

Abstract
Objective
To examine the long-term effects of Sur KO, SSTR5 KO, and double Sur/SSTR5 KO on insulin secretion and glucose regulation.
Summary Background Data
The sulfonylurea receptor (Sur) and somatostatin receptor type 5 (SSTR5) play an integral role in the regulatory pathways of the endocrine pancreas. Sur knockout (KO) and SSTR5 KO mice were generated in the authors’ laboratories and crossbred to generate Sur/SSTR5 KO mice. All mice were genotyped by Southern blotting and polymerase chain reaction analysis.
Methods
One-year-old Sur KO, Sur/SSTR5 KO, SSTR5 KO, and age-matched wild-type control mice underwent single-pass perfusion of isolated pancreata with low and high glucose concentration (n = 4–6/group). Another group of mice also underwent intraperitoneal glucose tolerance tests with 1.2 g glucose/kg body weight (n = 4/group per time point).
Results
Sur1 KO and Sur/SSTR5 KO mice had profoundly decreased insulin secretion in vitro, whereas SSTR5 KO had increased insulin secretion compared with wild-type mice. Sur1 KO and Sur/SSTR5 mice had increased glucose response in vivo compared with wild-type mice. Sur1 KO and Sur/SSTR5 KO mice exhibit glucose intolerance and SSTR5 KO mice show increased insulin response in vitro.
Conclusions
Sur1 KO causes glucose intolerance and SSTR5 KO causes increased insulin secretion. However, Sur/SSTR5 double ablation does not alleviate the diabetic state of the Sur1 KO.
The endocrine pancreas consists of the islets of Langerhans, which are in turn made up of several different cell types. The most common cell type in the islets are the beta cells, which make up 70% of the volume of the islet and are responsible for the secretion of insulin. Other cell types include the alpha cells, which produce glucagon, and the delta cells, which produce somatostatin. These two groups make up approximately 20% and 5%, respectively, of the volume of the islet. 1,2 Insulin secreted from the beta cell acts to regulate the blood glucose levels during fasting and fed states.
Somatostatin is an inhibitory peptide discovered in the early 1970s. 3 Further work showed it to be present in a variety of tissues, 4 and as techniques became more sophisticated, it was found in more tissues. 5–7 In the early 1990s, the receptors for somatostatin were isolated, cloned, and characterized. 8–10 The somatostatin receptors (SSTRs) were found to belong to a family of G protein-coupled receptors that affect potassium and calcium channels, cAMP, and protein phosphatases. 11 Further studies suggested that SSTR5 was a likely candidate to be involved with insulin inhibition and that it was present on the beta cell. 12–18 To further characterize the role of SSTR5 on insulin secretion, an SSTR5 knockout (KO) mouse model was created in our laboratory. These mice were viable and fertile and had a subtle aging phenotype in which there was an increased insulin secretion response to glucose stimulation in year-old mice.
ATP-sensitive potassium (KATP) channels consist of two different subunits: a sulfonylurea receptor (Sur), which belongs to the ATP-binding cassette family, which as a group function to transport substances across cell membranes, and an inwardly rectifying potassium channel (Kir). 19,20 Together these subunits form an ion channel (Kir) that is regulated by ATP binding to the Sur receptor, and this controls the flux of potassium out of the cell. The sulfonylurea receptor functions as the sensor for the channel and affects opening and closure, which sets the resting membrane of the cell below the threshold for calcium channel activation. 21 During the fed state, glucose is transported into the beta cell via the GLUT2 transporter and metabolized in the cell to ATP. This changes the ATP/ADP ratio in the cell and causes the KATP channel to close. This prevents the exit of potassium from the cell, raising the membrane potential and depolarizing the cell. Once the cell reaches the threshold for calcium channels, voltage-dependent calcium channels open and calcium is released into the cell. The influx of calcium into the cytosol allows calcium to bind to secretory granules, triggering exocytosis and the release of insulin. 22
To support these hypotheses, evidence obtained from Sur1 KO mice showed that the genetic ablation of Sur1 gene results in impaired glucose-stimulated insulin secretion at 3 months of age, characterized by a lack of first-phase insulin secretion and impaired second-phase secretion. These mice also show prolonged glucose levels after glucose stimulation. 23
The purposes of this study were to examine the long-term effects of SSTR5 KO, Sur KO, and Sur/SSTR5 double KO on insulin secretion and glucose levels, and to determine whether the increased secretion of insulin seen with the SSTR5 KO mice would alter the blunted insulin response of the Sur KO mice.
METHODS
Generation of a Double KO
SSTR5 KO mice were generated and maintained in our laboratory. Sur1 KO mice were obtained from Dr. Bryan’s laboratory (Department of Molecular and Cellular Biology, Baylor College of Medicine, Houston, TX) and were generated as described previously. 23 These mice were crossbred to generate a double heterozygous mouse (SSTR5 +/-, Sur1 +/-). The offspring were then inbred to develop the Sur/SSTR5 double homozygous KO.
Genotypic Screening
SSTR5 KO mice were screened routinely in our laboratory by Southern blotting. The subsequent generations were screened by polymerase chain reaction (PCR). PCR primers for the wild type (WT) were #1 ACA CCT AGC TGG AAT GCC TCA corresponding to bp 265–85 in mSSTR5, GI no. 2209142, and #2 CAG TAG GAG ACA GCA TTC corresponding to bp 546–28 in the same. PCR primers for the KO screen consisted of #1 AAG GCA GTC TGG AGC AT, corresponding to bp 2281–2297 in the PGK-Neo cassette, and #2 AAC CTG CGT GCA ATC CAT CTT, corresponding to bp 2806–2786. PCR amplification was carried out with Taq, primers, dNTPs, and MgCl2 for 5 minutes at 94°C, then 30 cycles of 94°C (1 minute), 60°C (1 minute), 72°C (1 minute), and a final 6 minutes at 72°C. Analysis on a 1% agarose gel revealed a band at 280 bp for the WT, 525 bp for the KO, and both for a heterozygous.
Sur1 mice were screened with PCR primers AGG TTG TTG GTG GAG GTC AG and CCA ACA CGA GCC TTG AAC TT, which produced a 524-bp band for the WT, whereas primers AGG TTG TTG GTG GAG GTC AG and CTG TCC ATC TGC ACG AGA CT produced a 350-bp band for the KO. PCR amplification was carried out with Taq, primers, dNTPs, and MgCl2 for 30 cycles of 62°C (30 seconds), 72°C (45 seconds), and 94°C (30 seconds).
Isolated Perfused Mouse Pancreas Model
The isolated perfused pancreas model has been successfully established in our laboratory and was used to assess in vitro insulin secretion. Mice were anesthetized with Ketamine/Xylocaine at a dose of 4 mL/kg (0.1 cc3/25 g) administered intraperitoneally. The steps involved in perfusing the isolated pancreas in situ are as follows: midline laparotomy followed by ligation of the distal colon. The duodenum was ligated distal to the sphincter of Oddi, approximately at the end of the C loop, and a PE 190 tube was placed proximal to this tie to allow for drainage of the stomach. The superior mesenteric artery was ligated en bloc with the small and large intestine and removed. The right renal vein and artery were ligated and a loose tie was placed around the portal vein and bile duct. The esophagus and left gastric artery were ligated, the splenic vessels were ligated, and the spleen was removed. The left renal vessels were ligated and the infrarenal aorta and inferior vena cava were ligated just proximal to the iliac vessels. The supraceliac aorta was dissected out and a loose tie was placed around it. Under the dissecting microscope, a loose tie was placed around the aorta and inferior vena cava distal to the renal vessels origin but proximal to the iliac ligature. The aorta was separated from the inferior vena cava to clear an area for aortotomy. The supraceliac aortic tie was tightened and the ischemic time began. An aortotomy was performed and a PE 50 cannula was placed in the aorta and secured. The perfusion solution was attached to the aortic cannula and perfusion was begun, thus ending the ischemic time. Attention was then turned to the portal vein. The bile duct was dissected off and the portal vein was incised and a PE 50 cannula placed to collect pancreatic effluent.
Single-pass perfusion was performed using a modified Krebs-Ringer’s buffer containing 3% dextran (Sigma, St. Louis, MO) and 0.5% bovine serum albumin containing 0.7 g/L glucose until the temperature reached 37°C. At that point, the next 5 minutes of effluent were discarded to allow for washout. After the washout period, portal vein effluent was collected for 4 minutes, and then the solutions were changed to a solution of 3 g/L glucose for a total of 30 minutes (stimulated). The perfusate was gassed with 95% O2/5% CO2 to achieve an O2 content of 400 and a pH of 7.4. The flow rate of the perfusate was 1 mL/min, and perfusate pressure was measured. The pancreas was maintained at 37°C using a heat lamp. Portal vein effluent was collected every minute and placed at −20°C for subsequent analysis using an insulin radioimmunoassay (RIA) kit (Linco Research, La Jolla, CA), which was carried out in duplicate.
Intraperitoneal Glucose Tolerance Test
Mice were divided into three groups (basal, 30 minutes, and 60 minutes) with four to six mice per group per time point and fasted for 16 hours. Mice that were to receive glucose (30-minute and 60-minute groups) were then injected with 1.2 g glucose/kg body weight intraperitoneally using a 10% dextrose solution (5 g dextrose, 0.45 g NaCl, and 50 mL dH2O). At set times from glucose injection (30 and 60 minutes), mice were then anesthetized with Avertin (2,2,2-tribromoethanol) at a dose of 16 mL/kg (0.4 mL/25 g). Once adequate anesthesia was obtained as judged by paw prick, a glass pipet was used to extract approximately 100 μL blood from the retroorbital venous plexus. The blood was then transferred to a serum separator tube (Becton-Dickinson) and stored on ice. The blood sample was spun down using a 4°C microfuge. Serum was transferred to a 200-μL tube and stored at −20°C. Glucose levels were determined in duplicate using a Beckman Glucose Analyzer 2. Insulin levels were measured in duplicate using a commercially available insulin RIA kit with internal standards and assay controls.
Immunohistochemistry Staining
Pancreata from WT, SSTR5 KO, and Sur/SSTR5 KO mice were removed and fixed in 10% formalin for 24 hours and embedded in paraffin. Tissue sections were cut and slides were deparaffinized in xylene five times for 5 minutes. Sections were hydrated gradually through graded alcohol. Slides were placed in a humidified chamber overlain with diluted antibodies against insulin, glucagon, or somatostatin (Vector Laboratory, CA) overnight at 4°C. The dilution factors for insulin, glucagon, and somatostatin were 75, 200, and 75, respectively. After washing with phosphate-buffered saline, sections were incubated with secondary antibody for 1 hour at room temperature and color was developed with substrate solutions.
Data Presentation and Statistical Analysis
Insulin data for the perfusion study is presented as mean ± standard error of the mean (ng/mL). Basal insulin secretion (minutes 1–5), and the glucose-stimulated first phase (minutes 6–10) and second phase (minutes 11–30) of insulin secretion were compared. Statistical analysis was by t test. Glucose and insulin data from the intraperitoneal glucose tolerance test (IPGTT) are presented as mean ± standard error of the mean in mg/dL and ng/dL, respectively. Basal and 30- and 60-minute glucose and insulin levels were compared. Statistical analysis was by t test.
RESULTS
Generation of Double KO Mice
Figure 1 shows a diagram of the generation for the Sur/SSTR5 double KO mice. The identification of SSTR5 KO and Sur1 KO mice by PCR is shown in Figure 2. The designing and sequence of the PCR primers were described above. The Sur/SSTR5 double KO animals were viable and fertile and appeared phenotypically normal. The mice showed no obvious changes in weight or behavior to distinguish them from their WT littermates.
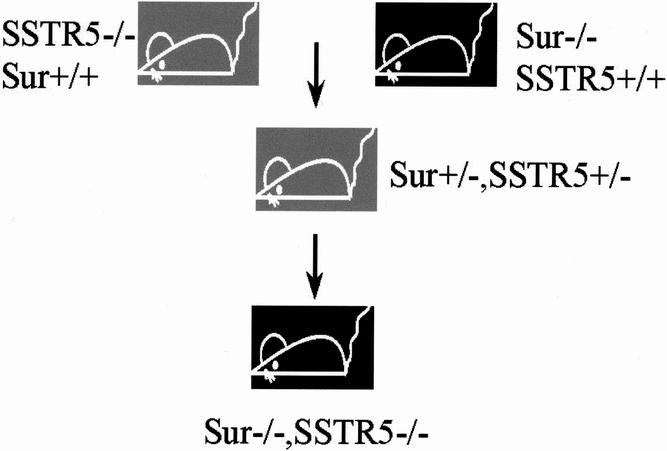
Figure 1. The generation of Sur/SSTR5 double knockout (KO) mice. SSTR5 KO and Sur1 KO mice were crossbred to yield double heterozygous animals. These animals were inbred and screened with polymerase chain reaction to detect mice that were homozygous null for both SSTR5 and for Sur1. These mice were then put into a separate colony to breed and age.
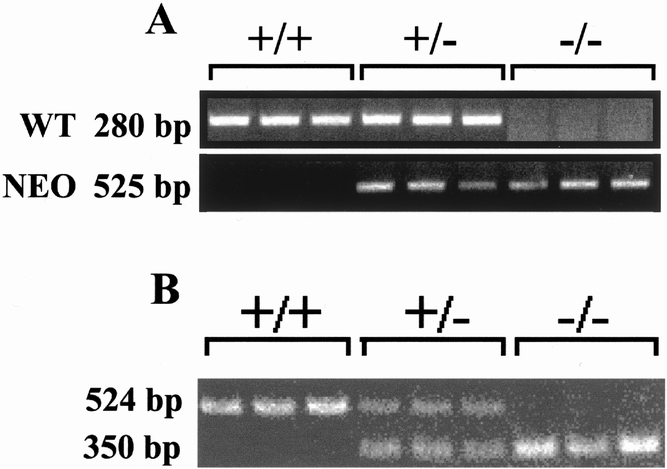
Figure 2. Polymerase chain reaction analysis of SSTR5 knockout (KO) mice and Sur1 KO mice. Tail DNA was combined with appropriate primers and underwent polymerase chain reaction amplification. Fifteen microliters was run on a 1% agarose gel with appropriate molecular markers. (A) SSTR5 primers result in a 280-bp band for the wild type, a 525-bp band for the KO, and both for a heterozygote. (B) Sur1 primers result in a 524-bp band for the wild type, a 350-bp band for the KO, and both for a heterozygote.
Isolated Perfused Mouse Pancreas Model
To compare insulin secretion patterns in these KO mice, year-old animals underwent single-pass perfusion. The comparison of the perfusion data are presented in Figure 3A. For basal, first-phase, and second-phase insulin secretion, the Sur and Sur/SSTR5 mice had a blunted insulin secretory pattern compared with both WT and SSTR5 KO mice alone.

Figure 3. Alterations in insulin secretion using the isolated perfused mouse pancreas model. Year-old SSTR5 knockout (KO), wild-type (WT), Sur1 KO, and Sur/SSTR5 double KO animals (n = 4–6 per group) underwent single-pass perfusion of isolated pancreata. (A) The means of these experiments are shown. At minute 5, the glucose solution was changed from 0.7 g/L to 3 g/L. (B) Statistical analysis of perfusion data. Basal corresponds to minutes 1 to 5, first phase to minutes 6 to 10, and second phase to minutes 11 to 30. Data are presented as mean ± standard error of the mean. Statistical analysis was conducted by t test. αP < .05, WT versus Sur1 KO at first and second phase, βP < .05, WT versus Sur/SSTR5 KO at first and second phase, ςP < .05 SSTR5 versus WT at basal and first and second phases, *P < .05, SSTR5 KO versus Sur1 KO at basal and first and second phases, #P < .05 SSTR5 KO versus Sur/SSTR5 at basal and first and second phases.
SSTR5 KO mice had a significantly higher basal (SSTR5 KO 0.485 ± 0.0619 ng/mL vs. WT 0.169 ± 0.0244 ng/mL), first-phase (SSTR5 KO 0.592 ± 0.0723 ng/mL vs. WT 0.311 ± 0.0370 ng/mL), and second-phase (SSTR5 KO 0.855 ± 0.0364 ng/mL vs. WT 0.447 ± 0.0454 ng/mL) insulin values compared with WT mice. There were no significant differences between Sur1 KO and Sur/SSTR5 KO mice at basal, first, and second phases of insulin secretion. WT mice were significantly higher than Sur1 KO and Sur/SSTR5 KO for first (WT 0.311 ± 0.0370 ng/mL vs. Sur1 KO 0.069 ± 0.0115 ng/mL vs. Sur/SSTR5 KO 0.085 ± 0.0088 ng/mL) and second phases (WT 0.447 ± 0.0454 ng/mL vs. Sur1 KO 0.085 ± 0.0066 ng/mL vs. Sur/SSTR5 KO 0.084 ± 0.0044 ng/mL) of insulin secretion. The summary of the statistical analysis of the perfusion study is presented in Figure 3B.
Intraperitoneal Glucose Tolerance Test
The results of the glucose levels are summarized in Figure 4A. After a 16-hour fast, Sur1 KO mice had significantly lower glucose levels when compared with WT, SSTR5 KO, and Sur/SSTR5 KO mice (WT 227 ± 6.4 mg/dL vs. Sur1 KO 155 ± 9.3 mg/dL vs. Sur/SSTR5 KO 245 ± 19.6 mg/dL vs. SSTR5 KO 225 ± 11.8 mg/dL). There were no significant differences between WT, SSTR5 KO, and Sur/SSTR5 KO mice at the basal time point. Thirty minutes after glucose injection, Sur1 KO and Sur/SSTR5 KO mice had elevated glucose levels compared with WT and SSTR5 KO controls. There were significant differences between Sur/SSTR5 KO mice compared with WT and SSTR5 KO mice (Sur/SSTR5 KO 469 ± 34.9 mg/dL vs. WT 172 ± 1.7 mg/dL vs. SSTR5 KO 163 ± 15.5 mg/dL). Sixty minutes after glucose injection, Sur1 KO and especially Sur/SSTR5 double KO mice continued to have elevated glucose levels. There were significant differences between Sur/SSTR5 KO and WT, Sur1 KO and SSTR5 KO controls (Sur/SSTR5 KO 482 ± 31.1 mg/dL vs. Sur1 KO 338 ± 35.0 mg/dL vs. WT 235 ± 21.6 mg/dL vs. SSTR5 KO 209 ± 21.4 mg/dL). There were also significant differences between Sur1 KO and WT and SSTR5 KO mice (Sur1 KO 338 ± 35.0 mg/dL vs. WT 235 ± 21.6 mg/dL vs. SSTR5 KO 209 ± 21.4 mg/dL).

Figure 4. Intraperitoneal glucose tolerance test (IPGTT). Mice were randomized to three different groups (basal, 30 minutes, and 60 minutes) and fasted overnight. The following day the basal group mice were anesthetized and had blood drawn from the retroorbital sinus. The 30- and 60-minute mice received an intraperitoneal injection of glucose at 1.2 mg/g body weight and at the appropriate time after glucose injection were anesthetized and underwent blood draw. (A) Glucose levels for IPGTT. αP < .05, WT versus Sur1 KO at basal and 60 minutes, βP < .05, SSTR5 versus Sur/SSTR5 KO at 30 and 60 minutes, ρP < .05, Sur/SSTR5 KO versus Sur1 KO at basal and 60 minutes, *P < .05, SSTR5 KO versus Sur1 KO at basal and 60 minutes, #P < .05, WT versus Sur/SSTR5 KO at 30 and 60 minutes. (B) Insulin levels for IPGTT. αP < .05, WT versus Sur/SSTR5 KO at basal and 30 minutes, βP < .05, WT versus Sur1 KO at 30 minutes, *P < .05, Sur/SSTR5 KO versus SSTR5 KO at basal.
The results of the insulin levels are summarized in Figure 4B. After overnight fasting, Sur/SSTR5 double KO mice had significantly lower insulin levels compared with WT and SSTR5 KO mice (Sur/SSTR5 0.21 ± 0.014 ng/mL vs. WT 0.31 ± 0.062 ng/mL and SSTR5 KO 0.34 ± 0.055 ng/mL). There were no significant differences between Sur1 KO and WT, Sur/SSTR5 KO, or SSTR5 KO mice at the basal time point. There were no significant differences between WT and Sur1 KO or SSTR5 KO mice at the basal time point. Thirty minutes after glucose injection, WT insulin levels were significantly higher than those in Sur1 KO and Sur/SSTR5 KO mice (WT 0.295 ± 0.0029 ng/mL vs. Sur1 KO 0.1922 ± 0.012 ng/mL and Sur/SSTR5 0.2190 ± 0.0154 ng/mL). At 30 minutes there were no other significant values; at 60 minutes there were no significant differences in insulin levels for any of the mice studied.
To further analyze the morphology of the islets of these Sur/SSTR5 double KO mice, an immunohistochemical analysis was performed; results are presented in Figure 5. Compared with WT mice there was no difference in islet morphology or insulin or somatostatin staining for SSTR5 KO and Sur/SSTR5 double KO mice. There was, however, an alteration in the glucagon staining in the islets of Sur/SSTR5 KO mice: the glucagon granules were present in the center of the islet rather than along the periphery, as seen in WT islets.
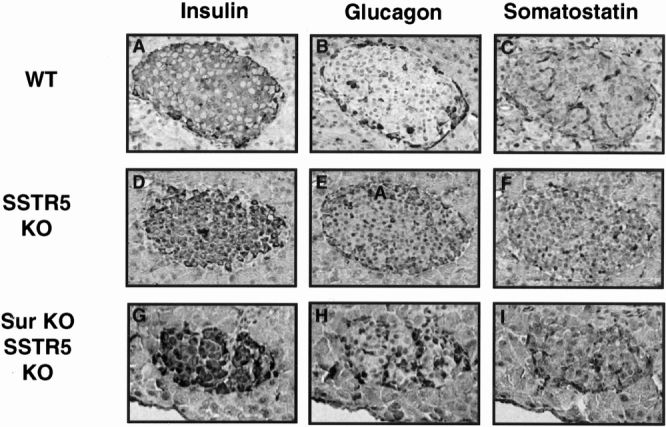
Figure 5. Immunohistochemical analysis of islet cells. Pancreata from wild type (WT), SSTR5 knockout (KO), and Sur/SSTR5 KO mice were processed for immunostaining using antibodies against insulin, glucagon, and somatostatin. To examine the morphology of the islet, serial sections were prepared from the same paraffin block and stained with different antibodies separately. There was no discernible difference in insulin and somatostatin between WT, SSTR5 KO, and Sur/SSTR5 KO islets. There was an alteration in glucagon staining in the Sur/SSTR5 KO islets.
DISCUSSION
Somatostatin receptors are G protein-coupled receptors and activate an extensive second messenger system. Somatostatin binding causes a conformational change within the receptor that activates G protein family members, which subsequently results in the release of the β subunit from γ and α subunits, and the α subunit, which in turn affects other molecules. SSTRs are linked to an inhibitory α subunit that activates adenylyl cyclase, K+ channels, and Ca2+ channels in a negative fashion. 11,24 The alpha subtypes of G proteins have been shown to be islet cell-specific, and their distribution is age- and species-dependent. 25 SSTRs have been shown to decrease the intracellular calcium concentration and to directly affect the voltage-gated channels to prevent Ca2+ influx. 26 SSTRs have also been linked to protein kinases and phosphatases, which act to regulate various downstream enzymes. 27
The crucial factor necessary for insulin release from the beta cell is the intracellular concentration of calcium. Substances such as hormones and peptides, including somatostatin, that affect calcium conductance will affect insulin secretion via alteration in intracellular concentration of calcium. 28 The binding of somatostatin with its receptors on the beta cell triggers multiple different second messenger systems, which result in the decreased calcium concentration in the cell and thereby inhibit insulin release. 22,29–32
Somatostatin inhibiting insulin secretion is mediated by the SSTRs present on beta cells. Among them, SSTR5 has been shown to be present on more than 80% of the beta cells, has been shown to colocalize with insulin, and has been linked to inhibition of insulin secretion in multiple studies. 12–18 Our perfusion results show that genetic ablation of the SSTR5 gene resulted in enhanced insulin secretion in year-old mice for basal, first phase, and second phase of insulin secretion, which is consistent with the loss of an inhibitory receptor. As previously shown, Sur1 KO mice lack KATP channels and therefore cannot regulate intracellular calcium levels. This causes markedly reduced insulin release from the beta cell. 23 In the Sur/SSTR5 double KO, we also show an insulin secretory pattern very similar to that of Sur1 KO mice. The data suggest that the KATP channel is the dominant factor in the regulation of insulin secretion. Although SSTR5 has been shown to interact with a G protein-coupled inward rectifying K+ channel (GIRK), this does not appear to be a dominant factor. 33 The addition of SSTR5 genetic ablation cannot overcome the Sur1 phenotype and does not result in an augmented insulin secretory pattern.
A similar pattern was also shown in the immunohistochemistry studies. Results showed that the islets of the SSTR5 KO animals are not significantly different from WT controls in terms of morphology or insulin, glucagon, or somatostatin staining. The Sur1 KO mice were reported to have alterations in the pattern of glucagon secretion with a central rather than peripheral distribution, whereas the insulin and somatostatin staining remained unchanged. The Sur/SSTR5 double KO mice also had changes in glucagon staining consistent with that of Sur1 ablated mice. In terms of insulin and somatostatin staining, there were no discernible changes in the Sur/SSTR5 double KO mice.
In addition, in vivo IPGTT results showed that SSTR5 KO mice had a slight increase in insulin secretion in response to glucose, but this was not significantly elevated over WT mice. Complimenting the perfusion studies, the Sur1 KO and Sur/SSTR5 KO mice had decreased insulin levels at basal, 30 minutes, and 60 minutes and did obtain significance at the basal and 30-minute time points. Previous data from the Sur1 KO study showed that these mice had a delayed insulin response to glucose stimulation and had a sustained high level of glucose after intraperitoneal injection. 23 Our objective was to determine whether the addition of SSTR5 gene ablation would alter the insulin or glucose levels at a later time point. In our study, the glucose levels for the Sur/SSTR5 double KO were markedly elevated and sustained for the entire 60-minute time course. Also at 60 minutes, the glucose levels for the Sur/SSTR5 KO mice were significantly higher than all other strains, including the Sur1 KO mice. These data suggest that the glucose intolerance exhibited by the Sur1 KO mice is exacerbated by the additional ablation of the SSTR5 gene.
In conclusion, a Sur/SSTR5 double KO mouse model was generated, and these mice showed significant phenotypic changes compared with WT and SSTR5 KO mice. The Sur/SSTR5 double KO also revealed alterations in islet cell structure compared with WT and SSTR5 KO mice. The Sur/SSTR5 mouse model represents a means by which to evaluate the KATP channel on the beta cell and how it interacts with somatostatin receptors.
Acknowledgments
The authors thank their colleagues working in Dr. DeMayo’s and Dr. Brunicardi’s laboratories for their support and technical assistance. We also thank Dr. J. Bryan for the use of the Sur1 KO mice for experimental and breeding purposes. Thanks also to Ms. Susie Lee for her constant assistance throughout this project.
Discussion
Dr. B. Mark Evers (Galveston, TX): This paper by Dr. Brunicardi provides a nice extension of previous studies by this group on the cellular mechanisms regulating pancreatic endocrine function and, specifically, glucose regulation. This group continues to provide us with a better understanding of the function of somatostatin on insulin and glucose regulation by the use of sophisticated gene ablation studies in transgenic animals. They demonstrate increased insulin secretion with ablation of the somatostatin receptor type 5 gene, an animal model that was developed in their laboratory. Ablation of the sulfonylurea receptor results in glucose intolerance and decreased insulin secretion. Crossing the two knockout strains, however, did not correct the glucose intolerance, suggesting that the effects of the somatostatin receptor are downstream of the potassium-ATP channels, which are lacking in the sulfonylurea receptor knockout mice. I have several questions for Dr. Brunicardi regarding this study. First, why do you think that the changes in insulin secretion noted in the somatostatin receptor knockout mice are only seen in the adult animals? I find it intriguing that these changes are not found earlier in the life of the mice, suggesting possible changes associated with aging in unmasking the alterations in insulin secretion. Have you had an opportunity to assess mice older than 1 year of age to see if the changes that you report are magnified with senescence? Second, your studies have primarily focused on the pancreas. Do you have information on whether the insulin sensitivity of peripheral tissues is altered in these knockout animals, or do you think that the changes are specifically localized to effects on pancreatic function? Third, is this increase in insulin secretion the result of increased insulin production? Alternatively, are the levels of peptides such as glucagon or somatostatin altered, which may prevent the inhibition of insulin secretion, thus resulting in enhanced insulin release? Finally, I am a bit concerned regarding the lack of phenotypic changes in these mice. If I understood correctly, the somatostatin receptor knockout mice do not show differences in weight, feeding characteristics, or early lethality. What do you then envision as the overall role of the somatostatin type 5 receptor in glucose and insulin regulation?
I enjoyed the paper very much and look forward to future extensions.
Dr. R. Daniel Beauchamp (Nashville, TN): I echo Dr. Evers’ congratulatory remarks. I have also a few questions about this model. I think these are very interesting physiologic models and I think that like any good set of experiments it raises more questions than it provides answers. It is interesting that you designate these as diabetic mice. In human diabetes there is a constellation of disease processes that occur that are typical of diabetes mellitus and that results in, if not treated, early death. And I just wonder if these mice actually have diabetes mellitus. In other words, do they have persistent elevations in blood glucose? Does it cause downstream pathologic effects on blood vessels and other tissues? Can you measure an altered hemoglobin A1C level, for example, in a mouse? I have never tried to do that. But I am sure that you would know. What is the role of the glucagon relocation in the islet in the altered glucose tolerance in these animals? Do you think that has a role? Or is it just a coincidental observation? With regard to the somatostatin knockout mice, do these mice ever get hypoglycemic? In other words, is the glucose level suppressed to the point where these mice become unconscious or have seizures at any point? Or if you stress them, does that occur? And if it does not occur, why do you think it does not occur? Because they have a significantly higher level of insulin even at basal levels, and when there is added glucose in the system their levels even go higher than the wild-type mice.
Again I congratulate you on your work and thank the Society for the privilege of the floor.
Dr. Alden H. Harken (Denver, CO): I would like to echo the observations of the previous discussants and say, Chuck, that was a beautifully presented paper. Even I understood what you were doing. We have been interested in the paradox that insulin-controlled diabetics have a lower, paradoxically lower, cardiovascular mortality than oral agent-treated diabetic patients. And we and others have explained that by virtue of the fact that the sulfonylurea agents which are usually used as oral agents are the mitochondrial inner membrane potassium channel blockers, and that prevents cardiomyocyte reprogramming or cardiomyocyte preconditioning. So we were tempted to use, or try to use, some of the sulfonylurea receptor blockers, like you so nicely explained. But in an effort to wildly oversimplify much of the technique that you have just nicely discussed, if you knock out a gene and you get a surviving animal, there are only a couple of options. That means that the gene was a vestigial unimportant gene, or the surviving animal was able to compensate by some other mechanism. How do you explain this? What was the other mechanism? What is the cell doing that permits it to survive, and apparently in a perfectly healthy state, in the absence of that sulfonylurea gene and the KATP channel inhibitor? Do you get an overexpression of the KATP channel? Do you see an increase in the calcium transients? That is the global question. The more focused question is, I was intrigued that your basal glucose levels were lower in the sulfonylurea knockouts; however, they were more glucose intolerant. Does that mean that there are totally different mechanisms controlling basal and challenged glucose levels?
Dr. M. Norman (Houston, TX): I would like to thank Dr. Beauchamp and Dr. Evers for their opinions and their insightful comments and their willingness to review this paper. In answer to Dr. Evers’ first question about the change in insulin secretion only in adult animals, previous work that we have done looking at the somatostatin 5 mice, initially when we created these animals, we were surprised to see that at a young age that they did not have any significant differences using the perfusion model, whereas at an older age they did have significant differences. That was shown in previous research, and then in the research that was presented here today we double-checked everything. What we have also been working on kind of on the side is looking at other somatostatin receptor knockout animal, somatostatin receptors, for the possibility that there could be compensation. Another possibility in terms of receptor models is the somatostatin 1 knockout mouse, which we have also looked at and which also showed differences in insulin secretion. So one of our thoughts with that we are currently working on to finalize and kind of tease out further, is whether there is a compensation in our redundant receptors. There are five receptors that are present in various somatostatin beta cells, so we are trying to figure out which is responsible for being able to compensate for the lack of one.
In answer to the second question about insulin sensitivity in peripheral tissue, that is a very good idea. We talked about doing that. But we haven’t gotten around to looking at insulin sensitivity in muscle or liver yet.
Next, whether it is a change in secretion versus a change in production. We have started to investigate that in looking at changes in glucagon or somatostatin. Briefly, currently right now we haven’t had any new data on that. We are starting to consider getting into looking at changes in glucagon or somatostatin at the islet level, basically isolating islets and isolating RNA from that and doing radioactive RT-PCR to see if there is any change at that level.
Finally, Dr. Beauchamp’s question whether these mice are diabetic and do they actually have a diabetes mellitus. I think Dr. Brunicardi had alluded to the possibility of pancreaticogenic diabetes where in these mice we have changed essentially the way the pancreas reacts and the way the islet reacts to glucose as an insulin secretagogue in that we have taken out the KTB channel which sets the rate and allows calcium in. By doing that, we have essentially reset the resting potential of the beta cell to a point where the calcium channels are in a much greater state of activation and in fact have shown differences in phenotype. I guess an aside to what Thuromont originally looked at was they were looking at—there is a process called persistent hyperinsulinemia-hypoglycemia where humans with this disorder which is caused by mutations of the sulfonylurea gene or some of the potassium and rectifying channels present with very high insulin levels and dangerously low levels of hypoglycemia, and frequently undergo a 99% pancreatectomy at a very young age. These mice, however, when they created a similar study, did not show that. And they are currently working with mice and not in humans.
In terms of the question of the role of glucagon in the islet and why it changes, the only speculation I can put to that is that with somatostatin there are negatively coupled gene protein receptors, whereas with glucagon there is a positive gene protein coupled receptor, so there is a small possibility that with different amounts of glucagon at the right place in the islet you could get changes in cyclo-GNP or potassium channels or calcium channels that would counteract the effects of altering these channels with either somatostatin receptor 5 or with the sulfonylurea changes. And then with the somatostatin receptor mice, do they get hyperglycemic? We have not done it with IPGTTs that we have done. I have fasted mice for anywhere from 12 hours to 24 hours and have not really seen any significant differences, at least in terms of their glucose levels, after a fast. Why that does not occur, again, we are still working with that to figure out essentially whether other receptors are able to compensate and whether any other changes in glucagon or stress hormones, cortisol, epinephrine, et cetera, that would allow the mice to compensate.
Finally, Dr. Harken’s comments about the basal glucose being lowered in the Sur knockout animals. That was an observation that was originally seen also in the original paper by the Joe Bryan lab and Victor Seagers. And one of their theories is that with the sulfonylurea receptors there are multiple different receptors, including there is also a sulfonylurea 2 and then there are different rectifying channels. These are present in different areas of the cells from myocardiac muscle, similar to vascular tissue. One possibility that they thought was that the changes in the sulfonylurea receptor could be affecting the muscle and that would compensate for the changes in basal glucose and that the muscle itself would be taking up more of the glucose rather than letting the pancreas see that.
I would like to thank the Association and Vice-President Carey and Secretary Townsend for this opportunity to present the work and the privilege of the floor.
Footnotes
Correspondence: F. Charles Brunicardi, MD, Professor and Chairman, Michael E. DeBakey Department of Surgery, Baylor College of Medicine, 6550 Fannin, Suite 1661, Houston, TX 77030.
E-mail: cbrunica@bcm.tmc.edu
Supported in part by NIH grant NIDDK R01-DK46441 (to F.C.B.).
Presented at the 113th Annual Session of the Southern Surgical Association, December 3–5, 2001, Hot Springs, Virginia.
Accepted for publication December 2001.
References
- 1.Junqueira L, Carneiro J, Kelley R. Basic histology. Norwalk, CT: Appleton & Lange, 1992.
- 2.Rhoades R, Tanner G. Medical physiology. Boston: Little, Brown and Company, 1995.
- 3.Brazeau P, Vale W, Burgus R, et al. Hypothalamic polypeptide that inhibits the secretion of immunoreactive pituitary growth hormone. Science 1973; 179: 77–79. [DOI] [PubMed] [Google Scholar]
- 4.Bruno J, Xu Y, Song J, Berelowitz M. Tissue distribution of somatostatin receptor subtype messenger ribonucleic acid in the rat. Endocrinology 1993; 133: 2561–2567. [DOI] [PubMed] [Google Scholar]
- 5.Kumar U, Laird D, Srikant C, et al. Expression of the five somatostatin receptor (SSTR1–5) subtypes in rat pituitary somatotrophes: Quantitative analysis by double-label immunofluorescence confocal microscopy. Endocrinology 1997; 138: 4473–4476. [DOI] [PubMed] [Google Scholar]
- 6.Kumar U, Ong W-Y, Patel S, et al. Cellular expression of the five somatostatin receptor subtypes (SSTR1–5) in rat hypothalamus: A comparative immunohistochemical analysis. Program Annual Meeting U.S. Endocrine Society, 1999.
- 7.Kumar U, Sasi R, Suresh S, et al. Subtype-selective expression of the five somatostatin receptors (hSSTR1–5) in human pancreatic islet cells: A quantitative double-label immunohistochemical analysis. Diabetes 1999; 48: 77–85. [DOI] [PubMed] [Google Scholar]
- 8.Yamada Y, Post S, Wang K, et al. Cloning and functional characterization of a family of human and mouse somatostatin receptors expressed in brain, gastrointestinal tract, and kidney. Proc Natl Acad Sci USA 1992; 89: 251–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yasuda K, Rens-Domiano S, Breder C, et al. Cloning of a novel somatostatin receptor, SSTR3, coupled to adenylate cyclase. J Biol Chem 1992; 267: 20422–20428. [PubMed] [Google Scholar]
- 10.Raynor K, O’Carroll A, Kong H, et al. Characterization of cloned somatostatin receptors SSTR4 and SSTR5. Mol Pharmacol 1993; 44: 385–392. [PubMed] [Google Scholar]
- 11.Reisine T, Bell G. Molecular biology of somatostatin receptors. Endocr Rev 1995; 16: 427–442. [DOI] [PubMed] [Google Scholar]
- 12.Rohrer S, Birzin E, Mosley R, et al. Rapid identification of subtype-selective agonists of the somatostatin receptor through combinatorial chemistry. Science 1998; 282: 737–740. [DOI] [PubMed] [Google Scholar]
- 13.Mitra S, Mezey E, Hunyady B, et al. Colocalization of somatostatin receptor sst5 and insulin in rat pancreatic beta-cells. Endocrinology 1999; 140: 3790–3796. [DOI] [PubMed] [Google Scholar]
- 14.Rossowski W, Coy D. Potent inhibitory effects of a type four receptor-selective somatostatin analog on rat insulin release. Biochem Biophys Res Commun 1993; 197: 366–371. [DOI] [PubMed] [Google Scholar]
- 15.Coy D, Rossowski W. Somatostatin analogues and multiple receptors: possible physiological roles. Ciba Foundation Symp 1995; 190: 240–254. [DOI] [PubMed] [Google Scholar]
- 16.Rossowski W, Coy D. Specific inhibition of rat pancreatic insulin or glucagon release by receptor-selective somatostatin analogs. Biochem Biophys Res Commun 1994; 205: 341–346. [DOI] [PubMed] [Google Scholar]
- 17.Zambre Y, Ling Z, Chen M, et al. Inhibition of human pancreatic islet insulin release by receptor-selective somatostatin analogs directed to somatostatin receptor subtype 5. Biochem Pharmacol 1999; 57: 1159–1164. [DOI] [PubMed] [Google Scholar]
- 18.Fagan S, Azizzadeh A, Moldovan S, et al. Insulin secretion is inhibited by subtype five somatostatin receptor in the mouse. Surgery 1998; 124: 254–259. [PubMed] [Google Scholar]
- 19.Aguilar-Bryan L, Clement J, Nelson D. Sulfonylurea receptors and ATP-sensitive potassium ion channels. Methods Enzymol 1998; 292: 732–744. [DOI] [PubMed] [Google Scholar]
- 20.Aguilar-Bryan L, Clement J, Gonzalez G, et al. Toward understanding the assembly and structure of KATP channels. Physiol Rev 1998; 78: 227–245. [DOI] [PubMed] [Google Scholar]
- 21.Babenko A, Gonzalez G, Aguilar-Bryan L, et al. Sulfonylurea receptors set the maximal open probability, ATP sensitivity and plasma membrane density of KATP channels. FEBS Lett 1999; 445: 131–136. [DOI] [PubMed] [Google Scholar]
- 22.Satin L. Localized calcium influx in pancreatic beta cells: its significance for Ca2+-dependent insulin secretion from the islets of Langerhans. Endocrine 2000; 13: 251–262. [DOI] [PubMed] [Google Scholar]
- 23.Seghers V, Nakazaki M, DeMayo F, et al. Sur1 knockout mice: A model for KATP channel-independent regulation of insulin secretion. J Biol Chem 2000; 275: 9270–9277. [DOI] [PubMed] [Google Scholar]
- 24.Raynor K, Wang H, Dichter M, et al. Subtypes of brain somatostatin receptors couple to multiple cellular effector systems. Mol Pharmacol 1991; 40: 248–253. [PubMed] [Google Scholar]
- 25.Emami S, Regnauld K, Ferrand N, et al. Stimulatory transducing systems in pancreatic islet cells. Ann NY Acad Sci 1998; 865: 118–131. [DOI] [PubMed] [Google Scholar]
- 26.Koch B, Blalock J, Schonbrunn A. Characterization of the cyclic AMP-independent actions of somatostatin in GH cells. I. An increase in potassium conductance is responsible for both the hyperpolarization and the decrease in intracellular free calcium produced by somatostatin. J Biol Chem 1988; 263: 216–225. [PubMed] [Google Scholar]
- 27.White R, Schonbrunn A, Armstrong D. Somatostatin stimulates Ca2+-activated K+ channels through protein dephosphorylation. Nature 1991; 351: 570–573. [DOI] [PubMed] [Google Scholar]
- 28.Aguilar-Bryan L, Bryan J. ATP-sensitive potassium channels, sulfonylurea receptors, and persistent hyperinsulinemic hypoglycemia of infancy. Diabetes Reviews 1996; 4: 336–346. [Google Scholar]
- 29.Ashcroft F. The yin and yang of the KATP channel. J Physiol 2000; 528: 405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fujii Y, Gonoi T, Yamada Y, et al. Somatostatin receptor subtype SSTR2 mediates the inhibition of high voltage activated calcium channels by somatostatin and its analogue SMS201–995. FEBS Lett 1994; 355: 117–120. [DOI] [PubMed] [Google Scholar]
- 31.Patel Y. Somatostatin and its receptor family. Front Neuroendo 1999; 20: 157–198. [DOI] [PubMed] [Google Scholar]
- 32.Roosterman D, Glassmeier G, Baumeister H, et al. A somatostatin receptor 1 selective ligand inhibits Ca2+ currents in rat insulinoma 1046–38 cells. FEBS Lett 1998; 425: 137–140. [DOI] [PubMed] [Google Scholar]
- 33.Kreienkamp H, Honck H, Richter D. Coupling of rat somatostatin receptor subtypes to a G-protein gated inwardly rectifying potassium channel (GIRK1). FEBS Lett 1997; 419: 92–94. [DOI] [PubMed] [Google Scholar]