

Abstract
Objective
To evaluate the effect of inhalation of aerosolized opsonized dead Escherichia coli on inflammatory pulmonary neutrophil (PMN) apoptosis, lung injury, and survival in a PMN-mediated lung injury model in vivo.
Summary Background Data
Neutrophils that have transmigrated into an inflammatory focus display increased functional capacity and delayed apoptosis, resulting in an increased capacity to injure normal host tissue. The authors have previously shown that E. coli induces PMN apoptosis in vitro.
Methods
Lung injury mediated by PMNs was established by aortic occlusion and reperfusion. Adult male Sprague-Dawley rats were randomized into four groups: sham ischemia–reperfusion (I/R) treated with intratracheal inhalation of aerosolized normal saline, I/R treated with aerosolized normal saline intratracheally, I/R treated with aerosolized opsonized dead E. coli intratracheally, and I/R treated with aerosolized opsonized dead E. coli and the caspase inhibitor zVAD-FMK intratracheally 5 minutes before reperfusion. Both systemic and bronchoalveolar lavage PMNs were isolated and apoptosis was quantified at 0, 6, 12, 18, and 24 hours. Lung injury parameters including wet/dry lung weight ratio, histology, myeloperoxidase activity, and protein content were also assessed. In addition, a survival study was performed, both in a prophylactic and in a therapeutic setting.
Results
Administration of aerosolized dead E. coli before the reperfusion injury induced pulmonary PMN apoptosis and reversed the delayed apoptosis evident in the I/R plus normal saline group. There was also a significant improvement in lung injury parameters as well as in survival, both prophylactically as well as therapeutically.
Conclusions
Directly modulating PMN cell death represents a novel mechanism for attenuating PMN-mediated lung injury and may ultimately benefit the outcome in patients with adult respiratory distress syndrome.
The process of programmed cell death, or apoptosis, is now known to play a major regulatory role in maintaining many biologic processes, not least of which is the inflammatory response. 1,2 Polymorphonuclear neutrophils (PMNs) are the most abundant circulating proinflammatory leukocytes and constitute the first line of defense against infectious agents or non-self substances that penetrate the body’s physical barriers. 3 Paradoxically, PMNs have a well-established potential to injure host tissues, and activated PMN-mediated endothelial cell damage has been implicated in the development of increased vascular permeability and the capillary leak syndrome during both adult respiratory distress syndrome (ARDS) and systemic inflammatory response syndrome (SIRS). 4 The human PMN is known to have a relatively short half-life in circulation, estimated to be 8 to 16 hours. This lifespan is short because circulating PMNs constitutively undergo apoptosis. For the normal resolution of an acute inflammatory reaction to occur, PMN apoptosis with subsequent ingestion by tissue macrophages is required, and this process plays a critical role in minimizing the autotoxic potential of this cell. 5 As PMNs undergo apoptosis, they lose cell surface adhesion molecules and their ability to secrete their intracellular granular contents. 6,7 PMNs that have left the circulation and transmigrated across the endothelial barrier into an inflammatory focus display both a delay in spontaneous apoptosis and an increased functional capacity. 8,9 A delay in the apoptotic program of activated PMNs results in the failure to terminate the acute inflammatory response, and this has been suggested as a precipitant of SIRS. 4 We have previously shown in an in vitro model that after the ingestion of opsonized Escherichia coli, PMNs undergo apoptosis in a dose-dependent manner, and this is mediated through the production of intracellular reactive oxygen species. 10 The fact that bacterial ingestion results in the death of the organism as well as the ingesting PMN is presumably a teleologic response aimed at minimizing host tissue injury and maximizing resolution of the inflammatory response. Inflammatory states not associated with an infectious etiology, however, appear to result in PMN transmigration and heightened activation, but delayed apoptosis. We hypothesized that the introduction of nonpathogenic organisms (i.e., dead E. coli) into a site of sterile inflammation would initiate PMN apoptosis, overriding inhibitory signals and ultimately resulting in resolution of the sterile inflammatory response.
METHODS
Ethical approval for all experiments involving animals was granted by the Department of Health and Children, according to the Cruelty to Animals Act, 1876.
Reagents
Neutrophil isolation medium (NIM-2) was obtained from Cardinal Associates (Santa Fe, NM). Sodium citrate, propidium iodide (PI), Triton X-100, EDTA, and Tris were purchased from Sigma Chemical Company (St. Louis, MO). Hanks balanced salt solution (HBSS), RPMI 1640, phosphate-buffered saline (PBS) without calcium and magnesium, fetal calf serum (FCS), penicillin, streptomycin sulfate, Fungizone, and glutamine were purchased from Gibco BRL (Paisley, Scotland). Opsonized dead E. coli (1.0 × 107/mL) and FITC-labeled opsonized dead E. coli (1.0 × 107/mL) were purchased from Orpegen (Heidelberg, Germany). The caspase inhibitor z-val-ala-asp (ome)-fluoromethylketone (zVAD-FMK) was purchased from Biomol (Plymouth, PA).
Rat Model of Acute Lung Injury
Adult male Sprague-Dawley rats weighing 250 to 400 g were obtained from the Biologic Services Unit, University College Cork, Ireland. PMN-mediated lung injury was established by infrarenal aortic occlusion for 30 minutes followed by reperfusion for 2 hours. Animals were randomized into one of four groups: sham ischemia–reperfusion (I/R) treated with intratracheal instillation of aerosolized normal saline, I/R treated with aerosolized normal saline intratracheally, I/R treated with aerosolized opsonized dead E. coli (1.0 × 107/mL), and I/R treated with aerosolized opsonized dead E. coli (1.0 × 107/mL) and zVAD-FMK at 10 μmol/kg body weight.
Animals were anesthetized using intraperitoneal thiopentone and maintained under anesthesia for the duration of the procedure using halothane inhalation. After anesthesia was induced, a 24-gauge intravenous cannula was inserted into the right external jugular vein for fluid and heparin administration. Core temperature was monitored for the duration of the procedure using a rectal temperature probe. Animals underwent a midline laparotomy and after systemic heparinization (400 units heparin per kg body weight), the infrarenal aorta was exposed and clamped using a microvascular clamp. In the control group, animals had their aorta exposed but not clamped. In the control and I/R plus normal saline groups, 1 mL of 0.9% saline was aerosolized into the trachea 5 minutes before aortic unclamping. In the third randomized group, 1 mL aerosolized opsonized dead E. coli (1.0 × 107/mL) was administered 5 minutes before aortic unclamping. In the fourth randomized group, 1 mL aerosolized opsonized dead E. coli and 1 mL aerosolized zVAD-FMK were administered 5 minutes before aortic unclamping. Aerosolization of the normal saline, the opsonized dead E. coli, and the zVAD-FMK was facilitated by the use of a miniature intratracheal aerosolizer (Penn-Century, Philadelphia, PA). After 2 hours of reperfusion, a midline sternotomy was performed and the left main bronchus clamped. Bronchoalveolar lavage (BAL) of the right lung was performed with 2 mL PBS containing 0.07 mol/L ethylene diamine tetra-acetic acid and repeated twice. The combined lavage of approximately 6 mL was centrifuged at 1,500 rpm for 20 minutes at 4°C, frozen at −20°C, and assessed subsequently for protein concentration.
Pulmonary Influx of Neutrophils
Myeloperoxidase is a heme-containing enzyme, specific to PMNs, and as such can be used as an indirect measure of tissue PMN infiltration. 11 The right ventricle was cannulated with a 25-gauge needle and the right pulmonary hilum was clamped. The pulmonary vasculature of the left lung was flushed with 50 mL PBS to clear the lung of intravascular PMNs. After weighing the left lung remnant (after removal of the lower lobe), the tissue was homogenized on ice in 10 mL 0.5% hexadecyltrimethyl ammonium bromide in 50 mmol/L potassium phosphate buffer at pH 6.0 frozen at −80°C for subsequent myeloperoxidase assessment. Myeloperoxidase activity was assessed as previously described. 11
Pulmonary Endothelial Permeability
The lower lobe of the left lung was removed and the wet/dry lung weight ratio was calculated after weighing the freshly harvested organ and heating it at 60°C in a gravity convection oven over a 72-hour period until the weight was constant. The supernatant of collected BAL fluid after centrifugation was assessed for protein content using a micro BCA protein assay reagent kit (Pierce, Rockford, IL).
Isolation of Rat Neutrophils
Rat whole blood obtained by cardiac puncture was collected in heparinized tubes. PMNs were purified using the isolation medium NIM-2 as previously described. 8 PMNs were washed with HBSS and resuspended in 2 mL erythrocyte lysing buffer for 5 minutes, then washed again. PMNs were counted and resuspended in complete RPMI medium at 1.0 × 106 cells/mL. PMNs transmigrated into the lungs after I/R were harvested by BAL with 6 mL PBS.
Neutrophil Apoptosis
Apoptosis of PMNs was quantified according to the percentage of cells with hypodiploid DNA by using the PI staining technique, as previously described. 10 Briefly, after centrifugation, PMNs (0.5 × 106 cells) in 17 × 100-mm polypropylene tubes (Falcon, Lincoln Park, NJ) were gently resuspended in 0.5 mL hypotonic fluorochrome solution (50 μg/mL PI, 3.4 mmol/L sodium citrate, 1 mmol/L Tris, 0.1 mmol/L EDTA, 0.1% Triton X-100) and incubated in the dark at 4°C for 2 hours before they were analyzed by a FACScan flow cytometer (Becton Dickinson). The forward and side scatter of PMN particles were simultaneously measured. The PI fluorescence of individual nuclei with an acquisition of fluorescence channel (FL) 2 was plotted against forward scatter, and the data were registered on a logarithmic scale. The minimum number of 5,000 events were collected and analyzed using Lysis II software. Apoptotic PMN nuclei were distinguished by their hypodiploid DNA content from the diploid DNA content of normal PMN nuclei. Cell debris was excluded from analysis by raising the forward threshold. All measurements were performed under the same instrument settings. PMN apoptosis was further confirmed by morphologic assessment using Wright’s Giemsa staining, as described by Martin et al. 12
Detection of Phagocytosis of E. coli
Neutrophils isolated from the BAL fluid of animals subjected to the I/R insult were incubated with heat-killed FITC-labeled E. coli at a ratio of 10:1 (E. coli:PMN) at 37°C in a shaking water bath for 45 minutes, as previously described. 10 PMNs were washed with cold PBS three times to remove nonphagocytosed bacteria. PMN phagocytosis of the opsonized dead E. coli was assessed flow cytometrically. PMN apoptosis after ingestion of E. coli was assessed by flow cytometry as mentioned above.
Histology
After the animals were killed, sections of the right upper lung lobe were placed in neutral buffered formalin. The sections were processed routinely using standard hematoxylin and eosin staining. These sections were evaluated for loss of normal lung architecture, edema, interstitial hemorrhage, and PMN infiltration.
Survival Study
To validate our in vitro results, we undertook a survival study. Briefly, animals were again randomized into one of the four groups: sham I/R treated with intratracheal instillation of aerosolized normal saline, I/R treated with aerosolized normal saline intratracheally, I/R treated with aerosolized opsonized dead E. coli, and I/R treated with aerosolized opsonized dead E. coli and zVAD-FMK 5 minutes before reperfusion. After 2 hours of reperfusion, animals were weaned off anesthesia and followed up for survival. In a separate set of experiments, the above experiments were repeated, but in this instance the aerosolized normal saline, E. coli, and E. coli + zVAD-FMK were administered to the animals 10 minutes after aortic unclamping. Once again, after 2 hours of reperfusion, animals were weaned off anesthesia and followed up for survival.
Statistical Analysis
All data are presented as the mean ± standard deviation of the mean. Statistical analysis was performed using analysis of variance. Differences were considered significant at P < .05.
RESULTS
Neutrophil Apoptosis
Time course studies of spontaneous apoptosis from PMNs isolated from the BAL of the animals were conducted using the technique described by Watson et al, 10 which assesses the percentage of cells with hypodiploid DNA. Flow cytometric analysis of PMN apoptosis was correlated with the rate of PMN apoptosis assessed by counting PMNs with morphologic changes characteristic of apoptosis (data not shown). A similar correlation has been observed in rat PMN apoptosis using the same flow cytometric technique and morphologic assessment of apoptosis. 13 The administration of aerosolized opsonized dead E. coli induced significant pulmonary PMN apoptosis in the BAL fluid at 6 and 18 hours compared with the group treated with intratracheal normal saline (Fig. 1). When the apoptosis inhibitor zVAD-FMK was administered along with the opsonized dead E. coli, the rate of pulmonary PMN apoptosis was similar to that in the I/R + normal saline group. No PMNs were retrieved from the lavage fluid of control animals, in keeping with previous studies. 14 Further, the number of PMNs retrieved from the BAL fluid of animals showed no significant intergroup difference.
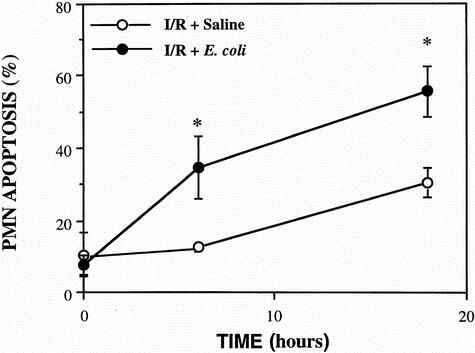
Figure 1. Spontaneous apoptosis in pulmonary neutrophils (PMNs) isolated from the bronchoalveolar lavage (BAL) fluid in the ischemia–reperfusion + saline, ischemia–reperfusion +Escherichia coli, and the zVAD-FMK and E. coli groups. PMNs isolated from the BAL fluid of all groups (n = 5) were cultured in complete RPMI 1640 medium at 37°C in 5% CO2 for 24 hours. PMN apoptosis was assessed at 0, 6, and 18 hours according to the percentage of cells with hypodiploid DNA by flow cytometry. Data are expressed as mean ± SD and are representative of five separate experiments. Significance was assessed with the I/R + saline group (*P < .05) and the zVAD-FMK and E. coli-treated group (†P < .05).
Time course studies of spontaneous apoptosis from PMNs isolated from whole venous blood were conducted using the technique described by Watson et al. 10 There were no significant differences between study groups, and the administration of aerosolized opsonized dead E. coli did not alter systemic PMN apoptosis (Fig. 2).
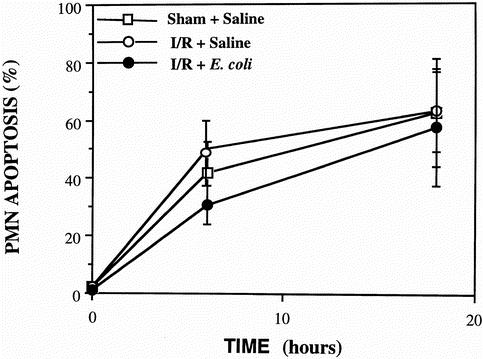
Figure 2. Spontaneous apoptosis in pulmonary neutrophils (PMN) isolated from venous blood in the sham + saline, ischemia–reperfusion + saline, and ischemia–reperfusion +Escherichia coli animals. PMNs isolated from whole blood (n = 5) were cultured in complete RPMI 1640 medium at 37°C in 5% CO2 for 24 hours. PMN apoptosis was assessed at 0, 6, and 18 hours according to the percentage of cells with hypodiploid DNA by flow cytometry. Data are expressed as mean ± SD and are representative of five separate experiments. There were no significant differences between groups.
Lung Injury Parameters
Pulmonary endothelial permeability, a marker of lung injury, can be assessed by the wet/dry lung weight ratio. Treatment with opsonized dead E. coli resulted in a significant reduction in the wet/dry lung weight ratio compared with the I/R + normal saline group (P < .03) (Fig. 3 A). In addition, the ratio was very similar in the E. coli and sham-operated groups (0.83 vs. 0.834), indicating a reversal in this ratio back to control levels in the dead E. coli group. Further, animals treated with both aerosolized zVAD-FMK and opsonized dead E. coli had a wet/dry lung weight ratio similar to that of the I/R + normal saline group.
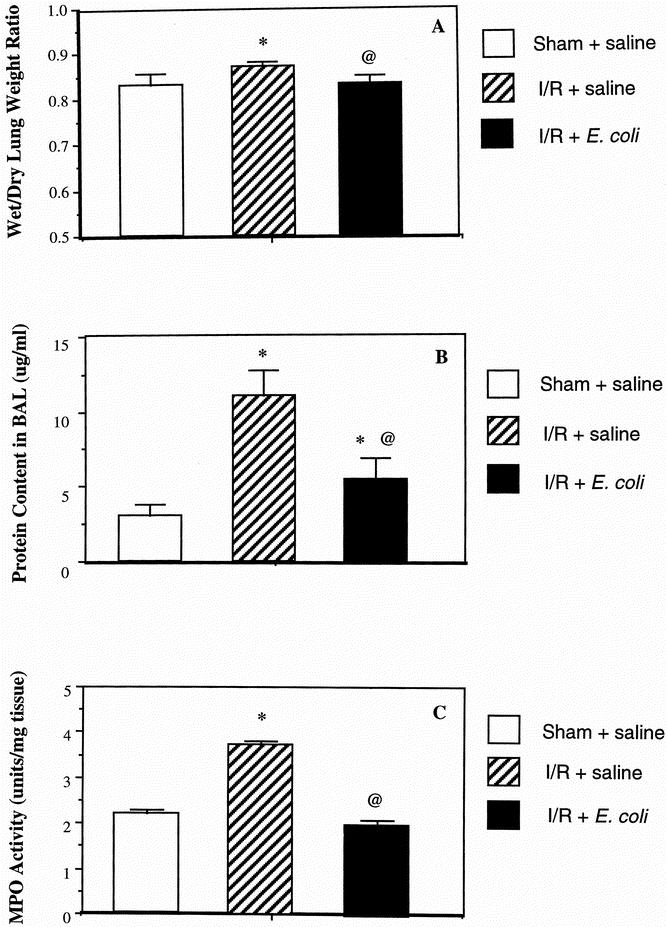
Figure 3. Improvement in lung injury parameters after treatment with aerosolized opsonized dead Escherichia coli. Wet/dry lung weight ratio (A) and bronchoalveolar lavage fluid protein content (B) were assessed as a measure of pulmonary endothelial permeability and therefore a marker of severity of lung injury. Myeloperoxidase activity (C) was also measured. Results are expressed as mean ± SD and are representative of five separate experiments. *P < .03 versus the sham + saline group; @P < .05 versus the ischemia–reperfusion + saline group; †P < .05 versus the ischemia–reperfusion + zVAD-FMK and E. coli groups.
Protein content in the BAL fluid is another measure of pulmonary endothelial permeability and as such is a marker for the severity of lung injury. There was a significant reduction in protein content in the E. coli-treated animals compared with the I/R plus normal saline group (P < .01), but it was still significantly greater than in the sham-operated group (P < .05) (see Fig. 3 B). Inhibiting apoptosis by the use of the caspase inhibitor zVAD-FMK resulted in a protein content not dissimilar to that in the I/R + normal saline animals.
As a measure of pulmonary PMN infiltration, myeloperoxidase was significantly decreased in the group treated with aerosolized opsonized dead E. coli compared with the I/R plus normal saline and the zVAD-FMK and opsonized dead E. coli groups (P < .05). Myeloperoxidase activity was similar in both the control and the E. coli-treated animals, indicating once again a reversal in lung injury seen in the I/R and saline group with the treatment of aerosolized dead E. coli (see Fig. 3 C).
Representative sections of lung tissue obtained from animals in the different groups and stained with standard hematoxylin and eosin revealed a marked loss of lung architecture, increased cellular infiltrate, marked hemorrhage, and edema in the I/R group treated with normal saline compared to the E. coli-treated group. In addition, animals treated with both aerosolized zVAD-FMK and opsonized dead E. coli had a histologic appearance very similar to that of the I/R + normal saline animals (Fig. 4).
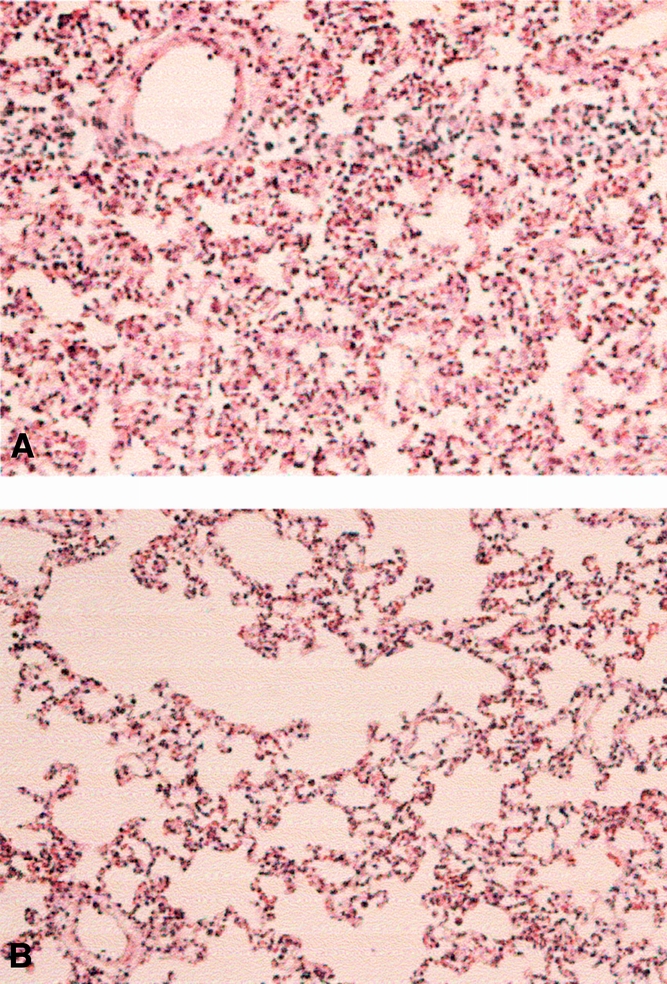
Figure 4. Histologic analysis of lung tissue from the ischemia–reperfusion (I/R) + saline, I/R +Escherichia coli, and I/R + zVAD-FMK and E. coli animals. Representative sections of lung tissue were obtained from the I/R + saline (A), I/R +E. coli (B), and zVAD-FMK and E. coli groups and stained with standard hematoxylin and eosin.
Neutrophil Ingestion of Opsonized dead E. coli
Figure 5 shows that the PMNs that have transmigrated into the lungs ingest the dead opsonized E. coli and subsequently undergo apoptosis.
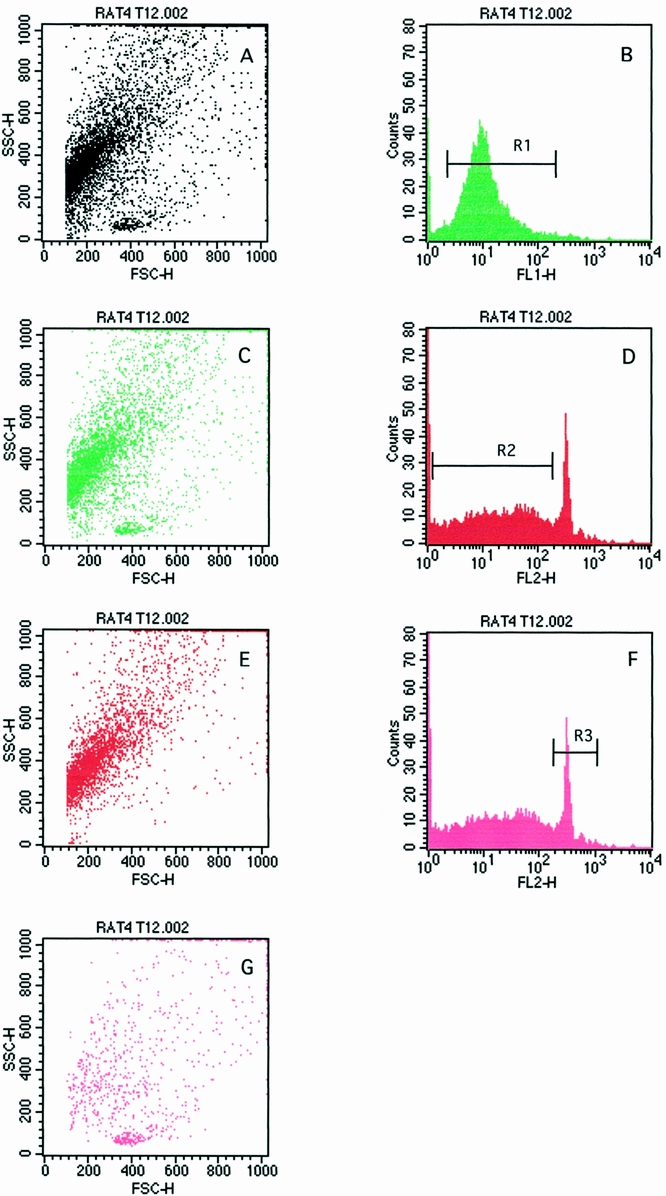
Figure 5. Pulmonary neutrophils (PMNs) undergo apoptosis after the ingestion of aerosolized opsonized dead Escherichia coli. (A1) Region 1 (R1) represents PMNs that have ingested FITC-labeled E. coli on an FL-1 histogram. (A2) Dot plot representation of PMN ingesting E. coli. (B1) Region 2 (R2) represents staining of apoptotic PMNs on an FL-2 histogram. (B2) Dot plot representation of apoptotic PMNs. (C1) Region 3 (R3) represents staining of normal PMNs on an FL-2 histogram. (C2) Normal PMN population on a dot plot.
Survival Study
To validate the efficacy of the treatment, a survival study was performed. As would be expected, there was a 100% survival rate in the sham-operated animals. This was reduced to 30% survival in the I/R plus normal saline group, but the survival rate was 80% in the dead E. coli-treated group, indicating a significant improvement in survival in the treated group (P < .05). Administering the caspase inhibitor zVAD-FMK along with the opsonized dead E. coli made no difference in survival compared with the I/R plus normal saline group (Fig. 6 A). One of the reasons why sepsis studies have failed over the years is that most of these studies have been prophylactic. We therefore conducted a second survival study to establish whether our treatment had a therapeutic benefit. In this instance, the aerosolized normal saline and opsonized dead E. coli as well as the caspase inhibitor were administered 10 minutes into the established reperfusion injury. Once again, as would be expected, there were no deaths in the sham-operated animals. This was once again reduced to 30% survival in the I/R plus normal saline group. Interestingly, after treatment with aerosolized opsonized dead E. coli 10 minutes into the established injury, there was a 70% survival rate, indicating a significant improvement in survival (P < .05), even in a therapeutic setting. Blocking the effect of the opsonized dead E. coli by the use of zVAD-FMK resulted in a significant decrease in survival compared with animals treated with opsonized dead E. coli alone (see Fig. 6 B).
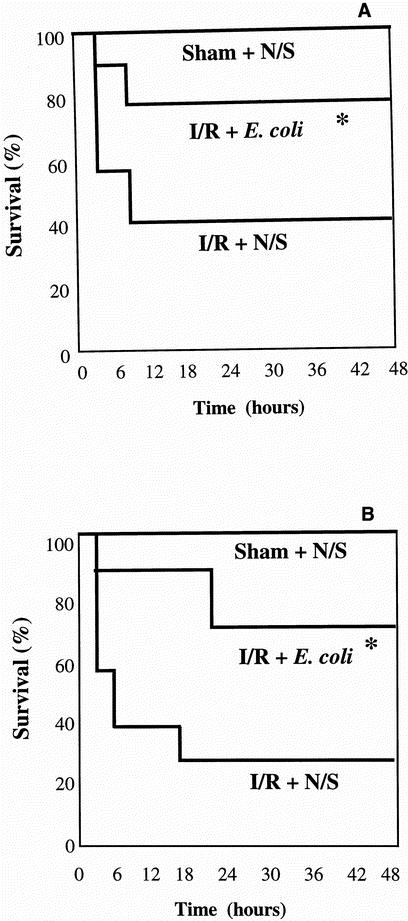
Figure 6. Improved survival after treatment with aerosolized opsonized dead Escherichia coli. After 30 minutes of infrarenal aortic occlusion, aerosolized saline, opsonized dead E. coli, or zVAD-FMK and E. coli was administered intratracheally followed by 2 hours of reperfusion (A). (B) The intratracheal administration of saline, dead E. coli, and zVAD-FMK and E. coli was done 10 minutes into the reperfusion injury. The results are representative of 10 separate experiments. The statistical significance were compared with the ischemia–reperfusion + saline group (*P < .05) and the ischemia–reperfusion + zVAD-FMK and E. coli group (†P < .05).
DISCUSSION
Although PMNs are primary effector cells with a beneficial role in host immune defense, they also play a critical role in the pathogenesis of certain pathologic conditions. It is now well recognized that apoptosis is a distinct mechanism by which PMNs die. 15 The cell undergoes shrinkage and zeiosis, the nucleus collapses, and the chromatin are cleaved into nucleosomal fragments. 16 This highly orchestrated form of cell death is in total contradistinction to necrosis, which results in a sudden, severe, uncontrolled cell lysis with subsequent release of its intracellular contents into the surrounding tissue space, potentially provoking an inflammatory response. Once PMNs have undergone apoptosis, certain cell surface alterations occur, with consequent rapid recognition and subsequent phagocytosis of these apoptotic bodies by tissue macrophages. 17,18 This macrophage phagocytosis process does not provoke the release of proinflammatory macrophage mediators, consequently limiting host tissue damage; in fact, this promotes resolution of the inflammatory response. 19 PMNs play a key role in the development of SIRS and multiple organ dysfunction syndrome. Because the lungs are the main target in these syndromes, with ARDS as the outcome, extensive research has been undertaken to prevent or mitigate ARDS. 20 The development of ARDS in the critically ill patient is associated with significant rates of death and complications. The pulmonary dysfunction in ARDS is largely secondary to PMN-mediated oxidant injury. 21
The major goal of this study was to determine whether the administration of aerosolized opsonized dead E. coli induces inflammatory pulmonary PMN apoptosis and whether this increased PMN apoptotic rate correlated with a diminution in lung injury and an overall improvement in survival. We clearly showed in our PMN-mediated lung injury animal model that the delayed PMN apoptosis in the lungs evident in the I/R plus normal saline animals was reversed in the animals treated with aerosolized opsonized dead E. coli. A variety of apoptotic signals induce cell death by activating cysteine proteases (caspases). Blocking caspase activity with the tetrapeptide inhibitor zVAD-FMK has a dramatic effect on apoptosis in several models. 22 We have shown a direct causal relationship between inducing pulmonary PMN apoptosis and a reduction in lung injury in that the administration of the apoptotic inhibitor zVAD-FMK at the time of E. coli ingestion mitigates the beneficial effects of administering opsonized dead E. coli alone.
Studies of PMN apoptosis during inflammatory responses in vivo have been few and far apart. The initial work by Lee et al 23 in rabbits with an inflammatory pneumonitis showed few apoptotic PMNs in the lungs. Others have found evidence of apoptotic PMNs in BAL fluid from infants with neonatal distress syndrome, 24 and Matute-Bello et al 25 showed that the proportion of apoptotic PMNs recovered from the lungs of patients with ARDS is low throughout the course of ARDS.
Little is known about the fate of PMNs in the airspaces of the lungs. In an in vivo study in humans, PMNs that have transmigrated across the endothelial barrier into the lungs in response to the chemotactic peptide leukotriene B4 display normal functional activity but lose some specific granules during transmigration. 26 Paradoxically, in ARDS, PMNs obtained from the lungs of these patients exhibit functional impairment, with reduced respiratory burst activity. 27
The in vivo relevance of our data needs to be interpreted carefully. Inducing inflammatory pulmonary PMN apoptosis by the administration of aerosolized opsonized dead E. coli is a significant observation. However, correlating this with a diminution in the severity of lung injury as assessed by myeloperoxidase activity (as a measure of PMN influx), pulmonary endothelial permeability (as assessed by both wet/dry lung weight ratio and BAL fluid protein concentration), and histologic analysis of representative lung sections is an even more significant finding. What is even more intriguing is that treatment of animals in our ARDS model with aerosolized opsonized dead E. coli resulted in a significant improvement in survival, not only in a prophylactic setting but also, and clinically of more importance, therapeutically. This concept with its implications has obvious clinical importance, not least because interventional immunotherapy has failed to significantly alter outcome in patients with SIRS and ARDS. 28
Directly modulating pulmonary PMN cell death represents a novel mechanism for attenuating PMN-mediated lung injury and may ultimately improve outcome in patients with ARDS.
Footnotes
Correspondence: Paul Redmond, FRCSI, Chairman, Department of Surgery, National University of Ireland, University College Cork/Cork University Hospital, Cork, Ireland.
E-mail: p.redmond@ucc.ie
Accepted for publication August 10, 2001.
References
- 1.Cohen JJ. Programmed cell death in the immune system. Adv Immunol 1991; 50: 55–85. [DOI] [PubMed] [Google Scholar]
- 2.Liles WC, Klebanoff SJ. Regulation of apoptosis in neutrophils: Fas track to death? J Immunol 1995; 155: 3289–3291. [PubMed] [Google Scholar]
- 3.Smith JA. Neutrophils, host defense, and inflammation: a double-edged sword. J Leukocyte Biol 1994; 56: 672–686. [DOI] [PubMed] [Google Scholar]
- 4.Lechin AE, Varon J. Adult respiratory distress syndrome (ARDS): the basics. J Emerg Med 1994; 12: 63–68. [DOI] [PubMed] [Google Scholar]
- 5.Cox G, Crossley J, Xing Z. Macrophage engulfment of apoptotic neutrophils contributes to the resolution of acute pulmonary inflammation in vivo. Am J Resp Cell Mole Biol 1995; 12: 232–237. [DOI] [PubMed] [Google Scholar]
- 6.Dransfield I, Stocks SC, Haslett C. Regulation of cell adhesion molecule expression and function associated with neutrophil apoptosis. Blood 1995; 85: 3264–3273. [PubMed] [Google Scholar]
- 7.Haslett C, Savil JS, Whyte MK, et al. Granulocyte apoptosis and the control of inflammation. Philos Trans R Soc Lond B Biol Sci 1994; 345: 327–333. [DOI] [PubMed] [Google Scholar]
- 8.Watson RWG, Rotstein OD, Nathens AB, et al. Neutrophil apoptosis is modulated by endothelial transmigration and adhesion molecule engagement. J Immunol 1997; 158: 945–953. [PubMed] [Google Scholar]
- 9.Jimenez MF, Watson WG, Parodo J, et al. Dysregulated expression of neutrophil apoptosis in the systemic inflammatory response syndrome. Arch Surg 1997; 132: 1263–1270. [DOI] [PubMed] [Google Scholar]
- 10.Watson RWG, Redmond HP, Wang JH, et al. Neutrophils undergo apoptosis following ingestion of Escherichia coli. J Immunol 1996; 156: 3986–3992. [PubMed] [Google Scholar]
- 11.O’Donovan DA, Kelly CJ, Abidh H, et al. Role of nitric oxide in lung injury associated with experimental acute pancreatitis. Br J Surg 1995; 82: 1122–1126. [DOI] [PubMed] [Google Scholar]
- 12.Martin SJ, Lennon A, Bonham AM, et al. Induction of apoptosis (programmed cell death) in human leukaemic HL-60 cells by inhibition of RNA or protein synthesis. J Immunol 1990; 145: 1895–1905. [PubMed] [Google Scholar]
- 13.Tsuchida H, Takeda Y, Takei H, et al. In vivo regulation of rat neutrophil apoptosis occurring spontaneously or induced with TNF-α or cycloheximide. J Immunol 1995; 154: 2403–2408. [PubMed] [Google Scholar]
- 14.Watson RWG, Rotstein OD, Parodo J, et al. Impaired apoptotic death signalling in inflammatory lung neutrophils is associated with decreased expression of interleukin-1 beta converting enzyme family proteases (caspases). Surgery 1997; 122: 163–172. [DOI] [PubMed] [Google Scholar]
- 15.Cohen JJ. Apoptosis. Immunol Today 1993; 14: 126–130. [DOI] [PubMed] [Google Scholar]
- 16.Golstein P, Ojcius DM, Young JD. Cell death mechanisms and the immune system. Immunol Rev 1991; 121: 29–35. [DOI] [PubMed] [Google Scholar]
- 17.Savill JS, Wyllie AH, Henson J, et al. Macrophage phagocytosis of ageing neutrophils in inflammation: programmed cell death in the neutrophil leads to its recognition by macrophages. J Clin Invest 1989; 83: 865–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rossi AG, McCutcheon JC, Roy N, et al. Regulation of macrophage phagocytosis of apoptotic cells by cAMP. J Immunol 1998; 160: 3562–3568. [PubMed] [Google Scholar]
- 19.Meagher LC, Savill JS, Baker A, et al. Phagocytosis of apoptotic neutrophils does not induce macrophage release of thromboxane B2. J Leuko Biol 1992; 52: 269–274. [PubMed] [Google Scholar]
- 20.Fujishima S, Aikawa N. Neutrophil-mediated tissue injury and its modulation. Intensive Care Med 1995; 21: 277–285. [DOI] [PubMed] [Google Scholar]
- 21.Davreux CJ, Soric I, Nathens AB, et al. N-acetyl cysteine attenuates acute lung injury in the rat. Shock 1997; 8: 432–428. [PubMed] [Google Scholar]
- 22.Eldadah BA, Yakovlev AG, Faden AI. The role of CED-3-related cysteine proteases in apoptosis of cerebellar granule cells. J Neurosci 1997; 17: 6105–6113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee A, Whyte MK, Haslett C. Inhibition of apoptosis and prolongation of neutrophil functional longevity by inflammatory mediators. J Leuko Biol 1993; 54: 283–288. [PubMed] [Google Scholar]
- 24.Grigg JM, Savill JS, Sarraf C, et al. Neutrophil apoptosis and clearance from neonatal lungs. Lancet 1991; 338: 720–722. [DOI] [PubMed] [Google Scholar]
- 25.Matute-Bello G, Liles WC, Radela F, et al. Neutrophil apoptosis in the acute respiratory distress syndrome. Am J Respir Crit Care Med 1997; 156: 1969–1977. [DOI] [PubMed] [Google Scholar]
- 26.Martin TR, Pistorese BP, Chi EY, et al. Effects of leucotriene B4 in the human lung: recruitment of neutrophils into the alveolar spaces without a change in protein permeability. J Clin Invest 1989; 84: 1609–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martin TR, Pistorese BP, Hudson LD, et al. The function of lung and blood neutrophils in patients with the adult respiratory distress syndrome: implications for the pathogenesis of lung infections. Am Rev Respir Dis 1991; 144: 251–252. [DOI] [PubMed] [Google Scholar]
- 28.Bone RC. Immunologic dissonance: a continuing evolution in our understanding of the systemic inflammatory response syndrome (SIRS) and the multiple organ dysfunction syndrome (MODS). Ann Intern Med 1996; 125: 680–687. [DOI] [PubMed] [Google Scholar]