

Abstract
Objective
To determine whether adjuvant postoperative active specific immunotherapy with a therapeutic polyvalent vaccine (PV) called Canvaxin can prolong survival following complete resection of melanoma metastatic to regional nodes (American Joint Committee on Cancer [AJCC] stage III melanoma).
Summary Background Data
Despite complete lymphadenectomy, 5-year overall survival (OS) for patients with melanoma metastatic to regional lymph nodes is only 20% to 50%, depending on the number of tumor-involved nodes. In 1984, the authors began phase II trials of Canvaxin PV as postsurgical adjuvant therapy for AJCC stage III melanoma.
Methods
Patients who received PV between 1984 and 1998 were compared with patients who did not receive PV postsurgical therapy between 1971 and 1998. The seven covariates recently defined by the AJCC Melanoma Staging Committee (number of metastatic nodes, palpable status, ulceration, age, primary site, pT stage, and gender) were included by Cox regression in a multivariate model of OS. A computerized program matched PV and non-PV patients by these covariates.
Results
Of 2,602 patients who underwent complete lymphadenectomy for AJCC stage III melanoma with regional nodal metastases and were followed up by the same team of oncologists between 1971 and 1998, 935 received PV and 1,667 did not. Median OS and 5-year OS were significantly higher in PV than non-PV patients (56.4 vs. 31.9 months and 49% vs. 37%, respectively;P = .0001). When the non-PV patients were matched by the four most significant covariates, 447 matched pairs were formed between patients seen before or after January 1, 1985, and the OS was not different between the two time periods (P = .789). However, when the PV patients were matched with non-PV patients by six covariates forming 739 pairs, the PV patients survived longer (P = .0001). Detailed analysis of the 1,505 patients who were seen or who began vaccine therapy within 4 months after lymphadenectomy, and who had more complete data on the seven prognostic covariates showed that median OS and 5-year OS were higher in 445 PV patients than in 1,060 non-PV patients: 70.4 versus 31 months and 52% versus 37%, respectively (P = .0001). Multivariate Cox regression analysis identified six significant prognostic factors: number of metastatic nodes, size of metastatic nodes, pT stage, ulceration, age, and PV therapy. PV therapy reduced the relative risk of death to 0.64 (95% confidence interval, 0.55–0.76) (P = .0001); sex and site of primary were of borderline significance.
Conclusions
This large single-institution study independently confirmed the significance of prognostic covariates in the new AJCC staging system. By using modern statistical methods that controlled for all known prognostic factors, it also demonstrated PV’s ability to significantly enhance OS. A multicenter phase III randomized trial is underway to validate the efficacy of PV as a postsurgical adjuvant.
The incidence of malignant melanoma of the skin is increasing throughout the world at a rate of approximately 5% per year. In the United States and Canada, melanoma has increased at a rate exceeding that of any other tumor except lung cancer in women. According to the American Cancer Society, this year an estimated 53,600 Americans will be diagnosed with melanoma, and 7,400 patients will die from this neoplasm. 1
With early diagnosis and adequate surgical treatment of primary melanoma, the 10-year overall cure rate is almost 90% for patients whose melanoma has a Breslow thickness of no more than 1.0 mm (American Joint Committee on Cancer [AJCC] stage I). 2 However, once melanoma has metastasized from the primary site to the regional nodes (AJCC stage III), the 5-year survival rate following complete lymphadenectomy is only 20% to 50%, depending on the number of involved nodes. 3 This dismal statistic reflects the progression of subclinical metastases remaining after the excision of all macroscopic disease.
The possibility of subclinical residual melanoma is a strong argument for postsurgical adjuvant systemic therapy. Indeed, postsurgical adjuvant therapy has proven effective in patients with breast cancer, colon cancer, and many other neoplasms. However, none of the many different adjuvant treatments evaluated for melanoma has yet proved to be unequivocally beneficial in influencing overall survival. 4 Postoperative administration of high-dose recombinant interferon alfa-2b can prolong disease-free survival, but its effect on overall survival has been inconsistent. 5,6 Moreover, up to 75% of patients develop level III or IV toxicity.
One of the most promising alternatives appears to be postoperative adjuvant active specific (vaccine) immunotherapy. Anecdotal reports describing spontaneous regression of metastatic melanoma implicate an immune response that might be modulated by vaccine stimulation, 7,8 particularly if the vaccine is administered postoperatively when the tumor burden is too low to exert an immunosuppressive effect.
The primary goal of our research during the past 40 years, first in animal models and later in man, has been to develop more effective methods for active specific immunotherapy of cancer. The early work with syngeneic tumors in mice and guinea pigs found that the most effective method to induce tumor regression was to immunize with irradiated tumor cells plus mycobacterial adjuvants. 9,10 The conceptual basis for active specific immunotherapy of melanoma derives from our original observations that the sera of melanoma patients contained antibodies reactive with membrane antigens in autologous melanoma cells. 11 We subsequently found that the intratumoral injection of cutaneous melanoma metastases with mycobacterial adjuvants resulted in systematic enhancement of active immunity, produced rising titers of antimelanoma antibodies, and caused regression of distant metastases. 7,12 These regressing distant metastases demonstrated intense lymphocytic infiltration on histopathological study.
Despite evidence for the immunogenicity of human melanoma, previous trials of early vaccine formulations by Morton et al, 13 Livingston et al, 14 Wallack et al, 15 and Kirkwood et al 16 have shown no consistent effect on the overall survival of patients receiving postoperative active immunotherapy. Our initial trials with active immunotherapy in the 1970s involved the intradermal injection of a vaccine composed of irradiated allogeneic melanoma cells of undefined antigenicity, mixed with bacille Calmette-Guerin (BCG). 13 Only one third of these patients developed high titers of antibodies reactive with melanoma-associated antigens, but these immune responders did exhibit prolonged survival. 17
In retrospect, our results were disappointing because we used a whole-cell vaccine formulation that did not express significant quantities of multiple immunogenic melanoma-associated antigens (MAAs). In 1984 we developed a new therapeutic polyvalent vaccine (PV) from three allogeneic melanoma cell lines known to contain high concentrations of six immunogenic MAAs: gangliosides GD2, GM2, and O-acetyl GD-3; lipoprotein M-TAA; and glycoproteins M-fetal antigen and M-urinary antigen. 2,18 We have since found that the melanoma cells of this PV (Canvaxin therapeutic cancer vaccine, CancerVax Corp., Carlsbad, CA) express more than 20 common MAAs. 19–46 One or more of these MAAs has been found in all tumor specimens from melanoma patients yet examined. We have demonstrated that the immune response induced by these antigens has prognostic significance not only in patients who receive Canvaxin PV, 25,39,47,48 but also in those patients not treated with PV. 49 This suggests that some of these MAAs are naturally immunogenic and can modulate the patient’s protective endogenous immune responses against melanoma.
The vaccine’s polyvalency is important. Because of its large number of defined immunogenic MAAs, PV’s ability to stimulate a cross-reactive immune response against the patient’s autologous tumor does not depend on any particular pattern of antigen expression in the patient’s melanoma cells. 50 Moreover, a polyvalent antigenic stimulus is more likely to induce effective polyvalent antibody and T-cell responses that can destroy tumor cells that might otherwise escape by antigen loss or downregulation of HLA. Finally, the wide array of HLA haplotypes in the vaccine provides an HLA haplotype allele match with 95% of melanoma patients. 51 Therefore, unlike specific HLA haplotype-directed peptide vaccines, Canvaxin PV does not depend on the patient’s MHC class type.
In 1984 we began phase II trials to evaluate the immunogenicity of Canvaxin PV therapy following complete resection of melanoma metastatic to regional lymph nodes (AJCC stage III). 18 In 1996, we reported that disease-free survival was significantly higher in 283 patients who received PV than in 1,474 historical control patients who did not receive PV (median survival 60 months vs. 35 months, 5-year survival rate 53% vs. 39%). 2 These findings were extremely encouraging but not definitive because we did not control for all important prognostic factors between the two groups. In the present study, we used modern statistical methods to determine the impact of PV therapy on overall survival (OS). Our hypothesis was that patients receiving PV would exhibit prolonged OS compared to patients treated with non-PV postsurgical therapies by the same team of surgical and medical oncologists. Although this was not a randomized study, we were able to minimize the risk of selection bias by statistically matching our populations according to prognostic factors independently identified by Balch et al 52 in a recent report of the AJCC Melanoma Staging Committee.
METHODS
Patients who had undergone complete surgical resection of regional nodal metastases (AJCC stage III melanoma) were identified by review of the John Wayne Cancer Institute’s (JWCI) melanoma database. This computerized database contains the records of all 10,583 patients seen by our staff since April 1971. Follow-up is complete to within 30 months of last follow-up or death for 94% of patients in the database. The database thus represents a 30-year prospective audit of results of therapy by JWCI staff.
Eligible patients either underwent complete lymphadenectomy by our group at the UCLA Division of Surgical Oncology (1971–1991) or JWCI (1991–1998), or they were referred to us for postoperative treatment/follow-up during the same period. Data collection for this study was complete through December 31, 2001, providing a minimum of 3 years on study. The postoperative absence of disease was confirmed by physical examination, laboratory tests, and radiologic studies. Before 1984, radiologic studies comprised chest x-rays and tomograms and brain and/or liver nuclear scans; after that time, computed tomography (CT) of the chest, abdomen, and pelvis, and CT or magnetic resonance imaging (MRI) of the brain was used.
The PV group was composed of patients who received Canvaxin PV in one of several postoperative adjuvant immunotherapy protocols that became available in or after September 1984. The decision to participate in a PV protocol was the individual patient’s choice. The control group was composed of patients who did not choose to receive adjuvant PV therapy or who were enrolled in postoperative adjuvant protocols utilizing BCG, previous vaccine formulations, or chemotherapy, or elected to receive no adjuvant therapy.
Canvaxin PV Protocols
Before initiation of therapy, all patients signed an informed consent, and all PV protocols were approved by the UCLA (1971–1991) or the joint JWCI–Saint John’s Health Center (1991–1998) Institutional Review Boards and in compliance with the Helsinki Declaration.
The first patient received PV in September 1984. PV protocols differed only in the use of different immunoadjuvants, the administration of which is described in a previous report. 53 The preparation and administration of PV also has been described in detail elsewhere. 18,53 Briefly, melanoma cells from the M10-V, M24-V, and M101-V cell lines were cultured in tissue culture medium RPMI 1640 and then harvested, washed, and pooled to obtain 25 × 106 viable total cells (8.3 × 106 cells per line). The cells were then irradiated with 150 Gy and cryopreserved in liquid nitrogen until they were thawed in preparation for administration. Each injection consisted of 25 × 106 viable cells as judged by vital dye staining. Before 1996, the three vaccine cell lines were cryopreserved separately; at the time of administration, they were thawed, washed, and combined. After 1996, the three melanoma cell lines were combined in the same vial before cryopreservation; contents of the vial were rapidly thawed at the time of administration. The vaccine was given intradermally every 2 weeks for three to five doses, then monthly for the remainder of the first year. After year 1 of PV therapy, the vaccine was administered every 3 months for four cycles, and thereafter at 6-month intervals. During the first two treatments, PV was given in conjunction with the Tice strain of BCG at a dose of 3-10 × 106 CFU per treatment. Clinical status was recorded prospectively for all patients until the time of death.
Statistical Analysis
Estimated melanoma-specific survival rates were obtained by the nonparametric Kaplan-Meier method. The log-rank test was used to determine differences in survival of patients from subgroups defined by different levels of risk factors, therapy, and time interval at which they entered the JWCI database. Multivariate analysis was performed by the Cox proportional hazards regression model. The relative importance of prognostic factors was based on the Wald test of the coefficient associated with the prognostic factors in the Cox model. A computerized matching of pairs of PV and control patients was carried out by a program that matched at a 1:1 ratio based on covariates selected for matching. P < .05 was considered significant. All statistical analyses were two-tailed and performed using SAS software (SAS Institute, Cary, NC). OS was defined as the time a patient remained alive after the date of lymphadenectomy for documented nodal-metastatic disease. Survival times were considered censored for patients who were alive at the last follow-up or who died without evidence of melanoma.
RESULTS
Between 1971 and 1998, 2,602 patients who had undergone regional lymphadenectomy for stage III melanoma were seen by JWCI staff. Of these, 935 patients, who were seen between September 1, 1984, and December 31, 1998, received postoperative adjuvant therapy with Canvaxin PV and were designated as the PV group. The remaining 1,667 patients, who were seen between April 1, 1971, and December 31, 1998, received no adjuvant therapy or non-PV adjuvant therapy and were designated as the control group.
Prognostic Factors
As shown in Table 1, prognostic factors identified as significant by the AJCC Melanoma Staging Committee 52 (i.e., number of metastatic nodes, palpable versus nonpalpable nodal metastases, and ulceration status) were well balanced for PV and control groups, except that the control group was slightly more likely to have nonpalpable nodal metastases, which is a more favorable prognostic factor. In addition, the control group had a larger proportion of patients with an unknown number of positive nodes, and the control group was more likely to have unknown data on ulceration status.
Table 1. DISTRIBUTION OF PROGNOSTIC FACTORS BETWEEN POLYVALENT VACCINE AND CONTROL GROUPS

* Most of these melanomas were mucosal.
The number of tumor-involved lymph nodes (P = .0001), the clinical status of these nodes (palpable or nonpalpable) (P = .0001), and the primary tumor’s stage (P = .0012) and ulceration status (P = .0002) were all significant prognostic factors for OS in both groups by univariate and multivariate analysis (Table 2). Sex was highly significant (P = .0001) by univariate analysis but borderline (P = .0557) by Cox proportional hazards when other covariates were considered in the multivariate model. The time of curative surgical resection (before or after January 1, 1985) did not influence OS (P = .9613) by Cox proportional hazards model. Patient age (P = .1489) and site of primary (P = .6108) were not significant.
Table 2. IMPACT OF PROGNOSTIC FACTORS ON OVERALL SURVIVAL

PV therapy was a highly significant prognostic factor by both univariate and multivariate analysis (P = .0002); when compared to the control group, the PV group had a reduced risk of death corresponding to a hazard ratio of 0.689. As shown in Figure 1, the entire PV group had a significantly (P = .0001) higher median OS (56.4 months vs. 31.9 months) and 5-year OS (49% vs. 37%). Similarly, when the PV and control groups were stratified and compared by the most important prognostic factors—that is, nodal size (palpable vs. nonpalpable) and number of involved nodes—OS was consistently higher in the PV group (Figs. 2 and 3).
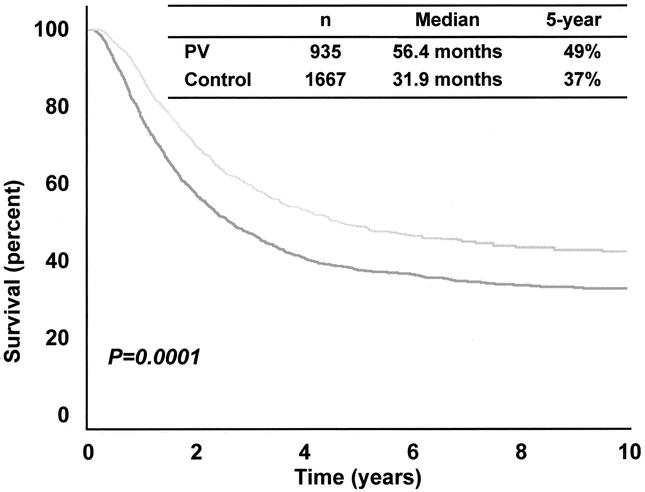
Figure 1. Overall survival of all PV and all control patients.

Figure 2. Overall survival of all PV and all control patients who had clinically palpable or nonpalpable lymph node metastases.

Figure 3. Overall survival of all PV and all control patients by tumor burden: one positive node, two or three positive nodes, or four or more positive nodes.
Consistency of Prognostic Factors and Prolonged Survival in the Vaccine Group Across Time
To examine the homogeneity of the entire patient population between 1971 and 1998, the control group was divided into two subgroups based on lymph node resection before (n = 1,001) or after (n = 666) January 1, 1985. As shown in Figure 4 (left), survival characteristics were almost identical for the two control subgroups: median OS 32.4 months versus 31.9 months, and 5-year OS 38 ± 2% versus 37 ± 1% before versus after January 1, 1985. However, the PV group exhibited significantly prolonged median OS (56.4 months) and higher 5-year OS (49 ± 2%) compared to control patients from either time period.

Figure 4. Overall survival according to time of treatment (before or after January 1, 1985). (Left) All PV and all control patients. (Right) 447 pairs of control patients matched by tumor burden, nodal status, primary T stage, and ulceration.
A computer program for randomized matching of control patients before and after January 1, 1985, identified 447 pairs matched by the four most important covariates: tumor burden (number of metastatic nodes), nodal size (palpable versus nonpalpable), primary T stage, and ulceration (Table 3; see Fig. 4). Median OS of these matched pairs was 29.8 months before 1985 and 32.5 months thereafter; 5-year OS was 33 ± 2% before 1985 and 38 ± 2% thereafter (Fig. 4, right). The 10-year survival of both groups was 29 ± 2%. Again, these differences were not significant (P = .7881). The covariates not used for matching (i.e., primary site, sex, and age) were balanced between the two time periods, as shown in Table 3.
Table 3. MATCHING OF CONTROL GROUP PATIENTS BY PROGNOSTIC FACTORS

* Most were mucosal primary sites.
Thus, there was no difference in the natural history of AJCC stage III melanoma in the control group as judged by OS based on chronological time of entry into JWCI’s melanoma database. Furthermore, when the important prognostic factors identified by Cox regression models were used for matching, control patients in the two time periods had similar OS. Therefore, enhanced survival of the PV patients, all but two of whom were seen after January 1, 1985, was unlikely to be due to differences in the time of therapy.
Overall Survival of PV and Control Groups Matched By Six Prognostic Factors
Because some other selection bias might have determined which patients received postoperative PV, we performed a computerized random matching of all PV and control patients from the entire group of stage III patients (see Table 1). Six prognostic factors were used in the match: number of positive nodes, nodal size (palpable versus nonpalpable), primary T stage, ulceration, sex, and age. The computer matched 739 pairs with the matching covariates shown in Table 4. As shown in Figure 5, the patients receiving PV had a significantly (P = .0001) higher median OS (55.3 months vs. 31.6 months), a higher 5-year OS (48.8 ± 2% vs. 36.8 ± 2%), and a higher 10-year OS (42 ± 2% vs. 31 ± 2%) compared to control patients matched by the same six covariates.
Table 4. CHARACTERISTICS OF 739 PAIRS OF POLYVALENT VACCINE AND CONTROL PATIENTS MATCHED BY SIX COVARIATES

* Most were mucosal primary sites.

Figure 5. Overall survival of 739 pairs of PV and control patients matched by six prognostic factors: number of positive nodes, nodal size, primary T stage, ulceration, sex, and age.
Overall Survival of PV and Control Groups Who Began Vaccine Within 4 Months of Lymphadenectomy
In order to perform an even more rigorous comparison of PV and control groups, we excluded any patient from Table 2 who was not seen by our staff or did not begin vaccine therapy within four months after lymphadenectomy. We then selected patients for analysis based upon available data for seven prognostic factors listed in Table 5. These factors included number of positive nodes, nodal size, primary T stage, ulceration, sex, site of primary, and age. These criteria were fulfilled in 445 PV patients and 1060 control patients. Table 5 shows the overall survival impact on this subset of patients of the various prognostic factors according to the Cox proportional hazards model. Postsurgical therapy with PV had a significant effect on OS (P = .0001) and it reduced the risk ratio of death due to melanoma to 0.64 (95% confidence interval; 0.55–0.76). The Kaplan Meier survival curves for PV and control patients were significantly different. Median OS and 5-year OS in all PV vs. all control patients were 70.4 vs. 31 months and 52% vs. 37%, respectivley (P = 0.0001) (data not shown).
Table 5. COX REGRESSION ANALYSIS OF EIGHT PROGNOSTIC FACTORS ON POLYVALENT VACCINE AND CONTROL PATIENTS

DISCUSSION
This study reports the first definitive analysis of the efficacy of adjuvant Canvaxin PV immunotherapy after resection of melanoma metastatic to the regional nodes. Multiple analyses of outcome data with diverse statistical methods showed that PV dramatically and consistently increased the OS of patients matched with a control group for all known prognostic factors and managed by the same team of surgical and medical oncologists at a tertiary melanoma referral center. Thus, our findings are consistent with the hypothesis that postoperative administration of PV prolongs survival.
This study would not have been possible without the large prospective melanoma database established 30 years ago by our group, or without consistent surgical guidelines for the workup and management of stage III melanoma throughout this time period. These guidelines allowed us to compare PV and control patients within and between time periods and thereby obtain a prospective audit of treatment results over the years.
Selection bias is always a concern when comparing groups of patients from nonrandomized phase II trials. Fortunately, the AJCC Melanoma Staging Committee’s recent identification and validation of important prognostic factors in stage III melanoma provided an opportunity to look for possible selection bias in the PV group. The similar distribution of these important prognostic factors between PV and control groups indicates the absence of a selection bias in this study. Also, there was no survival difference by time period for control patients matched by known prognostic factors. If more favorable patients had been selected for PV treatment, then the control patients seen after January 1, 1985, should have had a lower OS than similar patients seen during the earlier period. This type of comparison also rules out the possibility of selection bias introduced by the rapid evolution of scanning devices during the last 10 to 15 years. Increased detection of subclinical distant metastasis by routine CT and MR scanning could have eliminated patients who previously might have been enrolled in the PV protocol; if so, then OS would be higher for control patients treated more recently. However, the survival characteristics of control patients during the two time periods were nearly identical based on the overall population and by matched-pair analysis (see Fig. 4). Similarly, the PV patients showed improved survival independent of whether the control patients were selected from an earlier or more recent time period.
Other factors that should be considered as a possible source of bias include the thoroughness of lymphadenectomy and whether the surgical operation was performed by JWCI faculty or at an outside facility. However, the proportion of lymphadenectomies performed at JWCI (PV 32.5% vs. control 35.9%) versus an outside facility (PV 67.5% vs. control 64.1%) was similar between the two groups (P = .103 by chi-square test), and the number of resected lymph nodes as a surrogate marker for the completeness of lymphadenectomy 54 was equivalent between PV and control groups for inguinal (19.1 vs. 20.0), axillary (25.4 vs. 24.0), and cervical basins (15.9 vs. 17.0) (P = .28, 0.443, and 0.27, respectively, by Student t test). Performance status of patients entering a trial is always a concern, but AJCC stage III patients who have undergone a lymphadenectomy uniformly exhibit normal performance status; thus, performance status could not be the source of selection bias. The size of the nodal metastases (palpable or not palpable) and the number of metastatic nodes are the most important prognostic factors and the most obvious sources of potential selection bias. However, the PV and control groups were well balanced in relation to these factors, and this source could be excluded by the matched-pair analysis (see Table 4) and the survival characteristics of PV and control patients stratified by these factors (see Figs. 2–4).
Clinical trials for testing the efficacy of cancer vaccines are difficult because a vaccine has no direct cytotoxic effect on tumor cells. Instead, its therapeutic effects depend on the ability of its tumor-associated antigens to induce immune responses against cross-reactive antigens on the patient’s autologous tumor cells. 19 Thus, patients who do not develop cell-mediated immunity and/or antibody responses cannot derive any benefit from a therapeutic vaccine. A patient’s pretreatment tumor burden is of critical importance because the latency period for the induction of a maximum immune response is 8 to 16 weeks, during which the melanoma metastases are growing with an average doubling time of 30 to 40 days and likely to overwhelm the immune response generated by the vaccine. For this reason, patients with large tumor burdens are better served by surgical resection of the tumor mass or by agents that have an immediate cytotoxic effect on tumor cells. In melanoma, patients with in-transit disease represent the ideal population for studying a therapeutic vaccine because their tumor burden is small and visible. In fact, in-transit melanoma was the earliest clinical model used to test the efficacy of immunotherapy in man. In 1970, we first reported that injection of BCG into in-transit melanoma metastases induced regression of noninjected (15%) as well as injected (90%) lesions in the skin. 7 Similar observations of regression for intratumoral injections of melanoma metastatic to the bladder 55,56 eventually led to studies in bladder cancer and the FDA approval of intravesical BCG for the treatment of superficial bladder cancer. 57 Similarly, our group recently used the in-transit disease model to evaluate the efficacy of active immunotherapy with PV for small-volume evaluable disease. Objective responses to PV were evaluated in 54 patients with measurable in-transit disease. 53 The rate of clinical response was 17%, and seven patients had complete remission of all disease. The median duration of complete remission was 22 months, and three patients have remained in complete remission at a follow-up exceeding 10 years.
Although extensive analysis failed to identify any selection bias that could explain the improved survival in patients receiving postoperative adjuvant active immunotherapy with PV, we readily acknowledge that our findings are based on phase II trials of nonrandomized patients, not on a randomized clinical trial (RCT). The RCT is the gold standard for evaluation of new cancer therapies because the process of randomization should equally distribute important known (and unknown) prognostic factors between the two therapy groups and thereby avoid selection bias. However, an RCT is most practical in patients with measurable tumor burden. When metastatic tumor burden is easily measurable, clinical regression can be objectively evaluated, and expected overall survival is short enough to provide an alternative objective endpoint of therapeutic effectiveness. In melanoma, the ideal population for an RCT would be patients with in-transit melanoma. However, its rarity makes in-transit melanoma impractical for evaluation of PV in an RCT.
In the present study, our population of interest was patients who had undergone complete resection of melanoma metastatic to the regional lymph nodes. Not only does resection eliminate the use of objective tumor regression as an endpoint, but if resection is curative in a significant proportion of cases, then the number of patients and duration of follow-up must be extensive to detect significant differences in OS between study and control groups. Trials that are usually powered to detect a 10% or less improvement in OS may require several thousand patients and 6 to 10 years of follow-up. Although the effectiveness of postsurgical adjuvants in more common neoplasms such as breast and colon cancer has been demonstrated in such large trials, we were hesitant to embark on a long and expensive phase III trial of Canvaxin PV in melanoma until we had compelling evidence of activity from phase II trials. The search for effective immunomodulators as vaccine adjuvants and the difficulty in bioengineering the manufacturing process and quality control procedures for a viable whole-cell vaccine delayed the initiation of a phase III Canvaxin trial for many years; this delay allowed us to collect the data summarized in this paper. These data provide compelling evidence of PV’s efficacy as a postsurgical adjuvant.
The findings of our present study indicate that we have every reason to expect a positive result in the ongoing randomized phase III trial of adjuvant Canvaxin PV for stage III melanoma. We hope this trial will end our 40-year quest for effective active specific immunotherapy against melanoma. Now in its third year of accrual, the trial is a multicenter international randomized study of BCG plus PV versus BCG plus placebo as postsurgical adjuvant therapy for patients with resected AJCC stage III melanoma. The endpoints are overall and disease-free survival.
CONCLUSIONS
This large single-institution study has found that the OS of patients managed by the same team of surgical and medical oncologists was consistently and dramatically prolonged when postsurgical adjuvant therapy included Canvaxin PV. Using modern statistical methods and controlling for all known prognostic factors, we have been unable to find any selection bias that would explain the observed doubling in median survival and large reduction in hazard ratio for death due to melanoma. Thus, based on present knowledge we must conclude that PV has significant activity as a postsurgical adjuvant in AJCC stage III melanoma. We hope that the multicenter randomized phase III trial, sponsored by CancerVax Corporation, and which is ongoing at over 50 cancer centers in the United States, Europe, and Australia, will provide confirmation of these phase II results within the next few years.
This paper also presents the results from the largest single-institution series (n = 2,602) of AJCC stage III melanoma patients yet reported and provides independent confirmation of the prognostic features identified in the recently reported AJCC melanoma staging system. Although the AJCC staging system was based on patients whose lymph node metastases were present at the time of primary melanoma diagnosis (synchronous metastases), whereas our patients more often had delayed nodal metastases (asynchronous metastases), we have found the most important prognostic features (number of nodal metastases, size of nodal metastases, and ulceration of the primary) are similar for the two groups.
DISCUSSION
Dr. Marc C. Wallack (New York, NY): It has been a great honor for me to have been admitted to the American Surgical Association and also an honor to be asked to comment on the work of a surgical scientist whom I greatly admire. In this paper, which I have reviewed, Dr. Morton and his colleagues report results on the use of a polyvalent melanoma vaccine called Canvaxin which consists of three irradiated melanoma cell lines given incidentally with BCG as an adjuvant. Although Dr. Morton and his group are currently in the third year of a definitive randomized prospective Canvaxin adjuvant therapy trial, for this paper he has used his extraordinary computerized melanoma database of 10,583 patients seen by his staff since 1971 to create an outcome-based trial of AJCC stage III melanoma patients either treated with the vaccine after surgical lymphadenectomy versus those patients who chose surgery but elected not to participate in a trial but remained available for follow-up.
Of the 2,602 patients who underwent complete lymphadenectomy for stage III disease, median survival and 5-year overall survival were higher in the vaccine group, with a P value at .001. Furthermore, when the vaccine patients were matched with non-vaccine patients by six covariants mentioned in the paper, patients receiving the vaccine had a higher median and 5-year overall survival when compared to the computer-generated controls. Dr. Morton and his group are to be congratulated for the strengths of this paper, while we wait for the data from the randomized prospective trial. One, the same group of cancer scientists, Dr. Morton’s group, have been involved in this work from the beginning of these cancer vaccine trials, which started in 1984. Two, a clinical lab has been constructed at the John Wayne Cancer Institute so that the consistency of production of this vaccine is assured and closely monitored. Three, there has always been a clinical review of both the clinical and scientific data obtained from this trial to ensure scientific advancement. Four, a specifically noteworthy computerized patient-based database has been assembled which permits the preservation of data and also the presentation of this computer-generated trial, which continues to confirm the potential positive outcome of what we look for in a phase III randomized trial. And Number 5, and the most important, I think, is the passion and perseverance of this group led by Dr. Morton to work toward the completion of this phase III randomized trial.
Two questions, please, for Dr. Morton: Number 1, will we as vaccine investigators ever have a definitive laboratory test to show the biologic response modified by polyvaccine therapy prior to survival data being generated? And Number 2, a suggestion of whether or not an interferon group can be generated from your database, because many groups treated these patients with interferon, so that this group can be compared with the other two groups in the trial.
Presenter Dr. Donald l. Morton (Santa Monica, CA): In terms of immunologic monitoring, we have developed three tests that can be used during the first 4 months of vaccine immunotherapy. If the patient develops a positive skin test response and IgM antibodies to TA-90 (a glycoprotein antigen discovered in our laboratory) or to ganglioside GM2, survival increases by a factor of approximately threefold. The survival of patients who do not develop these immunologic responses to Canvaxin PV is similar to that of patients treated with surgery alone.
As far as interferon is concerned, the latest analysis of the interferon trials has shown that interferon prolongs disease-free survival but does not affect overall survival. In addition, interferon has considerable toxicity.
Dr. Steven A. Rosenberg (Bethesda, MD): Dr. Morton has been a pioneer in the field of immunotherapy for the past 30 years, and I am delighted he gave me a chance to review this paper before the meeting. The paper represents a heroic attempt to compare patients that received this Canvaxin vaccine to patients who did not receive it. It is not a randomized comparison, and although there were no factors identified that can explain the difference between the two groups, it is difficult in a nonrandomized comparison to be sure that one has taken account of all the relevant factors. The danger of retrospective, nonrandomized comparisons is illustrated clearly by a paper published last year in the Journal of Clinical Oncology. This paper presents data on two sequential trials conducted by cooperative cancer groups in the United States comparing no treatment in stage III melanoma patients to patients that received interferon. The #1684 trial was conducted between 1984 and 1990 and the #1690 trial between 1991 and 1995. The paper discusses the two no-treatment groups in the two sequential trials. Although the admission and eligibility criteria for both trials were identical, in the second trial, conducted 5 years after the first, the no-treatment group has a highly statistically significant improvement in survival (P < .001) compared to the no-treatment group in the first trial. Part of this improvement is a tribute to Dr. Morton, who devised a sentinel lymph node procedure which is in fact, in widespread use around this country which, in fact, can lead to stage migration. We now can detect patients with positive lymph nodes that might not have been found to have positive lymph nodes before. And if we add molecular PCR techniques to detect melanoma cells in lymph nodes, this kind of stage migration can become even more pronounced. So we do have to wait for a randomized trial of Canvaxin, and it is a further tribute to Dr. Morton that given these results, he has now instituted and is halfway through this randomized trial. I would ask him whether or not he thinks stage migration might account for part of the results he has seen in the present paper.
I am also interested in Dr. Morton’s approach to the randomized trial. You have definitively concluded in this paper that it is an advantage to receive Canvaxin. Yet in half of the patients in the randomized trial, they are not getting the vaccine. The no-vaccine group is getting BCG and there are multiple prospective randomized trials showing BCG is of no benefit in these patients. So how do you actually approach patients in the randomized trial since you seem convinced that the vaccine is indeed of benefit?
Dr. Donald L. Morton (Santa Monica, CA): I do not agree with Dr. Rosenberg’s interpretation of stage migration. Although both ECOG trials were for patients with AJCC stage IIB and stage III melanoma, patients in the first trial (ECOG 1684) underwent elective lymph node dissection and only 11% had negative nodes. Patients in the second trial (ECOG 1690) did not require elective lymph node dissection, and 25% of them had negative nodes. The apparent difference in survival can probably be explained by differences in the prognostic factors for the patients in these two trials. In regard to stage migration as a factor in the results of active immunotherapy, only 68 of the 935 patients (7.3%) in the Canvaxin PV group and 35 of the 1,667 patients (2.1%) in the control group underwent sentinel lymphadenectomy. Because these numbers represent a very small percentage of the total population, I do not believe that stage migration is relevant.
Why would we undertake a randomized trial when we already have such profound evidence of clinical efficacy? In fact, two regulatory agencies, one in Canada and the other in Australia, have agreed to accept a licensing application based upon this data from the John Wayne Cancer Institute. However, data from a single center are usually not adequate for many regulatory agencies, which require duplication of results at multiple sites. Therefore, the FDA has insisted on a multicenter randomized trial. The FDA makes the rules, and we must abide by them.
Dr. Timothy J. Eberlein (St. Louis, MO): Dr. Morton, first off I want to congratulate you. You have spent really the last 40 years trying to improve patient survival in melanoma. Obviously, other discussants have talked about the importance of waiting for the randomized prospective trial, so I am not going to ask about that and about some of the statistical evaluation of this manuscript. But I do want to ask a couple of things about the toxicity of the trial.
One of the points that you make in your paper is the comparison of the lack of toxicity using this polyvalent vaccine versus the interferon-alpha. So would you comment on the toxicity?
The second is that it is interesting that in the randomized prospective trial, you are utilizing BCG in both of the arms of the trial. What effect do you think the BCG is really playing, and how pivotal is that to the success of the polyvalent vaccine?
The third question is regarding the immune response. And again, maybe one of the issues that Dr. Rosenberg was trying to get at was the objective evidence, not relying on statistical evaluation, but objective evidence that the polyvalent vaccine is really playing a role. What immune response are you seeing in the patients who received the polyvalent vaccine either in the nonrandomized trial or in the randomized trial between the two arms?
Once again, thank you very much. We see the importance of maintaining an accurate prospective database.
Dr. Donald L. Morton (Santa Monica, CA): In regard to toxicity, the first two doses of Canvaxin PV are administered with BCG. Patients develop some ulcerations at the site of injection, which heal in about 6 to 8 weeks, and occasionally they develop low-grade fever and malaise. However, the symptoms of toxicity are minimal. Patients come to the clinic, receive the vaccine, and return to work. Compared to treatment with high-dose interferon, therapy with Canvaxin PV is a “walk in the park”—which is why patient compliance with the protracted vaccine regimen is extremely high.
This phase III multicenter trial represents the culmination of 40 years of research, during which I have learned not to underestimate or ignore any variable of potential prognostic significance. BCG is included in both arms of this randomized, double-blinded trial because that is the only way to separate its nonspecific immunostimulatory effect (if any) from the specific immune responses induced by the vaccine. Although not definitive, there is some evidence that BCG has immune activity. In the randomized trial undertaken by Cascinelli’s group (Cancer Immunol Immunother 1989; 28:282), adjuvant administration of BCG was associated with about a 10% enhanced survival compared to surgery alone. This increase was not statistically significant because the trials were powered for a larger difference.
Will the immune response induced by the vaccine correlate well with survival? Immune response to vaccine therapy is measured during the first 4 months, and survival is compared between immune responders and nonresponders. This allows us to determine whether the active agent is, in fact, the immune response generated by the vaccine. If the trial’s results show that immune response to the vaccine is a valid surrogate endpoint, then we will not need to wait 6 years for complete survival data to determine the outcome. Instead, we can look at improvements in terms of the immune response induced.
Footnotes
Supported in part by grant CA12582 from the National Cancer Institute and by funding from the Wayne and Gladys Valley Foundation (Oakland, CA) and the Harold McAlister Charitable Foundation (Los Angeles, CA).
Canvaxin and CancerVax are trademarks of CancerVax Corporation.
Presented at the 122nd Annual Meeting of the American Surgical Association, April 24–27, 2002, The Homestead, Hot Springs, Virginia.
Correspondence: Donald L. Morton, MD, John Wayne Cancer Institute, 2200 Santa Monica Boulevard, Santa Monica, CA 90404.
E-mail: mortond@jwci.org
Accepted for publication April 24, 2002.
References
- 1.Jemal A, Thomas A, Murray T, et al. Cancer statistics. CA Cancer J Clin 2002; 52: 23–47. [DOI] [PubMed] [Google Scholar]
- 2.Morton DL, Barth A. Vaccine therapy for malignant melanoma. CA Cancer J Clin 1996; 46: 225–244. [DOI] [PubMed] [Google Scholar]
- 3.Morton DL, Wanek L, Nizze JA, et al. Improved long-term survival after lymphadenectomy of melanoma metastatic to regional nodes. Analysis of prognostic factors in 1134 patients from the John Wayne Cancer Clinic. Ann Surg 1991; 214: 491–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barth A, Morton DL. Role of adjuvant therapy in melanoma management. Cancer 1995; 75: 726–734. [DOI] [PubMed] [Google Scholar]
- 5.Kirkwood JM, Ibrahim JG, Sondak VK, et al. High- and low-dose interferon alfa-2b in high-risk melanoma: first analysis of intergroup trial E1690/S9111/C9190. J Clin Oncol 2000; 18: 2444–2458. [DOI] [PubMed] [Google Scholar]
- 6.Lens MB, Dawes M. Interferon alfa therapy for malignant melanoma: a systematic review of randomized controlled trials. J Clin Oncol 2002; 20: 1818–1825. [DOI] [PubMed] [Google Scholar]
- 7.Morton DL, Eilber FR, Malmgren RA, et al. Immunological factors which influence response to immunotherapy in malignant melanoma. Surgery 1970; 68: 158–164. [PubMed] [Google Scholar]
- 8.Summer WC, Foraker AG. Spontaneous regression of human melanoma: clinical and experimental studies. Cancer 1960; 13: 79–81. [DOI] [PubMed] [Google Scholar]
- 9.Eagan RT, Sparks FC, Morton DL. Cancer immunotherapy. Ann Intern Med 1973; 78: 153–154. [DOI] [PubMed] [Google Scholar]
- 10.Eilber FR, Holmes EC, Morton DL. Immunotherapy experiments with a methylcholanthrene-induced guinea pig liposarcoma. J Natl Cancer Inst 1971; 46: 803–808. [PubMed] [Google Scholar]
- 11.Morton DL, Malmgren RA, Holmes EC, et al. Demonstration of antibodies against human malignant melanoma by immunofluorescence. Surgery 1968; 64: 233–240. [PubMed] [Google Scholar]
- 12.Morton DL, Eilber FR, Holmes EC, et al. BCG immunotherapy of malignant melanoma: summary of a seven-year experience. Ann Surg 1974; 180: 635–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morton DL, Eilber FR, Holmes EC, et al. Preliminary results of a randomized trial of adjuvant immunotherapy in patients with malignant melanoma who have lymph nodes metastases. Aust NZ J Surg 1978; 48: 49–52. [DOI] [PubMed] [Google Scholar]
- 14.Livingston PO, Wong GY, Adluri S, et al. Improved survival in stage III melanoma patients with GM2 antibodies: a randomized trial of adjuvant vaccination with GM2 ganglioside. J Clin Oncol 1994; 12: 1036–1044. [DOI] [PubMed] [Google Scholar]
- 15.Wallack MK, Sivanandham M, Balch CM, et al. Surgical adjuvant active specific immunotherapy for patients with stage III melanoma: the final analysis of data from phase III, randomized, double-blind, multicenter vaccinia melanoma oncolysate trial. J Am Coll Surg 1998; 187: 69–77. [DOI] [PubMed] [Google Scholar]
- 16.Kirkwood JM, Ibrahim JG, Sosman JA, et al. High-dose interferon alfa-2b significantly prolongs relapse-free and overall survival compared with the GM2-KLH/QS-21 vaccine in patients with resected stage IIb-III melanoma: results of intergroup trial E1694/S9512/C509801. J Clin Oncol 2001; 19: 2370–2380. [DOI] [PubMed] [Google Scholar]
- 17.Jones PC, Sze LL, Liu PY, et al. Prolonged survival for melanoma patients with elevated IgM antibody to oncofetal antigen. J Natl Cancer Inst 1981; 66: 249–254. [PubMed] [Google Scholar]
- 18.Morton DL, Foshag LJ, Hoon DSB, et al. Prolongation of survival in metastatic melanoma after active specific immunotherapy with a new polyvalent melanoma cell vaccine. Ann Surg 1992; 216: 463–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morton DL, Ollila DW, Hsueh EC, et al. Cytoreductive surgery and adjuvant immunotherapy: a new management paradigm for metastatic melanoma. CA Cancer J Clin 1999; 49: 101–116. [DOI] [PubMed] [Google Scholar]
- 20.Chi DD, Merchant RE, Rand R, et al. Molecular detection of tumor-associated antigens shared by human cutaneous melanomas and gliomas. Am J Pathol 1997; 150: 2143–2152. [PMC free article] [PubMed] [Google Scholar]
- 21.Gupta RK, Morton DL. Studies of a melanoma tumor-associated antigen detected in the spent culture medium of a human melanoma cell line by allogeneic antibody. III. Physicochemical properties. J Natl Cancer Inst 1984; 72: 83–92. [DOI] [PubMed] [Google Scholar]
- 22.Gupta RK, Morton DL. Studies of a melanoma tumor-associated antigen detected in the spent culture medium of a human melanoma cell line by allogeneic antibody. II. Immunobiologic characterization. J Natl Cancer Inst 1984; 72: 75–82. [DOI] [PubMed] [Google Scholar]
- 23.Gupta RK, Morton DL. Studies of a melanoma tumor-associated antigen detected in the spent culture medium of a human melanoma cell line by allogeneic antibody. I. Purification and development of a radioimmunoassay. J Natl Cancer Inst 1984; 72: 67–74. [DOI] [PubMed] [Google Scholar]
- 24.Hoon DS, Yuzuki D, Hayashida M, et al. Melanoma patients immunized with melanoma cell vaccine induce antibody responses to recombinant MAGE-1 antigen. J Immunol 1995; 154: 730–737. [PubMed] [Google Scholar]
- 25.Hsueh EC, Gupta RK, Qi K, et al. Correlation of specific immune responses with survival in melanoma patients with distant metastases receiving polyvalent melanoma cell vaccine. J Clin Oncol 1998; 16: 2913–2920. [DOI] [PubMed] [Google Scholar]
- 26.Huang SK, Okamoto T, Morton DL, et al. Antibody responses to melanoma/melanocyte autoantigens in melanoma patients. J Invest Dermatol 1998; 111: 662–667. [DOI] [PubMed] [Google Scholar]
- 27.Nishinaka Y, Ravindranath MH, Irie RF. Development of a human monoclonal antibody to ganglioside G(M2) with potential for cancer treatment. Cancer Res 1996; 56: 5666–5671. [PubMed] [Google Scholar]
- 28.Ravindranath MH, Morton DL, Irie RF. An epitope common to gangliosides O-acetyl-GD3 and GD3 recognized by antibodies in melanoma patients after active specific immunotherapy. Cancer Res 1989; 49: 3891–3897. [PubMed] [Google Scholar]
- 29.Ravindranath MH, Tsuchida T, Morton DL, et al. Ganglioside GM3: GD3 ratio as an index for the management of melanoma. Cancer 1991; 67: 3029–3035. [DOI] [PubMed] [Google Scholar]
- 30.Ravindranath MH, Amiri AA, Bauer PM, et al. Endothelial-selectin ligands sialyl Lewis(x) and sialyl Lewis(a) are differentiation antigens immunogenic in human melanoma. Cancer 1997; 79: 1686–1697. [DOI] [PubMed] [Google Scholar]
- 31.Sarantou T, Chi DD, Garrison DA, et al. Melanoma-associated antigens as messenger RNA detection markers for melanoma. Cancer Res 1997; 57: 1371–1376. [PubMed] [Google Scholar]
- 32.Tai T, Cahan LD, Tsuchida T, et al. Immunogenicity of melanoma-associated gangliosides in cancer patients. Int J Cancer 1985; 35: 607–612. [DOI] [PubMed] [Google Scholar]
- 33.Tai T, Paulson JC, Cahan LD, et al. Ganglioside GM2 as a human tumor antigen (OFA-I-1). Proc Natl Acad Sci USA 1983; 80: 5392–5396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tsuchida T, Saxton RE, Morton DL, et al. Gangliosides of human melanoma. J Natl Cancer Inst 1987; 78: 45–54. [DOI] [PubMed] [Google Scholar]
- 35.Okamoto T, Irie RF, Fujii S, et al. Anti-tyrosinase-related protein-2 immune response in vitiligo patients and melanoma patients receiving active-specific immunotherapy. J Invest Dermatol 1998; 111: 1034–1039. [DOI] [PubMed] [Google Scholar]
- 36.Morioka N, Kikumoto Y, Hoon DS, et al. A decapeptide (Gln-Asp-Leu-Thr-Met-Lys-Tyr-Gln-Ile-Phe) from human melanoma is recognized by cytotoxic T lymphocytes in melanoma patients. J Immunol 1994; 153: 5650–5658. [PubMed] [Google Scholar]
- 37.Takahashi T, Conforti A, Kikumoto Y, et al. Augmentation of IgM antibody to gp43 tumor-associated antigen-peptide by polyvalent melanoma cell vaccine. J Clin Immunol 1998; 18: 299–305. [DOI] [PubMed] [Google Scholar]
- 38.Tsuchida T, Saxton RE, Morton DL, et al. Gangliosides of human melanoma. Cancer 1989; 63: 1166–1174. [DOI] [PubMed] [Google Scholar]
- 39.Jones RC, Kelley M, Gupta RK, et al. Immune response to polyvalent melanoma cell vaccine in AJCC stage III melanoma: An immunologic survival model. Ann Surg Oncol 1996; 3: 437–445. [DOI] [PubMed] [Google Scholar]
- 40.Euhus DM, Gupta RK, Morton DL. Induction of antibodies to a tumor-associated antigen by immunization with a whole melanoma cell vaccine. Cancer Immunol Immunother 1989; 29: 247–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gupta RK, Morton DL. Immunochemical characterization of fetal antigen isolated from spent medium of a human melanoma cell line. J Natl Cancer Inst 1983; 70: 993–1004. [PubMed] [Google Scholar]
- 42.Wong JH, Gupta RK, Morton DL. Recovery of a cell surface fetal antigen and antibody in stage I melanoma patients. Cancer Immunol Immunother 1988; 27: 142–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wong JH, Xu SH, Gupta RK, et al. Tumor associated antigen immune complexes. Arch Surg 1990; 125: 187–191. [DOI] [PubMed] [Google Scholar]
- 44.Doi F, Chi DDJ, Charuworn BB, et al. Detection of beta-human chorionic gonadotropin mRNA as a marker for cutaneous malignant melanoma. Int J Cancer 1996; 65: 454–459. [DOI] [PubMed] [Google Scholar]
- 45.Cheresh DA, Reisfeld RA, Varki AP. O-acetylation of disialoganglioside GD3 by human melanoma cells creates a unique antigenic determinant. Science 1984; 225: 844–846. [DOI] [PubMed] [Google Scholar]
- 46.Wilson BS, Imai K, Natali PG, et al. Distribution and molecular characterization of a cell-surface and a cytoplasmic antigen detectable in human melanoma cells with monoclonal antibodies. Int J Cancer 1981; 28: 293–300. [DOI] [PubMed] [Google Scholar]
- 47.DiFronzo LA, Gupta RK, Essner R, et al. An enhanced humoral immune response correlates with improved disease-free and overall survival in American Joint Committee on Cancer stage II melanoma patients receiving adjuvant polyvalent vaccine. J Clin Oncol 2002; 20: 3242–3248. [DOI] [PubMed] [Google Scholar]
- 48.Barth A, Hoon DSB, Foshag LJ, et al. Polyvalent melanoma cell vaccine induces delayed-type hypersensitivity and in vitro cellular immune response. Cancer Res 1994; 54: 3342–3345. [PubMed] [Google Scholar]
- 49.Hsueh EC, Gupta RK, Yee R, et al. Does endogenous immune response determine the outcome of surgical therapy for metastatic melanoma? Ann Surg Oncol 2000; 7: 232–238. [DOI] [PubMed] [Google Scholar]
- 50.Chan AD, Morton DL. Active immunotherapy with allogeneic tumor cell vaccines: present status. Semin Oncol 1998; 25: 611–622. [PubMed] [Google Scholar]
- 51.Hoon DSB, Okamoto T, Wang H-J, et al. Is the survival of melanoma patients receiving polyvalent melanoma cell vaccine linked to the human leukocyte antigen phenotype of patients? J Clin Oncol 1998; 16: 1430–1437. [DOI] [PubMed] [Google Scholar]
- 52.Balch CM, Soong SJ, Gershenwald JE, et al. Prognostic factors analysis of 17,600 melanoma patients: validation of the American Joint Committee on Cancer melanoma staging system. J Clin Oncol 2001; 19: 3622–3634. [DOI] [PubMed] [Google Scholar]
- 53.Hsueh EC, Nathanson L, Foshag LJ, et al. Active specific immunotherapy with polyvalent melanoma cell vaccine in patients with in-transit melanoma metastases. Cancer 1999; 85: 2160–2169. [PubMed] [Google Scholar]
- 54.Chan AD, Essner R, Wanek LA, et al. Judging the therapeutic value of lymph node dissections for melanoma. J Am Coll Surg 2000; 191: 16–22. [DOI] [PubMed] [Google Scholar]
- 55.Silverstein MJ, DeKernion J, Morton DL. Malignant melanoma metastatic to the bladder: regression following intratumor injection of BCG vaccine. JAMA 1974; 229: 688. [PubMed] [Google Scholar]
- 56.DeKernion JB, Golub SH, Gupta RK, et al. Successful transurethral intralesional BCG therapy of a bladder melanoma. Cancer 1975; 36: 1662–1667. [DOI] [PubMed] [Google Scholar]
- 57.Lamm DL, DeHaven JI, Shriver J, et al. A randomized prospective comparison of oral versus intravesical and percutaneous bacillus Calmette-Guerin for superficial bladder cancer. J Urol 1990; 144: 65–67. [DOI] [PubMed] [Google Scholar]