

Abstract
Glycine is the major inhibitory neurotransmitter in the spinal cord and brain stem. Gephyrin is required to achieve a high concentration of glycine receptors (GlyRs) in the postsynaptic membrane, which is crucial for efficient glycinergic signal transduction. The interaction between gephyrin and the GlyR involves the E-domain of gephyrin and a cytoplasmic loop located between transmembrane segments three and four of the GlyR β subunit. Here, we present crystal structures of the gephyrin E-domain with and without the GlyR β-loop at 2.4 and 2.7 Å resolutions, respectively. The GlyR β-loop is bound in a symmetric ‘key and lock' fashion to each E-domain monomer in a pocket adjacent to the dimer interface. Structure-guided mutagenesis followed by in vitro binding and in vivo colocalization assays demonstrate that a hydrophobic interaction formed by Phe 330 of gephyrin and Phe 398 and Ile 400 of the GlyR β-loop is crucial for binding.
Keywords: gephyrin, glycine receptor, neuroreceptor anchoring, postsynaptic membrane, X-ray crystallography
Introduction
The glycine receptor (GlyR) is the main inhibitory neurotransmitter receptor in the spinal cord and brain stem. Binding of glycine results in channel opening and allows Cl− flux across the synaptic membrane. As a member of the ligand-gated ion channel superfamily, functional GlyRs have been generally believed to display a pentameric subunit assembly with an α3β2 stoichiometry in the adult organism; however, recently an alternative α2β3 stoichiometry was reported (Grudzinska et al, 2005). In contrast, prenatal GlyRs are predominantly α5-homopentamers. Up to date, four α-subunits (α1–α4) and one β-subunit have been identified, but it is not well understood how subunit diversity is related to the function and distribution of GlyRs (Lynch, 2004). For fast and precise signal transmission at inhibitory synapses, GlyRs as well as γ-aminobutyric acid type A receptors (GABAAR) are required to be highly concentrated at the postsynaptic membrane, a process which is dependent on the receptor-associated scaffolding protein gephyrin (Kneussel and Betz, 2000; Sheng and Pak, 2000; Legendre, 2001; Moss and Smart, 2001).
Gephyrin is a 93 kDa cytoplasmic protein, which was initially discovered through copurification with GlyRs from rat spinal cord (Pfeiffer et al, 1982). It consists of an N-terminal G-domain and a C-terminal E-domain, which are connected by a central region composed of ∼160 amino-acid residues. The two domains display significant similarities to the bacterial MogA and MoeA proteins, respectively, which are essential for molybdenum cofactor (Moco) biosynthesis. Strong experimental evidence has accumulated, which demonstrates that gephyrin is critical for GlyR clustering. Gephyrin colocalizes with GlyRs through the hydrophobic interaction between its E-domain and the large cytoplasmic loop connecting transmembrane (TM) segments 3 and 4 of the GlyR β-subunit (Kirsch et al, 1995; Kneussel et al, 1999; Schrader et al, 2004), which will be referred to as GlyR β-loop. Furthermore, a loss of gephyrin synthesis, either by antisense oligonucleotides in spinal cord neurons or by gene knockout in mice, prevents GlyR clustering (Kirsch et al, 1993; Feng et al, 1998). Although no direct interaction between gephyrin and GABAAR has been demonstrated, gephyrin also plays a crucial role in the stabilization of GABAAR clusters. Furthermore, gephyrin interacts with the microfilament components profilin, Mena/VASP and G-actin (Giesemann et al, 2003) as well as tubulin (Kirsch et al, 1991), and the simultaneous binding to GlyR and microtubules or the actin-based cytoskeleton forms the basis of the neuroreceptor-anchoring function of gephyrin.
In addition, a variety of other cellular components such as GABARAP, collybistin, RAFT1, and dynein light chains (Sabatini et al, 1999; Kins et al, 2000; Kneussel et al, 2000; Fuhrmann et al, 2002) also interact with gephyrin, although the physiological significance of these interactions is not well characterized at present. As expected from the primary sequence homology, gephyrin is also involved in Moco biosynthesis, specifically the last step of molybdenum insertion into the apo-cofactor. It was shown that gephyrin is able to reconstitute Moco biosynthesis in Moco-deficient bacteria, plants and mammalian cells (Stallmeyer et al, 1999), and a loss of gephyrin causes Moco deficiency in mice (Feng et al, 1998) and humans (Reiss et al, 2001). It remains unclear whether the described two functions of gephyrin are related to each other. Nevertheless, together they define gephyrin as a ‘moonlighting' protein. The gene encoding gephyrin is highly mosaic, and, as a result, its heterogeneity by alternative splicing could potentially contribute to its functional diversity (Meier et al, 2000; Rees et al, 2003), yet a correlation between the different functions and splice variants remains to be established.
The crystal structures of Escherichia coli MogA, the G-domains of gephyrin and the plant homologs Cnx1 and E. coli MoeA have been determined recently (Liu et al, 2000; Schwarz et al, 2001; Xiang et al, 2001). On the basis of these structural data, gephyrin was proposed to trimerize and dimerize via its G-domain and E-domain, respectively, and eventually assemble into a hexagonal lattice, which is proposed to connect inhibitory neuroreceptors to components of the cytoskeleton (Kneussel and Betz, 2000; Xiang et al, 2001). More recently, the crystal structure of gephyrin's E-domain in complex with a putative hexapeptide derived from GlyR β-loop suggested that the binding site of GlyR resides in subdomain IV of the E-domain (Sola et al, 2004), whereas our in vitro interaction assays suggested subdomain III instead (Schrader et al, 2004). This complex structure, however, was refined at a relatively low resolution of 3.25 Å, and neither the identity of the residues involved nor the details of the interaction could be identified. Here, we present the 2.4 Å crystal structure of gephyrin's E-domain in complex with the GlyR β-loop and the apo-structure of the E-domain at 2.7 Å. The cocrystal structure defines the interaction between GlyR and gephyrin in atomic detail. The contributions of selected residues to the binding have been probed by site-directed mutagenesis coupled with in vitro and in vivo binding studies providing insights into the molecular details of gephyrin-mediated clustering of glycine receptors.
Results and discussion
Structure determination of Geph-E alone and in complex with the GlyR β-loop
The E-domain of rat gephyrin (Geph-E, residues 318–736) was crystallized in space group P21212 containing one dimer per asymmetric unit. An initial model of Geph-E was built using a lower-resolution data set (3.3 Å) by a combination of a two-wavelength MAD experiment and molecular replacement using E. coli MoeA as the search model (Table I). Subsequently, a higher-resolution data set was obtained, and a final structure of Geph-E was determined at 2.7 Å resolution (Table II), resulting in an R-factor of 20.5% (Rfree=27.7%). The overall structure of Geph-E (Figure 1A) shows a high structural similarity to E. coli MoeA (Xiang et al, 2001), as indicated by an overall root mean square (r.m.s.) deviation of 2.02 Å, and is even more closely related to the recently published E-domain structure (Sola et al, 2004), with an overall r.m.s. deviation of 0.95 Å. Each Geph-E monomer consists of four subdomains (Figure 1A and B) as first described for MoeA, and two monomers interact to form an elongated dimer with subdomains II on opposite ends. A comparison of the two monomers reveals that the core structure (subdomains I, III and IV) is rather rigid, as reflected by an r.m.s. deviation of 0.7 Å for the Cα atoms, whereas subdomain II is not only oriented differently with respect to the core but also displays substantially higher r.m.s. deviations of 2.5 Å. This is probably due to its flexibility, which results in poorly defined electron density of subdomain II in one of the monomers.
Table 1.
Data collection and MAD statistics
| SeMet Geph-E |
||||
|---|---|---|---|---|
| Geph-E | Peak | Remote | Geph-E/GlyR β-loop | |
| Data collection | ||||
| Wavelength (Å) | 1.1 | 0.98021 | 0.96496 | 1.1 |
| Resolution limits (Å) | 50–2.7 | 50–3.5 | 50–3.5 | 50–2.4 |
| Number of reflections | 24 624 | 21 485 | 21 781 | 18 251 |
| Completeness | 0.99 (0.999) | 1.0 (0.999) | 1.0 (1.0) | 0.935 (0.77) |
| Mean redundancy | 4.8 | 5.8 | 3.8 | 5.0 |
| Rsyma | 0.10 (0.54) | 0.16 (0.43) | 0.17 (0.51) | 0.09 (0.56) |
| 〈I/σI〉b | 13.7 (2.6) | 21.5 (8.3) | 14.5 (4.9) | 21.5 (2.7) |
| Space group | P21212 | P21212 | P21212 | C2221 |
| Unit cell dimensions | ||||
| a, b, c (Å) | 108.8, 156.2, 51.4 | 109.0, 157.2, 51.5 | 109.0, 157.2, 51.5 | 51.3, 123.5, 155.0 |
| MAD | ||||
| Number of sites | 18 | |||
| Rcullisc | 0.86 | NA | ||
| FOMd |
|
0.2395 |
|
|
| aRsym=∑hkl∑i∣Ii−〈I〉∣/∑hkl∑iIi, where Ii is the ith measurement and 〈I〉 is the weighted mean of all measurements of I. | ||||
| b〈I/σI〉 indicates the average of the intensity divided by its standard deviation. Numbers in parentheses refer to the respective highest resolution data shell in each data set. | ||||
| cRcullis is the lack of closure divided by the absolute of the difference between FPH and FP for the anomalous differences. | ||||
| dFOM is the mean figure of merit at 4 Å resolution. | ||||
Table 2.
Refinement statistics
| Geph-E | Geph-E/GlyR β-loop | |
|---|---|---|
| Resolution limits (Å) | 20–2.7 | 20–2.4 |
| Number of reflections used in refinement | 23 473 | 17 248 |
| Number of protein/ solvent atoms | 6191/66 | 3298/150 |
| Rcrysta (Rfreeb) | 0.205 (0.277) | 0.192 (0.274) |
| Deviations from ideal values in | ||
| Bond distances (Å) | 0.022 | 0.019 |
| Bond angles (deg) | 1.60 | 1.59 |
| Torsion angles (deg) | 4.4, 35.6, 16.3 | 4.9, 37.9, 14.9 |
| Planar groups (Å) | 0.005 | 0.005 |
| DPI (Å)c | 0.39 | 0.31 |
| Ramachandran statistics (%)d | 84.8/12.2/2.6/0.4 | 91.0/7.8/0.8/0.3 |
| Overall average B-factor (Å2) |
62.1 |
40.5 |
| aRcryst=∑hkl∣∣Fo∣–∣Fc∣∣/∑hkl∣Fo∣, where Fo and Fc are the observed and calculated structure factor amplitudes. | ||
| bRfree is the same as Rcryst for 5% of the data randomly omitted from refinement. The number of reflections excludes the Rfree subset. | ||
| cDPI is the data precision index based on the free R factor as calculated by REFMAC. | ||
| dRamachandran statistics indicate the fraction of residues in the most favored, additionally allowed, generously allowed, and disallowed regions of the Ramachandran diagram as defined by PROCHECK. | ||
Figure 1.
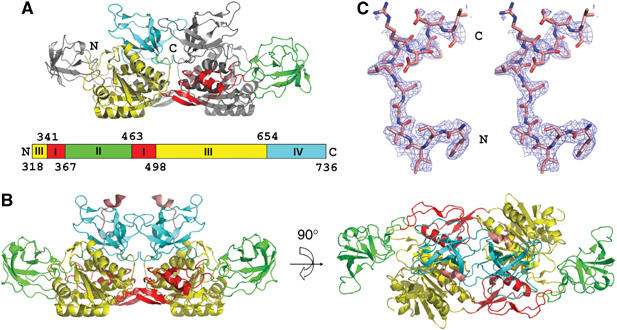
Overall structure of Geph-E and complex with the GlyR β-loop. (A) Ribbon diagram of the Geph-E apo-structure. The four subdomains of one monomer are color-coded as shown in the schematic representation (upper left), with the other monomer in gray. The N- and C-termini of Geph-E are indicated. The amino-acid numbering is based on the P1 splice variant (Prior et al, 1992). (B) Ribbon diagram of Geph-E in complex with the GlyR β-loop-derived peptide. The bound peptide is 13 residues long and colored in salmon. The orientation of the left view is identical to the apo structure, whereas the right view is rotated 90° relative to the left view around the horizontal axis. (C) Electron density of the GlyR β-loop. Stereo view of a simulated annealed omit map (contoured at 0.8 times the r.m.s. deviation) superimposed with the final model of the GlyR β-loop. The peptide is shown in atom-based color code (C: salmon, O: red, and N: blue) using a stick representation. The N- and C-termini of the peptide are indicated.
To characterize the interaction between Geph-E and GlyR in atomic detail, individually purified Geph-E and residues 378–426 of the GlyR β-loop were cocrystallized. Two different crystal forms belonging either to space group P212121 containing one Geph-E dimer and two peptides, or C2221 with one monomer and a single peptide were obtained under the same crystallization conditions. In the C2221 crystals, the functional unit again is a dimer, which is formed via a crystallographic twofold symmetry axis. Both structures were solved by molecular replacement using the apo-structure. An initial 3.0 Å data set of the P212121 crystal confirmed the presence of the peptide and revealed that ∼10 residues were well-ordered and their sequence could be unambiguously assigned already at this resolution. The complex structure was ultimately refined at 2.4 Å resolution to an R-factor of 19.2% (Rfree=27.4%) against a data set collected from a C2221 crystal (Tables I and II). In this structure 13 residues, corresponding to the segment from 398 to 410 of the GlyR β-loop, were found to be ordered (Figure 1C). Due to the higher resolution, the following discussion will focus on the crystallographic dimer from the C2221 crystal, which is displayed in Figure 1B.
The Geph-E/GlyR β-loop complex in the P212121 and C2221 crystals showed very similar structures (r.m.s. deviation of 0.88 Å), except for small changes in a few poorly defined loops. In addition, there are also no deviations between the two monomers in the P212121 form, indicating that the monomers in the dimer behave independently. This observation is in contrast to a published proposal (Sola et al, 2004), which suggests that the two Geph-E monomers display different affinities for the GlyR β-loop. Our previous biochemical studies have shown that each Geph-E monomer has two binding sites for the GlyR β-loop, one with high and the other with low affinity (Schrader et al, 2004). In our complex crystals prepared with a twofold excess of ligand, the lower-affinity binding site of Geph-E appears not to be occupied, as we cannot observe additional electron density corresponding to another bound peptide. The peptide derived from the GlyR β-loop does not display any secondary structure, with the exception of a short 310 helix formed by residues 406–410.
Although a minor shift in subdomain II was observed upon binding of the GlyR β-loop (r.m.s. deviation of 1.67 Å), no significant conformational changes in the core structure of the E-domain (r.m.s. deviations of 0.65 Å) were seen. Therefore, association of the GlyR β-loop into gephyrin can be primarily described as following a lock-and-key, rather than an induced-fit mechanism.
Details of the interface between Geph-E and the GlyR β-loop
The 13 residues of the GlyR β-loop adopt the shape of the letter C and fit tightly into a groove created by subdomains III and IV from one monomer and subdomain IV′ from the other monomer (Figure 2). The Geph-E/GlyR β-loop interface buries ∼950 Å2 of the total surface area, which constitutes ∼53% of the total peptide surface area. Complex formation is stabilized mainly by hydrophobic interactions, which contribute more than 60% to the buried interface, and also by seven direct hydrogen bonds. The primary contact region on Geph-E can be divided into four smaller segments (Figure 2C): a loop from subdomain IV, including residues 713–719 (L713–719), the central four-stranded β-sheet of subdomain IV, the first α-helix and several neighboring residues in subdomain III, and a loop in subdomain IV′ comprised of residues 682–685 (L682–685).
Figure 2.
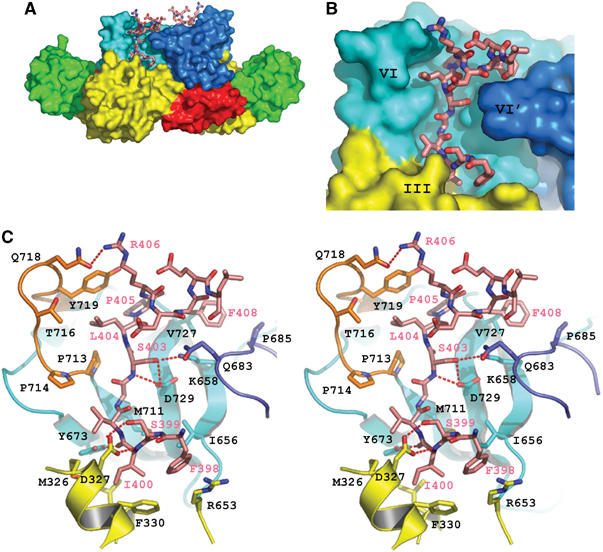
Detailed representation of Geph-E in complex with the GlyR β-loop. (A) Overall architecture of the complex. Subdomains of Geph-E are colored as in Figure 1, with the exception of subdomain IV′ from the second subunit, which is colored in dark blue. Both GlyR β-loops are shown in stick representation, utilizing the same color code as in Figure 1C. (B) Close-up view into the binding pocket. The GlyR β-loop is tightly packed in the cleft formed by subdomains III and IV from one monomer, as well as subdomain IV′ from the other monomer. (C) Detailed stereo view of the interactions between Geph-E and the GlyR β-loop. Left, right and lower walls of the peptide-binding pocket on Geph-E are displayed in orange, blue and yellow, respectively, and the bottom is colored in cyan. Geph-E is shown in ribbon representation and GlyR β-loop in stick representation. All residues in close proximity to the interface are highlighted and numbered (in black for Geph-E and magenta for the GlyR β-loop). Hydrogen bonds are shown as dotted red lines.
L713–719, which is not conserved in the MoeA homologs, creates the ‘left' wall of the binding pocket (as shown in Figure 2C), where Pro 713 and Tyr 719 make close contacts with Ser β403, Leu β404, Pro β405 and Arg β406 (the ‘β' indicates residues in the GlyR β-loop). To a smaller extent Pro 714 and Thr 716 also contribute to this interaction, while Gln 718 forms a hydrogen bond with Arg β406, which, however, should be treated with caution as the arginine side chain is only poorly defined in the electron density maps (Figure 1C). A recent study has demonstrated the importance of this loop for receptor binding via in vivo colocalization and in vitro interaction assays using the E-domain mutant in which L713–719 was replaced with that of MoeA (Sola et al, 2004). Based on a superposition of the loops in the MoeA and Geph-E/GlyR β-loop structures, the charged residues Glu 387 and Arg 390 of MoeA (corresponding to Pro 713 and Lys 715 of gephyrin, respectively) protrude into the peptide-binding site, which presumably interferes with peptide binding. In contrast, Lys 715 points into the opposite direction away from the peptide and is not involved in interactions. Lys 658, Tyr 673, Met 711 and Asp 729 are aligned across the β-sheet of subdomain IV and primarily form the bottom of the binding pocket, which also involves four hydrogen bonds: one between Ile β400 and Tyr 673, two between Ser β403 (main chain amide and side chain OH) and Asp 729 and one between the side chains of Ser β403 and Lys 658. Moreover, Ile 656 and Val 727 participate in hydrophobic interactions with Phe β398 and Phe β408, respectively, at either end of the bound peptide. The ‘lower' wall of the binding pocket is comprised of the first helix of subdomain III, as well as residues Leu 637 and Arg 653. Asp 327 forms two hydrogen bonds, one with the hydroxyl of Ser β399 and the other with the amide of Ile β400, which also makes hydrophobic contacts with Phe 330, Met 326 and Leu 637. In addition, the side chain of Arg 653 is parallel to the aromatic ring of Phe β398. Finally, L682–685 from the second monomer builds the ‘right' wall of the binding pocket, fitting into the inside of the C-shaped peptide. In particular, Gln 683 is the residue closest to the peptide, which is embraced by Ser β403 and Phe β408. In addition, Pro 685 and His 682 are engaged in weak nonpolar interactions with Phe β408 and Phe β398. The fact that the peptide binds into a groove formed by both monomers suggests that GlyR binding might affect Geph-E dimerization.
In vitro pulldown assays with mutants of gephyrin and the GlyR β-loop
In order to identify which of the residues in the binding interface are critical for the interaction between gephyrin and GlyR, nine Geph-E variants (D327A, F330A, R653A, I656A, Y673F, Q683A, P713E, P713A/P714A and D729A) and seven GlyR β-loop variants (βF398A, βS399A, βI400A, βS403A, βF408A, βF398A/I400A and βF398A/I400A/F408A) were generated by site-directed mutagenesis. With the exception of the Geph-E Y673F variant, all Geph-E and GlyR β-loop mutants were overexpressed and purified in the same manner as the wild-type (WT) proteins. Initially, the contribution of these residues was studied in pulldown assays using either WT GlyR β-loop and Geph-E mutants or GlyR β-loop variants and WT Geph-E.
The experiments with WT GlyR β-loop revealed that the F330A variant significantly reduces GlyR β-loop binding, while the P713E variant abolishes binding (Figure 3A). Interaction with P713A/P714A (PPAA) is also significantly abated, whereas the D327A, R653A, I656A, Q683A and D729A variants show similar levels of binding as the WT E-domain. Phe 330 is located in the first helix of subdomain III and is in the center of the lower wall of the binding pocket in close proximity to Phe β398. Loss of binding in the P713E variant was not unexpected, since not only a proline with its unique structural features is removed, but at the same time the negatively charged glutamate is introduced. Based on the MoeA structure, it is predicted to point towards the peptide-binding region and thus interferes with binding. In addition, similar results obtained with the double proline mutant PPAA may be explained by structural perturbations resulting from the replacement of the two proline residues, thus suggesting that Pro 713 and Pro 714 play an important role in maintaining L713–719 in a conformation that allows productive interactions with the GlyR β-loop. In contrast, the four hydrogen bonds formed by Asp 327 and Asp 729 are dispensable, and the other examined residues in the interface (Arg 653, Ile 656 and Gln 683) do not show a critical impact on binding either.
Figure 3.
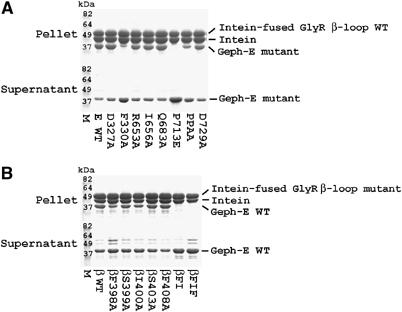
Pulldown assays with Geph-E and GlyR β-loop mutants. (A) Various Geph-E mutants were purified and co-precipitated with WT GlyR β-loop beads. WT Geph-E was also tested with WT GlyR β-loop beads as the control. Identical amounts of protein were applied to equal volumes of the beads. Bound protein in the pellet (upper panel) and unbound protein in the supernatant (lower panel) were subjected to SDS–PAGE analysis. The lane labeled M contains a molecular weight standard. (B) Cosedimentation of WT Geph-E with various GlyR β-loop mutants immobilized to the beads.
A second set of pulldown assays was performed to evaluate the GlyR β-loop variants. Previous colocalization assays revealed that only multiple, but not single, substitutions of hydrophobic side chains of GFP-49 (GlyR β-loop fused to a green fluorescent protein (GFP)) eliminated the interaction with gephyrin (Kneussel et al, 1999). In our current binding assays, surprisingly, the single substitution βF398A significantly impaired the interaction with Geph-E (Figure 3B). The βI400A substitution also showed a reduction in binding, albeit to a smaller extent, whereas βS403A and βF408A behaved like WT. The double (βF398A/I400A, βFI) and triple (βF398A/I400A/F408A, βFIF) variants containing βF398A completely abolished binding, which is thought to result from synergistic effects of βF398A and βI400A. Phe β398 and Ile β400 form a hydrophobic core in the lower binding wall together with Geph-E Phe 330, which, as outlined above, is also a major contributor to the strength of Geph-E/GlyR β-loop interaction. Analysis of the interactions involving either Asp 327 and Ser β399 or Asp 729 and Ser β403 through the respective single alanine substitutions revealed that the hydrogen bonds are not critical for this interaction.
Subsequently, the same mutations in Geph-E as well as the additional Y719A variant were introduced into full-length gephyrin, splice variant P2 (Prior et al, 1992), followed by in vitro binding assays to verify whether residues critical for Geph-E/GlyR β-loop interactions are indeed required for P2/GlyR β-loop complex formation. All P2 mutants were overexpressed and purified following the protocol for the WT protein, and pulldown assays using WT and variants of P2 and GlyR β-loop were performed as described above. The overall results are consistent with the binding studies involving only the E-domain (as seen in Figure 3 and Supplementary Figure S1), showing that F330A, P713E, βF398A, βFI and βFIF significantly reduce or abolish binding, while the PPAA, βS399A and βI400A variants considerably weaken the interaction. As described above, Tyr 673 forms a hydrogen bond with the main-chain carbonyl of Ile β400 via its phenolic OH-group. The importance of this hydrogen bond could not be tested in Geph-E, since the Geph-E Y673F variant could not be overexpressed and purified. Unexpectedly, deletion of this hydrogen bond by conversion of tyrosine into phenylalanine drastically reduced the interaction of full-length gephyrin and the GlyR β-loop, whereas disruption of the hydrogen bonds in the D327A and D729A variants did not significantly alter P2/GlyR peptide binding. In contrast, an additional point mutation in L714–719, Y719A, behaved just like WT. This demonstrates that among the observed hydrogen bonds only the one involving the side chain of Tyr 673 is critical to the binding interaction. Overall, there is good agreement of the results from the pulldown assays between the isolated E-domain and full-length gephyrin, thus suggesting that the cocrystal structure of the Geph-E/GlyR β-loop complex faithfully reproduces the interactions in the gephyrin/GlyR complex.
ITC studies of the Geph-E/GlyR interaction
For further confirmation of the pulldown assays and quantitative analyses, those Geph-E and GlyR β-loop variants, which showed significant effects in the pulldown assays, were subjected to isothermal titration calorimetry (ITC). To test gephyrin mutants, purified Geph-E WT and the F330A, P713E and PPAA variants as well as P2 WT and the Y673F variant were individually titrated with GlyR β-loop WT (Figure 4A). The resulting binding enthalpy was corrected for the heat of dilution, and the binding parameters were determined by curve fitting (Supplementary Table I). The dissociation constants and binding enthalpies obtained with WT Geph-E and P2 were very similar to those reported previously (Schrader et al, 2004); however, the binding stoichiometry of full-length gephyrin was reduced, which we attribute to the fact that the initial increase in enthalpy observed earlier (Schrader et al, 2004) was absent from the more recent experiments. While this initial phase was ignored for the curve-fitting procedure, it nevertheless led to an increase in the stoichiometry.
Figure 4.
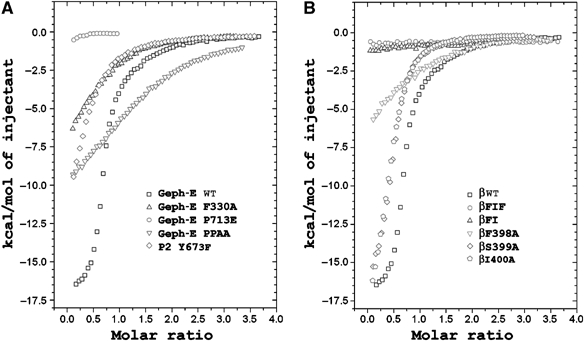
ITC experiments with Geph-E and GlyR β-loop variants. Overlaid binding isotherms of (A) Geph-E variants (WT, F330A, P713E and PPAA (P713A/P714A) and P2 Y673F titrated with WT GlyR β-loop and (B) WT Geph-E titrated with GlyR β-loop variants (WT, βF398A, βS399A, βI400A, βFI (βF398A/I400A) and βFIF (βF398A/I400A/F408A)). All experiments were performed under the same conditions and the measured binding enthalpies are plotted as a function of the molar ratio of GlyR β-loop to Geph-E. The P713E measurement was terminated earlier as no binding enthalpy could be detected. The binding parameters determined are summarized in Supplementary Table I.
Generally, the ITC results with the variants are in good agreement with the pulldown data. Specifically, the F330A and P2 Y673F showed one-site binding with dissociation constants (Kd) of ∼9.2 and ∼7.4 μM, respectively, which are about two orders of magnitude lower affinities than the WT (∼90–120 nM for the high-affinity binding site). The binding affinity of the PPAA variant was also greatly reduced (Kd 12 μM). At the same time, the binding stoichiometry of this variant was increased twofold, which we cannot explain currently. Finally, no signal was detected for the P713E variant as expected from the pulldown assays.
Subsequently, the contributions of residues in the GlyR β-loop were analyzed by titrating WT Geph-E separately with any of βF398A, βS399A, βI400A, βFI and βFIF variants. As observed for the Geph-E variants, the interaction patterns of GlyR β-loop mutants with Geph-E were also in agreement with the pulldown assays. In case of the βF398A, βS399A and βI400A variants, the data could be analyzed with a single binding site (Figure 4B and Supplementary Table I). The parameters of βF398A (including Kd 14.2 μM) were almost the same as Geph-E F330A, again demonstrating that the hydrophobic interface between these two residues is critical for complex formation. In case of the βS399A and βI400A variants, binding was lowered 15–20 times compared to WT (Kd 2 and 2.6 μM, respectively), while binding of the βFI and βFIF double and triple variants could not be detected by ITC. The result of the βS399A variant suggests that the hydrogen bond between its side chain and Asp 327 does contribute somewhat to the binding energy, at a level that cannot easily be detected in the pulldown assays.
Overall, it can be concluded that hydrophobic interactions are more important than hydrogen bonds, as demonstrated by the F330A and βF398A variants. These results are consistent with previous studies which identified multiple hydrophobic residues (Phe β398, Ile β400, Val β401, Leu β404, Phe β408 and Leu β410) as being crucial for interaction with Geph-E (Kneussel et al, 1999). However, the original suggestion that the gephyrin-binding region of GlyR β-loop forms an amphipathic α-helix is clearly incorrect, as demonstrated by the complex structure and also the double substitution (βFI), which is sufficient to abolish the GlyR β-loop/gephyrin interaction.
In vivo interaction studies with gephyrin and GlyR β-loop variants
In order to verify the in vitro data obtained from the pulldown and ITC experiments, we have performed colocalization experiments in human embryonic kidney 293 (HEK 293) cells with GFP-tagged gephyrin variants and red fluorescent protein (DsRed)-tagged GlyR β-loop variants, as described earlier (Kirsch et al, 1995). The fact that gephyrin transiently expressed in non-neuronal cells forms large intracellular aggregates (Figure 5A1) was used to develop a recruitment assay not only for the GlyR β-loop but also for other gephyrin interacting proteins (Meier et al, 2000; Giesemann et al, 2003). Single expression of WT GlyR β-loop showed a diffuse distribution with a strong signal in the nucleus due to the small size of the DsRed fusion protein (Figure 5A2). Coexpression of both WT gephyrin and WT GlyR β-loop resulted in complete redistribution of the GlyR β-loop into gephyrin clusters. Free GlyR β-loop is not detectable, neither in the nucleus nor in the cytoplasm.
Figure 5.
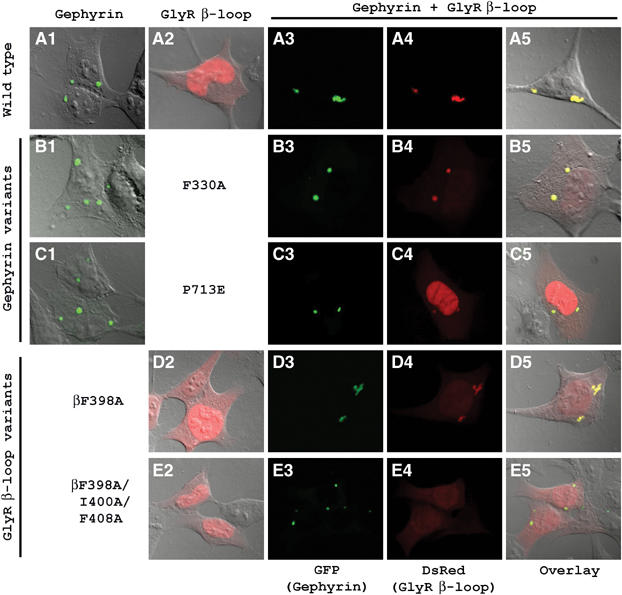
Transient expression of GFP-tagged gephyrin variants and DsRed-tagged GlyR β-loop variants in HEK 293 cells. HEK 293 cells were individually transfected with WT (A1), F330A (B1) and P713E gephyrin (C1) or WT (A2), F398A (D2) and F398A/I400A/F408A GlyR β-loop variants (E2). Coexpression with WT GlyR β-loop (A3–5, B3–5, C3–5) and WT gephyrin (A3–5, D3–5, E3–5) is shown in the corresponding panels with green gephyrin clusters (A3–E3), clustered or diffusely distributed GlyR β-loop (A4–E4) and the overlay of both images (A5–E5).
Subsequently, we expressed the gephyrin variants F330A and P713E, as well as the GlyR β-loop variants βF398A and βFIF in HEK 293 cells (Figure 5). Individual expression of gephyrin (Figure 5B1 and C1) and GlyR β-loop variants (Figure 5D2 and E2) showed no altered distribution as compared to the corresponding WT proteins. The F330A variant of gephyrin, which has a significantly reduced GlyR β-loop-binding affinity in the ITC experiments, is still able to recruit the GlyR β-loop in HEK 293 cells (Figure 5B3–5). However, there is also free GlyR β-loop detectable (Figure 5B5), reflecting a weaker interaction. In contrast, the P713E variant displays almost no colocalization with GlyR β-loop (Figure 5C3–5), which is found to be mainly localized in the nucleus and cytoplasm, as seen for individually expressed GlyR β-loop (Figure 5A2). The latter result is consistent with the ITC data, where almost no binding was detected. Similar to the gephyrin variants, the GlyR β-loop variants also reflect in their colocalization patterns the binding affinities determined by ITC for the purified proteins. βF398A (Figure 5D3–5) exhibits colocalization with gephyrin, yet a significant amount of diffuse fluorescence is present. Strong diffuse fluorescence in a noninteracting GlyR β-loop variant has already been observed in earlier experiments (Kneussel et al, 1999). In contrast, the triple mutant βFIF is mainly found in the cytoplasm and nucleus due to its dramatically reduced binding to gephyrin. Overall, the in vivo colocalization studies are in very good agreement with the pulldown and ITC measurements; however, variants that are significantly affected in in vitro experiments are still able to at least partially colocalize in HEK 293 cells. A closer examination of the fluorescence ratios in the respective clusters reveals even in the gephyrin F330A and the GlyR βF398A variants a subtle color change from yellow, indicating perfect colocalization, to yellow-green, which is consistent with a reduction of colocalized red fluorescence due to reduced GlyR β-loop binding.
Evolution of Geph-E functionality
As discussed earlier, gephyrin contains the G- and E-domains, which are homologous to the prokaryotic MogA and MoeA proteins, respectively. MogA and MoeA are involved in Moco biosynthesis in bacteria; however, in eukaryotes they always exist as fusion proteins such as gephyrin in vertebrates, cinnamon in the fruit fly and Cnx1 in plants. Compared to the homologs in nonvertebrate eukaryotes and prokaryotes, gephyrin in vertebrates has the additional function of anchoring inhibitory neurotransmitter receptors, such as GlyRs. As demonstrated here and elsewhere (Schrader et al, 2004; Sola et al, 2004), clustering of GlyRs via gephyrin is mediated via a direct interaction between Geph-E and GlyR β-loop. To delineate differences between the gephyrin homologs with dual function and those only involved in molybdenum cofactor biosynthesis, multiple sequence alignments of the corresponding E-domains were performed and the important residues for GlyR β-loop binding based on structural and mutational studies were highlighted (Figure 6). As expected, the alignment reveals a high sequence similarity throughout all species involving residues either related to the stabilization of the three-dimensional structure or the molybdenum cofactor biosynthetic function (Xiang et al, 2001). In addition, the alignment reveals that all residues identified in the crystal structure as being located in the Geph-E/GlyR β-loop binding interface are strictly conserved in vertebrates, but not in invertebrates. For example, the major contact residue Phe 330 is replaced with leucine in most cases, while the proline at position 714 is replaced with a variety of different residues, taking into account additional sequences not displayed in Figure 6. Tyr 673 is type-conserved with phenylalanine substitutions in all nonvertebrate sequences, suggesting that the aromatic phenyl ring is important for the structural integrity of the E-domain. However, since our binding studies have revealed that the Y673F variant showed a drastically impaired interaction with the GlyR β-loop, the phenolic OH-group is crucial for GlyR β-loop binding. Thus, only vertebrate gephyrins contain those residues that are required for the GlyR interaction, which is in good agreement with the fact that GlyRs have not been identified in invertebrates (Xue, 1998).
Figure 6.

Multiple sequence alignments of Geph-E homologs. Alignments were performed using the sequences of human, rat and chicken Geph-E, Xenopus laevis MGC83148, Drosophila melanogaster Cinnamon, Arabidopsis thaliana Cnx1, Caenorhabditis elegans Lin-46, and E. coli MoeA. Secondary structure elements of Geph-E are shown above the alignments, and amino acid numbers are listed preceding each sequence. Strictly conserved residues are labeled with white letters on red background, and type-conserved residues are colored red. Residues in contact with GlyR β-loop based on the cocrystal structure are shaded in yellow, and those, which are critical for interaction, are highlighted in green. Alignments were performed using ClustalW (http://www.ebi.ac.uk/clustalw) and were visualized with ESPript (http://espript.ibcp.fr).
Impact of GlyR β-loop interaction on gephyrin dimerization
Gephyrin displays a strong tendency to form higher-order oligomers in vitro (Schmitt et al, 1987) and in transfected cells (Kirsch et al, 1995). Based on the crystal structures of MogA/Geph-G and MoeA/Geph-E, which revealed trimer and dimers, respectively, a submembraneous hexagonal lattice of gephyrin was proposed (Kneussel and Betz, 2000; Xiang et al, 2001), which is supposed to facilitate the high-density accumulation of GlyRs in the postsynaptic membrane. However, the regulatory mechanism of this scaffolding process still remains to be deciphered. As revealed by the apo- and complex structures presented here, dimerization of Geph-E occurs independently of the GlyR β-loop interaction and no noticeable conformational change in Geph-E was observed upon peptide binding. Interestingly, the binding region for the GlyR β-loop is located at the Geph-E dimer interface, and is not confined to a single monomer. This may imply that binding of the GlyR β-loop influences dimerization of Geph-E by modulating the affinity between the Geph-E monomers. Specifically, on the basis of the crystal structure, one would predict that binding of the GlyR β-loop will promote dimerization of Geph-E. This observation suggests that dimerization of gephyrin is strengthened in the vicinity of the postsynaptic membrane, where the GlyR β-subunits are enriched, but it does not exclude the possibility that other factors such as phosphorylation could influence the stability of the hexagonal lattice.
Materials and methods
Crystallization and data collection
Cloning, site-directed mutagenesis, protein expression and purification are described in Supplementary Materials and methods. Irregular rod-shaped Geph-E and SeMet Geph-E crystals were obtained at 18°C by hanging drop vapor diffusion against a reservoir solution containing 0.1 M Tris–HCl, pH 7.5–8.5, 0.1–0.2 M NaAc and 25–30% PEG 4000 at protein concentrations of 1–2 mg/ml. Improved crystals were grown utilizing larger drop sizes and microseeding. For complex crystallization, Geph-E was incubated with GlyR β-loop at a molar ratio of 1:2 for 1 h. Very thin, plate-shaped crystals formed after 1–2 days after equilibrating against a reservoir solution containing 0–0.1 M Tris–HCl, pH 7.5, 0.1–0.2 M KSCN and 25–30% PEG 4000 at a protein concentration of 2 mg/ml. Crystals were transferred into their respective mother liquor containing 20–25% glycerol as cryoprotectant and flash cooled in liquid nitrogen. Diffraction data of Geph-E and the complex were collected at 100°K on beamline X26C at the National Synchrotron Light Source at Brookhaven National Laboratory at a wavelength of 1.1 Å on a Quantum IV ADSC CCD detector. Data from one SeMet-substituted Geph-E crystal were collected on beamline 19ID at the Advanced Photon Source at Argonne National Laboratory at λ=0.98021 Å and 0.96496 Å on a custom-built CCD detector.
Structure determination and refinement
All data were indexed, integrated and scaled with the HKL software (Otwinowski and Minor, 1997). Initial attempts to solve the structure of Geph-E by molecular replacement with a lower-resolution data set (3.3 Å) and the programs AMORE (Navaza, 1994) and MOLREP (Vagin and Teplyakov, 2000) utilizing truncated and full-length monomeric and dimeric forms of E. coli MoeA failed. On the other hand, neither SHELXD (Schneider and Sheldrick, 2002) nor ShakeNBake (Hauptman, 1997) were able to identify the positions of the anomalous scatterers in the MAD data set. At this point, the molecular replacement program PHASER (Storoni et al, 2004) became available and a search model consisting of domains I, III and IV or I and III could be located in the apo-structure. Using phases derived from the molecular replacement model, the positions of the anomalous scatterers could be identified. Phase refinement with MLPHARE (Otwinowski, 1991), followed by twofold averaging in DM (Cowtan and Zhang, 1999), led to a considerable improvement of the electron density maps and the MoeA model was converted into the Geph-E structure. During the model-building stage, the structure of Geph-E by Sola et al (2004) became available and was used to speed up the model-building process. The structure was initially refined at 3.3 Å resolution using CNS (Brunger et al, 1998) and REFMAC5 (Murshudov, 1997). After the 2.7 Å resolution data set became available, refinement was completed with REFMAC5, including TLS refinement in which each Geph-E monomer was divided into four TLS bodies according to its domain architecture.
The structure of the Geph-E–GlyRβ-loop complex was solved by molecular replacement with MOLREP utilizing a Geph-E monomer at a resolution of 4 Å. Clear density corresponding to at least 10 residues of the GlyR β-loop was already visible in the first electron density maps and assignment of the sequence was straightforward. The structure was refined at 2.4 Å resolution using REFMAC5, including refinement of TLS bodies as described for the apo-structure. All model building was carried out with the program O (Jones et al, 1991). Solvent molecules were added initially with ARP (Perrakis et al, 1999) and additional sites were identified manually. Secondary structure elements were assigned with PROMOTIF (Hutchinson and Thornton, 1996); the stereochemistry was analyzed with PROCHECK (Laskowski et al, 1993). Figures 1 and 2 were generated with PYMOL (DeLano Scientific, San Carlos, CA).
In vitro binding assays
For pulldown assays, either WT GlyR β-loop-intein fusion or variants with substitutions in the β-loop were immobilized on chitin beads. In all, 10–15 μl of chitin beads were incubated with 30 μM of purified Geph-E mutants or 20 μM of P2 mutants in a total volume of 30 μl of binding buffer (10 mM Tris–HCl, 250 mM NaCl, pH 8.0) at 4°C for 1 h. WT Geph-E and P2 were also applied to the assays as controls. The supernatant was collected after centrifugation at 1250 g for 5 min and beads were further washed three times with 1 ml of binding buffer, followed by SDS–PAGE analysis.
Based on the pulldown assays, selected mutants were further investigated by ITC. WT and variants of purified Geph-E (with the exception of P2 WT and Y673F) and GlyR β-loop were extensively dialyzed against identical buffer (10 mM Tris–HCl, 250 mM NaCl, 1 mM β-mercaptoethanol, pH 8.0) at 4°C, followed by filtration and degassing. In all, 300–400 μM of WT and mutant GlyR β-loops were titrated as the ligand into the sample cell containing 20–25 μM of Geph-E mutants and WT, respectively. A volume of 5 μl of ligand was added at a time with a total number of 50–55 injections, resulting in a final molar ratio of ligand to protein varying between 3:1 and 4:1. All experiments were performed using a VP-ITC instrument (MicroCal, Northampton, MA) at 25°C. Buffer-to-buffer titrations were performed as described above, so that the heat produced by injection, mixing and dilution could be subtracted prior to curve fitting. The binding enthalpy was directly measured, while the association constants (Ka) and stoichiometries (N) were obtained by data analysis using the ORIGIN software.
In vivo colocalization assays
HEK 293 cells were seeded on collagen-coated coverslips at 3 × 104 cells/3.5 cm2 dishes and grown in Dulbecco's modified Eagle medium (PAA Laboratories) containing 10% fetal calf serum at 37°C and 10% CO2. The cells were transfected 1 day after plating with FuGENE 6 transfection reagent (Roche) according to the manufacturer's instructions, using 1.5 μl Fugene and 0.5 μg DNA for single transfections with the constructs pEGFP-P2, pDsRed-GlyRβL, pEGFP-P2-F330A, pEGFP-P2-P713E, pDsRed-GlyRβL-F398A and pDsRed-GlyRβL-FIF. In cotransfection experiments, 0.5 μg pEGFP derivatives and 0.25 μg pDsRed derivatives were used. After incubation for 48 h, the transfected cells were washed with PBS and fixed for 20 min in 4% (w/v) paraformaldehyde in PBS. Coverslips with the adhered cells were washed again with PBS and water, and subsequently embedded on glass slides. Confocal laser-scanning microscopy was performed using a LSM 510 META (Carl Zeiss). Images were taken in the multi-track mode by using a laser excitation at 488 nm (GFP) and 543 nm (DsRed).
Supplementary Material
Supplementary Information
Acknowledgments
We thank A Chakraborty for initial help with crystallization, and Drs S Xiang and J Truglio (SUNY Stony Brook) for assistance during data collection, Dr S Illenberger and T Messerschmidt (TU Braunschweig) for help with cell culture experiments and Dr R Haensch (TU Braunschweig) for assistance with the confocal microscope. This work was supported by Deutsche Forschungsgemeinschaft Grant Schw759/2–4 to GS and National Institutes of Health Grant NS48605 to HS. Use of the National Synchrotron Light Source in Brookhaven National Laboratory and the Advanced Photon Source was supported by DOE and beamline X26C is supported in part by the State University of New York at Stony Brook and its Research Foundation. Coordinates of apo GephE (pdb entry 2FU3) and the complex (pdb entry 2FTS) have been deposited in the Protein Data Bank.
References
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL (1998) Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D 54: 905–921 [DOI] [PubMed] [Google Scholar]
- Cowtan KD, Zhang KYJ (1999) Density modification for macromolecular phase improvement. Prog Biophys Mol Biol 72: 245–270 [DOI] [PubMed] [Google Scholar]
- Feng G, Tintrup H, Kirsch J, Nichol MC, Kuhse J, Betz H, Sanes JR (1998) Dual requirement for gephyrin in glycine receptor clustering and molybdoenzyme activity. Science 282: 1321–1324 [DOI] [PubMed] [Google Scholar]
- Fuhrmann JC, Kins S, Rostaing P, El Far O, Kirsch J, Sheng M, Triller A, Betz H, Kneussel M (2002) Gephyrin interacts with Dynein light chains 1 and 2, components of motor protein complexes. J Neurosci 22: 5393–5402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giesemann T, Schwarz G, Nawrotzki R, Berhorster K, Rothkegel M, Schluter K, Schrader N, Schindelin H, Mendel RR, Kirsch J, Jockusch BM (2003) Complex formation between the postsynaptic scaffolding protein gephyrin, profilin, and Mena: a possible link to the microfilament system. J Neurosci 23: 8330–8339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grudzinska J, Schemm R, Haeger S, Nicke A, Schmalzing G, Betz H, Laube B (2005) The beta subunit determines the ligand binding properties of synaptic glycine receptors. Neuron 45: 727–739 [DOI] [PubMed] [Google Scholar]
- Hauptman HA (1997) Shake-and-bake: An algorithm for automatic solution ab initio of crystal structures. Methods in Enzymology 277: 3–13 [DOI] [PubMed] [Google Scholar]
- Hutchinson EG, Thornton JM (1996) PROMOTIF—a program to identify and analyze structural motifs in proteins. Protein Sci 5: 212–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones TA, Zou JY, Cowan SW, Kjeldgaard M (1991) Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A 47: 110–119 [DOI] [PubMed] [Google Scholar]
- Kins S, Betz H, Kirsch J (2000) Collybistin, a newly identified brain-specific GEF, induces submembrane clustering of gephyrin. Nat Neurosci 3: 22–29 [DOI] [PubMed] [Google Scholar]
- Kirsch J, Kuhse J, Betz H (1995) Targeting of glycine receptor subunits to gephyrin-rich domains in transfected human embryonic kidney cells. Mol Cell Neurosci 6: 450–461 [DOI] [PubMed] [Google Scholar]
- Kirsch J, Langosch D, Prior P, Littauer UZ, Schmitt B, Betz H (1991) The 93-kDa glycine receptor-associated protein binds to tubulin. J Biol Chem 266: 22242–22245 [PubMed] [Google Scholar]
- Kirsch J, Wolters I, Triller A, Betz H (1993) Gephyrin antisense oligonucleotides prevent glycine receptor clustering in spinal neurons. Nature 366: 745–748 [DOI] [PubMed] [Google Scholar]
- Kneussel M, Betz H (2000) Clustering of inhibitory neurotransmitter receptors at developing postsynaptic sites: the membrane activation model. Trends Neurosci 23: 429–435 [DOI] [PubMed] [Google Scholar]
- Kneussel M, Haverkamp S, Fuhrmann JC, Wang H, Wassle H, Olsen RW, Betz H (2000) The gamma-aminobutyric acid type A receptor (GABAAR)-associated protein GABARAP interacts with gephyrin but is not involved in receptor anchoring at the synapse. Proc Natl Acad Sci USA 97: 8594–8599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kneussel M, Hermann A, Kirsch J, Betz H (1999) Hydrophobic interactions mediate binding of the glycine receptor beta-subunit to gephyrin. J Neurochem 72: 1323–1326 [DOI] [PubMed] [Google Scholar]
- Laskowski RA, McArthur MW, Moss DS, Thornton JM (1993) Procheck: a program to check the stereochemical quality of protein structures. J Appl Crystallogr 26: 283–291 [Google Scholar]
- Legendre P (2001) The glycinergic inhibitory synapse. Cell Mol Life Sci 58: 760–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu MT, Wuebbens MM, Rajagopalan KV, Schindelin H (2000) Crystal structure of the gephyrin-related molybdenum cofactor biosynthesis protein MogA from Escherichia coli. J Biol Chem 275: 1814–1822 [DOI] [PubMed] [Google Scholar]
- Lynch JW (2004) Molecular structure and function of the glycine receptor chloride channel. Physiol Rev 84: 1051–1095 [DOI] [PubMed] [Google Scholar]
- Meier J, De Chaldee M, Triller A, Vannier C (2000) Functional heterogeneity of gephyrins. Mol Cell Neurosci 16: 566–577 [DOI] [PubMed] [Google Scholar]
- Moss SJ, Smart TG (2001) Constructing inhibitory synapses. Nat Rev Neurosci 2: 240–250 [DOI] [PubMed] [Google Scholar]
- Murshudov GN (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D 53: 240–255 [DOI] [PubMed] [Google Scholar]
- Navaza J (1994) AMoRe: an automated package for molecular replacement. Acta Crystallogr A 50: 157–163 [Google Scholar]
- Otwinowski Z (1991) Isomorphous replacement and anomalous scattering. In CCP4 Study Weekend Isomorphous Replacement and Anomalous Scattering, Wolf W, Evans, P, Leslie A (eds), pp 80–86. Warrington, UK: SERC Daresbury Laboratory [Google Scholar]
- Otwinowski Z, Minor W (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276: 307–326 [DOI] [PubMed] [Google Scholar]
- Perrakis A, Morris R, Lamzin VS (1999) Automated protein model building combined with iterative structure refinement. Nat Struct Biol 6: 458–463 [DOI] [PubMed] [Google Scholar]
- Pfeiffer F, Graham D, Betz H (1982) Purification by affinity chromatography of the glycine receptor of rat spinal cord. J Biol Chem 257: 9389–9393 [PubMed] [Google Scholar]
- Prior P, Schmitt B, Grenningloh G, Pribilla I, Multhaup G, Beyreuther K, Maulet Y, Werner P, Langosch D, Kirsch J, Betz H (1992) Primary structure and alternative splice variants of gephyrin, a putative glycine receptor–tubulin linker protein. Neuron 8: 1161–1170 [DOI] [PubMed] [Google Scholar]
- Rees MI, Harvey K, Ward H, White JH, Evans L, Duguid IC, Hsu CC, Coleman SL, Miller J, Baer K, Waldvogel HJ, Gibbon F, Smart TG, Owen MJ, Harvey RJ, Snell RG (2003) Isoform heterogeneity of the human gephyrin gene (GPHN), binding domains to the glycine receptor, and mutation analysis in hyperekplexia. J Biol Chem 278: 24688–24696 [DOI] [PubMed] [Google Scholar]
- Reiss J, Gross-Hardt S, Christensen E, Schmidt P, Mendel RR, Schwarz G (2001) A mutation in the gene for the neurotransmitter receptor-clustering protein gephyrin causes a novel form of molybdenum cofactor deficiency. Am J Hum Genet 68: 208–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatini DM, Barrow RK, Blackshaw S, Burnett PE, Lai MM, Field ME, Bahr BA, Kirsch J, Betz H, Snyder SH (1999) Interaction of RAFT1 with gephyrin required for rapamycin-sensitive signaling. Science 284: 1161–1164 [DOI] [PubMed] [Google Scholar]
- Schmitt B, Knaus P, Becker CM, Betz H (1987) The Mr 93 000 polypeptide of the postsynaptic glycine receptor complex is a peripheral membrane protein. Biochemistry 26: 805–811 [DOI] [PubMed] [Google Scholar]
- Schneider TR, Sheldrick GM (2002) Substructure solution with SHELXD. Acta Crystallogr D 58: 1772–1779 [DOI] [PubMed] [Google Scholar]
- Schrader N, Kim EY, Winking J, Paulukat J, Schindelin H, Schwarz G (2004) Biochemical characterization of the high affinity binding between the glycine receptor and gephyrin. J Biol Chem 279: 18733–18741 [DOI] [PubMed] [Google Scholar]
- Schwarz G, Schrader N, Mendel RR, Hecht HJ, Schindelin H (2001) Crystal structures of human gephyrin and plant Cnx1 G domains: comparative analysis and functional implications. J Mol Biol 312: 405–418 [DOI] [PubMed] [Google Scholar]
- Sheng M, Pak DT (2000) Ligand-gated ion channel interactions with cytoskeletal and signaling proteins. Annu Rev Physiol 62: 755–778 [DOI] [PubMed] [Google Scholar]
- Sola M, Bavro VN, Timmins J, Franz T, Ricard-Blum S, Schoehn G, Ruigrok RW, Paarmann I, Saiyed T, O'Sullivan GA, Schmitt B, Betz H, Weissenhorn W (2004) Structural basis of dynamic glycine receptor clustering by gephyrin. EMBO J 23: 2510–2519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stallmeyer B, Schwarz G, Schulze J, Nerlich A, Reiss J, Kirsch J, Mendel RR (1999) The neurotransmitter receptor-anchoring protein gephyrin reconstitutes molybdenum cofactor biosynthesis in bacteria, plants, and mammalian cells. Proc Natl Acad Sci USA 96: 1333–1338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storoni LC, McCoy AJ, Read RJ (2004) Likelihood-enhanced fast rotation functions. Acta Crystallogr D 60: 432–438 [DOI] [PubMed] [Google Scholar]
- Vagin A, Teplyakov A (2000) An approach to multi-copy search in molecular replacement. Acta Crystallogr D 56: 1622–1624 [DOI] [PubMed] [Google Scholar]
- Xiang S, Nichols J, Rajagopalan KV, Schindelin H (2001) The crystal structure of Escherichia coli MoeA and its relationship to the multifunctional protein gephyrin. Structure (Cambridge) 9: 299–310 [DOI] [PubMed] [Google Scholar]
- Xue H (1998) Identification of major phylogenetic branches of inhibitory ligand-gated channel receptors. J Mol Evol 47: 323–333 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information