed] [Google Scholar]
- Fang G (2002) Checkpoint protein BubR1 acts synergistically with Mad2 to inhibit anaphase-promoting complex. Mol Biol Cell 13: 755–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang G, Yu H, Kirschner MW (1998) The checkpoint protein MAD2 and the mitotic regulator CDC20 form a ternary complex with the anaphase-promoting complex to control anaphase initiation. Genes Dev 12: 1871–1883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraschini R, Beretta A, Sironi L, Musacchio A, Lucchini G, Piatti S (2001) Bub3 interaction with Mad2, Mad3 and Cdc20 is mediated by WD40 repeats and does not require intact kinetochores. EMBO J 20: 6648–6659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habu T, Kim SH, Weinstein J, Matsumoto T (2002) Identification of a MAD2-binding protein, CMT2, and its role in mitosis. EMBO J 21: 6419–6428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagan RS, Sorger PK (2005) Cell biology: the more MAD, the merrier. Nature 434: 575–577 [DOI] [PubMed] [Google Scholar]
- Hardwick KG (2005) Checkpoint signalling: Mad2 conformers and signal propagation. Curr Biol 15: R122–R124 [DOI] [PubMed] [Google Scholar]
- Hardwick KG, Johnston RC, Smith DL, Murray AW (2000) MAD3 encodes a novel component of the spindle checkpoint which interacts with Bub3p, Cdc20p, and Mad2p. J Cell Biol 148: 871–882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell BJ, McEwen BF, Canman JC, Hoffman DB, Farrar EM, Rieder CL, Salmon ED (2001) Cytoplasmic dynein/dynactin drives kinetochore protein transport to the spindle poles and has a role in mitotic spindle checkpoint inactivation. J Cell Biol 155: 1159–1172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyt MA, Totis L, Roberts BT (1991) S. cerevisiae genes required for cell cycle arrest in response to loss of microtubule function. Cell 66: 507–517 [DOI] [PubMed] [Google Scholar]
- Hwang LH, Lau LF, Smith DL, Mistrot CA, Hardwick KG, Hwang ES, Amon A, Murray AW (1998) Budding yeast Cdc20: a target of the spindle checkpoint. Science 279: 1041–1044 [DOI] [PubMed] [Google Scholar]
- Kallio M, Weinstein J, Daum JR, Burke DJ, Gorbsky GJ (1998) Mammalian p55CDC mediates association of the spindle checkpoint protein Mad2 with the cyclosome/anaphase-promoting complex, and is involved in regulating anaphase onset and late mitotic events. J Cell Biol 141: 1393–1406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Lin DP, Matsumoto S, Kitazono A, Matsumoto T (1998) Fission yeast Slp1: an effector of the Mad2-dependent spindle checkpoint. Science 279: 1045–1047 [DOI] [PubMed] [Google Scholar]
- Li R, Murray A (1991) Feedback control of mitosis in budding yeast. Cell 66: 519–531 [DOI] [PubMed] [Google Scholar]
- Luo X, Fang G, Coldiron M, Lin Y, Yu H, Kirschner MW, Wagner G (2000) Structure of the mad2 spindle assembly checkpoint protein and its interaction with cdc20. Nat Struct Biol 7: 224–229 [DOI] [PubMed] [Google Scholar]
- Luo X, Tang Z, Rizo J, Yu H (2002) The Mad2 spindle checkpoint protein undergoes similar major conformational changes upon binding to either Mad1 or Cdc20. Mol Cell 9: 59–71 [DOI] [PubMed] [Google Scholar]
- Luo X, Tang Z, Xia G, Wassmann K, Matsumoto T, Rizo J, Yu H (2004) The Mad2 spindle checkpoint protein has two distinct natively folded states. Nat Struct Mol Biol 11: 338–345 [DOI] [PubMed] [Google Scholar]
- Michaelis C, Ciosk R, Nasmyth K (1997) Cohesins: chromosomal proteins that prevent premature separation of sister chromatids. Cell 91: 35–45 [DOI] [PubMed] [Google Scholar]
- Morrow CJ, Tighe A, Johnson VL, Scott MI, Ditchfield C, Taylor SS (2005) Bub1 and aurora B cooperate to maintain BubR1-mediated inhibition of APC/CCdc20. J Cell Sci 118: 3639–3652 [DOI] [PubMed] [Google Scholar]
- Musacchio A, Hardwick KG (2002) The spindle checkpoint: structural insights into dynamic signalling. Nat Rev Mol Cell Biol 3: 731–741 [DOI] [PubMed] [Google Scholar]
- Nasmyth K (2005) How do so few control so many? Cell 120: 739–746 [DOI] [PubMed] [Google Scholar]
- Pinsky BA, Biggins S (2005) The spindle checkpoint: tension versus attachment. Trends Cell Biol 15: 486–493 [DOI] [PubMed] [Google Scholar]
- Poddar A, Stukenberg PT, Burke DJ (2005) Two complexes of spindle checkpoint proteins containing Cdc20 and Mad2 assemble during mitosis independently of the kinetochore in Saccharomyces cerevisiae. Eukaryot Cell 4: 867–878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sattler M, Schleucher J, Griesinger C (1999) Heteronuclear multidimensional NMR experiments for the structure determination of proteins in solution employing pulsed field gradients. Prog NMR Spectrosc 34: 93–158 [Google Scholar]
- Shah JV, Botvinick E, Bonday Z, Furnari F, Berns M, Cleveland DW (2004) Dynamics of centromere and kinetochore proteins; implications for checkpoint signaling and silencing. Curr Biol 14: 942–952 [DOI] [PubMed] [Google Scholar]
- Shannon KB, Canman JC, Salmon ED (2002) Mad2 and BubR1 function in a single checkpoint pathway that responds to a loss of tension. Mol Biol Cell 13: 3706–3719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman F (2002) Getting started with yeast. Methods Enzymol 350: 3–41 [DOI] [PubMed] [Google Scholar]
- Sironi L, Mapelli M, Knapp S, Antoni AD, Jeang K-T, Musacchio A (2002) Crystal structure of the tetrameric Mad1–Mad2 core complex: implications of a ‘safety belt' binding mechanism for the spindle checkpoint. EMBO J 21: 2496–2506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sironi L, Melixetian M, Faretta M, Prosperini E, Helin K, Musacchio A (2001) Mad2 binding to Mad1 and Cdc20, rather than oligomerization, is required for the spindle checkpoint. EMBO J 20: 6371–6382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudakin V, Chan GK, Yen TJ (2001) Checkpoint inhibition of the APC/C in HeLa cells is mediated by a complex of BUBR1, BUB3, CDC20, and MAD2. J Cell Biol 154: 925–936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Z, Bharadwaj R, Li B, Yu H (2001) Mad2-independent inhibition of APC-Cdc20 by the mitotic checkpoint protein Bub1R. Dev Cell 1: 227–237 [DOI] [PubMed] [Google Scholar]
- Wassmann K, Benezra R (1998) Mad2 transiently associates with an APC/p55Cdc complex during mitosis. Proc Natl Acad Sci USA 95: 11193–11198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wassmann K, Liberal V, Benezra R (2003) Mad2 phosphorylation regulates its association with Mad1 and the APC/C. EMBO J 22: 797–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia G, Luo X, Habu T, Rizo J, Matsumoto T, Yu H (2004) Conformation-specific binding of p31(comet) antagonizes the function of Mad2 in the spindle checkpoint. EMBO J 23: 3133–3143 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures