

Abstract
Objective
To describe a genotype–phenotype correlation in MEN2 families with RET germline mutations of codons 790/791 and discuss options for the therapeutic management of gene carriers.
Summary Background Data
Heredity of MEN2 syndromes is caused by a heterozygous germline mutation in the RET protooncogene. Rare mutations of codons 790/791 associated with incomplete penetrant MEN2A/FMTC phenotype were reported in five families, contraindicating the prophylactic thyroidectomy for the genetically affected children.
Methods
Forty-five patients with a putative sporadic MTC were screened for RET germline mutations by direct DNA sequencing. Family members of identified index cases underwent genetic analysis. Gene carriers were examined clinically and biochemically, and all gene carriers underwent prophylactic thyroidectomy.
Results
Five index patients were identified, four of whom harbored mutations in codons 790/791 and one in codon 634. In the kindreds, four L790F carriers and one Y791F carrier were detected. The thyroid gland histology of L790F carriers revealed medullary thyroid carcinoma in two patients (aged 29 and 50 years) and C-cell hyperplasia in two additional patients (aged 9 and 16 years). The Y791F carrier had a normal histology.
Conclusions
Codon 790/791 mutations had diverse penetrance. Whereas prophylactic thyroidectomy in children is a justifiable approach for codon 790 mutation carriers, the indication for thyroidectomy should depend on the clinical course of codon 791 carriers.
Multiple endocrine neoplasia type 2 (MEN2) is an autosomal dominant inherited tumor syndrome comprising medullary thyroid carcinoma (MTC) as an obligatory feature. MEN2 is divided into three subtypes in accordance to the remaining tissues involved. Familial medullary thyroid carcinoma (FMTC) is characterized by an isolated MTC occurrence, while MEN2A cases have an association of pheochromocytoma and primary hyperparathyroidism in approximately 50% and 15% to 30%, respectively. MEN2B has a similar disease phenotype with an earlier onset of MTC, and rarely hyperparathyroidism. In addition, MEN2B individuals have a marfanoid habitus, abnormal facies, mucosal neuromas, hyperplasia of the enteric nerve plexi, and in some cases delayed puberty. 1
The RET protooncogene on 10q11.2 has been recognized as the susceptibility locus for MEN2. This gene is involved in the development of neural crest cell lineages by coding for a membrane-bound receptor tyrosine kinase. Several functional ligands of RET have been demonstrated: the glial cell line-derived neurotrophic factor (GDNF), the neurturin (NRTN), the artemin (ARTN), and the persephin (PSPN). These ligands, in association with the corresponding members of the GDNF family receptor alpha (GFRα-1–4), bind to the extracellular RET receptor domain, inducing a homodimerization of RET molecules and a specific activation of the intracellular tyrosine kinase domain. 2–5
Germline missense mutations of RET protooncogene have been detected in each one of six cysteine codons within exon 10 (codons 609, 611, 618, and 620) and exon 11 (codons 630 and 634), in about 95% of MEN2A and in up to 85% of FMTC families. 1,6–8 The substitution of one of these highly conserved cysteine residues results in RET monomers capable of constitutive dimerization in the absence of functional RET ligands. 9,10 In addition to these common cysteine codon mutations, some rare noncysteine mutations located within the intracellular tyrosine kinase domain of RET have been described. 11,12 The effects of these mutations are unclear, but the E768D (exon 13) and V804L (exon 14) mutations also increase the transforming activity, possibly by altering the substrate specificity or the ATP-binding capacity of RET, thus modifying the kinase activity. 13
Because of the assumed complete penetrance of MTC in MEN2 families and the reliability of genetic analysis for MEN2-associated RET germline mutations, predictive testing has become a routine procedure in the management of MEN2 kindreds. Such tests allow the presymptomatic identification of individuals at risk, permitting prophylactic thyroidectomy before C-cell carcinoma occurs. 1,14,15 According to the reported de novo mutation rate of up to 9% in MEN2A/FMTC, and a sometimes unknown family history, the genetic analysis should be expanded on all patients with an apparently sporadic MTC to exclude a hereditary form of this tumor or to identify index cases of new MEN2 kindreds. 16
In this study we have investigated a population of apparently sporadic MTC patients and revealed RET germline mutations in codons 790 and 791 in four of five cases. We were particularly interested in the genotype–phenotype correlation in MEN2 kindreds with codons 790/791 germline mutations of RET protooncogene. Mutations of these two codons have been previously reported and were described as associated with incomplete penetrance in MEN2A/FMTC phenotype. 12 Similarly to these data, our identified at-risk subjects were biochemically analyzed, but we have additionally performed a prophylactic thyroidectomy due to the diverse penetrance pattern found in the present study.
METHODS
Patient Population
We have analyzed 45 patients with seemingly sporadic MTC for mutations of the RET protooncogene. Written informed consent was obtained from all patients. None of them had any family history of the disease. Moreover, additional features of MEN2 in this population such as pheochromocytoma or primary hyperparathyroidism were excluded by clinical and biochemical examination. After genetic counseling, family members of new identified MEN2 index cases were screened genetically to identify mutation carriers. Subjects at risk were examined for clinical and biochemical symptoms of MEN2, and all mutation carriers underwent subsequent prophylactic thyroidectomy. Histologic examination of the resected thyroid gland was performed by pathologists from the Department of Pathology, Technische Universität Dresden.
Biochemical Evaluation
Preoperative basal and provocative calcitonin tests were performed in all mutation carriers. Plasma calcitonin levels were determined by a radioimmunoassay (IBL, Hamburg, Germany) before and at 1, 2, 5, 10, 15, and 20 minutes after pentagastrin provocation, which consisted of a single intravenous injection of pentagastrin (0.5 μg/kg; pentagastrin injection BP, Cambridge Laboratories, UK) immediately followed by intravenous calcium gluconate administration (0.02 mL/kg of a 10% solution; Braun Melsungen AG, Germany). Additionally, the serum concentrations of calcium and parathyroid hormone, as well as the 24-hour urinary excretion of catecholamines, vanillylmandelic acid, and homo-vanillylmandelic acid, were determined.
DNA Analysis
Genomic DNA was obtained from leukocytes from peripheral venous blood samples isolated by standard protocols. All exons of the RET protooncogene harboring known MEN2-associated mutations were analyzed, including exons 10, 11, and 13 to 16. Exons were amplified from genomic DNA using the primers and methodology described by Ceccherini et al. 17 for exons 11 and 16, and by Mulligan et al. 18 for exons 10 and 13. To amplify the remaining exons we generated new primer pairs (exon 14: sense 5′-TGTGTCCACCCCCTTACTCATTGG3′; antisense 5′-CGTGGTGGGTCAGGGTGT GG3′; exon 15: sense 5′-CCCCCGGCCCAGGTCTCAC3′; antisense 5′-GCTCCACTAATCTTCGGTATCTTT3′). Fifty nanograms of DNA was amplified in a Perkin-Elmer 9600 thermocycler (Perkin Elmer Applied Biosystems, Weiterstadt, Germany) in a volume of 25 μL containing 0.1 μmol/L of each oligonucleotide primer, 1.5 mmol/L MgCl2, and 0.75 U Taq polymerase (Invitrogene, Berlin, Germany). Thermocycling conditions consisted of an initial denaturation at 94°C for 5 minutes, followed by 35 cycles of 30 seconds denaturation at 94°C, 30 seconds annealing (exons 10, 15, and 16 at 55°C, and exons 11, 13, and 14 at 60°C), and 30 seconds extension at 72°C, completed by a final extension step for 7 minutes at 72°C. The amplified DNA was analyzed on 1% agarose gel and purified with the Qiagen Quickspin kit (Qiagen, Hilden, Germany). The sequence analysis of these DNA fragments was carried out with a direct DNA-sequencing approach using the Thermo Sequenase Fluorescent Cycle Sequencing kit (Amersham Pharmacia Biotech, Freiburg, Germany). The sequencing primers were identical to the PCR primers with an additional Cy5 labeling, allowing sequence analysis on A.L.F. express devices (Amersham Pharmacia Biotech, Freiburg, Germany).
We also investigated the genotype distribution of polymorphisms of codons 45 (exon 2), 125 (exon 3), 432 (exon 7), 691 (exon 11), 769 (exon 13), 836 (exon 14),and 904 (exon 15) of the coding region of the RET protooncogene in the index patients with codon 790/791 RET germline mutations. The seven investigated exons were amplified from genomic DNA using primers and methodology described previously. 19 All analyzed polymorphisms created or obstructed a restriction site of an endonuclease, namely Eag I, Mbo II, Bsm I, Ban I, Taq I, Alu I, and Rsa I. 17 Genotypes were determined by digestion of the PCR product and electrophoresis on a polyacrylamide gel (codons 45, 125, and 432) or by direct sequencing (codons 691, 769, 836, and 904).
RESULTS
A RET germline mutation was found in 5 patients among a population of 45 patients with an apparently sporadic MTC. Only one of these five patients harbored a C634F (CTG → TTG) mutation, representing one of the well-documented and assumed completely penetrant cysteine codon changes of exons 10 and 11; in the other four cases, mutations of RET protooncogene were detected in exon 13 codons 790 and 791. We have found the same heterozygous mutation L790F in three unrelated patients and the Y791F mutation in another (Fig. 1). To exclude that the latter two mutations represent rare genomic variants, 117 healthy blood donors were screened for both transversions, but in none of the 234 control chromosomes was either of the two variants detected. Analyzing the genotypes of seven RET polymorphisms, we have proven that the detected codon L790F mutations involve different RET haplotypes, excluding a possible founder effect (Fig. 2). After genetic counseling of the involved families, we performed predictive genetic testing with regard to the identification of MEN2 gene carriers. The MTC of the index person harboring one of the common MEN2 mutations (C634F) was diagnosed clinically at the age of 54, although the history concerning his parents was unknown. The brother and the niece of the index patient were identified as gene carriers, and the histologic evaluation of the removed thyroid glands revealed MTC in both patients.
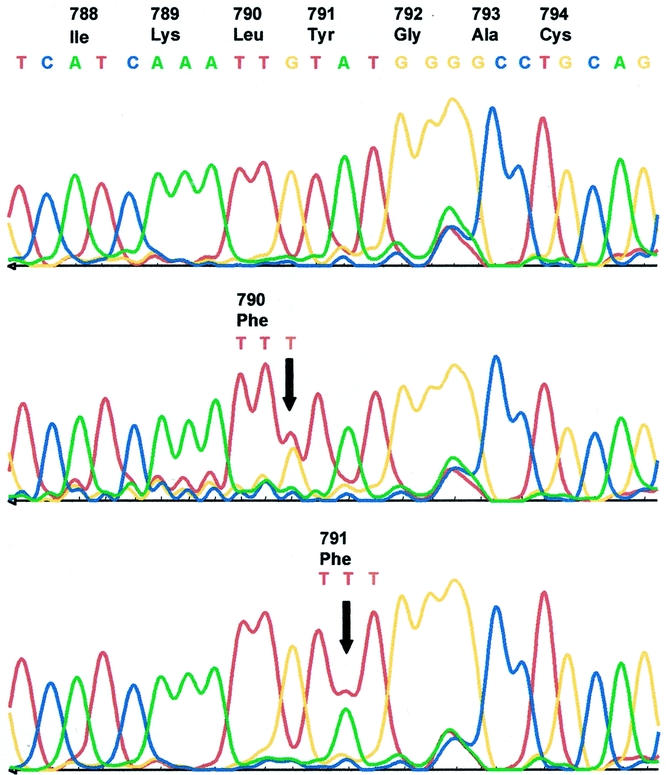
Figure 1. Sequence analysis of amplified genomic DNA from RET protooncogene exon 13 shows the reference sequence of a normal control individual (upper panel), the codon 790 mutation (TTG → TTT) detected in families A, B, and C (middle panel), and the codon 791 mutation (TAT → TTT) detected in family D. Both transversions result in a substitution of phenylalanine. The positions of heterozygous mutation are indicated by arrows.

Figure 2. Pedigrees of MEN 2A/FMTC families with mutations in exon 13 codons 790/791 of the RET protooncogene. Index cases of families are characterized by the genotypes of RET protooncogene polymorphisms of exons 2 (codon 45 GCG → GCA), 3 (codon 125 GTC → GTA), 7 (codon 432 GCG → GCA), 11 (codon 691 GGT → AGT), 13 (codon 769 CTT → CTG), 14 (codon 836 AGC → AGT), and 15 (codon 904 TCC → TCG). W, wild-type allele (underlined); p, polymorphic allele.
Besides the well-described common c634 mutations, we were particularly interested in the genetic and clinical analysis of the families with detected rare RET germline mutations. In kindred A, a multifocal MTC without lymph node metastasis was diagnosed in the 34-year-old index person, and genetic testing revealed a RET germline mutation codon L790F (TTG → TTT). Although her mother and son were not carriers, the mutation was detected in the 9-year-old daughter, but the clinical and biochemical examination yielded no pathologic findings. In contrast, the histologic examination of the prophylactically removed thyroid gland revealed a multifocal nodular C-cell hyperplasia (Table 1).
Table 1. CLINICAL AND BIOCHEMICAL DATA OF INDEX CASES AND PATIENTS AT RISK FOR MEN 2A/FMTC WHO INHERITED A CODON 790/791 MUTATION

ND, not determined; CEA, serum carcinoembryonic antigen concentration, normal <4 ng/mL; PTH, serum parathyroid hormone concentration, normal 10–72 pg/mL; Ca, serum calcium concentration, normal range; MTC, medullary thyroid carcinoma; CCH, C-cell hyperplasia.
Basal calcitonin level normal <20 pg/mL.
Index cases of the kindreds A–D are in bold.
Normal 2.08–2.60 mmol/L.
The index case of a second family (kindred B) harbored the same codon L790F mutation (TTG → TTT) and had a MTC at age 35. In addition, we have identified three gene carriers in this kindred. Two female patients, ages 50 and 29, had multiple intrathyroidal nodes confirmed by ultrasound. Whereas in the older patient we detected elevated values of basal and provoked calcitonin, the younger one had only a pathologic provoked test. In both patients we performed a total thyroidectomy with lymph node dissection of the cervicocentral compartment, and the histologic evaluation revealed a multifocal MTC without lymph node involvement. The third gene carrier of this kindred was a 16-year-old boy with normal ultrasound examination of the thyroid gland and a normal calcitonin test. Due to his family history, he and his parents agreed to thyroidectomy, which revealed a C-cell hyperplasia histologically.
The third index case with an L790F mutation (TTG → TTT) was a woman who was diagnosed with a bilateral MTC with primary lymph node metastases at age 62. All members of the kindred could be excluded as gene carriers (kindred C). In another case, a 45-year-old man had an invasive MTC with primary lymph node metastasis and repeated operative interventions due to tumor recurrence. Genetic analysis revealed a Y791F mutation (TAT → TTT; kindred D). One of his two daughters was identified as a gene carrier but showed a normal clinical and biochemical profile. There were no irregular findings in the histologic analysis of the prophylactically removed thyroid gland.
Due to the discrepancy between genotype and phenotype within this family, we analyzed the tumor from the index patient for somatic genetic alterations of the RET protooncogene, which revealed an additional M918T mutation (ATG → ACG). In contrast, we excluded the occurrence of this somatic mutation in the tumors of all other index cases, as well as all gene carriers described in this study.
Neither the identified index cases nor the gene carriers from the above-described families showed clinical or biochemical evidence of hyperparathyroidism or pheochromocytoma at the time of thyroidectomy. In regard to postoperative morbidity, we did not observe a palsy of the recurrent laryngeal nerve, and only the 9-year-old girl suffered from a transitory hypoparathyroidism, which was treated with calcium substitution for 3 months. Histologic evaluation of the thyroid gland removed from this girl revealed two intrathyroidal parathyroidal glands, which may be the cause of this transitory hypoparathyroidism.
DISCUSSION
Germline missense mutations of the RET protooncogene are found in one of six cysteine codons of exons 10 and 11 associated with a nearly complete penetrance of MTC in gene carriers, in 95% of MEN2A families, and in up to 85% FMTC families. 1,14,15 It has been suggested that prophylactic total thyroidectomy be performed in early childhood in gene carriers of MEN2A/FMTC families, since in almost all cases the tumor spread is restricted to the thyroid gland under the age of 6, and the radical surgery is the only accepted curable approach to MTC, particularly in the hereditary setting, when MTC occurs often multifocally. 14,15,20
Mutations in codons 790 and 791 of RET exon 13 have been described in 5 of 181 German MEN2A/FMTC families as new hot spots leading to hereditary MTC and pheochromocytoma, 12 besides other rare RET germline mutations associated with MEN2A/FMTC. In contrast to the low detection rate of codon 790/791 germline mutations in MEN2 kindreds (2.8%), the same study detected 35 germline mutations in 305 patients with seemingly sporadic MTC (11.5%), involving codons 790/791 in 11 of 35 cases (31.4%). These findings, in addition to the described variable expression, suggest a lower penetrance of these mutations compared with the common MEN2 mutations of exons 10 and 11. 12
In our study, we analyzed 45 patients with an apparently sporadic MTC for RET germline mutations that revealed a similar mutation rate of 11.1%, but four of the five new index cases harbored a mutation in codons 790/791. The screening of 234 chromosomes of 117 healthy blood donors did not reveal any genomic variants in codons 790/791, confirming the results of Berndt et al. 12 and implicating an etiological relevance of these mutations for the pathogenesis of MEN2. The assumption of a possible founder effect in the German population concerning these mutations was ruled out due to different ethnic descent of the investigated kindreds. 12 Our data support this hypothesis on the basis of haplotype analysis comprising seven RET polymorphisms. In particular, we demonstrated that the three detected L790F mutations involved at least two different RET haplotypes.
The analysis of the phenotypic expression suggested that codon 790/791 mutations might be associated with low penetrance and might generate a mild thyroid disease phenotype when compared to that of the more common MEN2 mutations. Notably, the age of clinical MTC manifestation in the index cases of our study ranged from 34 to 62 years, similarly to that reported by Berndt et al. 12 In contrast to the latter study, which described a kindred in which three of seven gene carriers (L790F; TTG → TTT) had both MTC and pheochromocytoma, we did not detect any clinical features of pheochromocytoma or hyperparathyroidism in any of our patients.
In three families with the L790F mutation we identified four gene carriers who underwent total thyroidectomy, revealing either an invasive MTC or a nodular C-cell hyperplasia on histologic evaluation. Two patients with a C-cell hyperplasia (aged 9 and 16 years) had normal basal and provoked calcitonin level, while a 50-year-old woman with multifocal invasive MTC had elevated basal and provoked calcitonin levels. Likewise, her daughter, who also had an invasive MTC at age 29 years, had a positive stimulated calcitonin test. The only gene carrier harboring a Y791F mutation also had normal calcitonin tests as well as a normal histology after thyroidectomy. According to these data, the determination of basal and provoked calcitonin does not serve as a parameter to discriminate between the different stages of C-cell pathomorphology. We therefore confirm the findings of Hinze et al. 21 that elevated basal calcitonin levels were restricted to MTC, while normal plasma levels after pentagastrin provocation were found only in patients with C-cell hyperplasia. However, increased provoked calcitonin levels were detected in 8 of 11 patients with a C-cell hyperplasia investigated by Hinze et al. Similar observations were reported by Hotz et al. 22 Moreover, a false-positive response to pentagastrin stimulation has been described in mutation-negative members of MEN2A families, 23 while Dralle et al. reported several patients with MTC and negative basal and provoked calcitonin tests. 15 Since the prophylactic operation should be aimed toward removing the thyroid gland before malignant transformation into a MTC occurs, basal and provoked calcitonin levels are not reliable markers for the indication of prophylactic thyroidectomy.
In contrast to the strong association of the L790F germline mutation with MTC or C-cell hyperplasia in our study, the only gene carrier of family D (harboring the Y791F TAT → TTT germline mutation) had a normal histology after a prophylactic thyroidectomy at age 29. Based on this finding and the poor clinical course of the index person with recurrent lymph node metastases, we analyzed his tumor for additional somatic RET mutations and detected the most common M918T (ATG → ACG) somatic mutation of sporadic MTCs. We have excluded the occurrence of this somatic mutation in all other index cases, as well as in the MTCs of the gene carriers. The coexistence of germline and somatic mutations in a familial cancer syndrome caused by a dominant-acting oncogene has been reported only by Marsh et al. in three cases of MEN2-associated MTCs and in one C-cell hyperplasia. 24 In our data, as well as in that of Marsh et al., it was not possible to determine if the germline and the somatic mutations were located on different alleles. Interestingly, in this context the same Y791F RET mutation was described in association with a sporadic HSCR phenotype without MTC. 25
Based on the variable phenotype associated with the Y791F mutation, and the finding that only three of eight identified Y791F gene carriers (described by Berndt et al. 12 and our study) showed clinical or histologic manifestation of MTC, we do not support prophylactic thyroidectomy in early childhood in such cases. We do, however, suggest clinical and biochemical follow-up of gene carriers until the third decade of life or until further symptoms of MTC, when a total thyroidectomy should be performed (the youngest patient with MTC associated with a c791 mutation was 21 years old). In contrast, c790 mutations appear to possess a considerable potential for transformation, comparable to the c804 mutations studied by Frohnauer and Decker. 26 All seven c790 gene carriers of our study, as well as nine clinically affected individuals of 13 gene carriers reported by Berndt et al., 12,27 had a pathologic C-cell histology, albeit limited familial data. The youngest affected patient underwent prophylactic thyroidectomy at age 9. Therefore, the data suggest that c790 gene carriers should be treated similarly to those with the frequent MEN2A/FMTC mutations. Based on the higher incidence of codon 790/791 RET germline mutations than expected, we conclude that the screening of these codons should always be performed in the genetic testing of MTC patients.
Footnotes
Correspondence: Guido Fitze, MD, Department of Pediatric Surgery, University of Technology Dresden, Fetscherstrasse 74, D-01307 Dresden, Germany.
E-mail: guido.fitze@mailbox.tu-dresden.de
Accepted for publication February 21, 2002.
References
- 1.Eng C, Mulligan LM. Mutation of the RET proto-oncogene in the multiple endocrine neoplasia type 2 syndromes, related sporadic tumours, and Hirschsprung disease. Hum Mutat 1997; 9: 97–109. [DOI] [PubMed] [Google Scholar]
- 2.Trupp M, Arenas E, Fainzilber M, et al. Functional receptor for GDNF encoded by the c-RET proto-oncogene. Nature 1996; 381: 785–789. [DOI] [PubMed] [Google Scholar]
- 3.Lin LFH, Doferty DH, Lile JD, et al. GDNF: a glial cell line-derived neurotrophic factor for midbrain dopaminergic neurons. Science 1993; 260: 1130–1132. [DOI] [PubMed] [Google Scholar]
- 4.Kotzbauer PT, Lampe PA, Heuckeroth RO, et al. Neurturin, a relative of glial cell line-derived neurotrophic factor. Nature 1996; 384: 467–470. [DOI] [PubMed] [Google Scholar]
- 5.Baloh RH, Tansey MG, Lampe PA, et al. Artemin, a novel member of the GDNF ligand family, supports peripheral and central neurons and signals through the GFRα3-RET receptor complex. Neuron 1998; 21: 1291–1302. [DOI] [PubMed] [Google Scholar]
- 6.Mulligan LM, Kwok JBJ, Healey CS, et al. Germline mutations of the RET proto-oncogene in multiple endocrine neoplasia type 2A. Nature 1993; 363: 458–460. [DOI] [PubMed] [Google Scholar]
- 7.Donis-Keller H, Dou S, Chi D, et al. Mutations in the RET proto-oncogene are associated with MEN2A and FMTC. Hum Mol Genet 1993; 2: 851–856. [DOI] [PubMed] [Google Scholar]
- 8.Hofstra RM, Landsvater RM, Ceccherini I, et al. A mutation in the RET proto-oncogene associated with multiple endocrine neoplasia type 2B and sporadic medullary thyroid carcinoma. Nature 1994; 367: 375–376. [DOI] [PubMed] [Google Scholar]
- 9.Borrello MG, Smith DP, Pasini B, et al. RET activation by germline MEN2A and MEN2B mutations. Oncogene 1995; 11: 2419–2427. [PubMed] [Google Scholar]
- 10.Santoro M, Carlomagno F, Romano A, et al. Activation of RET as a dominant transforming gene by germline mutations of MEN 2A and MEN 2B. Science 1995; 267: 381–383. [DOI] [PubMed] [Google Scholar]
- 11.Bolino A, Schuffenecker I, Luo Y, et al. RET mutations in exons 13 and 14 of FMTC patients. Oncogene 1995; 10: 2415–2419. [PubMed] [Google Scholar]
- 12.Berndt I, Reuter M, Saller B, et al. A new hot spot for mutations in the RET protooncogene causing familial medullary thyroid carcinoma and multiple endocrine neoplasia type 2A. J Clin Endocrinol Metab 1998; 83: 770–774. [DOI] [PubMed] [Google Scholar]
- 13.Pasini A, Geneste O, Legrand P, et al. Oncogenic activation of RET by two distinct FMTC mutations affecting the tyrosine kinase domain. Oncogene 1997; 15: 393–402. [DOI] [PubMed] [Google Scholar]
- 14.Wells SA, Chi DD, Toshima K, et al. Predictive DNA testing and prophylactic thyroidectomy in patients at risk for multiple endocrine neoplasia type 2A. Ann Surg 1994; 200: 237–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dralle H, Gimm O, Simon D, et al. Prophylactic thyroidectomy in 75 children and adolescents with hereditary medullary thyroid carcinoma: German and Austrian experience. Wold J Surg 1998; 22: 744–751. [DOI] [PubMed] [Google Scholar]
- 16.Schuffenecker I, Ginet N, Goldgar D, et al. Prevalence and parental origin of de novo RET mutations in multiple endocrine neoplasia type 2A and familial medullary thyroid carcinoma. Le Groupe d’Etude des Tumeurs a Calcitonine. Am J Hum Genet 1997; 60: 233–237. [PMC free article] [PubMed] [Google Scholar]
- 17.Ceccherini I, Hofstra RMW, Luo Y, et al. DNA polymorphisms and condition for SSCP analysis of the 20 exons of the ret proto-oncogene. Oncogene 1994; 9: 3025–3029. [PubMed] [Google Scholar]
- 18.Mulligan LM, Eng C, Attie T, et al. Diverse phenotype associated with exon 10 mutations of the RET proto-oncogene. Hum Mol Genet 1994; 3: 2163–2167. [DOI] [PubMed] [Google Scholar]
- 19.Fitze G, Schreiber M, Kuhlisch E, et al. Association of RET protooncogene codon 45 polymorphism with Hirschsprung disease. Am J Hum Genet 1999; 65: 1469–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gimm O, Dralle H. C-cell cancer: prevention and treatment. Langenbeck’s Arch Surg 1999; 384: 16–23. [DOI] [PubMed] [Google Scholar]
- 21.Hinze R, Holzhausen HJ, Gimm O, et al. Primary heredity medullary thyroid carcinoma: C-cell morphology and correlation with preoperative calcitonin levels. Virchows Arch 1998; 433: 203–208. [DOI] [PubMed] [Google Scholar]
- 22.Hotz HG, Runkel NSF, Frank-Raue K, et al. Prophylactic thyroidectomy in MEN IIA: does the calcitonin level correlate with tumor spread? Langenbeck’s Arch Surg 1998; 383: 170–173. [DOI] [PubMed] [Google Scholar]
- 23.Marsh DJ, McDowall D, Hyland VJ, et al. The identification of false-positive response to the pentagastrin stimulation test in RET mutation negative members of MEN 2A families. Clin Endocrinol 1996; 44: 213–220. [DOI] [PubMed] [Google Scholar]
- 24.Marsh DJ, Andrew SD, Eng C, et al. Germline and somatic mutations in an oncogene: RET mutations in inherited medullary thyroid carcinoma. Cancer Res 1996; 56: 1241–1243. [PubMed] [Google Scholar]
- 25.Seri M, Yin L, Barone V, et al. Frequency of RET mutations in long- and short-segment Hirschsprung disease. Hum Mutat 1997; 9: 243–249. [DOI] [PubMed] [Google Scholar]
- 26.Frohnauer MK, Decker RA. Update on the MEN 2A c804 RET mutation: Is prophylactic thyroidectomy indicated? Surgery 2000; 128: 1052–1058. [DOI] [PubMed] [Google Scholar]
- 27.Machens A, Gimm O, Hinze R, et al. Genotype–phenotype correlation in hereditary medullary thyroid carcinoma: Oncological features and biochemical properties. J Clin Endocrinol Metab 2001; 86: 1104–1109. [DOI] [PubMed] [Google Scholar]