

Abstract
Objective
To investigate use of transcriptional regulatory elements (promoters) for tumor-associated antigens to achieve HSV-1 replication preferentially in cells that overexpress the tumor-associated antigens.
Summary Background Data
An important advantage of replicating viruses for cancer therapy is their ability to simultaneously destroy tumor cells by replication and release progeny virion to infect and destroy adjacent cancer cells. This strategy requires regulation of the viral life cycle to obtain robust replication in neoplastic cells and minimize replication in nonneoplastic cells.
Methods
Promoters for the human carcinoembryonic antigen (CEA) and MUC1/DF3 tumor-associated antigens were characterized and cloned into HSV-1 mutants as heterologous promoters regulating expression of two different HSV-1 genes. Viral replication in tumor cells and cytotoxicity was quantified with in vitro assays. Antineoplastic efficacy was characterized in a flank tumor xenograft model.
Results
Several CEA promoters were cloned and characterized using luciferase reporter assays. The most specific promoter was used to construct and isolate two different HSV-1 mutants in which critical genes are regulated by this promoter (ICP4 and γ134.5). Similarly, the promoter for the DF3/MUC1 tumor-associated antigen was cloned into a third HSV-1 mutant such that it regulates expression of γ134.5. Regulation of ICP4 expression by the CEA promoter during HSV-1 infection overly attenuates viral replication. Regulation of γ134.5 expression by either the CEA promoter or the MUC1/DF3 promoter during HSV-1 infection modulates viral replication, with preferential replication in cells that overexpress the corresponding tumor-associated antigen. A single intratumoral inoculation of an HSV-1 mutant with the MUC1/DF3 promoter regulating γ134.5 expression results in significant antineoplastic activity in MUC1-positive pancreatic carcinoma xenografts as compared to mock inoculation.
Conclusions
Promoters for tumor-associated antigens may be incorporated into the HSV-1 genome to regulate HSV-1 replication. The choices of HSV-1 gene and tumor-associated promoter are important determinants of success of this strategy. Because of its preferential replication in MUC1-positive tumors, an HSV-1 mutant with the MUC1/DF3 promoter regulating γ134.5 expression will undergo further examination as a novel cancer therapy agent.
Most cancer gene therapies using viruses have focused on replication-defective adenovirus, adeno-associated virus, retrovirus, vaccinia virus, and herpes simplex virus 1 (HSV-1). When using such replication-defective viruses, antineoplastic effects are achieved by delivery and expression of therapeutic transgenes. Although replication-defective viruses are used in a majority of current cancer gene therapy trials, 1 this approach has important drawbacks. Therapeutic transgene delivery is nonselective: both normal and cancer cells are infected. Replication-defective viruses are also unable to spread progeny virion to cells that were not initially infected.
An alternative strategy exploits viral replication for tumor destruction, whereby infection of tumor cells by virus leads to cell destruction and simultaneous release of progeny virion that can infect adjacent tumor cells. 2 In this strategy, antineoplastic efficacy is dependent on viral replication; accordingly, it is important to maintain robust viral replication in neoplastic cells and simultaneously attenuate viral replication in nonneoplastic cells. Replication-conditional viruses can also deliver therapeutic transgenes to improve antineoplastic efficacy beyond that achieved by replication alone. 3 Tumor infection with replication-conditional viruses leads to longer transgene expression and better distribution throughout the tumor compared to tumor infection with replication-defective viruses. 4 Several viruses have been examined for their utility as replication-conditional oncolytic viruses, including HSV-1, 5 vaccinia virus, 6 Newcastle disease virus, 7 adenovirus, 8 and reovirus. 9
We have previously demonstrated preferential replication of an HSV-1 mutant hrR3 in tumors rather than normal tissue following intravascular administration. 10,11 hrR3 replication and oncolysis are attenuated in normal or quiescent cells because it is deficient in expression of viral ribonucleotide reductase (infected cell protein 6 [ICP6]). 12 The selectivity of hrR3 for liver tumors is related to significantly higher expression of ribonucleotide reductase and higher intracellular nucleotide pools in most tumors compared to surrounding normal tissues. 13 However, there are disadvantages to developing ICP6-defective HSV-1 mutants for clinical studies. ICP6-defective HSV-1 mutants are most effective against replicating cells, and accordingly quiescent cancer cells may be less susceptible to viral oncolysis. In addition, some virus may reach sites outside the liver despite regional administration, 14 and normal tissues with high replicative activity may be susceptible to infection and cytolysis by an ICP6-defective HSV-1 mutant. The normal tissues that are at greatest risk are those with high replicative activity, such as gastrointestinal mucosa and bone marrow.
We have looked to manipulate viral genes that are critical for robust HSV-1 replication but, unlike viral ribonucleotide reductase, have no cellular homologues and whose function is poorly complemented by normal cells. The immediate-early ICP4 gene product is required for HSV-1 replication in cell culture. 15 Cells do not efficiently complement HSV-1 deficiency of ICP4, and accordingly, ICP4-defective HSV-1 mutants display markedly attenuated replication. The ICP4 gene is therefore a good candidate gene to regulate to achieve replication preferentially in cancer cells. Another good candidate gene to regulate is γ134.5, which is also required for robust HSV-1 replication. The γ134.5 gene product interacts with a cellular protein phosphatase to dephosphorylate elongation initiation factor 2α (eIF-2α) and permit cellular (and viral) protein translation to proceed. 16,17 Regulation of γ134.5 expression has been demonstrated by others to be an effective strategy to regulate HSV-1 lytic replication. 18
We examined a strategy in which ICP4 expression and γ134.5 expression are regulated by heterologous transcriptional regulatory elements for tumor-associated antigens. We first examined transcriptional regulatory elements for carcinoembryonic antigen (CEA) and MUC1/DF3 because they are overexpressed in a wide variety of epithelial cancers. We isolated HSV-1 mutants in which either ICP4 or γ134.5 expression is regulated by transcriptional regulatory elements for CEA and MUC1.
METHODS
Cells and Viruses
Vero African Green Monkey kidney cells and SW620, HT29, NCIH508, LS174T, and Lovo human colon carcinoma cells were obtained from American Type Culture Collection (Manassas, VA). E5 cells (ICP4-transformed) and d 120 virus 15 were provided by David Knipe (Harvard Medical School, Boston, MA). MC26 mouse colon carcinoma cells were obtained from the National Cancer Institute Tumor Repository (Frederick, MD). A375 human melanoma cells were provided by Isaiah Fidler (M.D. Anderson Cancer Center, Houston, TX). MCF-7 cells were provided by Donald Kufe (Dana-Farber Cancer Institute, Boston, MA). SW1990 and CAPAN2 human pancreatic carcinoma cells were provided by Andrew Warshaw (Massachusetts General Hospital). Primary human hepatocytes were prepared as previously described. 11 HUVEC cells were obtained from Cell Applications, Inc. (San Diego, CA). HSV-1 viruses F strain and R3616 were provided by Bernard Roizman (University of Chicago, IL), and MGH1 was provided by E. Antonio Chiocca (Massachusetts General Hospital). hrR3, F strain, CEAγ34.5, CEAICP4, and DF3γ34.5 were propagated and titered on Vero cells, and d 120 was propagated and titered on E5 cells. Heat inactivation of virus was performed as described. 19
Cloning the CEA 5′ Flanking Region and Construction of Plasmids for Homologous Recombination
The recombining plasmid pKpX2Δ-CEAICP4afpI was constructed as follows. A 440-base pair fragment of the human CEA 5′ upstream sequence between −335 and +109 relative to the translational start site was PCR-amplified from the genomic DNA of the HT29 human colon carcinoma cell line using oligonucleotides (forward: 5′-TGTTGGATCCCATCCCACCTTCCCAGAGCC-3′; reverse: 5′-GGAGCGGCCGTGGTCTCTGCTGTCTGCTCT-3′) and designated PCEA1. The amplification product was cloned into pCRII (Invitrogren, Carlsbad, CA) to create pCRIICEAP21. To improve the strength and specificity of this transcriptional regulatory sequence, the sequence between nucleotides −150 and −199 was multimerized using a method identical to that reported by Richards et al. 20 Four copies of this sequence are inserted into the Eco 72I-Avr II site of pCRIICEAP21 to create the construct pCRIICEAP4x, and this putative CEA transcriptional regulatory sequence is designated PCEA2. The 2.1-kb sequence between −6.1 kb and −4.0 kb upstream of the transcriptional start site of the CEA gene (CEALF) was PCR-amplified from Lovo cell DNA using oligonucleotides (forward: 5′-TGGGGAATTCTGTAGACTTT-3′; reverse: 5′-CCTCCCGGGTTCAAGCAATT-3′). The amplification product was digested with Sma I and Eco RI and cloned into the plasmid pBS.SR (Stratagene, La Jolla, CA) to obtain the construct pBS.CEALF. The Eco RV-Hind III CEAP4x fragment was excised from pCRIICEAP4x and cloned into pBS.CEALF such that the CEA promoter containing four copies of the −89 to −40 sequence is located downstream and in reverse orientation relative to the CEALF sequence to create pBS.CEALFCEAP4x. This putative CEA transcriptional regulatory sequence is designated PCEA3. The 4.1-kb SalI-Mse I fragment of pGH108 (provided by Gary Hayward) containing the ICP4 coding sequence was subcloned into pBS.CEALFCEAP4x downstream of the PCEA3 sequence to obtain pBS.CEAICP4. The 6.8-kb Xba I-Sph I fragment containing the PCEA3 sequence and ICP4 coding sequence was excised from pBS.CEAICP4 and subcloned into the shuttle plasmid pSP72 (Promega) to create pSPCEAICP4. The PCEA3-ICP4 fragment was then excised as a Xba I-Xho I fragment from pSPCEAICP4 and subcloned into pcDNA3.1 (Invitrogen) to generate pcDNACEAICP4. cDNA encoding AutoFluoroescence protein (AFP) was excised from pQBI25-fC1 plasmid (QUANTUM Biotechnologies, Carlsbad, CA) with Spe I and Not I and inserted into pcDNA3.1. The resulting expression cassette, with the cytomegalovirus (CMV) promoter upstream and the polyA tail downstream of the AFP gene, was excised as a Pme I fragment and cloned into the Stu I site of pKpX2 (provided by E. Antonio Chiocca, Harvard Medical School, Boston, MA), which contains the ICP6 gene, to produce pKpX2-AFP. This plasmid was digested with Bbv CI and Bsi WI, thereby deleting 145 base pairs from the 5′ portion of the ICP6 coding sequence, generating the construct pKpX2Δ-AFP. The 7.0-kb Pme I CEAICP4 fragment of pcDNACEAICP4 was subcloned into the Eco RV site of pKpX2Δ-AFP to create the recombining plasmid pKpX2Δ-CEAICP4afpI.
The recombining plasmid pKpX2Δ-CEAγ34.5R was constructed as follows. cDNA encoding the BGH polyA tail was excised from the plasmid pQBI25-fC1 as a Nru I-Pvu II fragment and cloned into the Eco RV site of pLitmus29 (New England Biolabs, Beverly, MA) to create pLitmuspolyA. The 2.7-kb Hind III-Xba I CEALFCEAP4x fragment was excised from pSPCEAICP4 and subcloned into pLitmuspolyA to generate pLitmusCEApolyA. The Nco I-Sac I fragment of pDF3 (provided by Donald Kufe, Dana-Farber Cancer Institute, Boston, MA) containing the γ134.5 coding sequence was subcloned into pcDNA3.1 to obtain pcDNAγ34.5. The γ134.5 gene was excised from this plasmid as a Hind III fragment and subcloned into pLitmusCEApolyA to create pLitmusCEAγ34.5polyA. The CEAγ34.5polyA fragment was then excised with Bgl II-Sna BI, treated with Klenow fragment, and cloned into the Eco RV site of pKpX2Δ-AFP to obtain the recombining plasmid pKpX2Δ-CEAγ34.5R.
Luciferase Reporter Assays
We created luciferase reporter constructs with PCEA1, PCEA2, and PCEA3 and compared their ability to regulate gene expression relative to the SV40 promoter in hepatocytes and several types of human cells in which we have quantified CEA expression by Northern blot. We also compared these promoters with the strongest and most specific CEA transcriptional regulatory elements constructed by Richards and Huber, PCEA167 (provided by Brian Huber, Glaxo Wellcome Inc., Research Triangle Park, NC). 20 Cells were transfected with a plasmid containing the SV40 promoter driving luciferase expression or one of the putative CEA transcriptional regulatory sequences driving luciferase expression. Cells were transfected using LipofectAMINE/PLUS reagent (GIBCO/BRL, Gaithersburg, MD) according to the manufacturer’s directions. Protein lysates were derived from the harvested cells 48 hours later and analyzed for luciferase expression according to the manufacturer’s directions (Promega, Madison, WI). Differences in transfection efficiency among different cell lines were accounted for by normalization using luminometer readings from lysates prepared from cells transfected with the SV40 promoter driving luciferase expression.
Isolation and Characterization of Viral Mutants
To create the HSV-1 mutant CEAICP4, Bsp HI-linearized pKpX2Δ-CEAICP4afpI and d 120 DNA were cotransfected into E5 cells with LipofectAMlNE/PLUS. E5 cell lysates were then freeze-thawed three times to release infectious virus and replated onto fresh E5 cells with an agarose overlay. Recombinant viruses were identified as green fluorescent plaques under fluorescence microscopy and were isolated and plaque-purified four times on E5 cells. CEAICP4 virus stocks were then prepared by infection of E5 cells and titered by standard plaque assay in duplicate.
To create the recombinant CEAγ34.5 virus, Bsp HI-linearized pKpX2Δ-CEAγ34.5R and R3616 DNA were cotransfected into Vero cells using LipofectAMINE/PLUS reagent as described by the manufacturer. Vero cell lysates were then freeze-thawed three times to release infectious virus and replated onto Vero cells with an agarose overlay. Recombinant viruses were identified as green fluorescent plaques under fluorescence microscopy and were isolated and plaque-purified three times on Vero cells. CEAγ34.5 virus stocks were then prepared by infection of Vero cells and titered by standard plaque assay in duplicate.
Isolation and characterization of the HSV-1 mutant DF3γ34.5 is described in a separate manuscript (in preparation).
Southern Blot Analysis
Viral DNA was isolated after lysis of infected E5 cells with 0.5% SDS and proteinase K (500 μg/mL) by repeated phenol-chloroform extraction and ethanol precipitation. DNA was digested with Pst I, Nru I, or Bam HI, separated by agarose gel electrophoresis, and transferred to a nylon membrane (Amersham Corp., Arlington Heights, IL). Probes to ICP4 (3.2-kb Hinc II fragment of pGH108), ICP6 (0.7-kb Bam HI fragment of pKpX2), CEA promoter (0.6-kb Eco RV-Hind III fragment of pBSCEAP), CEALF (0.4-kb Nde I fragment of pBSCEALF), or γ134.5 were labeled, hybridized to the membrane, and detected with an ECL (enhanced chemiluminescence) system (Amersham Corp.) as described by the manufacturer.
Viral Replication and Cytotoxicity Assays
Viral replication assays were performed as described. 13 Briefly, 3 × 106 cells were infected with 6 × 106 plaque-forming units (pfu) of virus for 2 hours, at which time unadsorbed virus was removed by washing with a glycine-saline solution (pH 3.0). Forty hours after infection the supernatant and cells were harvested, exposed to three freeze-thaw cycles to release virions, and titered on Vero cells. Viral cytotoxicity assays were performed as described. 10 Briefly, cells were plated onto 96-well plates at 5,000 cells per well for 36 hours. Virus was added at multiplicity of infection (moi) values ranging from 0.0001 to 10 and incubated for 6 days. The number of surviving cells was quantitated using a colorimetric MTT assay. Tests were performed in quadruplicate.
Animal Studies
Athymic BALB/c (nu/nu) mice were obtained from Charles River Labs (Wilmington, MA). Animal studies were performed in accordance with policies of the Massachusetts General Hospital Subcommittee on Research Animal Care. Fifty-cubic-millimeter fragments of CAPAN2 tumors were inoculated into the flanks of mice, and 7 days later 1 × 108 pfu virus was inoculated directly into the tumors. Tumor sizes were measured every 5 days.
RESULTS
Functional analysis of CEA 5′ flanking region
We selected transcriptional regulatory elements (promoters) for the MUC1 and CEA genes to regulate lytic HSV-1 replication. The 5′ flanking region that regulates transcription of the MUC1 gene has been characterized, 21 and we selected a sequence that has been used to successfully regulate adenoviral replication. 22 5′ flanking sequences for the CEA gene have not been used previously to regulate viral replication, and we therefore analyzed sequences upstream of the CEA gene for specificity of transcriptional regulation. We engineered four different luciferase reporter constructs containing CEA transcriptional regulatory elements (PCEA1, PCEA2, PCEA3, and PCEA167) and compared their ability to regulate luciferase expression (relative to the SV40 promoter) in hepatocytes and several human colon carcinoma cell lines in a transient expression assay (Fig. 1A). We also performed Northern blot analysis (data not shown) to confirm differences in CEA expression between these cell lines that have been previously reported. 20

Figure 1. Analysis of CEA promoter and enhancer. (A) Schematic diagram of CEA gene transcriptional regulatory elements cloned for this study. Enhancer sequences reside in the 2.9-kb Aat II-Hind III fragment and the 2.1-kb Eco RI-Sma I fragment. The hatched arrows show the location of PCR primers used to amplify sequences between −335 and +109 relative to the transcriptional start site. The open arrows show the location of PCR primers used to amplify the enhancer sequence between the Eco RI and Sma I sites. PCEA2, PCEA3, and PCEA167 contain four copies of a sequence between −40 and −89. (B) Comparison of luciferase reporter activity between different CEA gene transcriptional regulatory elements in hepatocytes and colon carcinoma cell lines with either high or low CEA expression.
PCEA3 provides for very low levels of transcriptional activation in human hepatocytes and, in comparison, provides for 40-fold higher expression in CEA-positive NCIH508 cells (see Fig. 1B). Although PCEA1, PCEA2, and PCEA167 produced higher levels of transcription in CEA-positive cells, we chose to use PCEA3 to regulate HSV-1 gene expression because it produces the lowest levels of transcriptional activation in hepatocytes, and its performance by that measurement is superior to that of PCEA167. The specificity of PCEA3 was also demonstrated in mouse hepatocytes, which did not activate luciferase transcription based on PCEA3.
Many of the HSV immediate-early gene products are transcriptional activators, which may nonspecifically activate the CEA promoter. However, we observed that luciferase transcription regulated by the CEA promoter is not spuriously increased 3, 6, or 24 hours after HSV infection (data not shown).
Construction of HSV-1 Mutants
Based on these data we selected the PCEA3 and DF3 promoters to regulate HSV-1 lytic replication. ICP4 is an immediate-early gene product that regulates most β and γ genes and is critical for HSV-1 replication. 15 We confirmed that replication of an ICP4-defective HSV-1 mutant (d 120) is attenuated by three to five log orders compared to wild-type F strain in HT29 human colon carcinoma cells. Accordingly, ICP4 is a candidate HSV-1 gene to regulate with a tumor-associated promoter such as PCEA3. γ134.5 is an HSV-1 gene product that promotes dephosphorylation of eIF-2α and is necessary for robust HSV-1 replication. 17,18 We confirmed that replication of a γ134.5-defective HSV-1 mutant (R3616) is attenuated by one to two log orders compared to wild-type F strain. Accordingly, γ134.5 is also a candidate HSV-1 gene to regulate with a tumor-associated promoter.
We constructed three HSV-1 mutants in an attempt to regulate viralreplication by the CEA promoter and the DF3 promoter (Table 1, Fig. 2):
Table 1. HSV-1 MUTANTS

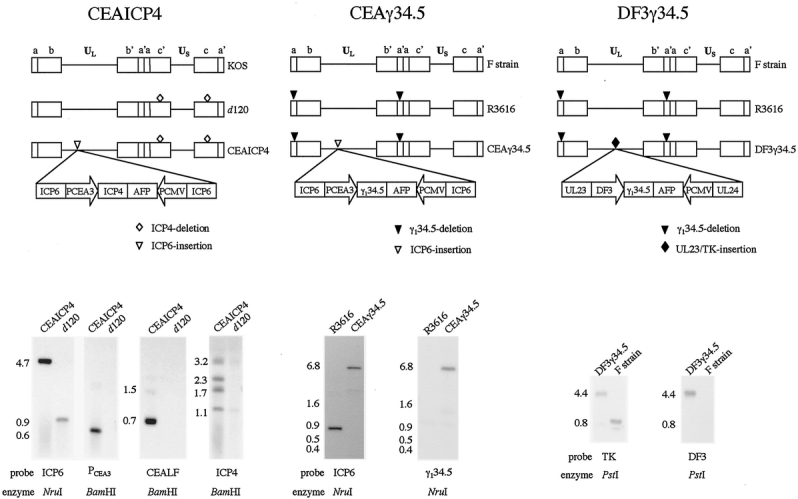
Figure 2. Schematic diagrams and Southern blot analyses of the three HSV-1 mutants. Southern blot analysis of CEAICP4 and d 120 DNA digested with Nru I using an ICP6 probe revealed the presence of the transgene insertion into the ICP6 gene locus. The 900-bp fragment observed in d 120 is expected in the absence of homologous recombination, whereas the 4.7-kb fragment observed in CEAICP4 results from integration of transgene sequences into the ICP6 gene locus. The presence of the CEA promoter construct as well as of the upstream CEA flanking sequence was confirmed by hybridization of Bam HI-digested DNA with probes to PCEA3 and CEALF. CEAICP4 contains the expected 0.6-kb fragment in the case of the PCEA3 probe and the expected 0.7-kb and 1.5-kb fragments in the case of the CEALF probe, none of which are present in d 120 DNA. Lastly, the presence of the correct ICP4 fragments in CEAICP4 was confirmed by hybridization of Bam HI-digested DNA with the 3.2-kb Hinc II fragment of ICP4 as probe. CEAICP4 maintains the ICP4 gene deletions of the d 120 backbone (1.1-kb and 3.2-kb fragments) and in addition contains the ICP4 transgene insertion into the ICP6 locus, resulting in two new fragments (1.7-kb and 2.3-kb) hybridizing to an ICP4 probe. Southern blot analysis of R3616 and CEAγ34.5 DNA digested with Nru I using an Nru I fragment of ICP6 as a probe reveals the expected 888-bp band in R3616 and the expected 6.7-kb band in CEAγ34.5 that results from transgene insertion into the ICP6 locus. Nru I-digested R3616 and CEAγ34.5 DNA examined with a γ134.5 probe reveals no hybridization in R3616 as expected and a 6.7-kb band in CEAγ34.5 as a result of homologous recombination of γ134.5 into the ICP6 gene locus. Southern blot analysis of F strain and DF3γ34.5 DNA digested with Pst I using a thymidine kinase (TK) probe reveals a 0.8-kb band of the native TK gene locus in F strain but a 4.4-kb band in DF3γ34.5 as a result of homologous recombination, with insertion of γ134.5 sequences into the TK gene locus. When hybridized with a DF3 promoter sequence probe, only the expected 4.4-kb band is observed in DF3γ34.5 DNA, and as expected no band is observed in F strain DNA.
CEAICP4 is missing both native copies of ICP4, with a single copy of this gene recombined into the ICP6 (viral ribonucleotide reductase) locus under the regulation of PCEA3. This mutant is therefore completely defective in ICP6 expression, and ICP4 expression is regulated by a CEA promoter.
CEAγ34.5 is missing both native copies of the γ134.5 gene, with a single copy of this gene recombined into the ICP6 gene locus under the transcriptional regulation of PCEA3. This mutant is therefore completely defective in ICP6 expression, and γ134.5 expression is regulated by a CEA promoter.
DF3γ34.5 is missing both copies of the γ134.5 gene, with a single copy of this gene recombined into the thymidine kinase (TK) gene locus under transcriptional regulation of a DF3 promoter. This mutant is therefore completely defective in TK expression, and γ134.5 expression is regulated by a DF3 promoter. The correct genotypes of these viruses were confirmed by Southern blot analysis (see Fig. 2).
Regulation of Viral Replication
CEAICP4 is completely deficient in viral ribonucleotide reductase and expresses ICP4 under the control of a CEA promoter (PCEA3). We compared replication of CEAICP4 with that of a mutant completely defective in ICP4 expression (d 120) and a mutant defective in viral ribonucleotide reductase but with wild-type ICP4 expression (hrR3). In Vero African Green Monkey kidney cells, d 120 replication is attenuated by five to six log orders compared to the control virus hrR3 that is deficient in only ICP6 (Fig. 3A). The CEA promoter is not activated in these cells, and CEAICP4 replication is as attenuated as d 120 replication. In contrast, CEAICP4 and d 120 replicate three to five log orders more robustly in E5 cells, which are ICP4-transformed and therefore complement any deficiency in ICP4 expression. In two of four human colon carcinoma cells lines, CEAICP4 replication was two log orders greater than that of d 120; however, disappointingly the overall level of replication was markedly attenuated compared to the control hrR3 virus. In addition, CEAICP4 replication was essentially undetectable in the Lovo and LS174T CEA-positive cell lines. These results suggest that in the absence of viral ribonucleotide reductase, regulation of ICP4 expression by a heterologous CEA promoter may regulate HSV-1 replication. But the overall magnitude of viral replication is quite attenuated compared to a virus deficient in only ICP6, and is probably inadequate for effective viral oncolysis in a clinical setting.
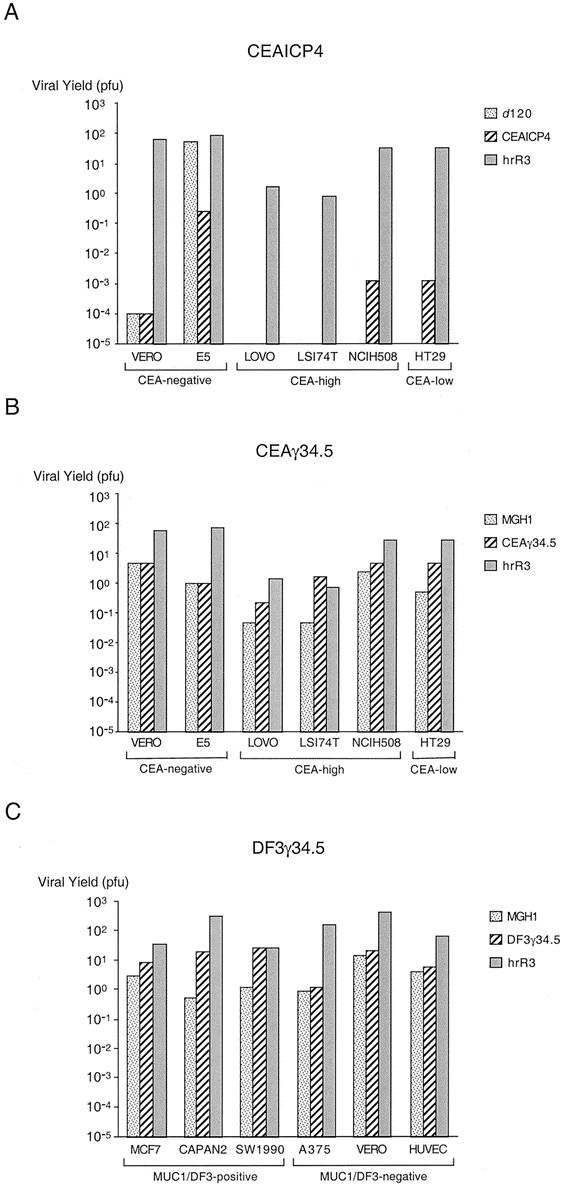
Figure 3. Comparison of replication by HSV-1 mutants. CEAICP4 (A) and CEAγ34.5 (B) replication were assessed in a single-step burst assay in CEA-negative cell lines as well as CEA-low and CEA-high cell lines. DF3γ34.5 replication (C) was assessed as a single-step burst assay in MUC1-negative and MUC1-positive cell lines.
We next examined replication of CEAγ34.5 in the same cell lines. This HSV-1 mutant is deficient in ICP6 expression, and γ134.5 expression is regulated by a CEA promoter (PCEA3). We compared replication of CEAγ34.5 with that of a mutant completely defective in γ34.5 and viral ribonucleotide reductase expression (MGH1) and a mutant defective in viral ribonucleotide reductase but with wild-type γ34.5 expression (hrR3). In Vero cells and E5 cells, MGH1 and CEAγ34.5 replication are 10-fold to 100-fold less than hrR3, which maintains wild-type γ134.5 expression (see Fig. 3B). CEAγ34.5 replication is approximately one log order greater than that of MGH1 in all of the colon carcinoma cell lines, and the overall level of replication is several log orders greater than that of CEAICP4. While CEAγ34.5 replication was observed to be always greater than that of MGH1, CEAγ34.5 replication did not vary (relative to MGH1 replication) as expected in the cell lines with high CEA expression (LS174T, NCIH508, and Lovo) compared to the cell line with low CEA expression (HT29).
We next examined replication of DF3γ34.5 in several cell lines. We used a different panel of cells lines that we have examined for MUC1 expression by fluorescence activated cell sorting (data not shown). MCF7 human breast carcinoma cells, CAPAN2 human pancreatic carcinoma cells, and SW1990 human pancreatic carcinoma cells overexpress MUC1, whereas MUC1 is not detectable on A375 human melanoma cells, Vero cells, and HUVEC cells. Analogous to our examination of CEAγ34.5, we used MGH1 and hrR3 as control HSV-1 viruses. DF3γ34.5 replication in the MUC1-negative cells (A375, Vero, HUVEC) was as attenuated as that of the MGH1 virus, which is defective in both γ134.5 and ICP6 expression (see Fig. 3C). In contrast, DF3γ34.5 expression was as robust as that of hrR3 in MUC1-positive SW1990 cells and one to two log orders greater than MGH1 in all of the MUC1-positive cells. These results suggest that the DF3 promoter regulates viral replication by appropriate transcriptional regulation of γ134.5.
DF3γ34.5 Cytotoxicity In Vitro and Inhibition of Pancreatic Cancer Xenografts
Of the three HSV-1 mutants that we constructed, we selected the DF3γ34.5 HSV-1 mutant for further study because its overall level of replication is relatively robust in vitro and correlates with MUC1 expression. We examined its ability to induce cytopathic effects in vitro against both MUC1-positive and MUC1-negative cells. As expected based on replication assay results, DF3γ34.5-induced cytotoxicity in MUC1-negative A375 and HUVEC cells was attenuated compared to hrR3 (Fig. 4A). And as expected from the replication assay results, DF3γ34.5-induced toxicity against Vero African Green Monkey kidney Vero cells was similar to that of hrR3. DF3γ34.5-induced cytotoxicity in the three MUC1-positive cancer cell lines was similar to that of hrR3. These current results are therefore promising because we have previously demonstrated in animal models that hrR3 has very significant antineoplastic activity.

Figure 4. Figure 4. Cytotoxicity and anti-tumor activity of DF3γ34.5. (A) MUC1-negative cells (A375, Vero, and HUVEC) and MUC1-positive cells (MCF7, CAPAN2, and SW1990)were infected with increasing moi values of DF3γ34.5, MGH1, or hrR3. Cell survival was measured 6 days later. (B) CAPAN2 tumors growing on the flanks of BALB/c (nu/nu) mice were treated with a single intratumoral inoculation of 1 x 108 pfu DF3g34.5 or mock-infected media and tumor volumes were subsequently measured.
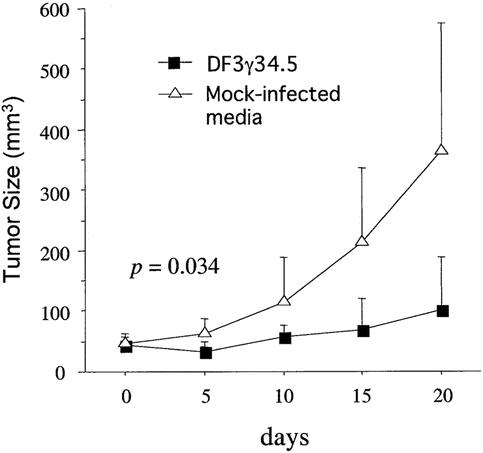
Figure 4. Continued
We examined the ability of DF3γ34.5 to inhibit flank tumor growth by implantation of MUC1-positive CAPAN2 cells into the flanks of nude mice. These tumors were treated with a single direct intratumoral inoculation of 1 × 108 pfu DF3γ34.5 or heat-inactivated DF3γ34.5. Tumors injected with heat-inactivated virus grew rapidly, whereas tumor growth was significantly inhibited following DF3γ34.5 injection (Fig. 4B).
DISCUSSION
Viruses are naturally suited for gene therapy applications. Over several millennia they have evolved efficient processes to evade host defenses, infect cells, deliver their genes and express them, reproduce their genome, and produce progeny virion. Most gene therapy research has followed a paradigm in which the viruses are genetically modified to render them replication-defective, such that they are capable of only transgene delivery and not replication. This therapeutic strategy is predicated on the belief that viral replication is undesirable as it may produce unwanted toxicity. A different paradigm involves destruction of tumor cells by viral replication (viral oncolysis). In this strategy, it is critical to augment viral replication in neoplastic cells and markedly attenuate replication in normal cells. This is most commonly achieved by modification of the viral genome.
Destruction of tumor cells by viral replication is an efficient process that differs in several important aspects from delivery of cytotoxic agents. The first is that the process of viral replication is exceedingly efficient, with tumor cell destruction observed following entry of as little as a single infectious virion into a tumor cell. Second, because progeny virion are liberated as a product of lytic replication, the maximum dose is greater than the input dose. Third, cell-to-cell propagation of infectious virion may be a more efficient mechanism of intratumoral dissemination compared to intravascular dissemination. High interstitial pressures within a tumor, combined with dysregulation of tumor neovasculature, limit distribution of traditional agents throughout a tumor. 23 Finally, the kinetics of tumor cell destruction are rapid, with cell destruction observed within 16 to 24 hours.
Safety issues are important to address in the design of replication-conditional viruses used to infect humans for purposes of tumor destruction. The potential for serious toxicity is evidenced by the recent death of a patient in a clinical trial following administration of a replication-defective adenovirus into the hepatic artery. 24 The ideal replication-conditional virus for cancer therapy should have several important properties. The virus should not integrate into the cellular genome to minimize the risk of transformation. The virus should replicate preferentially in neoplastic cells rather than in normal cells, and in the case of replication in normal cells, the virus should not cause serious medical illness. An effective antidote should be readily available. HSV-1 mutants meet these criteria. Including DF3γ134.5 in the current study, we have observed that several HSV-1 mutants replicate preferentially in neoplastic cells compared to normal cells. 12,14,25 HSV-1 does not integrate into the cellular genome. HSV-1 rarely causes serious medical illness, even though a large percentage of the population has been exposed to the virus. 26 Acyclovir is an effective antiherpetic agent that is commonly used to limit unwanted HSV-1 spread. CEAICP4 and CEAγ34.5 are sensitive to acyclovir, but DF3γ34.5 is resistant to acyclovir because of its defective TK expression. Despite the availability of other antiherpetic agents to which these TK-defective viruses should be sensitive, they are not suitable for clinical trials without repairs of the TK gene. We selected this locus for homologous recombination because of the ease with which recombinants can be selected with ganciclovir, and we are mainly interested in demonstrating proof-of-principals.
An important focus of our research has been to regulate lytic HSV-1 replication with use of a tumor-associated promoter. The CEA promoter that we constructed for our studies clearly regulates transcription of a luciferase reporter gene in transient transfection assays. However, this promoter does not efficiently regulate HSV-1 replication by regulation of ICP4. It is possible that the cis-acting elements in the ICP6 gene locus interfere with its transcriptional regulatory function, although another HSV-1 mutant has been reported in which a heterologous promoter functioned appropriately in this locus. 18 Others have recombined heterologous promoters into the HSV-1 thymidine kinase locus and have observed strong and specific transcriptional regulation. 27 Alternatively, the strength of PCEA3 may be insufficient to express sufficient levels of ICP4. We selected this particular transcriptional regulatory element based on its specificity (i.e., less “leaky”) as a strategy to reduce replication in CEA-negative cells. Our decision to use the most specific promoter was also based on our desire to minimize the risk that HSV-1 immediate early gene products—which are all transcriptional activators—would spuriously activate the CEA promoter. Perhaps one of the other CEA promoters that we characterized that has stronger transcriptional activity (but also less specificity) would produce more robust HSV-1 replication if used to regulate ICP4 expression. Finally, the kinetics of transcriptional regulation also may play an important role in regulation of ICP4 or γ134.5 expression. In comparison to the native ICP4 promoter, perhaps PCEA3 does not activate gene transcription rapidly enough to support robust lytic replication.
Previous studies have demonstrated that the DF3 gene is overexpressed in nearly 80% of human breast carcinomas, 28 and that expression of this gene is regulated at the transcriptional level. 29 This gene is also overexpressed in pancreas, 30 lung, 31 and ovarian cancers. 32 The MUC1 promoter that we used in the construction of DF3γ34.5 has been previously characterized. 21 Based on transient transfection assays, the region within the first 618 bases upstream of the transcriptional start site contains the regulatory sequences necessary for DF3 transcription in MUC1-positive MCF7 cells. Kurihara et al used a DF3 promoter to regulate lytic replication of an adenovirus mutant through its transcriptional regulation of E1A expression. 22 This genetically engineered adenovirus (Ad.DF3-E1) replicates throughout breast cancer xenografts in mice following direct intratumoral administration, and it inhibits growth of these tumors. We chose the same DF3 promoter sequence to regulate γ134.5 expression in the HSV-1 mutant DF3γ34.5. In addition, γ134.5 gene expression and HSV-1 replication have been regulated by a cell cycle-dependent B-myb promoter in the HSV-1 mutant Myb34.5. 18 We have extended these results in our construction of DF3γ34.5 by demonstrating preferential HSV-1 replication of DF3γ34.5 in MUC1-positive cells and inhibition of tumor growth. The magnitude of tumor growth inhibition is similar to that observed with the oncolytic adenovirus mutant Ad.DF3-E1 and the oncolytic HSV-1 mutant Myb34.5.
The principal advantage of DF3γ34.5 compared to R3616 (completely defective in γ134.5 expression) is the potential for reduced toxicity by virtue of attenuated replication in normal cells. Although we have demonstrated that the LD50 of DF3γ34.5 is indeed higher than that of R3616 in mice following tail vein injection (manuscript in preparation), mice are not the ideal species for preclinical toxicology testing because they do not accurately replicate the course of HSV-1 infection observed in humans. At present, Aotus (owl) monkeys are considered the species of choice for preclinical toxicology studies of HSV-1. 33 These New World monkeys are exquisitely sensitive to HSV-1 encephalitis, although the neuropathology associated with Aotus herpetic encephalitis is different from that observed in humans.
In MUC1-positive carcinoma cells, DF3γ34.5 replication is as robust as that of hrR3 (defective in only ICP6). This is an important result, as we have previously demonstrated that hrR3 treatment of liver metastases by intravascular administration enhances survival in a model of diffuse liver metastases. 14 We have previously demonstrated that the antineoplastic efficacy observed with HSV-1 treatment of tumors is not dependent on T-cell immunity, and it is dependent on viral replication. 12 Moreover, we have previously demonstrated that preexisting antibodies to HSV-1 do not reduce the observed antineoplastic efficacy. This is an important observation because as many as 80% of some patient populations have been previously exposed to HSV-1. 26
In summary, we have demonstrated that HSV-1 lytic replication can be controlled by regulation of γ134.5 gene expression by the DF3 promoter. Preferential replication of this HSV-1 mutant in MUC1-positive cells produces significant antineoplastic activity against flank tumor xenografts, suggesting that this strategy has potential for development of oncolytic viruses to treat cancers that overexpress specific antigens. The choices of promoter and HSV-1 gene to regulate are critical to the success of this strategy. 34,35
Acknowledgments
The authors appreciate the assistance of Dr. Donald Kufe for providing the DF3 promoter, Dr. Brian Huber for providing PCEA167, and Drs. David Knipe and E. Antonio Chiocca for insightful discussions.
DISCUSSION
Dr. Douglas L. Fraker (Philadelphia, PA): The systemic treatment of cancer relies on a differential effect normal cell versus neoplastic cells, or so-called therapeutic index. Clearly with available systemic agents they are woefully inadequate, as we get almost no durable responses, and new strategies are needed.
Dr. Tanabe has worked for years on developing the herpes virus as a new strategy with a hopefully better therapeutic index, and his work presented today really is built on that principle. It combines some very detailed and sophisticated studies both in terms of viral replication, cell biology and gene expression, and molecular techniques to make these constructs, and Dr. Tanabe is to be commended for his elegant work.
However, there are clearly problems in terms of selectivity, even with all of these very specialized constructs. And I have several questions, mostly in that regard.
First, even in the luciferase reporter assays, the degree of difference that you showed between CEA-expressing lines and nonexpressing lines varies with the different constructs you used. Some of them were only a 50% increase; some of them were several-fold increase. One would, I hope, with this type of strategy to get a several log-order increase in expression. And I want to know what the different degree of expression is in terms of CEA in these lines, and why wasn’t there a more amplified system difference, even in the luciferase reporter assays? This clearly carried over in the viral replication assays, as again there was not the degree of difference that one would want to see between CEA-expressing and CEA-nonexpressing lines. What is the reason after making these constructs where virus replication is dependent activation and expression of CEA?
The second question: Do we have any knowledge of the factors that influence the transcription of CEA? In other words, is there another strategy to add to this one to get a differential expression to somehow manipulate and enhance CEA expression even transiently to get a more selective effect against tumor cells?
The third question is: What is the normal cell expression of both CEA and MUC1? Clearly, we all realize that CEA levels have a normal range up to 2.5 or higher in smokers. And will this have unexpected toxicities with this selective approach?
Finally, if any of these herpes gene therapy strategies are going to make it to clinical trials, how is the problem of immunity that we all have after being exposed to various forms of herpes viruses going to impact on the efficacy of this therapy, especially when it depends on replication of virus to enhance the therapeutic delivery?
Presenter Dr. Kenneth K. Tanabe (Boston, MA): Concerning the differences in expression of CEA between the different cell lines, they range from 10- to 100-fold differences between the high and low CEA-expressing cell lines.
You also asked about normal cellular expression of these tumor-associated antigens. Indeed, normal cells, including hepatocytes and epithelial cells, do express some CEA and some MUC1 and therefore may to some degree support viral replication. We are looking to develop viruses that replicate preferentially in the tumor cells rather than the normal cells, but I don’t think we will be able to completely shut off replication in the normal cells.
Our data demonstrate that the choice of the promoter and the choice of the herpes gene are both critical determinants to the success of this strategy. There are several other herpes genes that we could try to regulate with this same CEA promoter. Concerning the CEA promoter itself, it is possible that a better strategy would have been to choose a promoter that has stronger transcriptional activity. We chose one that was the most specific in order to minimize replication in cells that don’t express CEA. In other words, we chose the least leaky promoter. Depending on the herpes gene that is being regulated, the kinetics with which a promoter regulates gene transcription are presumably critically important. However, at this time, we are left determining the best combination empirically.
I should also point out that in an aim to find effective strategies to maximize viral replication in neoplastic cells and attenuate replication in normal cells, we can employ other strategies. For example, we have demonstrated that deletion of the viral ribonucleotide reductase gene enhances replication 1,000-fold in liver metastases compared to normal liver. And now we show that use of a tumor-associated promoter can achieve another 10- to 100-fold increase in cancer cells relative to normal cells.
Presumably, by combining these strategies we can further increase the ratio of replication in cancer cells compared to normal cells. However, ultimately the safety of these viruses requires examination in clinical trials.
Dr. H. Richard Alexander, Jr. (Bethesda, MD): I would like to echo Dr. Fraker’s comments and compliment you on what really has been a very sustained and credible laboratory effort in the use of the herpes simplex virus for cancer therapy.
Dr. Fraker did ask some questions that I also had concerns about, specifically the CEA promoter and its lack of specificity in the high versus low CEA expressing cells. I think you have addressed that, but am also concerned as to the use of herpes simplex virus in clinical trials when there is such a high prevalence of preexisting antibodies that many of us would have. I would like to give you another opportunity to address that issue.
I would like to ask one other question. In your last figure, which was the in vivo data, you showed the efficacy of your recombinant herpes virus in an MUC1-positive pancreatic xenograft. The leap of faith that you are asking us to make is that this is a specific effect in an MUC1-positive tumor. So I presume that you have looked at this in an MUC-negative tumor line and would like to ask you what those data show. Also, have any mixing studies been done to see whether or not there is a substantial bystander effect which would be relevant to any clinical application of this in terms of looking at a population of cells that have MUC-positive and MUC-negative expression? So if you could answer this question and also address the issue of preexisting host immunity I would be most grateful. Thank you for the privilege of reviewing the manuscript.
Dr. Kenneth K. Tanabe (Boston, MA): Thank you, Dr. Alexander. Taking your last question first, the bystander effect. Certainly by mixing cells that have been infected with herpesvirus with cells that have not been infected there will be bystander killing, because there will be liberation of progeny virion that will infect adjacent cells. So in in vitro mixing studies, there will be a bystander effect.
You both have addressed the issue of preexisting antibodies, which is an important point. All of the mice used in these experiments came from a good ZIP code. They were presumably seronegative for herpes. There are some patient populations in the United States where as many as 80% have antibodies to herpesvirus. Therefore, we have also performed experiments using mice that have been vaccinated such that they have neutralizing antibodies to herpesvirus. The antineoplastic effect that we observe is the same whether or not the mice have been previously immunized.
Concerning the issue of whether this virus works in MUC1-negative tumors, we have further characterized this virus in MUC1-negative tumors. As expected, a single inoculation of this virus in the MUC1-negative tumors does provide some tumor regression, but the effect is less than that observed in MUC1-positive tumors.
These data indicate that the virus does replicate some in the MUC1-negative tumors, but, more importantly, the issue is whether the virus will replicate in MUC1-negative or MUC1 low-normal cells and cause toxicity. Toxicity studies in mice with this virus suggest that the biodistribution of this virus is more restricted and the toxicity is less than that of viruses that express γ34.5 constitutively. But again, these data need to be validated in clinical trials.
Dr. David C. Allison (Toledo, OH): The authors are to be congratulated on a technical tour de force in the construction of viral vectors driven by tumor-specific promoters. This is really good work. Although this approach could, and hopefully will, lead to specific anticancer effects, all recognize several hurdles remain.
One area I am interested in is that cancer cells are often chromosomally and genetically unstable, with varying genetic compositions even among several different cells of the same cancer. Thus, the escape of even one cancer cell in 1,000 or less from driving the specifically targeted promoter could defeat such a therapeutic strategy.
Along this line, I have two questions. First, in the cancer lines you have tested, what percentage of the cells are actually driving your promoters? Second, are these promoters only active in cancer cells, or will you have to look at other type of normal cells in addition to hepatocytes to determine whether these promoters are also active in noncancerous tissues?
Dr. Kenneth K. Tanabe (Boston, MA): I think that the issue of herpes-resistant cells resulting from chromosomal instability is a fascinating one. I proposed to the NIH that we should study this question, and they thought it was fairly ridiculous. So we have not addressed that question.
What percentage of cells express CEA? We have analyzed it by both Northern blot and FACS analysis, and we observe a standard distribution where cell populations two standard deviations below the mean express a lot less CEA than cell populations two standard deviations above the mean. This brings us back to Dr. Alexander’s question, “Is there a bystander effect?” With this approach we are indeed hoping for some degree of bystander effect to destroy all cells.
These viruses are very difficult to test in animal models. I can put a human tumor into a nude mouse, administer a herpesvirus, and destroy all the tumor cells and foolishly congratulate myself. Yet it may be that the only thing that I am really doing is using a virus that preferentially infects human cells with the human CEA promoter compared to murine cells. Therefore, the issues of replication in normal cells and toxicity are difficult to address with presently available animal models. Clinical trials are required to determine toxicity. Until then, we are trying to examine replication of this virus in as many normal tissues as possible.
Footnotes
Supported by NIH grants CA76183, GM07035, DK43352, and CA71345; the Claude E. Welch Research Fellowship; the Marshall K. Bartelett Research Fellowship; and the Carl Ockerbloom Research Fund.
The first two authors contributed equally to this manuscript.
Presented at the 122nd Annual Meeting of the American Surgical Association, April 24–27, 2002, The Homestead, Hot Springs, Virginia.
Correspondence: Kenneth K. Tanabe, MD, Division of Surgical Oncology, Massachusetts General Hospital, Cox Bldg. 626, Boston, MA 02114.
E-mail: ktanabe@partners.org
Accepted for publication April 24, 2002.
References
- 1.Rosenberg SA, Blaese RM, Brenner MK, et al. Human gene marker/therapy clinical protocols. Hum Gene Ther 2000; 11: 919–979. [DOI] [PubMed] [Google Scholar]
- 2.Martuza RL. Conditionally replicating herpes vectors for cancer therapy. J Clin Invest 2000; 105: 841–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nakamura H, Mullen JT, Chandrasekhar S, et al. Multimodality therapy with a replication-conditional herpes simplex virus 1 mutant that expresses yeast cytosine deaminase for intratumoral conversion of 5-fluorocytosine to 5-fluorouracil. Cancer Res 2001; 61: 5447–5452. [PubMed] [Google Scholar]
- 4.Ichikawa T, Chiocca EA. Comparative analyses of transgene delivery and expression in tumors inoculated with a replication-conditional or -defective viral vector. Cancer Res 2001; 61: 5336–5339. [PubMed] [Google Scholar]
- 5.Kooby DA, Carew JF, Halterman MW, et al. Oncolytic viral therapy for human colorectal cancer and liver metastases using a multi-mutated herpes simplex virus type-1 (G207). FASEB J 1999; 13: 1325–1334. [DOI] [PubMed] [Google Scholar]
- 6.Puhlmann M, Grant M, Brown CK, et al. Thymidine kinase-deleted vaccinia virus expressing purine nucleoside phosphorylase as a vector for tumor-directed gene therapy. Hum Gene Ther 1999; 10: 649–657. [DOI] [PubMed] [Google Scholar]
- 7.Sinkovics JG, Horvath JC. Newcastle disease virus (NDV): brief history of its oncolytic strain. J Clin Virol 2000; 16: 1–15. [DOI] [PubMed] [Google Scholar]
- 8.Bischoff JR, Kirn DH, Williams A, et al. An adenovirus mutant that replicates selectively in p53-deficient human tumor cells. Science 1996; 274: 373–376. [DOI] [PubMed] [Google Scholar]
- 9.Coffey MC, Strong JE, Forsyth PA, et al. Reovirus therapy of tumors with activated ras pathway. Science 1998; 282: 1332–1334. [DOI] [PubMed] [Google Scholar]
- 10.Carroll NM, Chiocca EA, Takahashi K, et al. Enhancement of gene therapy specificity for diffuse colon carcinoma liver metastases with recombinant herpes simplex virus. Ann Surg 1996; 224: 323–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yoon SS, Carroll NM, Chiocca EA, et al. Cancer gene therapy using replication-competent herpes simplex virus type 1. Ann Surg 1998; 228: 366–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goldstein DJ, Weller SK. Herpes simplex virus type 1-induced ribonucleotide reductase activity is dispensable for virus growth and DNA synthesis: isolation and characterization of an ICP6 lacZ insertion mutant. J Virol 1988; 62: 196–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yoon SS, Nakamura H, Carroll NM, et al. An oncolytic herpes simplex virus type 1 selectively destroys diffuse liver metastases from colon carcinoma. FASEB J 2000; 14: 301–311. [PubMed] [Google Scholar]
- 14.Nakamura H, Kasuya H, Mullen JT, et al. Regulation of Herpessimplex virus γ1 34: 5: expression and oncolysis of diffuse liver metastases by Myb34.5. J Clin Invest 2002; 109: 871–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deluca NA, McCarthy AM, Schaffer PA. Isolation and characterization of deletion mutants of herpes simplex virus type 1 in the gene encoding immediate-early regulatory protein ICP4. J Virol 1985; 56: 558–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chou J, Roizman B. The gamma 1(34.5) gene of herpes simplex virus 1 precludes neuroblastoma cells from triggering total shutoff of protein synthesis characteristic of programed cell death in neuronal cells. Proc Natl Acad Sci USA 1992; 89: 3266–3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.He B, Gross M, Roizman B. The gamma(1)34.5 protein of herpes simplex virus 1 complexes with protein phosphatase 1alpha to dephosphorylate the alpha subunit of the eukaryotic translation initiation factor 2 and preclude the shutoff of protein synthesis by double-stranded RNA-activated protein kinase. Proc Natl Acad Sci USA 1997; 94: 843–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chung RY, Saeki Y, Chiocca EA. B-myb promoter retargeting of herpes simplex virus gamma 34.5 gene-mediate virulence toward tumor and cycling cells. J Virol 1999; 73: 7556–7564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lelie PN, Reesink HW, Lucas CJ. Inactivation of 12 viruses by heating steps applied during manufacture of a hepatitis B vaccine. J Med Virol 1987; 23: 297–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Richards CA, Austin EA, Huber BE. Transcriptional regulatory sequences of carcinoembryonic antigen: identification and use with cytosine deaminase for tumor-specific gene therapy. Hum Gene Ther 1995; 6: 881–893. [DOI] [PubMed] [Google Scholar]
- 21.Abe M, Kufe D. Characterization of cis-acting elements regulating transcription of the human DF3 breast carcinoma-associated antigen (MUC1) gene. Proc Natl Acad Sci USA 1993; 90: 282–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kurihara T, Brough DE, Kovesdi I, et al. Selectivity of a replication-competent adenovirus for human breast carcinoma cells expressing the MUC1 antigen. J Clin Invest 2000; 106: 763–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jain RK. Delivery of molecular medicine to solid tumors. Science 1996; 271: 1079–1080. [DOI] [PubMed] [Google Scholar]
- 24.Marshall E. Clinical trials: gene therapy death prompts review of adenovirus vector. Science 1999; 286: 2244–2245. [DOI] [PubMed] [Google Scholar]
- 25.Pawlik TM, Nakamura H, Yoon SS, et al. Oncolysis of diffuse hepatocellular carcinoma by intravascular administration of a replication-competent, genetically engineered herpesvirus. Cancer Res 2000; 60: 2790–2795. [PubMed] [Google Scholar]
- 26.Becker TM, Lee F, Daling JR, et al. Seroprevalence of and risk factors for antibodies to herpes simplex viruses, hepatitis B, and hepatitis C among southwestern Hispanic and non-Hispanic white women. Sex Transm Dis 1996; 23: 138–144. [DOI] [PubMed] [Google Scholar]
- 27.Miyatake S, Iyer A, Martuza RL, et al. Transcriptional targeting of herpes simplex virus for cell-specific replication. J Virol 1997; 71: 5124–5132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kufe D, Inghirami G, Abe M, et al. Differential reactivity of a novel monoclonal antibody (DF3) with human malignant versus benign breast tumors. Hybridoma 1984; 3: 223–232. [DOI] [PubMed] [Google Scholar]
- 29.Abe M, Kufe D. Transcriptional regulation of DF3 gene expression in human MCF-7 breast carcinoma cells. J Cell Physiol 1990; 143: 226–231. [DOI] [PubMed] [Google Scholar]
- 30.Metzgar RS, Rodriguez N, Finn OJ, et al. Detection of a pancreatic cancer-associated antigen (DU-PAN-2 antigen) in serum and ascites of patients with adenocarcinoma. Proc Natl Acad Sci USA 1984; 81: 5242–5246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jarrard JA, Linnoila RI, Lee H, et al. MUC1 is a novel marker for the type II pneumocyte lineage during lung carcinogenesis. Cancer Res 1998; 58: 5582–5589. [PubMed] [Google Scholar]
- 32.Friedman EL, Hayes DF, Kufe DW. Reactivity of monoclonal antibody DF3 with a high molecular weight antigen expressed in human ovarian carcinomas. Cancer Res 1986; 46: 5189–5194. [PubMed] [Google Scholar]
- 33.Meignier B, Martin B, Whitley RJ, et al. In vivo behavior of genetically engineered herpes simplex viruses R7017 and R7020. II. Studies in immunocompetent and immunosuppressed owl monkeys (Aotus trivirgatus). J Infect Dis 1990; 162: 313–321. [DOI] [PubMed] [Google Scholar]
- 34.Goldstein DJ, Weller SK. Factor(s) present in herpes simplex virus type 1 infected cells can compensate for the loss of the large unit of the viral ribonucleotide reductase: characterization of an ICP6 deletion mutant. Virology 1988; 166: 41–51. [DOI] [PubMed] [Google Scholar]
- 35.Kramm CM, Chase M, Herrlinger U, et al. Therapeutic efficiency and safety of a second-generation replication-conditional HSV1 vector for brain tumor gene therapy. Hum Gene Ther 1997; 8: 2057–2068. [DOI] [PubMed] [Google Scholar]