

Abstract
Objective
To evaluate the incidence of posttransplant lymphoproliferative disease (PTLD) and the risk factors and the impact of this complication on survival outcomes in a large cohort of liver transplant recipients at a single institution.
Summary Background Data
Liver transplantation has been accepted as a therapeutic option for patients with end-stage liver disease since 1983, in large part due to the availability and reliance on the use of nonspecifically directed immunosuppression. However, as predicted and subsequently verified in 1968, an increased incidence of certain de novo malignancies has been observed, particularly with regards to lymphoid neoplasms. While many reports have confirmed and clarified the nature of PTLD, the literature is fraught with conflicting experience and outcomes with PTLD.
Methods
Four thousand consecutive patients who underwent liver transplants between February 1981 and April 1998 were included in this analysis and were followed to November 2001. The effect of recipient age at the time of transplant, recipient gender, diagnosis, baseline immunosuppression, grading of PTLD, and association with Epstein-Barr virus were compared. The causes of death were also examined. Treatment for PTLD varied over the 20-year period, but all included massive reduction or elimination of baseline immunosuppression.
Results
The 1-year patient survival for liver transplant patients with PTLD was 85%, while the overall patient survival for the entire cohort was 53%. The actuarial 20-year survival was estimated at 45%. The overall median time to PTLD presentation was 10 months, and children had an incidence of PTLD that was threefold higher than adults. Patient survival was better in children, in patients transplanted in the era of tacrolimus immunosuppression, in patients with polymorphic PTLD, and in those with limited disease. Interestingly, neither the presence or absence of Epstein-Barr virus nor the timing of PTLD presentation appeared to influence overall patient survival. Patients transplanted for alcohol-related liver disease had a similar incidence of PTLD but had a higher risk of mortality.
Conclusions
While PTLD continues to pose problems in patients receiving liver transplants, improvements in patient survival have been observed over time. While it is too early to assess the impact of new advances in prophylaxis, diagnosis, and treatment, such approaches are based on an increased knowledge of the pathophysiology of PTLD.
Transplantation of solid organs has been successful in large part due to the development of immunosuppressive regimens that have controlled the recipient’s immune system from rejecting the allograft. By suppressing recipient T lymphocytes with cyclosporin or tacrolimus or reversing rejection with antilymphocyte agents such as ATGAM or OKT3, rejection has become a rare cause of allograft loss. 1 However, the penalty for the nonspecific nature of immunosuppression is the susceptibility of the recipient to the development of opportunistic infections (including viral, fungal, and protozoal organisms), as well as the increased risk of developing malignancies. 2
Posttransplant lymphoproliferative disease (PTLD) can be considered, for the most part, an opportunistic infectious complication that arises after transplantation, usually involving the Epstein-Barr virus (EBV). 3 Lymphoid tumors were first described in transplant patients in 1968 and were called “reticulum cell sarcomas”; a subgroup of these was termed “pseudolymphomas” in recognition of their ability to undergo regression after reduction of immunosuppression. 4–6 PTLD describes a heterogeneous group of lymphoproliferative diseases ranging from benign polyclonal B-cell proliferation, as seen in acute EBV infections (e.g., mononucleosis), to a relatively malignant monoclonal lymphomatous lesion. In addition, the spectrum of presentations varies from localized to disseminated involvement, and nodal to extranodal, including the allograft organ itself. 7,8
The risk factors and incidence of PTLD, as well as outcomes after the development of this complication in liver transplantation (LTX), are not clearly appreciated, in part due to variations in the study population, changing definitions of PTLD, improved detection methods, and a higher index of suspicion. The current study assessed the incidence of PTLD, the risk factors, and the impact of this complication on survival outcomes in a large cohort of LTX recipients at a single institution.
METHODS
The study subjects were the first 4,000 consecutive patients who underwent LTX since the inception of the program at the University of Pittsburgh (February 1981 to April 1998) and have been described elsewhere. 1 Briefly, this group of patients received a total of 4,947 allografts. Nine hundred twenty-two patients in our overall LTX experience were excluded from analysis because they were transplanted at the VA Medical Center, received combined liver and intestinal allografts, or did not have a minimum of 3 years of follow-up. The study populations were analyzed based on the age of the recipient at the time of transplant (i.e., adult vs. pediatric) and into two timeframes (based on the routine use of cyclosporin or tacrolimus). The demographics of the patients studied with respect to immunosuppression, age, and follow-up time are shown in Table 1. The follow-up between patients on cyclosporin was significantly longer than for tacrolimus (P < .01).
Table 1. PATIENT DEMOGRAPHICS
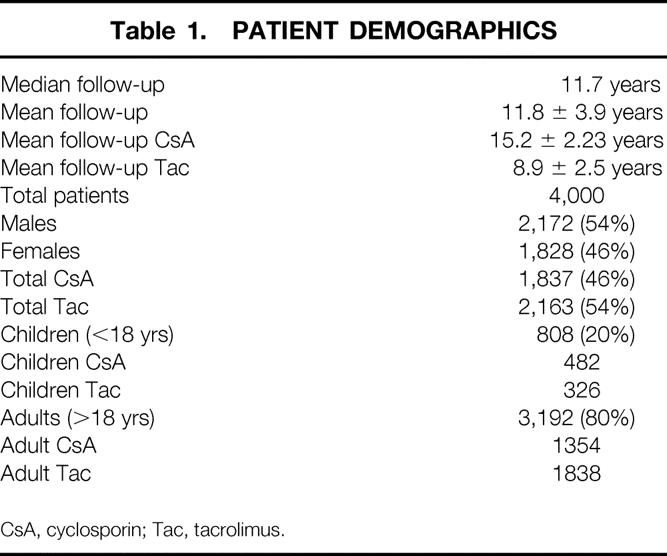
CsA, cyclosporin; Tac, tacrolimus.
A single experienced transplant pathologist (M.N.), in a blinded fashion, reviewed all PTLD specimens that were available from the beginning of the program. Since 1991, in situ hybridization with probes to detect the EBER-1 gene was added to document the presence or absence of EBV in PTLD specimens. 9 The scoring system used was adapted from the Society for Hematopathology Workshop held in 1995, which defined the spectrum of recognized PTLD. 10 The characteristics used to define reactive or early lesions, polymorphic PTLD, and lesions that appeared to represent lymphomas or hematopoietic neoplasms, including a category for lesions such as plasmacytomas, T-cell rich B-cell lymphomas, and T-cell lymphomas under the umbrella term of PTLD are shown in Table 2.
Table 2. CLASSIFICATION SYSTEM FOR PTLD
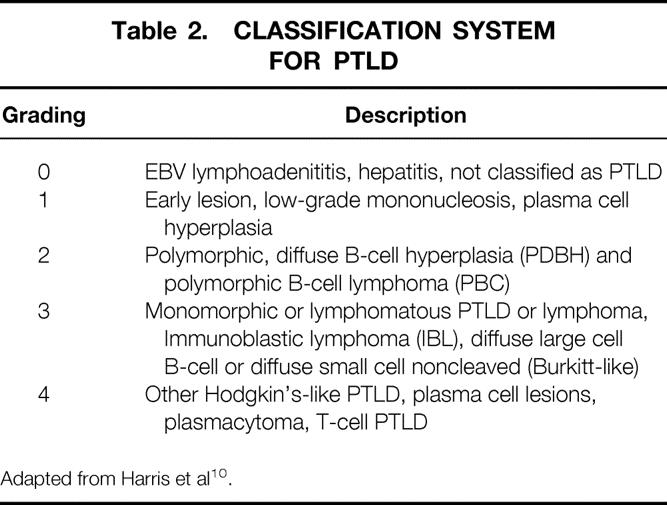
Adapted from Harris et al 10.
All patient information was collected prospectively and entered into the Thomas Starzl Transplantation Institute Electronic Data Interface for Transplantation (EDIT), which stores demographics, laboratory tests, medications, pathology, and other relevant clinical information by interfacing with all hospital information systems and also includes manually entered data from external sources. Data for analysis were rendered anonymous by stripping it of unique patient identifiers by an “honest broker,” according to the requirements of the exempt University of Pittsburgh Institutional Review Board-approved protocol (IRB#020177).
Statistical Analysis
Kaplan-Meier estimates were used to calculate survival curves. Differences in survival curves were compared using log-rank statistics. Differences in proportions were tested using the chi-square test (or Fisher exact test). P < .05 was considered statistically significant.
RESULTS
Incidence of PTLD
Of the 4,000 LTX patients studied, 170 (4.3%) were found to have PTLD; 10 of these cases were only diagnosed or confirmed at autopsy. 11 The incidence of PTLD was significantly higher in children (9.7%) compared to adults (2.9%) (P < .01). Although the overall incidence of PTLD was similar between patients receiving cyclosporin and tacrolimus, the incidence of PTLD in children was higher under tacrolimus than cyclosporin (12.6% vs. 7.7%, P = .06), while the incidence of PTLD in adults was no different based on immunosuppression (3.1% with cyclosporin vs. 2.6% with tacrolimus). The difference in pediatric PTLD incidence was in part due to a significantly higher early mortality in the cyclosporin group versus the tacrolimus group (32% vs. 15% at 3 years post-LTX, respectively), thus reducing the at-risk population. 12 The diagnosis-associated incidence of PTLD is shown in Table 3. The higher rate of PTLD in the “metabolic” and “biliary atresia” groups reflects the preponderance of children for these two indications.
Table 3. INCIDENCE OF PTLD BASED ON GROUPING FOR LTX INDICATIONS
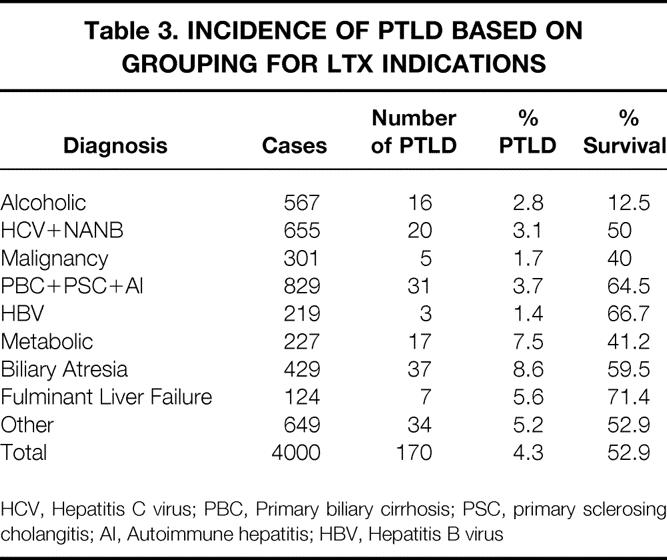
HCV, Hepatitis C virus; PBC, Primary biliary cirrhosis; PSC, primary sclerosing cholangitis; AI, Autoimmune hepatitis; HBV, Hepatitis B virus
Timing of PTLD
The overall median time to development of PTLD was 10 months. This was significantly shorter in children compared to adults (8.1 months vs. 15 months, P = .02). In adults, cyclosporin was associated with a shorter time to PTLD development compared to tacrolimus (6.1 months vs. 23.6 months, P = .01), while the opposite was true for children (27 months cyclosporin vs. 4.7 months tacrolimus). Figure 1 demonstrates the wide range in the timing to developing PTLD.
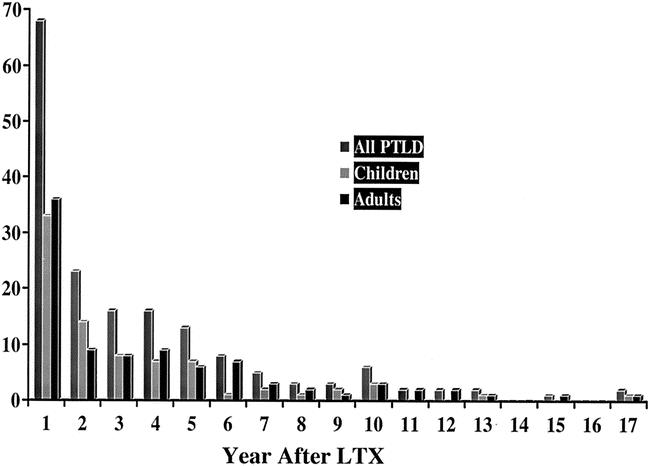
Figure 1. Incidence of PTLD as a measure of Time after LTX. Y-axis represents actual number of cases.
Presentation of PTLD
In hematologic malignancies, staging of disease correlates with treatment response and survival, but little data exist in the area of PTLD. Table 4 summarizes the number of patients with single- or multiple-site involvement with PTLD. A slightly greater number of patients presented with single-site involvement (58%) compared to multiple sites (41%) (P = NS), and this was no different among the age or immunosuppressive categories. The locations of PTLD are shown in Table 5. Lymph nodes were the most predominant site involved (35%); in children this was higher (41%) compared to adults (31%). Gastrointestinal involvement was seen in 25%, while the liver and spleen was involved in 16% of cases. CNS involvement was present in only 4% of cases.
Table 4. EXTENT OF PTLD INVOLVEMENT—SINGLE VS. MULTIPLE SITES*
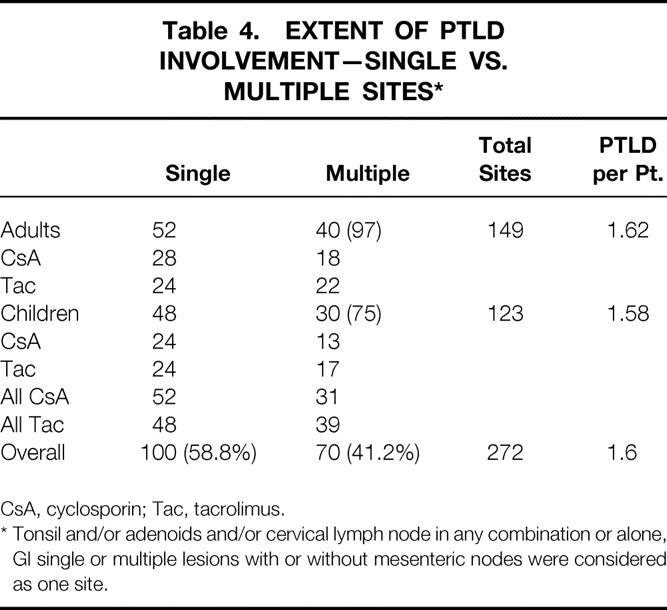
CsA, cyclosporin; Tac, tacrolimus.
* Tonsil and/or adenoids and/or cervical lymph node in any combination or alone, GI single or multiple lesions with or without mesenteric nodes were considered as one site.
Table 5. LOCATION OF PTLD INVOLVEMENT

CsA, cyclosporin; Tac, tacrolimus.
Grading of PTLD
Using the grading system adopted by the Society for Hematopathology and revised by the European-American Lymphoma Group, 10 148 of the 170 cases of PTLD were classified. As shown in Table 6, 12% were considered as “early lesions” grade I, 35% were classified as “PTLD–polymorphic” grade II, 43% were categorized as “PTLD–monomorphic” grade III, and 10% were classified as “PTLD–other” grade IV. While there were no notable differences in the grading based on type of immunosuppression, there was a notable difference between adults and children. Sixty-eight percent of pediatric PTLD cases were classified as grades I or II, while 70% of adult PTLD cases were classified as grades III or IV (P < .01). Two cases in grade IV were T-cell PTLD.
Table 6. GRADING OF PTLD

CsA, cyclosporin; Tac, tacrolimus.
* Using criteria described by Harris et al 10 —a total of 148 cases available.
Role of EBV
With the availability of probes to detect the EBV-encoded small RNA, EBV can be detected in paraffin-fixed specimens using in situ hybridization. 9 EBER was positive in 80% of samples, while EBER was not detected in 20% of the 104 PTLD samples studied. Of interest was that 98% of pediatric PTLD cases were EBER-positive, while only 68% of adult PTLD cases were EBER-positive.
Patient Survival
The actuarial patient survival rates for entire population of PTLD patients at 1, 5, 10, 15 and 20 years were 85%, 69%, 55%, 47%, and 45% respectively, as shown in Figure 2. While there was a numerical difference in survival, with women having a better survival than men, this was only evident at 10 years after PTLD diagnosis and did not reach statistical significance. Long-term survival rates for pediatric patients with PTLD were better than for adults (60% pediatric at 15 years, compared to 39% for adults) but did not quite reach significant difference (P = .06) (Fig. 3). As a reflection of both the impact of improvements over time and with immunosuppression, survival in the tacrolimus group was significantly better than for cyclosporin (60% vs. 40% by 12 years) (P < .02) (Fig. 4). Other factors that appeared to have a positive effect on survival included grade I PTLD versus grades II to IV PTLD (P = .04) (Fig. 5) and single site versus multiple site (P < .02) (Fig. 6). No effect of EBER positivity or time to development of PTLD was apparent on patient survival (data not shown). As noted in Table 3, patients transplanted for alcohol-related liver disease, who developed PTLD, had a higher risk of dying.
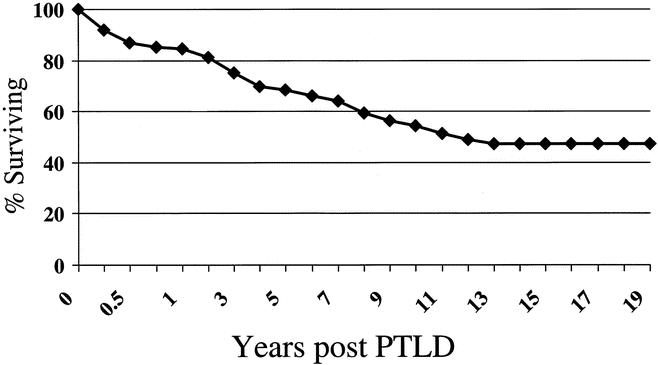
Figure 2. Overall survival after PTLD diagnosis.
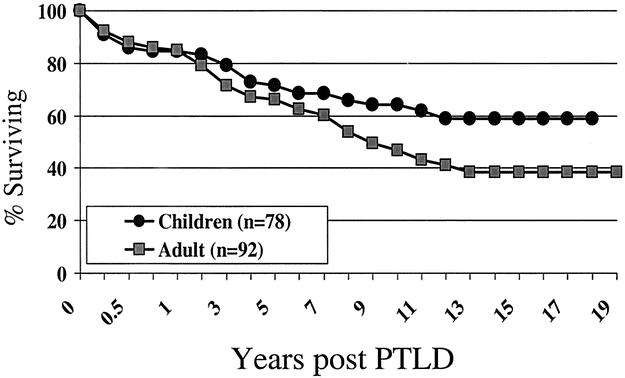
Figure 3. Survival after PTLD diagnosis - age effect.
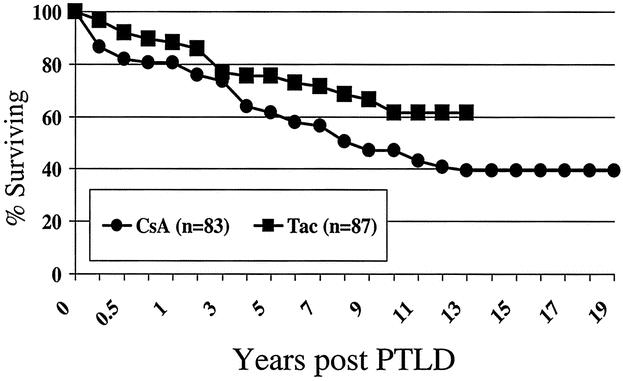
Figure 4. Survival after PTLD diagnosis - effect of baseline immunosuppressive agent.
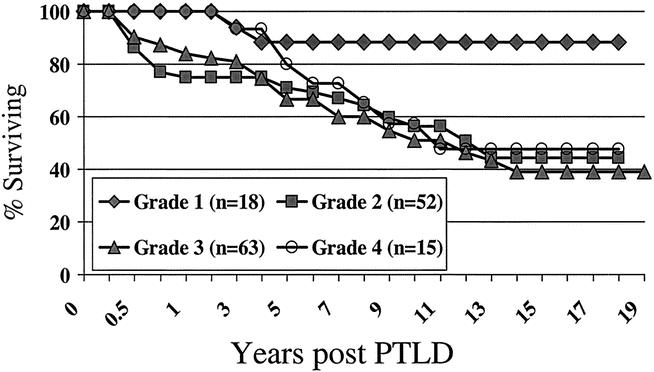
Figure 5. Survival after PTLD diagnosis - effect of grade.
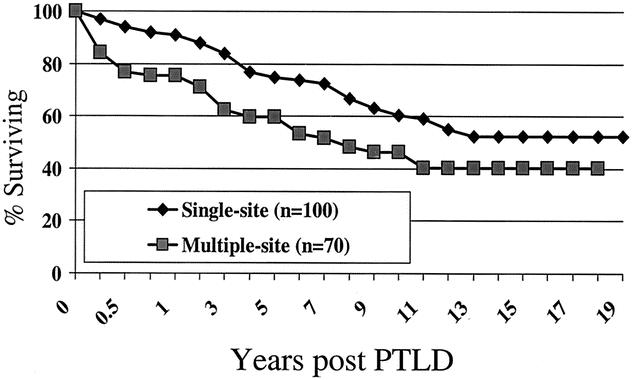
Figure 6. Survival after PTLD diagnosis - effect of disease dissemi-nation.
Causes of Death
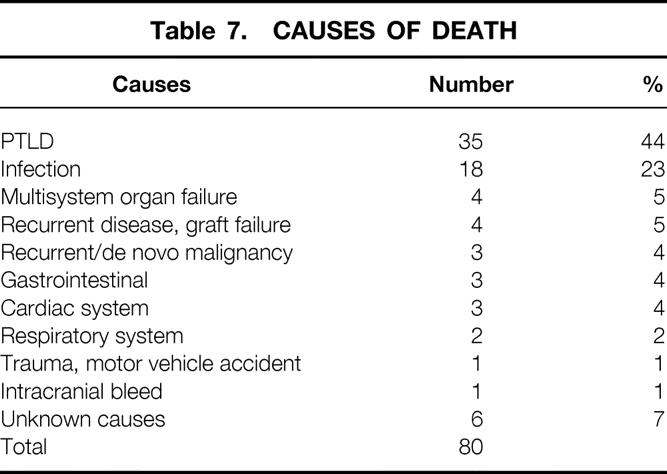
A total of 80 patients (47.1%) died during the entire follow-up period. The causes of death are shown in Table 7. PTLD was thought to be the major contributing cause of death in 44% (n = 35) patients with PTLD. Infection and multisystem organ failure accounted for the second most common causes of death, representing 28% of all deaths. This was followed by recurrent or de novo cancers (5%) and recurrent disease (5%).
Table 7. CAUSES OF DEATH
Treatment
As one would expect, treatment for PTLD varied considerably over the 20-year period. However, the mainstay of treatment was immunosuppressive drug reduction or discontinuation in all cases where this could be documented (the exceptions are patients in whom the diagnosis of PTLD was not made before death). Other treatments included antiviral therapy (e.g., acyclovir or ganciclovir) in 82/122 cases (67%), followed by chemotherapy or radiotherapy (31%), surgery (17%), and more recently the use of anti-B-cell monoclonal antibodies (11%). Immunomodulation or immunotherapy was attempted in 6% of cases.
DISCUSSION
The overall incidence of PTLD in LTX patients is estimated to be 2% to 3% overall. 2,8,13,14 However, there are populations of LTX recipients that can be identified as “high-risk” patients. These include patients with lack of previous EBV infection (i.e., EBV seronegative); pediatric transplant recipients; and those who receive antilymphocyte antibodies. These risk factors are at least additive, so that the pediatric LTX transplant recipient who is EBV seronegative and requires antilymphocyte antibodies is at extremely high risk (PTLD risk up to 30%). 8,15–17 Other risk factors for PTLD have been implicated, such as hepatitis C virus (HCV) coinfection in liver transplant recipients. 18,19 We did not observe this correlation in our patients: the incidence of PTLD in the HCV-positive group was 3.01% versus 2.85% of HCV-negative adult LTX recipients.
Before 1981, lymphoid tumors in transplant patients were uniformly referred to as immunoblastic sarcomas. That year, Frizzera et al from the University of Minnesota examined tumors from a small number of renal transplant recipients. 20 They observed several forms of lymphoproliferation that had not been previously described and applied the term “polymorphic” to emphasize the heterogeneity in size and shape of the tumor cells. Ancillary studies showed the tumors to comprise B lymphocytes. They also stressed that the behavior of PTLD could not be reliably predicted by pathologic studies alone. In 1988, we reported our experience with PTLD observed in the University of Pittsburgh transplant population. We were unable to discern any significant difference in the clinical behavior of the two types of polymorphic lesions and combined them under the heading “polymorphic PTLD.” In contrast, lesions that resembled typical non-Hodgkin’s lymphomas were recognized as a variant of PTLD and the term “monomorphic PTLD” was introduced to distinguish these lesions from polymorphic PTLD. 7 As shown in this study, we were unable to show an association between poorer outcomes and more advanced grades of PTLD, although patients with “early” lesions appeared to have a better survival rate. However, it has been suggested that further categorization of PTLD is possible based on combined pathologic and molecular features—specifically, that more recalcitrant tumors have a monomorphic histology, are monoclonal, and contain rearrangements of the c-myc protooncogene. 21 We did not routinely perform analysis for chromosomal rearrangements in our experience, but we and others have clearly shown that gene analysis can help to assess clonality and possibly prognosis. 21–24 Broader application of molecular techniques may help to distinguish lower-risk from higher-risk PTLD for purposes of treatment.
Treatment of PTLD is one of the most controversial areas in solid organ transplantation. 12 The lack of a clear consensus in the management of patients with established PTLD stems, in part, from the limited understanding of its pathogenesis, the lack of characterization of PTLD, and the specific immune defects associated with PTLD. Nevertheless, based on single-center reports, four major areas of treatment should be considered: reduction of immunosuppression; chemo- and biologic therapy; anti-B-cell monoclonal antibody therapy; and cell-based therapies. We believe that the comparatively good outcomes for PTLD that are reported here, and that span the 20-year existence of this program, hinge on the principle of recovery of the recipient’s immune response, leading to modulation of the PTLD, generally by reducing or eliminating immunosuppression. 6 Our current algorithm for treatment in documented or suspected PTLD is the initial intervention of reduction in immunosuppression. However, how much reduction, for how long, and how to predict the response to such reduction is unknown. Regression of monoclonal and polyclonal lesions following reduction of the dose of immunosuppression ranges from 23% to 50%. 6,12 Potential consideration for the level of immunosuppression reduction should assess the following: the severity of illness, the location and presentation of PTLD, the length and type of immunosuppressive therapy, and the immunohistochemical and molecular characterization of the PTLD. At the ASTP/ASTS Workshop on PTLD, 12 the consensus in critically ill patients with extensive disease was to decrease prednisone to a maintenance dose of 7.5 to 10 mg/d, with all other immunosuppression stopped. If there is no response or the response is not adequate within 7 to 21 days, then more aggressive interventions should be considered. In addition, in the less critically ill patient with limited disease, the initial management strategy should include reduction of cyclosporin or tacrolimus and prednisone by at least 50%, while azathioprine or MMF should be discontinued. After a 14-day trial of decreased immunosuppression, a further decrease of 25% can be considered. Should clinical urgency or failure of “conservative” therapy develop, chemotherapy using a lymphoma protocol has generally been adopted. 12,25–27 Alternatively, promising results using anti-B-cell monoclonal antibodies have been reported, beginning with the initial results of Fischer et al using a combination of anti-CD21 and anti-CD24 monoclonal antibodies. 28 The availability of anti-CD20 monoclonal antibody, which targets most B cells, has been shown to have promising preliminary results. 29,30
The results of this approach reveal that short-term and long-term patient survival rates were not as dismal as reported in other series. 14,26 Nevertheless, the decline in survival was approximately 6% per year in the adult PTLD population and 4% per year in the pediatric PTLD population, worse than that observed in the LTX group as a whole (declines of approximately 3.5% and 1.5% per year, respectively). 1 The fact that PTLD contributed to 44% of deaths in patients afflicted with PTLD highlights the need for advances in the prophylaxis, detection, and treatment of PTLD, as well as a better understanding of other risk factors associated with this disease. We also note that while LTX patients transplanted for alcoholic liver disease had similar risks for PTLD, their long-term mortality rate was significantly higher (87%) compared to 50% in nonalcoholic LTX patients. It may be that the reported karyotypic chromosomal lymphocyte aberrations associated with ethanol 31 may potentiate the known effect of EBV in causing chromosomal translocation. 8
Green et al at the University of Pittsburgh have initiated a study examining the use of high-titer anti-EBV intravenous immunoglobulin for EBV-associated PTLD in high-risk pediatric recipients. 32 McDiarmid et al from UCLA have suggested that the use of intravenous ganciclovir in high-risk EBV-positive donors to EBV-negative recipients may prevent subsequent PTLD. 33
Rowe et al examined the utility of EBV viral loads as a means to monitor patients at risk for PTLD. 34,35 Others have also suggested that EBV monitoring can be useful in high-risk transplant recipients. 36 The Montpellier group has suggested that detection of persistent monoclonal immunoglobulins in LTX recipients was associated with a 23% incidence of PTLD. 37 Certainly, if PTLD can be detected preemptively or while still in an “early” stage, the outcomes would likely be better.
Cell-based immunotherapy holds promise for the treatment of PTLD; however, the use of adoptive cellular immunotherapy using IL-2-stimulated lymphokine activated killer (LAK) cells was associated with a response rate of only 30% in refractory PTLD. 38 Targeted approaches using EBV-specific T-effector cells have been used in patients with PTLD following bone marrow transplantation since, in these patients, although the PTLD arises from donor B cells, the EBV-specific effector T cells can be obtained from the donor, who is not under the effect of immunosuppression. 39 Similar approaches to generate EBV-specific cytolytic T cells (CTL) in high-risk EBV-seropositive transplant recipients has also shown promise as a means to preemptively treat patients with an elevated EBV viral load. 40 In the case of an EBV-seronegative solid organ transplant recipient, there are significant technical limitations in priming ex vivo EBV-specific CTL that may limit their expansion and subsequent clinical use. However, recent advances in understanding the biology of antigen-presenting cells and especially growth factors may make this approach possible in the future for treating solid organ transplant recipients. 41
There is increasing recognition that EBV-negative PTLD is a separate entity: these tumors tend to arise later in the posttransplant course 42,43 and have been reported to have a higher frequency of c-myc rearrangements. 44 Although we have seen an increasing detection of EBV-negative PTLD, 45 as noted in this study, we did not detect any impact on clinical outcomes, recognizing that this entity appears to be mostly restricted to adult transplant recipients. Thus far, no etiologic agent has been found.
DISCUSSION
Dr. Paul S. Russell (Boston, MA): I think this is a very interesting subject. And you will recall that in the early days our colleagues over in the Brigham Hospital had experience with cancers developing in the transplanted kidney of donor origin from patients who had mammary carcinoma, for example, and so on. And I know a couple of those that I knew rather well had evidence of regional metastasis with lymph nodes in the pelvis, and with reduction of immunosuppression they cleared entirely. And of course at the time it wasn’t clearly in everyone’s mind just exactly how all these things were working. They thought they had a new treatment for cancer.
Well, I think in a way, there is a very interesting situation you have here. These, as you said, are very largely of recipient origin, and yet the reduction of immunosuppression will clear those cells in a number of cases. That is a very interesting thing to me. And it implies that at least some of the patients have enough in the way of new antigenic material associated with those malignant cells so that the normal recipient cells can actually clear them.
You have said here that you think that more individualized treatment might have a future, and I agree with that. I wonder if you have any evidence yet as to selection of patients who have evidence of in vitro reactivity of the normal recipient cells to the malignant recipient cells as a harbinger of whether or not reduction of immunosuppression will work. Because there are these different groups of patients, and I am sure you are right, that you have to sort them out.
Presenter Dr. John J. Fung (Pittsburgh, PA): Thank you, Dr. Russell. I think all of those comments are appropriate to the discussion. We know that the Epstein-Barr virally transformed B cells express a number of proteins that can be considered tumor-specific antigens. The concept of augmenting an immune response against those antigens to clear virally infected cells therefore makes sense. We were surprised that EBV-negative PTLDs would not have responded as well to reduction of immunosuppression because of a paucity of virally expressed proteins. However, as shown here, reduction in immunosuppression with or without ancillary treatments has been effective, suggesting that other PTLD tumor-specific antigens may be important in immunomodulatory approaches to EBV-negative PTLD.
The concept of using immunoassays to ascertain whether a given recipient has immunocompetence against a given tumor is a very attractive one. There are studies that have attempted to quantify cytolytic T-cells responses, while others have looked at T-cell cytokine profiles in these patients to correlate this to clinical response.
In my mind, one of the important lessons to be learned from this experience is that the residual immunity in these patients on a calcineurin inhibitor is greater than is currently believed.
Therefore, the principal approach to PTLD should be reduction or elimination of immunosuppression, which will lead to reduction or clearance of PTLD.
Dr. Stephen T. Bartlett (Baltimore, MD): I want to congratulate Dr. Fung on an excellent review of a very large number of liver transplant recipients with the longest follow-up reported. Dr. Fung makes some interesting points regarding the impact of immunosuppression and recipient age on the risk of development of PTLD.
One of the risk factors described in the literature for PTLD is the use of potent antilymphocyte antibodies. Increased dose duration and repeated courses of antilymphocyte therapy within a short time interval substantially increase the risk. This has been primarily attributed to OKT3, but the other preparations have been shown to increase risk as well.
Has your center’s immunosuppression protocol changed over the 20 years, and have you looked at the need for antilymphocyte antibodies as a function of time? If so, has the incidence of PTLD changed? I can imagine an exaggerated use of antilymphocyte therapy 10 years ago in a setting where hepatitis C recurrence was much more likely to be interpreted as rejection, leading, in turn, to increasing dosages of immunosuppression that now seem to be inappropriate as recurrent HCV is better understood.
A second question is related to survival. With modern treatment protocols, including the anti-CD20 monoclonal antibody, have you analyzed survival following PTLD in the modern era instead of the whole 20-year period? And do you think it would likely change?
There is strong evidence, as you pointed out in discussion, that the development of PTLD is strongly related to EBV. Your results support what has been reported in the literature, that the majority of PTLD specimens contain DNA or EBV-specific proteins. The profound impact of pretransplant EBV serum negativity on the risk of PTLD has been emphasized by our group by Dr. Susan Keay; other authors have reported this as well. This would probably account for the increased rate of PTLD in children, as you just pointed out, since more children are likely to be EBV naive at a young age. Have you considered special strategies in seronegative children to decrease the risk of EBV-related PTLD?
The last point I would like to address is graft loss associated with PTLD. Have you had to retransplant some of those patients? If so, how did you manage them? Have you needed to resort to surgical resection of PTLDs that are obstructing the portal triad? With the new high-dose thymoglobulin protocol that Dr. Starzl has introduced in Pittsburgh, are you seeing an increased incidence of PTLD?
Dr. John J. Fung (Pittsburgh, PA): The immunosuppressive protocols have changed in our 20-year history of liver transplantation from cyclosporine and steroids to OKT3, cyclosporine and steroids, to tacrolimus and steroids, to tacrolimus, mycophenolate and steroids, and now with tacrolimus and thymoglobulin without steroids. So it is obvious that the results that we showed are not intended to necessarily suggest that cyclosporine or tacrolimus has benefits over the other. OKT3 was used in about 12% of the patients in the cyclosporine era and only 2% in the tacrolimus era. Whether that was associated with the development of PTLD was not analyzed.
The role of hepatitis C as a risk factor for PTLD was examined, although hepatitis C was not a defined etiologic agent until 1991. The surrogate diagnosis of non-A/non-B hepatitis was substituted for HCV in the cyclosporine era. In both tacrolimus and cyclosporine patients we did not see a relationship between hepatitis C and PTLD.
The possibility of infecting or vaccinating an EBV-seronegative candidate has been discussed with Dr. Epstein. Unfortunately, there has not been an appropriate EBV vaccine developed. While deliberate infection with EBV has been entertained by some centers, I am not aware of any center reporting on this approach. We have been involved in a trial of hyperimmune antibody infusion as prophylaxis for PTLD; this trial has been conducted at the Children’s Hospital of Pittsburgh. The trial is ongoing, so it is inappropriate to state whether this is a valid approach or not; we hope to analyze these results in a year.
We had four patients in this group that required retransplantation due to rejection during treatment of their PTLD, while there were four other patients that died from a combination of recurrent disease and chronic rejection in this group.
We have not yet seen any increase of PTLD under the current tacrolimus and thymoglobulin protocol, which is the single, high-dose thymoglobulin infusion with low-dose monotherapy tacrolimus in the postoperative period.
With respect to the anti-CD20 monoclonal antibody, Rituximab, we have begun to utilize this for PTLDs that are not responsive to reduction of immunosuppression, or for those lesions that are located in critical areas, such as the CNS. There tends to be excellent initial response to treatment, but it is critical to significantly lower the level of immunosuppression; otherwise, one risks recurrence of PTLD. Of course, it is essential to test the PTLD ahead of time to verify that it is CD20-positive.
Dr. John S. Najarian (Minneapolis, MN): I enjoyed the paper very much. It is a tremendous experience. I just have one question. In any of these patients who have negative EBV serum titers, have you considered the use of antiviral therapy? Since EBV is so ubiquitous throughout the entire population, are you willing to say that it is going to be in many of the organs we transplant? So under those circumstances, would it be worthwhile to use something like acyclovir? We have done so in our kidney transplant patients, and we virtually do not see PTLD anymore. Is it possible that acyclovir could be used in your liver patients as well?
Dr. John J. Fung (Pittsburgh, PA): The nucleoside analogs have been used empirically for PTLD treatment, in large part due to the contributions from the University of Minnesota. Sue McDermott from UCLA has reported that the acyclovir prophylaxis has reduced PTLD in their high-risk pediatric liver transplant population. As I mentioned before, we have embarked on a different approach to prophylaxis, which is the use of Cytogam, which also has high titers of anti-EBV antibodies. We are currently conducting a blinded study involving pediatric liver transplant recipients using Cytogam for EBV prophylaxis. While we do not have yet the results of this study, we do have pilot data that suggested that high-risk EBV-seronegative children do benefit from the use of anti-EBV antibodies as prophylaxis.
Footnotes
Presented at the 122nd Annual Meeting of the American Surgical Association, April 24–27, 2002, The Homestead, Hot Springs, Virginia.
Correspondence: John Fung, MD, PhD, Thomas E. Starzl Transplantation Institute, Fourth Floor Falk Clinic, 3601 Fifth Avenue, University of Pittsburgh, Pittsburgh, PA 15213.
E-mail: fungjf@msx.upmc.edu
Accepted for publication April 24, 2002.
References
- 1.Jain A, Reyes J, Kashyap R, et al. Long-term survival after liver transplantation in 4,000 consecutive patients at a single center. Ann Surg 2000; 232: 490–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fung JJ, Jain A, Kwak EJ, et al. De novo malignancies after liver transplantation: a major cause of late death. Liver Transplant 2001; 7: S109–S118. [DOI] [PubMed] [Google Scholar]
- 3.Hanto DW, Frizzera G, Purtilo DT. Clinical spectrum of lymphoproliferative disorders in renal transplant recipients and evidence for the role of Epstein-Barr virus. Cancer Res 1981; 41: 4253. [PubMed] [Google Scholar]
- 4.Starzl TE. Discussion of Murray JE, Wilson RE, Tilney NL, et al. Five years’ experience in renal transplantation with immunosuppressive drugs: Survival, function, complications, and the role of lymphocyte depletion by thoracic duct fistula. Ann Surg 1968; 168: 416–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Geis WP, Iwatsuki S, Molnar Z, et al. Pseudolymphoma in renal allograft recipients. Arch Surg 1978; 113: 461–466. [DOI] [PubMed] [Google Scholar]
- 6.Starzl TE, Porter KA, Iwatsuki S, et al. Reversibility of lymphomas and lymphoproliferative lesions developing under cyclosporin-steroid therapy. Lancet 1984; 1: 583–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nalesnik MA, Jaffe R, Starzl TE, et al. The pathology of post-transplant lymphoproliferative disorders in the setting of cyclosporine A-prednisone immunosuppression. Am J Pathol 1988; 133: 173–192. [PMC free article] [PubMed] [Google Scholar]
- 8.Nalesnik MA, Makowka L, Starzl TE. The diagnosis and treatment of post transplant lymphoproliferative disorders. Curr Probl Surg 1988; 25: 367–472. [DOI] [PubMed] [Google Scholar]
- 9.Randhawa PS, Jaffe R, Demetris AJ, et al. Expression of Epstein-Barr virus-encoded small RNA (by the EBER-1 gene) in liver specimens from transplant recipients with post-transplantation lymphoproliferative disease. N Engl J Med 1992; 327: 1710–1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Harris NL, Ferry JA, Swerdlow SH. Posttransplant lymphoproliferative disorders: summary of Society for Hematopathology Workshop. Semin Diagn Path 1997; 14: 8–14. [PubMed] [Google Scholar]
- 11.Collins MH, Montone KT, Leahey AM, et al. Autopsy pathology of pediatric posttransplant lymphoproliferative disorder. Pediatrics 2001; 107: E89. [DOI] [PubMed] [Google Scholar]
- 12.Jain A, Mazariegos G, Kashyap R, et al. Pediatric liver transplantation. A single-center experience spanning 20 years. Transplantation 2002; 73: 941–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Paya CV, Fung JJ, Nalesnik MA, et al. Epstein-Barr virus-induced posttransplant lymphoproliferative disorders. ASTS/ASTP EBV-PTLD Task Force and the Mayo Clinic Organized International Consensus Development Meeting. Transplantation 1999; 68: 1517–1525. [DOI] [PubMed] [Google Scholar]
- 14.Ben-Ari Z, Amlot P, Lachmanan SR, et al. Posttransplant lymphoproliferative disorder in liver recipients: characteristics, management, and outcome. Liver Transplant Surg 1999; 5: 184–191. [DOI] [PubMed] [Google Scholar]
- 15.Ho M, Jaffe R, Miller G. The frequency of Epstein-Barr virus infection and associated lymphoproliferative syndrome after transplantation and its manifestations in children. Transplantation 1988; 45: 719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cacciarelli TV, Reyes J, Jaffe R, et al. Primary tacrolimus (FK506) therapy and the long-term risk of post-transplant lymphoproliferative disease in pediatric liver transplant recipients. Pediatr Transplant 2001; 5: 359–364. [DOI] [PubMed] [Google Scholar]
- 17.Younes BS, McDiarmid SV, Martin MG, et al. The effect of immunosuppression on posttransplant lymphoproliferative disease in pediatric liver transplant patients. Transplantation 2000; 70: 94–99. [PubMed] [Google Scholar]
- 18.Hezode C, Duvoux C, Germanidis G, et al. Role of hepatitis C virus in lymphoproliferative disorders after liver transplantation. Hepatology 1999; 30: 775–778. [DOI] [PubMed] [Google Scholar]
- 19.McLaughlin K, Wajstaub S, Marotta P, et al. Increased risk for posttransplant lymphoproliferative disease in recipients of liver transplants with hepatitis C. Liver Transplant 2000, 6: 570–574. [DOI] [PubMed] [Google Scholar]
- 20.Frizzera G, Hanto DW, Gajl-Peczalska KJ. Polymorphic diffuse B-cell hyperplasias and lymphomas in renal transplant recipients. Cancer Res 1981; 41: 4262. [PubMed] [Google Scholar]
- 21.Locker J, Nalesnik MA. Molecular genetic analysis of lymphoid tumors arising after organ transplantation. Am J Pathol 1989; 135: 977–987. [PMC free article] [PubMed] [Google Scholar]
- 22.Kaplan MA, Ferry JA, Harris NL, et al. Clonal analysis of posttransplant lymphoproliferative disorders, using both episomal Epstein-Barr virus and immunoglobulin genes as markers. Am J Clin Pathol 1994; 101: 590–596. [DOI] [PubMed] [Google Scholar]
- 23.Chadburn A, Cesarman E, Knowles DM. Molecular pathology of posttransplant lymphoproliferative disorders. Semin Diagn Pathol 1997; 14: 15–26. [PubMed] [Google Scholar]
- 24.Cesarman E, Chadburn A, Liu YF, et al. BCL-6 gene mutations in posttransplantation lymphoproliferative disorders predict response to therapy and clinical outcome. Blood 1998; 92: 2294–2302. [PubMed] [Google Scholar]
- 25.McCarthy M, Ramage J, McNair A, et al. The clinical diversity and role of chemotherapy in lymphoproliferative disorder in liver transplant recipients. J Hepatol 1997; 27: 1015–1021. [DOI] [PubMed] [Google Scholar]
- 26.Glez-Chamorro A, Jimenez C, Moreno-Glez E, et al. Management and outcome of liver recipients with post-transplant lymphoproliferative disease. Hepato-Gastroenterology 2000; 47: 211–219. [PubMed] [Google Scholar]
- 27.Gallego-Melcon S, Sanchez de Toledo J, Martinez V, et al. Non-Hodgkin’s lymphoma after liver transplantation: response to chemotherapy. Med Pediatr Oncol 1996; 27: 156–159. [DOI] [PubMed] [Google Scholar]
- 28.Fischer A, Blanche S, Le Bidois J, et al. Anti-B cell monoclonal antibodies in the treatment of severe B-cell lymphoproliferative syndrome following bone marrow and organ transplantation. N Engl J Med 1991; 324: 1451–1456. [DOI] [PubMed] [Google Scholar]
- 29.Zompi S, Tulliez M, Conti F, et al. Rituximab (anti-CD20 monoclonal antibody) for the treatment of patients with clonal lymphoproliferative disorders after orthotopic liver transplantation: a report of three cases. J Hepatol 2000; 32: 521–527. [DOI] [PubMed] [Google Scholar]
- 30.Milpied N, Vasseur B, Parquet N, et al. Humanized anti-CD20 monoclonal antibody (Rituximab) in post-transplant B-lymphoproliferative disorder: a retrospective analysis on 32 patients. Ann Oncol 2000; 11: 113–116. [PubMed] [Google Scholar]
- 31.Huttner E, Matthies U, Nikolova T, et al. A follow-up study on chromosomal aberrations in lymphocytes of alcoholics during early, medium, and long-term abstinence. Alcohol Clin Exp Res 1999; 23: 344–348. [PubMed] [Google Scholar]
- 32.Green M, Reyes J, Webber S, et al. The role of antiviral and immunoglobulin therapy in the prevention of Epstein-Barr virus infection and post-transplant lymphoproliferative disease following solid organ transplantation. Transplant Infect Dis 2001; 3: 97–103. [DOI] [PubMed] [Google Scholar]
- 33.McDiarmid SV, Jordan S, Kim GS, et al. Prevention and preemptive therapy of posttransplant lymphoproliferative disease in pediatric liver recipients. Transplantation 1998; 66: 1604–1611. [DOI] [PubMed] [Google Scholar]
- 34.Rowe DT, Qu L, Reyes J, et al. Use of quantitative competitive PCR to measure Epstein-Barr virus genome load in the peripheral blood of pediatric transplant patients with lymphoproliferative disorders. J Clin Microbiol 1997; 35: 1612–1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Green M, Cacciarelli TV, Mazariegos GV, et al. Serial measurement to Epstein-Barr viral load in peripheral blood in pediatric liver transplant recipients during treatment for posttransplant lymphoproliferative disease. Transplantation 1998; 66: 1641–1644. [DOI] [PubMed] [Google Scholar]
- 36.Vajro P, Lucariello S, Migliaro F, et al. Predictive value of Epstein-Barr virus genome copy number and BZLF1 expression in blood lymphocytes of transplant recipients at risk for lymphoproliferative disease. J Infect Dis 2000; 181: 2050–2054. [DOI] [PubMed] [Google Scholar]
- 37.Pageaux GP, Bonnardet A, Picot MC, et al. Prevalence of monoclonal immunoglobulins after liver transplantation: relationship with posttransplant lymphoproliferative disorders. Transplantation 1998; 65: 397–400. [DOI] [PubMed] [Google Scholar]
- 38.Nalesnik MA, Rao AS, Furukawa H, et al. Autologous lymphokine-activated killer cell therapy of Epstein-Barr virus-positive and -negative lymphoproliferative disorders arising in organ transplant recipients. Transplantation 1997; 63: 1200–1205. [DOI] [PubMed] [Google Scholar]
- 39.Rooney CM, Smith CA, Ng CY, et al. Use of gene-modified virus-specific T lymphocytes to control Epstein-Barr-virus-related lymphoproliferation. Lancet 1995; 345: 9–13. [DOI] [PubMed] [Google Scholar]
- 40.Comoli P, Labirio M, Basso S, et al. Infusion of autologous Epstein-Barr virus (EBV)-specific cytotoxic T cells for prevention of EBV-related lymphoproliferative disorder in solid organ transplant recipients with evidence of active virus replication. Blood 2002; 99: 2592–2598. [DOI] [PubMed] [Google Scholar]
- 41.Metes D, Storkus WJ, Zeevi A, et al. Use of autologous dendritic cells loaded with apoptotic LCL for ex vivo generation of specific CTL from the PBMC of EBV (-) individuals. Transplant Proc 2001; 33: 441. [DOI] [PubMed] [Google Scholar]
- 42.Leblond V, Davi F, Charlotte F, et al. Posttransplant lymphoproliferative disorders not associated with Epstein-Barr virus: a distinct entity? J Clin Oncol 1998; 16: 2052–2059. [DOI] [PubMed] [Google Scholar]
- 43.Nelson BP, Nalesnik MA, Bahler DW, et al. Epstein-Barr virus-negative post-transplant lymphoproliferative disorders: a distinct entity? Am J Surg Pathol 2000; 24: 375–385. [DOI] [PubMed] [Google Scholar]
- 44.Dotti G, Fiocchi R, Motta T, et al. Epstein-Barr virus-negative lymphoproliferate disorders in long-term survivors after heart, kidney, and liver transplant. Transplantation 2000; 69: 827–833. [DOI] [PubMed] [Google Scholar]
- 45.Nalesnik MA. Clinicopathologic characteristics of post-transplant lymphoproliferative disorders. Recent Results Cancer Res 2002; 159: 9–18. [DOI] [PubMed] [Google Scholar]