

Abstract
Objective
To quantify the various components of splanchnic free fatty acid and very-low-density lipoprotein-triacylglycerol (VLDL-TAG) metabolism in order to gain insight into the mechanisms responsible for the development of fatty liver in severely burned patients, and to determine if decreasing free fatty acid availability by use of propranolol could potentially reduce hepatic fatty acid accumulation.
Summary Background Data
Hepatic fat accumulation results from an imbalance between fatty acid uptake, oxidation, and release via VLDL-TAG. Fatty acid delivery is accelerated in burn patients because of stimulated lipolysis. Since propranolol decreases lipolysis, it should also decrease hepatic fatty acid uptake and thus TAG synthesis.
Methods
Stable isotope-labeled tracers and regional catheterization enabled quantification of various parameters of lipid metabolism across the splanchnic bed in severely burned patients. The acute effects of propranolol treatment were studied in all patients, and in a subgroup of patients the chronic (3 weeks) effects of propranolol were assessed.
Results
The rate of splanchnic uptake of palmitate was 1.68 ± 1.3 μmol/kg/min, whereas the rates of oxidation and VLDL-TG secretion were only 0.12 ± 0.11 and 0.003 ± 0.02 μmol/kg/min, respectively. Propranolol significantly reduced palmitate delivery, and thus palmitate uptake, without significantly affecting oxidation or VLDL-TAG secretion. Thus, palmitate storage was reduced from 1.53 ± 1.30 μmol/kg/min without propranolol to 0.76 ± 0.58 μmol/kg/min after propranolol.
Conclusions
Hepatic fat storage in burn patients is due to low rates of both fatty acid oxidation and VLDL-TAG secretion. Propranolol can decrease hepatic fat storage by limiting fatty acid delivery.
Enlarged livers are commonly observed following burn injury, often resulting in a doubling of liver weight within 15 to 30 days after burn. At least some of the increase in liver size is due to increased hepatic triglyceride (TAG). 1 Animal studies have shown that fatty liver increases the risk for infectious complications, 2,3 and in human patients liver size may increase sufficiently to impede ventilation. Whereas fatty liver is presumed to be related to the nature of nutritional support, the actual mechanism responsible for net fat deposition in the liver of burned patients is not known. 4
Hepatic fat accumulation results from an imbalance between fatty acid uptake, oxidation, and release via very-low-density lipoprotein (VLDL)-triacylglycerols (TAG). Alterations in any or all of these factors could be responsible for the development of fatty liver. Since lipolysis is stimulated after burn injury to a far greater extent than the requirement for fatty acid oxidation, 5 it is reasonable to presume that accelerated hepatic fatty acid uptake resulting from increased free fatty acid (FFA) availability in plasma is the principal initiating factor. However, it is also possible that fatty acids synthesized de novo could contribute to hepatic fat accumulation. Although we recently showed that de novo fatty acid synthesis is not likely a major source of VLDL-TAG in burn patients, 6 we have also found that de novo fatty acid synthesis was stimulated in normal volunteers when hypercaloric amounts of carbohydrate were given for several days. 7 Another possibility is that in burn patients fatty acids taken up by the liver are channeled away from oxidation and towards TAG synthesis. This possibility is suggested by the fact that hyperglycemia/hyperinsulinemia, which is often present in burn patients, inhibits hepatic fatty acid oxidation, but not uptake. 8 Finally, it is possible that the principal defect leading to fatty liver after burn is that the normal packaging and secretion of hepatic TAG as VLDL-TAG are disrupted.
The principal aim of our study was to quantify the various components of splanchnic fatty acid and VLDL-TAG metabolism in order to gain insight into the mechanisms responsible for the development of fatty liver in severely burned patients. Thus, we have quantified fatty acid uptake, oxidation, and secretion in VLDL-TAG across the splanchnic bed in severely burned patients. Our study was further aimed at determining if decreasing FFA availability by use of propranolol could potentially reduce hepatic fatty acid accumulation. Because continuous β-blockade may cause tachyphylaxis, 9 we studied both the acute and long-term effects of propranolol medication on splanchnic fatty acid metabolism.
METHODS
Patients
Tracer infusion studies were performed in eight patients who were admitted to the Burn Unit of the University of Texas Medical Branch and/or the Shriners Burn Hospital in Galveston, Texas. The protocol was approved by the Institutional Review Board of the University of Texas Medical Branch.
Materials
U-13C potassium palmitate (98%+ enriched) and [2,2-2H] potassium palmitate (97% enriched) were purchased from Cambridge Isotope Laboratories (Andover, MA). Human albumin 5% solution was purchased from Baxter Healthcare Corporation (Glendale, CA). Vivonex (15% of calories as free amino acids, 82% as carbohydrate and 3% as fat) came from T.E.N. (Sandoz Nutrition Corporation, Minneapolis, MN) and indocyanine green from Akron (Buffalo Grove, IL).
Experimental Design
Each patient was studied twice and thus served as his or her own control. The patients were randomly allocated to the propranolol group (n = 4) or to the control group (n = 4). The propranolol group received propranolol treatment throughout 3 weeks between the two studies, whereas the control group did not receive any treatment. Propranolol dosage was approximately 2 mg/kg/d, given orally every 4 hours. The dose was titrated to achieve a 25% decease in heart rate from the subject’s own 24-hour average heart rate immediately before drug treatment. Heart rate and blood pressure were monitored continuously throughout the study. Propranolol was given as scheduled during operative procedures.
During both studies, the patients were fed a carbohydrate-rich enteral diet (Vivonex T.E.N), which was infused continuously via nasogastric and duodenal catheters, providing approximately 1.4 times as many calories as the measured resting energy expenditure. The concentration of Vivonex (J/mL) was individually adjusted so that there were no clinical symptoms such as diarrhea. A comparable low-fat/high-carbohydrate diet has been previously shown to eliminate the influx of exogenous fat via absorption 10 so that the FFA and the TAG that appear in plasma under these dietary conditions are from endogenous sources. All studies were performed in the fed state.
To coordinate each study with the clinical treatment, the first study was done 7 days after the first grafting procedure, immediately before the second grafting procedure. A femoral arterial line, a central trilumen venous line, and a hepatic vein catheter via the femoral vein by fluoroscopy were placed on the study day. A lead glove was placed over the genitalia before the procedure. After patient preparation, the right common femoral vein was punctured and a 6 French sheath was inserted. Through this sheath, a straight 5 French catheter with several side holes near its tip was manipulated into the right or middle hepatic vein. This catheterization was performed by using a deflecting-tip 0.035-inch guidewire within the straight catheter. After the catheter was positioned into the hepatic vein, a digital venogram was performed to verify placement, and both the sheath and catheter were infused with heparinized saline to maintain patency. The position of the catheter was confirmed again by a plain-view abdominal x-ray immediately after the end of the study. A short, straight 4 French catheter connected to a pressurized flush setup was then placed retrograde into the right common femoral artery. After both catheters and the sheath had been sutured in place, a sterile transparent dressing was used to cover the vascular entry sites.
The patients received a continuous (0.04 μmol/kg/min), nonprimed infusion of U-13C palmitate for 8 hours during the initial study. The bicarbonate pool was primed using NaH13CO3 (25 μmol/kg). After 4 hours, the patients received a bolus of propranolol (1.5 mg/kg), and then a continuous (0.04 μmol/kg/min), nonprimed infusion of [2,2-2H] palmitate was started. Thus, all subjects were studied in the basal (fed) state, and then during the acute response to propranolol. Both tracers were infused through a central venous catheter using calibrated syringe pumps (Harvard Apparatus, Natick, MA). Blood samples were drawn from a peripheral artery and femoral vein throughout the isotopic tracer infusion. Blood flow was determined twice (at 135 and 375 minutes) using a constant infusion of indocyanine green dissolved in 0.9% saline. The dye was infused through the femoral artery catheter at the rate of 0.5 mg/min for 55 minutes, and blood samples were taken after 40, 45, 50, and 55 minutes simultaneously from the hepatic vein and a peripheral vein.
The second study was performed after 3 weeks with or without (control subjects) propranolol treatment. The second study lasted only 4 hours and was done either with (propranolol group) or without (control group) propranolol infusion.
Determination of Acetate Correction Factor
On a different occasion, a primed (45 μmol/kg), 6-hour continuous infusion (1.5 μmol/kg/min) of [1,2-13C] acetate was performed in the fed state to determine the “acetate correction factor” (i.e., the acetate-carbon recovery rate) for use in the calculation of fatty acid oxidation rate. 11 It was not necessary to determine acetate carbon recovery across the splanchnic region in this study, because we have recently shown that acetate recovery across the splanchnic region is similar to whole body acetate recovery. 12
Analysis of Samples
FFA and VLDL-TAG
Blood samples were placed into 10-mL Vacutainers containing disodium EDTA for the determination of the isotopic enrichment of palmitate bound to VLDL-TAG and albumin-bound free plasma palmitate. All samples were placed on ice. Plasma was separated by centrifugation shortly after sampling. Samples for VLDL isolation were stored at 4°C until they were processed the next morning.
TAG concentration in plasma was determined photospectrometrically at 540 nm (Sigma Diagnostics Inc., St. Louis, MO). VLDL was isolated from 3 mL of plasma by overlaying the plasma with a density equal to 1.006 solution (0.9% NaCl) as described in Aarsland et al. 6 TAG in the VLDL suspension was isolated by TLC, hydrolyzed to FFA, and derivatized to FA methyl esters. Palmitate and total FFA concentrations in plasma were determined by gas chromatography (model 5890; Hewlett-Packard Co., Palo Alto, CA) using heptadecanoic acid as an internal standard. Isotopic enrichment of palmitate was determined by GCMS (model 5992; Hewlett-Packard Co.) in the electron impact ionization mode for the ultimate computation of the tracer/tracee ratio. 6
Blood Flow
Blood samples were placed into 4-mL Vacutainers containing SST gel and clot activator. Plasma was separated by centrifugation shortly after sampling. Dye concentration was determined spectrophotometrically at 805 nm. Splanchnic blood flow was determined as described previously. 8
Blood CO2
Blood samples for CO2 analysis were collected into prechilled tubes containing sodium heparin. Blood CO2 concentration was measured immediately using 965 Ciba Corning CO2 analyzer, and the remaining blood samples were kept frozen until further analysis. For the determination of blood CO2 enrichment, 5 to 10 mL of hyperphosphoric acid was added to 1 mL of blood in a sealed tube to release the CO2. The 13C-to-12C ratio in the headspace was then determined using a V.G. isotope ratio mass spectrometer.
Calculations
The plasma palmitate appearance rate was calculated by means of the steady-state equation. 13 The palmitate and CO2 kinetic parameters for the splanchnic region were calculated using femoral artery and hepatic venous measurements and splanchnic blood flow as described in Sidossis et al. 8
The fractional release of plasma palmitate into VLDL-TAG was calculated from the increase in the enrichment of palmitate in VLDL-TAG over time (%/h) divided by the enrichment of palmitate in the femoral artery, since we found that the latter was similar to that of the intrahepatic precursor pool (unpublished data). The amount of palmitate released into VLDL-TAG was calculated by multiplying the fractional release of plasma palmitate into VLDL-TAG by the VLDL-TAG plasma pool (i.e., arterial VLDL-TAG concentration times plasma volume calculated using the equation from Diraison and Beylot). 14
Statistical Analysis
Results are reported as means ± SE. The effects of propranolol on the various parameters were calculated using a two-tailed Student t test. Significance was set at the 0.05 level.
RESULTS
Subject Characteristics
The patient characteristics are given in Table 1. Their ages ranged from 5.8 to 56.5 years and their body weight from 17.3 to 97.3 kg. The burn size ranged from 56% to 99% of total body surface, of which 41% to 99% was third degree. The subjects from the propranolol group were slightly younger and lighter than those from the control group, but the extent of the burn injury was similar between the two groups. The first study was performed 23 ± 20 days after injury (5–65 days) and the second study 22 ± 7 days after the first (10–31 days). Despite the wide range of ages, responses were similar in all patients.
Table 1. PATIENT CHARACTERISTICS
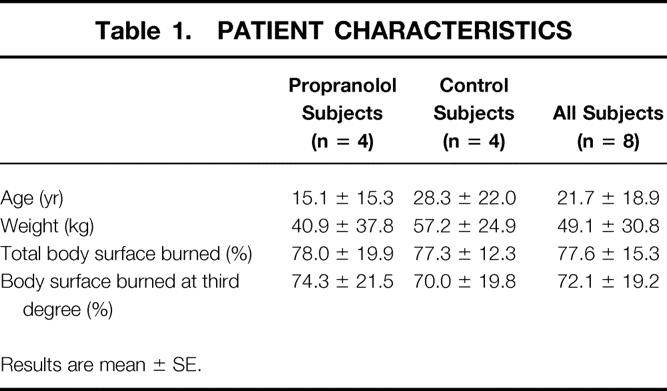
Results are mean ± SE.
Metabolism of Fatty Acids in the Splanchnic Bed in Initial Study
Splanchnic plasma flow and femoral artery and hepatic venous concentrations of FFA and TAG are given in Table 2. Splanchnic inflow (Fin) of palmitate averaged 2.77 ± 2 μmol/kg/min, and the fractional extraction of palmitate was 59.7 ± 12.6%. Therefore, the rate of uptake of palmitate by the splanchnic bed averaged 1.68 ± 1.39 μmol/kg per minute. Palmitate uptake was closely correlated with both Fin and Fout (r > 0.99, P < .001).
Table 2. ACUTE RESPONSE TO PROPRANOLOL
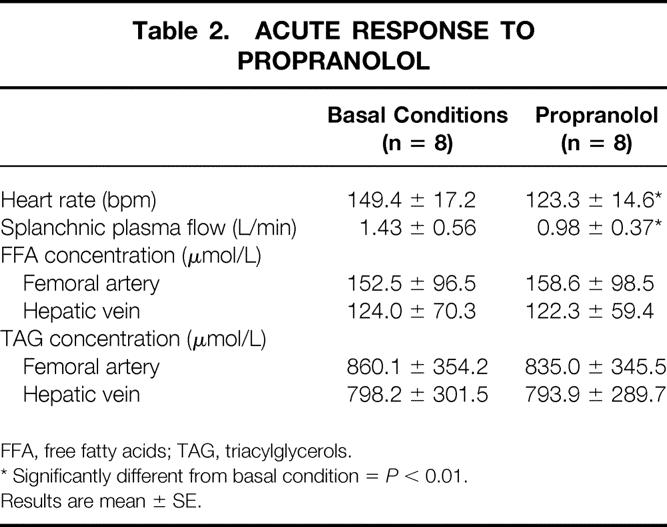
FFA, free fatty acids; TAG, triacylglycerols.
* Significantly different from basal condition =P < 0.01.
Results are mean ± SE.
The splanchnic palmitate oxidation rate was 0.12 ± 0.11 μmol/kg per minute, while the rate of plasma palmitate appearing in plasma VLDL-TAG was only 0.03 ± 0.02 μmol/kg per minute. The latter result was due to a very low fractional synthetic rate of VLDL-TAG (5.9 ± 3.7%/h). Only 3.8 ± 4.1% of the plasma palmitate taken up by the splanchnic bed was released into the plasma in VLDL-TAG. Therefore, the plasma palmitate was stored within the splanchnic bed at a rate of 1.53 ± 1.30 μmol/kg per minute. This represents approximately 0.45 ± 0.38 mg/kg per minute or 0.65 ± 0.55 g/kg per day of TAG stored within the splanchnic bed.
Acute Changes in the Splanchnic Metabolism of Fatty Acids With Propranolol
The main cardiovascular effects of propranolol were a 17% decrease in heart rate and a 31% decrease in splanchnic plasma flow (P < .01 vs. control) (Table 2). The metabolic results are highlighted in Figure 1. The femoral artery and hepatic venous concentrations of FFA and TAG did not change significantly, despite a 13% decrease in whole body palmitate appearance rate (P < .05 vs. control). Splanchnic inflow of palmitate fell to 2.10 ± 1.87 μmol/kg/min (−25%, P < .05 vs. control). The fractional extraction of palmitate did not change significantly (51.2 ± 12.2%), so the uptake of palmitate by the splanchnic bed was 0.96 ± 0.72 μmol/kg per minute (−43%, P < .05 vs. control). Palmitate uptake was closely correlated with both Fin and Fout in both the basal condition and during propranolol administration (r > 0.99, P < .001).
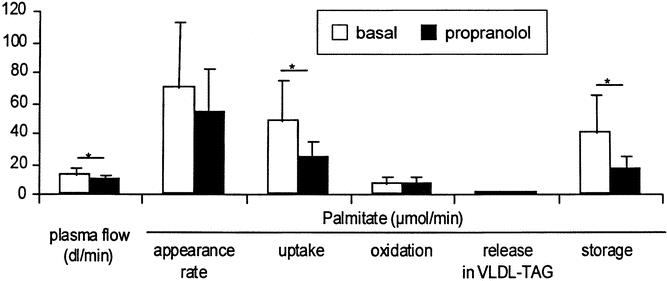
Figure 1. Metabolic responses to propranolol in initial study. Asterisk indicates a significant difference between parameters indicated (P < .05). Appearance rate is the whole body appearance rate. Uptake, oxidation, release, and storage all refer to the splanchnic bed. Plasma flow is the hepatic vein plasma flow (i.e., splanchnic plasma flow).
The splanchnic palmitate oxidation rate and the rate of plasma palmitate reincorporation into VLDL-TAG were not significantly altered by changes in FFA availability (0.16 ± 0.21 and 0.04 ± 0.04 μmol/kg per minute, respectively). The fractional synthetic rate of VLDL-TAG was also unchanged by propranolol (5.1 ± 3.6%/h, P = NS vs. control). Although splanchnic FFA uptake was decreased by propranolol, the percentage of plasma palmitate taken up by the splanchnic bed and released into the VLDL-TAG slightly increased to 7.9 ± 10.4% (P = NS vs. control).
Because of the decrease in splanchnic FFA uptake and the maintenance of VLDL-TAG secretion, the rate of plasma palmitate stored within the splanchnic bed decreased to 0.76 ± 0.58 μmol/kg per minute during propranolol administration (P < .05 vs. control). This represents approximately 0.22 ± 0.17 mg/kg per minute or 0.32 ± 0.24 g/kg per day of palmitate stored as TAG within the splanchnic bed. The amount of palmitate stored within the splanchnic bed was closely correlated to the rate of palmitate uptake (r = 0.96, P < .001, Fig. 2).
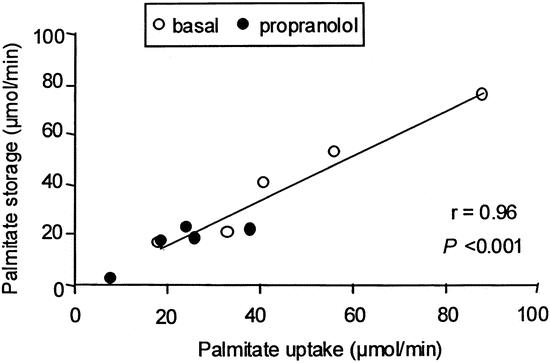
Figure 2. Correlation between splanchnic palmitate uptake and splanchnic palmitate storage in the initial study.
Long-Term Alterations in Splanchnic Metabolism of Fatty Acids With Propranolol Treatment
Body weights of patients changed over time. Therefore, to avoid confusion due to changes in body weight between the two studies, results are now expressed in μmol/min (Tables 3 and 4).
Table 3. EFFECT OF PROPRANOLOL TREATMENT ON PHYSIOLOGICAL PARAMETERS

* Significantly different from initial study =P < .05.
Results are mean ± SE.
Table 4. EFFECT OF PROPRANOLOL TREATMENT ON METABOLIC PARAMETERS

FFA, free fatty acids; TAG, triacylglycerols; VLDL, very-low-density lipoprotein.
* Significantly different from initial study =P < .05.
Initial study was in the basal (fed) state, and the second study was either at the end of 3 weeks of propranolol treatment (propranolol group) or again in the basal (fed) state.
Results are mean ± SE.
Chronic treatment with propranolol maintained a 17% decrease in heart rate and a 30% decrease in plasma flow (P < .05 vs. control). The femoral artery and hepatic venous concentrations of FFA and TAG remained constant. Whole body palmitate appearance rate was unchanged from the initial value, whereas splanchnic Fin of palmitate was decreased by 37% as compared to the initial value (P = .08 vs. control). The fractional extraction of palmitate remained at 55.5 ± 28.2% (P = NS vs. control). Hence, the rate of uptake of palmitate by the splanchnic bed was reduced by 40%, but the difference did not reach the level of significance (P = .16 vs. control). Similarly, the rate of net storage was further reduced by the 3 weeks of propranolol treatment.
The palmitate oxidation rate and the rate of plasma palmitate reincorporation into VLDL-TAG were not significantly altered. Hence, the rate of plasma palmitate stored within the splanchnic bed was decreased by 41%.
Splanchnic Metabolism of Fatty Acids in Control Patients Not Given Propranolol
After 3 weeks without propranolol treatment, heart rate was reduced 8% below the initial value (P < .05 vs. control), but plasma flow was similar (P = NS vs. initial value). The femoral artery and hepatic venous concentrations of FFA increased slightly (P = NS vs. control) and those of TAG remained constant. The whole body palmitate appearance rate increased by 140% (P < .05 vs. control), and splanchnic Fin of palmitate increased (45%), but not significantly. The fractional extraction of palmitate decreased to 45.7 ± 8.5% (−29%, P < .05 vs. initial study), so that the net uptake of palmitate by the splanchnic bed remained unchanged, as did the rate of palmitate oxidation (P = NS vs. control).
The rate of plasma palmitate reincorporation into VLDL-TAG was more than doubled except in one subject (+172%, P = .10 vs. control). However, the rate of plasma palmitate stored within the splanchnic bed was similar to that calculated in the initial study (P = NS vs. control).
DISCUSSION
Our principal aim was to determine the basis for the storage of fatty acids within the liver of severely burned patients. We studied the patients while they received nutritional support consisting of a high-carbohydrate diet, since this is their normal clinical state. Our data indicate that whereas the release of fatty acids into the blood was low in this condition, presumably due to the inhibitory effect of hyperinsulinemia on lipolysis, plasma fatty acids were nonetheless taken up from the blood and stored within the liver due to limitations in both fatty acid oxidation and VLDL-TAG secretion. Coupled with a likely increase in the rate of de novo synthesis of fatty acids from the high- carbohydrate intake, 6 fatty acid storage during this treatment could likely be clinically important. When delivery of fatty acids to the liver was reduced by propranolol treatment, fatty acid uptake was reduced and there was a corresponding fall in the rate at which TAG was stored in the liver.
The accumulation of TAG in the liver results from an imbalance between the uptake of fatty acids and the oxidation and release of fatty acids into the plasma as VLDL-TAG. Whereas we found that splanchnic fatty acid uptake was proportional to fatty acid delivery, splanchnic fatty acid oxidation and VLDL-TAG secretion rates were unresponsive to changes in fatty acid availability. Because the rate of fatty acid oxidation was close to the suppressed rate observed in healthy adults during euglycemia-hyperinsulinemia, 8 it is likely that the low values we observed in the burn patients were also due to the inhibitory effect of the ingested glucose and resultant insulin response. On the other hand, the depressed VLDL-TAG secretion rate was likely a direct consequence of the burn injury. Indeed, our previous studies in burn patients showed that, with or without insulin and glucose nutritional support, the VLDL-TAG secretion rate was reduced by more than 80% in burn patients as compared with healthy subjects. 6,15
The combination of inhibited fatty acid oxidation and depressed VLDL-TAG secretion meant that a large percentage (approximately 85%) of splanchnic uptake of fatty acids was stored. This corresponds to a rate of 0.3 g/kg per day of palmitate stored in the splanchnic bed as TAG, or approximately 0.9 g/kg per day of TAG. We have previously reported that average liver weight increases at a rate of approximately 1 to 2 g/kg per day in severely burned children, 1 and analysis of livers at autopsy suggests that most of the weight gain is due to fat accumulation. Thus, our tracer-determined value for fat accumulation in the current study is reasonable when considered in relation to the rate of liver weight gain generally observed in similar patients. Further, the percent of palmitate uptake that is re-esterified into VLDL-TAG that we calculated previously in burned patients using an entirely different approach relying on 13C-acetate as a tracer ranged from 7% to 14% of total palmitate uptake, depending on the clinical circumstance, which is comparable to the value we observed in the current study.
The impairment of TAG secretion from the liver may be the principal mechanism responsible for the increased hepatic TAG content. Under normal conditions, an increased fatty acid availability also causes enhanced TAG esterification within the liver, but this is balanced by an increased rate of VLDL-TAG secretion, 16 so that TAG accumulation does not occur. Several factors could potentially limit the secretion of VLDL-TAG in burn patients. A deficiency in apo-B or MTP, two proteins essential for the packaging of VLDL, is one, since a general protein catabolic state exists after burn, 17 and a deficiency in these proteins lends to fatty liver. 18 Another possibility is that choline is deficient after burn injury. Choline deficiency also causes fatty liver, 19,20 and we found that the plasma choline concentration was significantly lower in burn patients (3.96 ± 0.50 μmol/L) compared with healthy subjects (8–12 μmol/L) (unpublished results). Choline is a precursor of phosphatidylcholine synthesis, which is an important component of VLDL, so choline deficiency may be limiting VLDL-TAG secretion in burn patients.
Despite the decrease in VLDL-TAG secretion rate in the burn patients, the plasma concentration of VLDL-TAG was elevated in comparison with healthy subjects. 15 This was due to an impaired peripheral clearance. It is possible that the impaired peripheral clearance indirectly contributed to the reduction in VLDL-TAG secretion, if the latter is responsive to the prevailing plasma concentration of VLDL-TG. The impaired VLDL-TAG clearance may result from a reduced level of apoproteins (C-II, C-III, A-I) necessary for TAG metabolism. 21 Another possibility is that a decreased activity of the enzyme lipoprotein lipase (LPL), which hydrolyzes the circulating TAG to provide fatty acids to peripheral tissues, was responsible for decreased VLDL-TAG clearance. Decreased LPL activity has been reported in adipose and muscle tissues of gram-negative septic rats and critically ill patients. 22–25 Similarly, deficient LPL activity was observed in postheparin plasma of infected rabbits with elevated VLDL-TAG concentration. 26 However, Verschoor et al found that VLDL-TAG clearance was unrelated to changes in adipose LPL activity in rats, 27 and no study has directly addressed the effect of burn on LPL activity. Thus, although a decrease in clearance would be expected to follow from the decreased activity of LPL, the role for changes in LPL activity in our study remains speculative.
Our study revealed a previously unrecognized general characteristic of splanchnic fatty acid metabolism. We found that fatty acid uptake by the splanchnic bed is a function of fatty acid delivery (i.e., flow times concentration), regardless of whether concentration changes. In other words, the rate of blood flow is a major determinant of splanchnic fatty acid uptake. The control of FFA uptake by blood flow has been barely discussed previously despite an abundant literature on fat metabolism during exercise or ischemia. Our data demonstrate that in severely burned patients, FFA extraction by the splanchnic bed is primarily determined by the amount of FFA delivered per unit of time rather than the plasma FFA concentration itself or by the fate of FFA within the splanchnic bed.
We pooled the results from subjects ranging in age from 5 to 56 years. Our justification was that the primary endpoint analysis was a paired design in which each subject served as his or her own control. Further, we have studied both adults and children with severe burns previously and found no differences in either the rate of appearance of fatty acids (8.22 ± 2.86 and 8.30 ± 1.71 mmol/kg per day for adults and children, respectively) or the rate of secretion of VLDL-TAG (19 ± 65 vs. 165 ± 138 μmol/kg per day for adults and children, respectively). 1,6 Further, we found no relation between age and the primary endpoint values in the current study. Therefore, although a more homogenous subject pool in terms of age might have been desirable, it is unlikely that our conclusions were affected by the wide range of age we examined.
The major clinical implication of our findings is that the storage of fatty acids within the splanchnic bed is closely related to the plasma fatty acid availability. As delivery of fatty acids to the liver increases, uptake increases proportionately, yet oxidation is not responsive to changes in uptake. Fatty acids are therefore channeled into hepatic TAG, the release of which into the blood (via VLDL-TAG) is inhibited in burn patients. Our results therefore suggest that, to at least some extent, fat accumulation in the liver is part of the metabolic response to burn injury, irrespective of the diet. Even during the conditions of suppressed lipolysis studied in the current investigation, VLDL-TAG secretion could not keep pace with splanchnic uptake of plasma FFA. Any further stimulation of hepatic TAG formation would be expected to result in even greater TAG storage. Hence, whereas the high carbohydrate feeding used in this study inhibits hepatic fatty acid oxidation and stimulates de novo lipogenesis, 6,15 substitution of fat for carbohydrate would not likely improve the situation. This is because a greater proportion of fat in the diet would increase hepatic fatty acid uptake and thus storage. Fatty acid deposition in the liver would be expected to occur even during fasting in burn patients, since the plasma FFA levels are markedly elevated above normal in postabsorptive burn patients. 5 It is thus not surprising that histologic examination showed that the amount of fat storage in the liver of critically ill patients was unrelated to the nature of nutritional support, but was primarily a function of the seriousness of the illness. 4 This presumably reflects that the extent of stimulation of lipolysis is related to the seriousness of the illness.
Because it may not be possible to avoid fatty livers in burn patients receiving nutritional support, pharmacological management may be necessary to preserve the integrity of the liver. Our results showed that a decrease in splanchnic fatty acid availability caused by propranolol was associated with a decrease in splanchnic fatty acid storage. Further, whereas it might have been expected that patients would have developed tachyphylaxis to propranolol, 9 3 weeks of propranolol treatment caused even further reductions in fatty acid availability and reduced TAG storage in the liver as compared to untreated counterparts. The major effect of propranolol in the current study was a decrease in splanchnic blood flow. An inhibition of lipolysis might also have been expected, 28 but this probably did not occur because lipolysis was already highly inhibited by the insulin response to the high-carbohydrate diet. The consequence of the already low rate of lipolysis in this circumstance was that the reduced inflow of fatty acids into the splanchnic bed in the initial study was due entirely to a reduced blood flow. Further, 3 weeks of propranolol treatment maintained a depressed rate of lipolysis, whereas the latter was doubled in the time-control patients. We conclude, therefore, that propranolol treatment may benefit burn patients by decreasing hepatic TAG storage. Other benefits of propranolol treatment in burn patients have been previously reported in terms of cardiovascular response 29 and muscle protein metabolism. 30
In conclusion, in burn patients receiving a high-carbohydrate diet, fatty acid storage within the splanchnic bed is substantial and proportional to plasma fatty acid availability, because both fatty acid oxidation and release into VLDL-TAG are highly depressed. An additional decrease in splanchnic fatty acid availability using propranolol is associated with a decrease in fatty acid storage. The propranolol effect persisted after 3 weeks of medication. Therefore, propranolol may benefit burn patients with regard to hepatic TAG storage.
Footnotes
Correspondence: Robert R. Wolfe, PhD, Shriners Burns Hospital, 815 Market Street, Galveston, TX 77550.
E-mail: rwolfe@utmb.edu
Supported by NIH grants DK34817 and GM56687 and grant 8490 from Shriners Hospitals for Children.
Accepted for publication December 4, 2001.
References
- 1.Aarsland A, Chinkes D, Wolfe RR, et al. Beta-blockade lowers peripheral lipolysis in burn patients receiving growth hormone. Rate of hepatic very low density lipoprotein triglyceride secretion remains unchanged. Ann Surg 1996; 223: 777–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hirsch RL, McKay DG, Travers RI. Hyperlipidemia, fatty liver and bromsulfophthalein retention in rabbits injected with bacterial endotoxin. J Lipid Res 1976; 5: 563–568. [PubMed] [Google Scholar]
- 3.Yang SQ, Lin HZ, Lane MD, et al. Obesity increases sensitivity to endotoxin liver injury: Implications for the pathogenesis of steatohepatitis. Proc Natl Acad Sci USA 1997; 94: 2557–2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wolfe BM, Walker BK, Shawl DB. Effect of total parenteral nutrition on hepatic histology. Arch Surg 1988; 123: 1084–1090. [DOI] [PubMed] [Google Scholar]
- 5.Wolfe RR, Herndon DN, Jahoor F, et al. Effect of severe burn injury on substrate cycling by glucose and fatty acids. N Engl J Med 1987; 317: 403–408. [DOI] [PubMed] [Google Scholar]
- 6.Aarsland A, Chinkes DL, Sakurai Y, et al. Insulin therapy in burn patients does not contribute to hepatic triglyceride production. J Clin Invest 1998; 101: 2233–2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aarsland A, Wolfe RR. Hepatic secretion of VLDL fatty acids during stimulated lipogenesis in men. J Lipid Res 1998; 39: 1280–1286. [PubMed] [Google Scholar]
- 8.Sidossis LS, Mittendorfer B, Walser E, et al. Hyperglycemia-induced inhibition of splanchnic fatty acid oxidation increases hepatic triacylglycerol secretion. Am J Physiol 1998; 275: E798–E805. [DOI] [PubMed] [Google Scholar]
- 9.Klein S, Peters EJ, Holland OB, et al. Effect of short- and long-term beta-adrenergic blockade on lipolysis during fasting in humans. Am J Physiol 1989; 257: E65–E73. [DOI] [PubMed] [Google Scholar]
- 10.Grundy SM. Biliary lipids, gallstones and treatment of hyperlipidaemia. Eur J Clin Invest 1979; 9: 179–180. [DOI] [PubMed] [Google Scholar]
- 11.Sidossis LS, Coggan AR, Gastaldelli A, et al. A new correction factor for use in tracer estimations of plasma fatty acid oxidation. Am J Physiol 1995; 269: E649–656. [DOI] [PubMed] [Google Scholar]
- 12.Mittendorfer B, Sidossis LS, Walser E, et al. Regional acetate kinetics and oxidation in human volunteers. Am J Physiol 1998; 274: E978–E983. [DOI] [PubMed] [Google Scholar]
- 13.Wolfe RR. Radioactive and Stable Isotope Tracers in Biomedicine. Principles and Practice of Kinetic Analysis. New York: Wiley-Liss Inc., 1992.
- 14.Diraison F, Beylot M. Role of human liver lipogenesis and reesterification in triglycerides secretion and in FFA reesterification. Am J Physiol 1998; 274: E321–E327. [DOI] [PubMed] [Google Scholar]
- 15.Sidossis LS, Mittendorfer B, Chinkes D, et al. Effect of hyperglycemia-hyperinsulinemia on whole body and regional fatty acid metabolism. Am J Physiol 1999; 276: E427–E434. [DOI] [PubMed] [Google Scholar]
- 16.Hopkins PN, Williams RR. A simplified approach to lipoprotein kinetics and factors affecting serum cholesterol and triglyceride concentrations. Am J Clin Nutr 1981; 34: 2560–2590. [DOI] [PubMed] [Google Scholar]
- 17.Wolfe RR. Alterations in protein metabolism due to the stress of injury and infection. In: Protein and Amino Acids. Washington DC: National Academy Press; 1999: 279–284.
- 18.Marcos E, Mazur A, Cardot P, et al. Serum apolipoprotein B and A-I and naturally occurring fatty liver in dairy cows. Lipids 1990; 25: 575–577. [DOI] [PubMed] [Google Scholar]
- 19.Nolan JP, Vilayat M. Endotoxin and the liver. I. Toxicity in rats with choline deficient fatty livers. Proc Soc Exp Biol Med 1968; 129: 29–31. [DOI] [PubMed] [Google Scholar]
- 20.Yamamoto A, Sano M, Isozaki M. Studies on phospholipid metabolism in choline deficient fatty liver. J Biochem (Tokyo) 1969; 65: 85–91. [PubMed] [Google Scholar]
- 21.Vega GL, Grundy SM. Pathogenesis of hypertriglyceridemia: implications for coronary heart disease and therapy. Adv Exp Med Biol 1988; 243: 311–326. [DOI] [PubMed] [Google Scholar]
- 22.Alvares C, Ramos A. Lipids, lipoproteins, and apoproteins in serum during infection. Clin Chem 1986; 32: 142–145. [PubMed] [Google Scholar]
- 23.Grundy SM. Hypertriglyderidemia: mechanisms, clinical significance, and treatment. Med Clin North Am 1982; 66: 519–535. [DOI] [PubMed] [Google Scholar]
- 24.Lanza-Jacoby S, Lansey SC, Cleary MP, et al. Alterations in lipogenic enzymes and lipoprotein lipase activity during gram-negative sepsis in the rat. Arch Surg 1982; 117: 144–147. [DOI] [PubMed] [Google Scholar]
- 25.Liao W, Floren CH. Endotoxin, cytokines, and hyperlipidemia. Scand J Gastroenterol 1993; 28: 97–103. [DOI] [PubMed] [Google Scholar]
- 26.Rouzer CA, Cerami A. Hypertriglyceridemia associated with Trypanosoma brucei brucei infection in rabbits: role of defective triglyceride removal. Mol Biochem Parasitol 1980; 2: 31–38. [DOI] [PubMed] [Google Scholar]
- 27.Verschoor L, Chen YD, Reaven GM. In search of a relationship between physiologically-induced variations in adipose tissue lipoprotein lipase activity and very low-density lipoprotein kinetics in normal rats. Metabol 1982; 31: 499–503. [DOI] [PubMed] [Google Scholar]
- 28.Wolfe RR, Herndon DN, Peters EJ, et al. Regulation of lipolysis in severely burned children. Ann Surg 1987; 206: 214–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Minfree PK, Barrow RE, Desai M, et al. Improved myocardial oxygen utilization following propranolol infusion in adolescents with post-burn hypermetabolism. J Pediatr Surg 1989; 24: 806–810. [DOI] [PubMed] [Google Scholar]
- 30.Herndon DN, Hart DW, Wolf SE, et al. Reversal of catabolism after severe burn: The effect of long-term β-blockade. N Engl J Med 2001; 345 (17): 1223–1229. [DOI] [PubMed] [Google Scholar]