

Abstract
Objective
To analyze the involvement of connective tissue growth factor (CTGF) in the transforming growth factor-β (TGF-β) pathway during acute necrotizing pancreatitis (ANP) in humans and rats.
Summary Background Data
Connective tissue growth factor is involved in several fibrotic diseases and has a critical role in fibrogenesis and tissue remodeling after injury.
Methods
Normal human pancreas tissue samples were obtained through an organ donor program from five individuals without a history of pancreatic disease. Human ANP tissues were obtained from eight persons undergoing surgery for this disease. In rats, ANP was induced by intraductal infusion of taurocholate. The expression of CTGF was studied by Northern blot analysis, in situ hybridization, and immunohistochemistry in both human and rat pancreatic tissue samples.
Results
Northern blot analysis revealed enhanced CTGF mRNA expression in human ANP tissue samples compared with normal controls. In addition, a concomitant increase in TGF-β1 was present. By in situ hybridization, CTGF mRNA was localized in the remaining acinar and ductal cells and in fibroblasts. In regions of intense damage adjacent to areas of necrosis, CTGF mRNA signals were most intense. Inflammatory cells were devoid of any CTGF mRNA signals. By immunohistochemistry, CTGF protein was localized at high levels in the same cell types as CTGF mRNA. In ANP in rats, concomitantly enhanced mRNA levels of CTGF, TGF-β1, and collagen type 1 were present, with a biphasic peak pattern on days 2 to 3 and day 7 after induction of ANP.
Conclusions
These data indicate that CTGF participates in tissue remodeling in ANP. The expression of CTGF predominantly in the remaining acinar and ductal cells indicates that extracellular matrix synthesis after necrosis is at least partly regulated by the remaining pancreatic parenchyma and only to a minor extent by inflammatory cells. Blockage of CTGF, a downstream mediator of TGF-β in fibrogenesis, might be useful as a target to influence and reduce fibrogenesis in this disorder.
Acute pancreatitis is an episodic inflammation of the pancreas that can develop as a mild form (edematous pancreatitis) or as a severe form (necrotizing pancreatitis). Inflamed pancreatic tissue, surviving the primary damage, can heal with a “restitutio ad integrum” after edematous pancreatitis. In contrast, in the case of necrotizing pancreatitis, recovery is often associated with fibrosis and scarring. Patients with necrotizing pancreatitis have in general a more severe clinical course, and intensive care treatment and surgery are often required. 1
The reparative process after acute inflammation of the pancreas is characterized by cell proliferation as well as synthesis and transient deposition of extracellular matrix. 1 In fact, after acute necrotizing pancreatitis (ANP), the necrotic areas are sealed off by granulation tissue, which mainly consists of collagen fibers. In addition, a coordinated release of inflammatory mediators and growth factors by activated platelets and endothelial cells is postulated to contribute to mesenchymal cell recruitment and proliferation. Among these early cellular products, platelet-derived growth factor (PDGF), fibroblast growth factor (FGF), and transforming growth factor-beta (TGF-β) are the major candidates that initiate and afterward support fibroblast proliferation and chemotactic activity, resulting in the replacement of necrosis and formation of a scar. 2–5
A previous study in human ANP tissues reported that TGF-β and its signaling receptors are overexpressed in a concomitant fashion with collagen type 1 mRNA in the remaining parenchyma, suggesting that these growth factors play a critical role in pancreatic tissue remodeling and in the fibrotic repair of the necrotic areas. 5 In addition, similar results were reported in rat acute edematous pancreatitis, where TGF-β upregulation has been described. 6–9 Expression levels of TGF-β mRNA were biphasically increased, with an initial early peak probably related to the acute pancreatic damage and inflammatory cell infiltration, and a second peak probably related to the intense extracellular matrix synthesis and tissue repair. 6–9
A recent report studying concomitant overexpression of connective tissue growth factor (CTGF), a novel peptide that exhibits PDGF-like chemotactic and mitogenic activities for mesenchymal cells, and TGF-β1 and collagen type 1 in patients with chronic pancreatitis showed that CTGF may play a central role in fibrogenesis during chronic pancreatic damage. 10 In addition, TGF-β1 is at present the only known inducer of CTGF in human tissue, and several studies have confirmed that CTGF is a downstream component of the TGF-β signaling cascade that stimulates extracellular matrix synthesis in several fibrotic disorders. 11–13 However, TGF-β1 is a multifunctional peptide, and its expression in the pancreas has been found in different cell types, such as islet cells, inflammatory cells, and acinar cells. In agreement with their multifunctional capacity, different mediators for specific biologic actions are induced by TGF-β. 14 Connective tissue growth factor represents a more selective peptide that plays a unique role in fibrogenesis in various disorders, and this peculiarity makes CTGF a potential target for new therapy that might regulate and finally modulate fibrogenesis in various human disorders. 10,15,16
In view of these observations, in the present study we analyzed CTGF in human ANP and in a rat model of ANP to further evaluate its potential involvement in fibrogenesis in these disorders.
METHODS
Patients and Tissue Collection
Normal pancreatic tissue samples were obtained from five patients (four women, one man; mean age 35 years [range 27–47]) through an organ donor program in which other organs were taken for organ transplantation. Necrotizing pancreatitis tissues were collected from eight individuals (five women, three men; mean age 51 years [range 37–60]) undergoing surgery for ANP. The mean Ranson score of the patients with ANP was 4.6 (range 3–6). Pancreatic surgery was performed a median of 12.8 days (range 2–35) after the onset of the acute attack. The etiology of ANP was gallstones in four patients, complication of endoscopic retrograde cholangiopancreatography in one patient, and hyperlipemia in one patient. In two patients no specific etiology could be established (Table 1). Histopathologic analysis revealed no morphologic differences between the different etiologies.
Table 1. CLINICAL DATA OF PATIENTS WITH ACUTE NECROTIZING PANCREATITIS

CTGF, connective tissue growth factor; ERCP, endoscopic retrograde cholangio-pancreatography; TGF-β1, transforming growth factor β1.
Pancreatic tissue samples of patients with ANP were taken at the border between necrotic and nonnecrotic regions. Immediately on surgical removal, normal pancreas and ANP tissue samples were fixed in paraformaldehyde for 12 to 24 hours and were paraffin-embedded for histologic analysis. Simultaneously, a randomly selected part of the removed tissue sample that was destined for RNA extraction was snap-frozen in the operating room in liquid nitrogen and maintained at −80°C until use.
This study was approved by the Human Subjects Committee of the University of Bern.
Rats
Approval for the experiments was obtained from the Animal Research Committee of the Canton Bern, Switzerland. Sixty-six male Wistar rats weighing 240 to 280 g were kept in single cages exposed to a light/dark cycle with free access to chow and water. After an overnight fast, the animals destined for pancreatitis induction or sham treatment were preanesthetized with ether, followed by intramuscular injection of phenobarbital (Nembutal sodium solution, Abbott Laboratories, Chicago, IL) at a dosage of 2 mg/kg and buprenorphine (Temgesic, Essex Chemie, Luzern, Switzerland) at a dosage of 6 μg/kg to induce anesthesia and analgesia, respectively. To induce ANP, the biliopancreatic duct was cannulated transmurally with a 27-gauge needle after a midline laparotomy, as reported previously. 6 The bile duct was clamped with a bulldog clamp proximal to the pancreas at the hilum of the liver and 2% taurocholate solution (sodium taurocholate, Sigma, St. Louis, MO) was infused under constant pressure (30 cm H20) at the rate of 0.1 mL/100 g body weight, as described previously. 6 After the administration of taurocholate, the bile duct was unclamped and the needle removed. The abdominal wall was closed in two running layers. Pancreatic tissue samples (n = 6) were obtained from untreated rats serving as the control group. Sham group animals (n = 6) received intraductal infusion of NaCl 0.9% instead of 2% sodium taurocholate. Animals with sodium taurocholate infusion (six per time point) were killed 4 hours, 8 hours, 1, 2, 3, 4, 5, 6, or 7 days after the end of the taurocholate infusion and the whole pancreatic gland was removed. Tissue samples were taken as in humans at the border between necrotic and nonnecrotic pancreatic regions and randomly fixed for histology or frozen in liquid nitrogen.
Probe Synthesis
The CTGF probe consisted of a 600-bp EcoRI/PstI fragment of human CTGF cDNA (kindly provided by Dr. Barry S. Oemar, Cardiovascular Research Laboratory, Institute of Physiology, University of Zürich, Switzerland). 13 TGF-β1 cDNA, TGF-β2 cDNA, and TGF-β3 cRNA probes were derived from rat clones and have been described previously. 5,6,10 In addition, a human TGF-β1 cDNA probe was used for the Northern blot experiments. 5 The collagen cDNA probe consisted of a 1.8-kb EcoRI fragment of human fibroblast type 1 collagen cDNA (ATCC, Rockville, MD). 10 The amylase cDNA probe consisted of a 545-bp EcoRI/Sau3AI fragment of human amylase cDNA (ATCC). 10 A 190-bp BamHI/BamHI fragment of mouse 7S cDNA was used to verify equivalent RNA loading in the Northern blot experiments. 17–19 For Northern blot analysis, the cDNA probes (CTGF, TGF-β1, TGF-β2, collagen type 1, amylase, and 7S) and the cRNA probe (TGF-β3) were radiolabeled with [α-32P]dCTP and [α-32P]CTP, respectively, as previously reported. 17–19 For in situ hybridization analysis, sense and antisense CTGF cRNA probes were labeled with digoxigenin, as previously reported. 10,17–19
Northern Blot Analysis
Total RNA was extracted by the guanidine isothiocyanate method, size-fractionated on 1.2% agarose/1.8 mol/L formaldehyde gels, and stained with ethidium bromide for verification of RNA integrity and loading equivalency, as previously reported. 10,17–21 The RNA was electrotransferred onto nylon membranes and cross-linked by UV irradiation. The filters were then prehybridized, hybridized, and washed under conditions appropriate for specific cDNA and cRNA probes, as previously described. 17–21
Membranes were exposed at −80°C to Fuji x-ray films with intensifying screens, and the intensity of the radiographic bands was quantified by a computerized video system and Image-Pro Plus 3.0 software (Media Cybernetics, Silver Spring, MD). All membranes were rehybridized with the 7S cDNA probe to assess equivalent RNA loading and transfer, as previously reported. 5,6,10,17–21 Data are expressed as the ratio of the CTGF mRNA signals divided by the corresponding 7S signals.
In Situ Hybridization
In situ hybridization was performed as previously reported. 10,17–21 Briefly, normal pancreas and human ANP tissue samples were fixed in paraformaldehyde and paraffin-embedded. For each sample, several consecutive tissue sections were analyzed. The tissue sections (4 μm) were deparaffinized, rehydrated with 1× phosphate-buffered saline (PBS) and incubated in 0.2 mol/L HCl for 20 minutes at room temperature. After the slides were rinsed in 2× sodium chloride/sodium citrate buffer (SSC), the sections were treated with proteinase K for 15 minutes at 37°C. After acetylation, postfixation with 4% paraformaldehyde in PBS, and washing in 2× SSC, the samples were prehybridized and hybridized overnight. After hybridization, excess probe was removed by washing in 2× SSC and by RNase treatment. After washing for 20 minutes in 2× SSC at 65°C and for 20 minutes in 0.2× SSC under the same stringent conditions, the tissue sections were incubated with an antidigoxigenin antibody conjugated with alkaline phosphatase (Roche Diagnostics, Rotkreuz, Switzerland). For color reaction, 5-bromo-4-chloro-3-indolyl phosphate and nitro blue tetrazolium were used.
Immunohistochemistry
Paraffin-embedded tissue sections (2–4 μm in thickness) were subjected to immunostaining using the Dako Envision + System (DAKO Diagnostics AG, Zürich, Switzerland). Tissue sections for each tissue sample were deparaffinized with xylene and rehydrated through graded alcohol into distilled water. The sections were microwaved in 10 mmol/L citrate buffer (pH 6) for 3 minutes on the high setting and for 15 minutes on the medium setting. Slides when then washed in Tris buffered saline (5 mmol/L Tris-HCI, 0.3 mol/L NaCI2, pH 7.4). Endogenous peroxidase activity was quenched by incubating the slides in 0.03% hydrogen peroxide and sodium azide, followed by washing in Tris buffered saline. The sections were then incubated overnight at 4°C with affinity-purified polyclonal anti-CTGF IgG (2.5 μg/mL) diluted in 0.05 mol/L Tris-HCl buffer containing 1% bovine serum albumin. These antibodies were raised in rabbits against residues 81 to 94 of hCTGF, which is a nonconserved domain among other CTGF-related molecules. The purified antibody has been previously validated for immunohistochemistry in other tissues. 22,23 Bound antibody was detected with a streptavidin-biotin-horseradish peroxidase (HRP) system (DAKO Diagnostics AG) in which slides were successively incubated with biotinylated antirabbit IgG, streptavidin-HRP, and 3-3′ diaminobenzidine (DAB). Slides were counterstained with Mayer’s hematoxylin. To ensure antibody specificity, control slides were incubated either in the absence of primary antibody or with a nonspecific IgG antibody. In neither case was immunostaining detected. All slides were analyzed by two independent observers unaware of patient status; any differences were resolved by joint review and consultation with a third observer.
Statistical Analysis
Results are expressed as mean and range. For statistical analysis the Mann-Whitney test was used. Significance was defined as P < .05.
RESULTS
Northern Blot Analysis
Humans
Northern blot analysis was initially performed to determine the expression levels of CTGF and TGF-β1 mRNA in both normal pancreas and human ANP tissue samples. Very low levels of CTGF mRNA (transcript size approximately 2.4 kb) and TGF-β1 mRNA (transcript size 2.4 kb) were observed in the normal pancreatic tissue samples (Fig. 1). In contrast, in all ANP tissue samples there was a marked increase in CTGF and TGF-β1 mRNA expression levels. Densitometric analysis of the Northern blots indicated that compared with the normal pancreas, ANP tissue samples had on average a 16-fold (P < .001) increase in the CTGF mRNA levels and an 11-fold (P < .05) increase in the TGF-β1 mRNA levels.

Figure 1. Northern blot analysis of connective tissue growth factor (CTGF) and transforming growth factor-β1 (TGF-β1) mRNA in normal human pancreas (lanes 1–4) and human acute necrotizing pancreatitis tissue samples (lanes 5–12). In the acute necrotizing pancreatitis samples, concomitantly enhanced CTGF and TGF-β1 mRNA expression levels were present compared with the normal controls. 7S RNA was used to assess equivalent RNA loading.
Rats
Low levels of CTGF, TGF-β1, TGF-β2, TGF-β3, and collagen type 1 mRNA were present in normal rat pancreas samples obtained from both the sham and the control groups (Fig. 2). In contrast, CTGF, TGF-β1, TGF-β2, TGF-β3, and collagen type 1 mRNA levels were markedly upregulated in the ANP rats. Both CTGF and TGF-β1 showed a coordinated biphasic expression pattern after taurocholate infusion. The level of CTGF mRNA was already upregulated in the pancreas 8 hours after pancreatitis induction. The highest CTGF and TGF-β1 mRNA expression levels were detectable between day 2 and day 3 and returned progressively to control values at days 4 to 6. A second increase in CTGF and TGF-β1 was observed at day 7. At day 2, densitometric analysis of the Northern blot signals revealed 20-fold and 8-fold increases in CTGF and TGF-β1 mRNA levels, respectively (P < .01). The difference in the expression levels for CTGF and TGF-β1 between normal and ANP again reached significance on day 7. Comparable results were found for TGF-β3 and collagen type 1 mRNA expression, with 4-fold and 3.8-fold increases at day 4 and day 7, respectively, for TGF-β3 (P < .01) and 9.7-fold and 11-fold increases, respectively, at day 3 and 7 (P < .01) for collagen type 1. The highest TGF-β2 mRNA levels were present at day 6, with a 4.4-fold increase (P < .01) compared with the corresponding control samples. As a marker of acinar cell damage, amylase gene expression was also evaluated in the rat pancreas. In contrast to CTGF, TGF-β1, TGF-β2, TGF-β3, and collagen type 1 mRNA expression, amylase mRNA expression levels were markedly decreased already 4 hours after pancreatitis induction, followed by a gradual return to control values. Densitometric analysis of the Northern blot signals revealed a 14-fold decrease in the amylase mRNA levels 4 hours after taurocholate infusion compared with the normal controls (P < .01).

Figure 2. Northern blot analysis of connective tissue growth factor (CTGF), transforming growth factor-β1 (TGF-β1), TGF-β2, TGF-β3, collagen type 1, and amylase mRNA gene expression in control normal pancreas (NP; lanes 1 and 2) compared with rats with acute necrotizing pancreatitis (lanes 3–11). Concomitant high values of CTGF, TGF-β1, and collagen type 1 mRNA expression were present on days 2 to 3 and again on day 7. Amylase mRNA expression showed initially marked reduction with a progressive recovery. 7S RNA was used to assess equivalent RNA loading.
In Situ Hybridization in Humans
In situ hybridization was performed to localize the exact sites of CTGF mRNA production in the normal and ANP tissue samples in humans.
In the normal pancreas, faint CTGF mRNA signals (Fig. 3) were found in some smooth muscle and endothelial cells of small and medium-sized arteries. Acinar cells and ductal cells in the normal pancreas exhibited no CTGF mRNA signals. In contrast, ANP samples showed strong CTGF mRNA in situ hybridization signals. Very intense CTGF mRNA signals were present primarily in the remaining acinar and ductal cells, especially in those parts adjacent to the necrotic areas and in ductal cells. In addition, fibroblasts localized in areas with a high degree of pancreatic damage and necrosis exhibited high CTGF mRNA expression. Inflammatory cells were devoid of any CTGF mRNA in situ hybridization signals. In situ hybridization experiments using the DIG-labeled sense probe corresponding to the antisense probe failed to produce any signal.
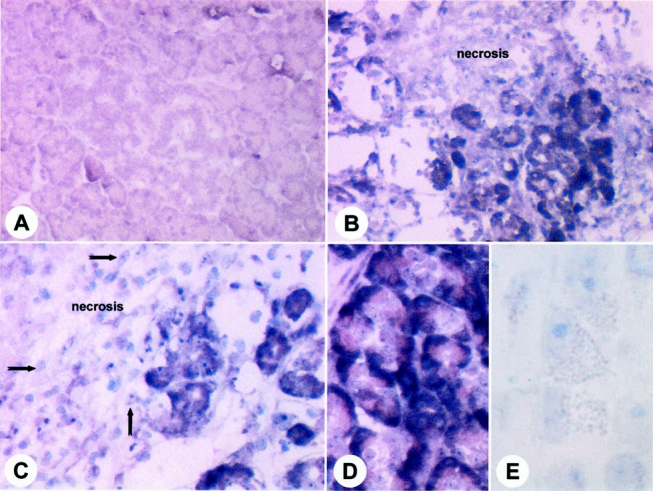
Figure 3. In situ hybridization of connective tissue growth factor (CTGF) mRNA expression in human tissue sections of normal pancreas (A) and acute necrotizing pancreatitis (B, C, D) samples. In acute necrotizing pancreatitis tissue sections, CTGF mRNA signals were mainly present in remaining acinar cells and in fibroblasts, especially in those areas adjacent to the necrosis (B, C). Inflammatory cells were devoid of CTGF mRNA signals (C, arrows). In situ hybridization experiments using the DIG-labeled sense probe corresponding to the antisense probe failed to produce hybridization signals (E). Original magnification ×200 (A–C); ×400 (D, E).
Immunohistochemistry in Humans
Immunohistochemistry was performed to localize CTGF protein in human ANP tissue samples (Fig. 4). In the normal pancreas, weak CTGF immunoreactivity was present in islet cells and in a few cells of small pancreatic ducts. Acinar cells were devoid of any CTGF immunoreactivity. Further, in normal pancreatic tissue samples, endothelial cells showed faint CTGF immunoreactivity. In contrast, ANP tissue sections exhibited, in general, intense CTGF immunoreactivity. Very intense CTGF immunoreactivity was present in the remaining acinar cells and in the ductal cells, and these findings were more prominent in the tissue areas adjacent to the necrosis. In addition, fibroblasts and endothelial cells exhibited moderate to strong CTGF immunoreactivity. In contrast, the inflammatory cells were completely devoid of CTGF immunoreactivity.

Figure 4. Immunohistochemical analysis of connective tissue growth factor (CTGF) in human tissue sections of normal pancreas (A) and acute necrotizing pancreatitis (B, C, D) samples. In acute necrotizing pancreatitis tissue sections, CTGF immunoreactivity was mainly present in the cells of the small ducts and in all remaining acinar cells, especially in those areas adjacent to the necrosis (B, C, D, arrows). i, islet; d = duct. Original magnification ×100 (A, B); ×200 (C); ×400 (D).
DISCUSSION
Pancreatic tissue regeneration after ANP is a central event in the natural history of this disease. Although it is well documented from autopsy studies that pancreatic necrosis is replaced by granulation tissue and subsequently by connective tissue, the molecular mechanisms that mediate these changes are not well understood, mostly because of the lack of adequate human tissue material from these patients. 5–7,24,25
In the present report we evaluated the potential role of CTGF during pancreatic repair after ANP in humans. In addition, to better understand the human findings, the time course of CTGF mRNA expression was studied in relation to TGF-β in a standardized rat model of ANP. We determined that in human ANP, CTGF mRNA is upregulated and its expression is closely associated with the expression of TGF-β1, as reported in other diseases where fibrogenesis is a dominant event. 10,11,13 In the animal model of ANP, both CTGF and TGF-β1 mRNA expression showed a biphasic peak pattern, with highest levels of expression occurring at day 2 after pancreatitis induction, followed by a reduction to the normal level at days 4 to 6 and a second increase at day 7. Similar results were obtained in the human samples, with all ANP tissue specimens showing strong overexpression of CTGF mRNA. In addition, the source of CTGF mRNA and protein in human ANP was identified mainly in the remaining ductal cells, in acinar cells, and in fibroblasts present in areas adjacent to the necrosis. Inflammatory cells did not exhibit CTGF mRNA signals.
Based on the present results, we may postulate that TGF-β and CTGF are both activated during pancreatic regeneration by the nonnecrotic remaining exocrine pancreatic parenchyma, and that tissue repair and remodeling in ANP is at least in part mediated by paracrine and autocrine release of CTGF. The intense CTGF expression in the areas adjacent to the necrosis, but not in inflammatory cells, suggests that the remaining exocrine pancreatic parenchyma itself regulates tissue repair and extracellular matrix deposition. Our results in human ANP are supported by the demonstrated coordinated gene expression of CTGF, TGF-β1, and collagen type 1 in the well-established taurocholate infusion model of ANP in rats. This animal model exhibits histologic damage comparable to that seen in human ANP. The biphasic peak pattern of CTGF, TGF-β1, and collagen type 1 in rat pancreatitis is suggestive of an ongoing up- and downregulation of this system after pancreatic damage has occurred. In the animal model of ANP, CTGF mRNA expression was upregulated already 8 hours after pancreatitis induction, whereas TGF-β mRNA upregulation was not yet evident. These observations indicate that CTGF is rapidly activated on the transcriptional level and are consistent with the immediate early gene aspect of CTGF induction by TGF-β.
Moreover, the present results are consistent with the hypothesis that the growth stimulatory effects of TGF-β on connective tissue cells are indirectly mediated by induction of autocrine growth factors such as PDGF-like peptides. 26 Our data strongly suggest that CTGF is the candidate or at least is a major mediator for TGF-β action. It has been proposed that an adequate balance between profibrotic peptides, such as CTGF and TGF-β, and fibrinolysis inducers is required for adequate tissue repair, with an equal replacement of damaged parenchyma and necrosis by extracellular matrix. 27 In fact, upregulation of the urokinase plasminogen activator (uPA) and its receptor, which activate proteolysis in the remaining parenchyma during human ANP, has been reported previously. 24 Therefore, activation of proteolytic factors in the remaining pancreatic parenchyma during the course of ANP in humans might create a milieu that enhances tissue lysis, thereby accelerating the removal of necrotic tissue. Urokinase plasminogen activator is a well-known activator of latent TGF-β. Therefore, the increased levels of TGF-β that occur in a coordinated matter with increased uPA expression might result from the enhanced catalytic conversion of its precursors by uPA. Activated TGF-β might then stimulate formation of extracellular matrix, granulation tissue, and fibrogenesis. TGF-β may also in turn induce plasminogen activator inhibitor 1, thereby downregulating this proteolytic system, which favors fibrogenesis by decreasing extracellular matrix turnover. 24
At present, modulation of CTGF levels or inhibition of its functions in vivo is not possible. The receptor to which CTGF binds and by which this molecule exerts its fibrosis-inducing effects has not been identified. Therefore, in vivo CTGF blocking studies—for example, in animals with pancreatitis—cannot be performed but would be of great interest. However, our data indicate that the taurocholate pancreatitis model would be useful to evaluate anti-CTGF effects because findings were similar to those made in humans.
In conclusion, our data show that expression of CTGF is induced in both human and rat ANP, indicating that this fibrosis-causing protein is derived from the remaining pancreatic parenchyma. Our findings suggest that pancreatic fibrosis after ANP is regulated by the remaining exocrine pancreatic parenchyma itself and only to a minor degree by inflammatory cells. Our data also support the critical role for TGF-β and CTGF in pancreatic repair and tissue remodeling after acute pancreatic damage. TGF-β may act through fibrinolytic factors, mainly presented by the uPA-dependent system, or by activation of profibrotic factors, including FGF, PDGF, and the more selective fibrogenesis stimulator CTGF. Taken together, these observations suggest that modalities that modulate or suppress excessive CTGF expression or action in vivo should be the aims of further studies to determine whether they may attenuate the extent of pancreatic fibrosis that occurs after ANP.
Footnotes
Correspondence: Helmut Friess, MD, Department of General Surgery, University of Heidelberg, 3M Nevenheimer Feld, 110 D-69120 Heidelberg, Germany.
E-mail: helmut_friess@med.uni-heidelberg.de
Accepted for publication April 5, 2001.
References
- 1.Ammann RW, Muellhaupt B. Progression of alcoholic acute to chronic pancreatitis. Gut 1994; 35: 552–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Menke A, Yamaguchi H, Giehl K, et al. Hepatocyte growth factor and fibroblast growth factor are over-expressed after cerulein-induced acute pancreatitis. Pancreas 1999; 18: 28–33. [DOI] [PubMed] [Google Scholar]
- 3.Ebert M, Yokoyama M, Ishiwata T, et al. Alteration of fibroblast growth factor and receptor expression after acute pancreatitis in humans. Pancreas 1999; 18: 240–246. [DOI] [PubMed] [Google Scholar]
- 4.Ludwig CU, Menke A, Adler G, et al. Fibroblasts stimulate acinar cell proliferation through IGF-I during regeneration from acute pancreatitis. Am J Physiol 1999; 276: G193–198. [DOI] [PubMed] [Google Scholar]
- 5.Friess H, Zhao L, Riesle E, et al. Enhanced expression of TGF-βs and their receptors in human acute pancreatitis. Ann Surg 1998; 227: 95–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Riesle E, Friess H, Wagner M, et al. Enhanced expression of TGF-βs following acute edematous pancreatitis in rats suggests a role in pancreatic repair. Gut 1997; 40: 73–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Müller-Pillasch F, Gress TM, Yamaguchi H, et al. The influence of transforming growth factor beta 1 on the expression of genes coding for matrix metalloproteinases and tissue inhibitors of metalloproteinases during regeneration from cerulein-induced pancreatitis. Pancreas 1997; 15: 168–175. [DOI] [PubMed] [Google Scholar]
- 8.Menke A, Yamaguchi H, Gress TM, et al. Extracellular matrix is reduced by inhibition of transforming growth factor beta 1 in pancreatitis in the rat. Gastroenterology 1997; 113: 295–303. [DOI] [PubMed] [Google Scholar]
- 9.Konturek PC, Dembinski A, Warzexha Z, et al. Comparison of epidermal growth factor and transforming growth factor-beta 1 expression in hormone-induced acute pancreatitis in rats. Digestion 1998; 59: 110–119. [DOI] [PubMed] [Google Scholar]
- 10.di Mola FF, Friess H, Martignoni ME, et al. Connective tissue growth factor is a regulator for fibrosis in human chronic pancreatitis. Ann Surg 1999; 230: 63–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Franzier K, Williams S, Kothapalli D, et al. Stimulation of fibroblast cell growth, matrix production, and granulation tissue formation by connective tissue growth factor. J Invest Dermatol 1996; 107: 404–411. [DOI] [PubMed] [Google Scholar]
- 12.Grotendorst GR, Okochi H, Hayashi N. A novel transforming growth factor β response element controls the expression of the connective tissue growth factor gene. Cell Growth Differ 1996; 7: 469–480. [PubMed] [Google Scholar]
- 13.Oemar BS, Werner A, Garnier J-M, et al. Human connective tissue growth factor is expressed in advanced atherosclerotic lesions. Circulation 1997; 95: 831–839. [DOI] [PubMed] [Google Scholar]
- 14.Yamanaka Y, Friess H, Büchler M, et al. Synthesis and expression of transforming growth factor β1, β2 and β3 in the endocrine and exocrine pancreas. Diabetes 1993; 42: 746–756. [DOI] [PubMed] [Google Scholar]
- 15.Igarashi A, Nashiro K, Kikuchi K, et al. Significant correlation between connective tissue growth factor gene expression and skin sclerosis from patients with systemic sclerosis. J Invest Dermatol 1995; 105: 280–284. [DOI] [PubMed] [Google Scholar]
- 16.Igarashi A, Okochi H, Bradham DM, et al. Regulation of connective tissue growth factor gene expression in human skin fibroblasts during wound repair. Mol Biol Cell 1993; 4: 637–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Friess H, Zhu ZW, di Mola FF, et al. Nerve growth factor and its high-affinity receptor in chronic pancreatitis. Ann Surg 1999; 230: 615–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Friess H, Lu Z, Andrén-Sandberg Å, et al. Moderate activation of the apoptosis inhibitor bcl-xL worsens the prognosis in pancreatic cancer. Ann Surg 1998; 228: 780–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhu Z, Friess H, di Mola FF, et al. Nerve growth factor expression correlates with perineural invasion and pain in human pancreatic cancer. J Clin Oncol 1999; 17: 2419–2428. [DOI] [PubMed] [Google Scholar]
- 20.Friess H, Guo XZ, Berberat P, et al. Reduced KAI1 expression in pancreatic cancer is associated with lymph node and distant metastases. Int J Cancer 1998; 79: 349–355. [DOI] [PubMed] [Google Scholar]
- 21.Friess H, Cantero D, Graber H, et al. Enhanced urokinase plasminogen activation in chronic pancreatitis suggests a role in its pathogenesis. Gastroenterology 1997; 113: 904–913. [DOI] [PubMed] [Google Scholar]
- 22.Brigstock DR, Steffen CL, Kim GY, et al. Purification and characterization of novel heparin-binding growth factors in uterine secretory fluids. Identification as heparin-regulated Mr 10,000 forms of connective tissue growth factor. J Biol Chem 1997; 272: 20275–20282. [DOI] [PubMed] [Google Scholar]
- 23.Steffen CL, Ball-Mirth DK, Harding PA, et al. Characterization of cell-associated and soluble forms of connective tissue growth factor (CTGF) produced by fibroblast cells in vitro. Growth Factors 1998; 15: 199–213. [DOI] [PubMed] [Google Scholar]
- 24.Friess H, Duarte R, Kleeff J, et al. The plasminogen activator/plasmin system is up-regulated after acute necrotizing pancreatitis in human beings. Surgery 1998; 124: 79–86. [PubMed] [Google Scholar]
- 25.Van Laethem JL, Robberecht P, Resibois A, et al. Transforming growth factor beta promotes development of fibrosis after repeated courses of acute pancreatitis in mice. Gastroenterology 1996; 110: 576–582. [DOI] [PubMed] [Google Scholar]
- 26.Battegay EJ, Raines EW, Seifert RA, et al. TGF-β induces bimodal proliferation of connective tissue cells via complex control of an autocrine PDGF loop. Cell 1990; 63: 515–524. [DOI] [PubMed] [Google Scholar]
- 27.Gress TM, Müller-Pillasch F, Lerch MM, et al. Balance of expression of genes coding for extracellular matrix proteins and extracellular matrix degrading proteases in chronic pancreatitis. Z Gastroenterol 1994; 32: 221–225. [PubMed] [Google Scholar]